
ABSTRACT
Background: Limited clinical data exists to assess differences between various infant pneumococcal conjugate vaccine schedules. In this trial, we evaluated immunogenicity of the 10-valent pneumococcal non-typeable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV) administered using 3 different immunization schedules in HIV unexposed-uninfected infants in South Africa.
Methods: In this phase III, open, single‐center, controlled study (clinicaltrials.gov: NCT00829010), 300 infants were randomized (1:1:1) to 1 of 3 PHiD-CV schedules: 3-dose priming and booster (3 + 1); 3-dose priming without booster (3 + 0); or 2-dose priming and booster (2 + 1). The booster was administered at 9–10 months of age. immune responses were assessed up to 21 months after primary vaccination.
Results: Post‐priming antibody levels tended to be lower in the 2 + 1 group. At 6 months post‐priming, antibody concentrations and opsonophagocytic activity titers were within similar ranges after 2‐ or 3‐dose priming. Robust increases were observed pre‐ to post‐booster in the 3 + 1 and 2 + 1 groups.
Conclusions: PHiD-CV was immunogenic when administered in different schedules. Post‐booster responses suggest effective immunological priming with both 2‐ and 3‐dose primary series and support administration of the booster dose at 9–10 months of age.
1. Introduction
Pneumonia is responsible for 2500 child deaths every day and therefore remains the leading infectious cause of death among children younger than 5 years, despite a 47% reduction over the last 15 years [Citation1,Citation2]. The main cause of severe pneumonia among children in developing countries is Streptococcus pneumoniae, which is also responsible for upper respiratory tract infection and invasive pneumococcal disease (IPD) [Citation1,Citation3]. Pneumococcal conjugate vaccines (PCVs) are key in preventing and reducing pneumococcal infections, including childhood pneumonia. The World Health Organization (WHO) recommends PCVs to be included in infant immunization programs worldwide and especially in countries with high childhood mortality, according to either a 3 + 0 or a 2 + 1 schedule. The choice of the schedule depends among others on local pneumococcal epidemiology, predicted vaccination coverage, and logistics. Countries also need to balance the need for a booster dose versus cost-effectiveness and programmatic constraints [Citation4,Citation5].
The South African immunization program introduced the 7-valent PCV (PCV7) in April 2009 and replaced it by the 13-valent PCV (PCV13) in May 2011. While funding of the program precluded introduction of a 3 + 1 schedule, the observed lack of sustained protection against IPD in HIV-infected children indicated the need for a booster dose [Citation6]. Moreover, waning of protection against serotype 1 has been observed for a 9-valent PCV administered according to a 3 + 0 schedule, further supporting the use of a vaccination schedule with a booster dose [Citation7]. Hence, the South African immunization program has adopted a 2 + 1 vaccination schedule given at the age of 6 weeks, 14 weeks, and 9 months [Citation6,Citation8]. This schedule enhances the number of children likely to receive their booster dose; according to South African estimates for 2015, 95% of children received their booster PCV dose and 97% their first measles vaccine dose at 9 months of age, while fewer children (84%) received their second measles vaccine dose at 18 months of age [Citation9].
The 10-valent pneumococcal non-typeable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV) was licensed in South Africa in June 2010 for active immunization against the 10 serotypes included in the vaccine. In addition, protection against the vaccine-related serotype 19A was recognized by the European Medicine Agency in 2015 [Citation10]. PHiD-CV was previously shown to be immunogenic and effective against pneumococcal diseases when administered to children following different 3 + 1 schedules, all including a booster dose at the age of 11–18 months [Citation11–Citation17], as well as following a 2 + 1 schedule (2-dose primary series at 3 and 5 months of age followed by a booster dose at 11–12 months) [Citation11–Citation13,Citation15–Citation17]. In a resource-limited setting in Nepal, the use of a 2 + 1 PHiD-CV schedule with primary doses at 6 and 14 weeks and a booster at age 9 months improved antibody persistence through early childhood without compromising antibody responses in early infancy (i.e. postprimary vaccination) relative to a 3 + 0 PHiD-CV schedule with primary doses at 6, 10, and 14 weeks [Citation18].
We have previously reported the immunogenicity and safety of PHiD-CV when given as a 3 + 1 schedule in HIV-infected, HIV-exposed-uninfected, and HIV-unexposed-uninfected infants in South Africa [Citation19]. Here, we present immunogenicity and safety of PHiD-CV when administered according to various schedules (3 + 1, 3 + 0, or 2 + 1) to South African HIV-unexposed-uninfected infants, including the evaluation of a PHiD-CV booster dose at 9 months of age.
2. Patients and methods
2.1. Study design and participants
This phase III, open, single-center, partially randomized, controlled study was conducted in South Africa between February 2009 and June 2012. For HIV-unexposed-uninfected group, eligible participants were infants 6–10 weeks of age at first vaccination, without any known or suspected health problems that would contraindicate initiation of routine childhood immunizations. Mothers and infants were confirmed as HIV-uninfected by HIV enzyme-linked immunosorbent assay (ELISA) after 24 weeks of gestation for the mother (testing performed within the Prevention Mother-To-Child Transmission program) and at visit 1 for the infant. Details on HIV testing were reported previously [Citation19].
The study was conducted according to the principles of Good Clinical Practice and the Declaration of Helsinki, and with the approval of an independent ethics committee (Wits Human Research Ethics Committee). Written informed consent was obtained from the parent(s) or legally acceptable representative(s) of each child before any study-specific procedures. An independent data-monitoring committee provided oversight of the study by assessing potential treatment harm and reviewing all-cause mortality and morbidity.
This study has been registered at www.clinicaltrials.gov (NCT00829010). A protocol summary is available at www.gsk-clinicalstudyregister.com (study ID: 111634).
2.2. Study objectives
The primary objective evaluating immune responses to PHiD-CV given as a 3 + 1 schedule to HIV-infected, HIV-exposed-uninfected, and HIV-unexposed-uninfected infants was reported elsewhere [Citation19]. Here, we present secondary objectives related to different vaccination schedules administered to HIV-unexposed-uninfected infants. These include evaluation of PHiD-CV immunogenicity 1 month following primary vaccination and following a booster dose given at age 9–10 months; antibody persistence at the age of 9–10 months (i.e. approximately 6 months after 2- or 3-dose priming) and at the age of 24–27 months (i.e. approximately 15 months post-booster, or approximately 21 months after priming without booster); and PHiD-CV reactogenicity and safety.
Other secondary objectives, including immunogenicity of the coadministered vaccines and assessment of nasopharyngeal carriage and salivary samples, will be reported elsewhere.
2.3. Treatment allocation and study vaccines
Infants were randomized (1:1:1) into three groups to receive a 3-dose primary vaccination course at 6, 10, and 14 weeks of age, with a booster dose at 9–10 months of age (3 + 1 group), 3 primary doses at 6, 10, and 14 weeks of age, without a booster (3 + 0 group), or a 2-dose primary vaccination course at 6 and 14 weeks of age, followed by a booster dose at 9–10 months of age (2 + 1 group) (). Treatment allocation at the investigator site was performed using a central internet randomization program (SBIR). A randomization list generated at GSK Biologicals, Rixensart, Belgium, using a standard statistical analysis system (SAS) program was used to number the vaccines. A randomization blocking scheme was used to ensure that balance between schedules was maintained. Due to the different vaccination schedules, the study was conducted in an open manner; however, GSK laboratory technicians were blinded to the vaccination schedule group allocation.
Figure 1. Study design
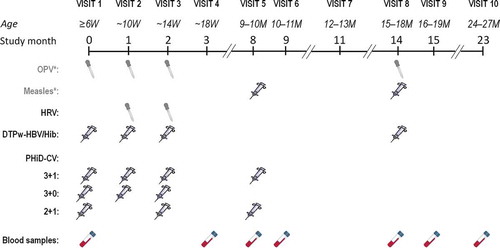
Each dose of PHiD-CV (Synflorix, GSK, Belgium) consists of 1 μg S. pneumoniae capsular polysaccharide for serotypes 1, 5, 6B, 7F, 9V, 14, and 23F, and 3 μg for serotype 4, each individually conjugated to non-typeable H. influenzae protein D, and 3 μg of capsular polysaccharide of serotypes 18C and 19F conjugated to tetanus and diphtheria toxoids, respectively.
Coadministered vaccines included diphtheria–tetanus–whole cell pertussis–hepatitis B vaccine combined with lyophilized H. influenzae type b tetanus-conjugate vaccine (DTPw-HBV/Hib; Tritanrix-HepB/Hib, GSK, Belgium) administered at 6, 10, and 14 weeks and 15–18 months of age and live oral attenuated human rotavirus vaccine (HRV; Rotarix, GSK, Belgium), administered at 10 and 14 weeks of age. As HRV was included after protocol amendment (December 2008), some children did not receive the vaccine through participation in the study. PHiD-CV and DTPw-HBV/Hib were administered intramuscularly in the right and left thigh, respectively. Additionally, as part of the national immunization program, oral polio vaccine (OPV) (at 6, 10, 14 weeks and 15–18 months) and measles vaccine (at 9–10 and 15–18 months) were also administered but were not considered as study vaccines ().
2.4. Immunogenicity assessment
For serological assessment of PHiD-CV responses (reported here), blood samples were taken at the ages of 6 weeks, 18 weeks, 9–10 months, 10–11 months, and 24–27 months (). Immunogenicity assessment of the coadministered vaccines will be reported elsewhere.
Antipneumococcal IgG antibodies against vaccine serotypes and vaccine-related serotypes 6A and 19A were quantified using GSK vaccines’ 22F ELISA (cutoff: 0.05 µg/mL). Immune response was described in terms of percentages of children with IgG concentrations ≥0.2 µg/mL (equivalent to antibody concentrations ≥0.35 µg/mL measured by the non-22F ELISA of the WHO reference laboratory [Citation20]).
S. pneumoniae opsonophagocytic activity (OPA) against vaccine and vaccine-related pneumococcal serotypes was measured by a pneumococcal killing assay using an HL-60 cell line (cutoff titer: 8) [Citation21]. Results are presented as the reciprocal of the dilution of serum (opsonic titer) able to sustain 50% killing of pneumococci under assay conditions.
IgG antibodies to the H. influenzae protein D were measured by an ELISA assay developed by GSK, with the nonlipidated protein D as coating material. Concentration of specific protein D antibodies was determined using a standard reference serum. The cutoff of the assay was 100 ELISA units (EL.U)/mL.
2.5. Safety assessment
Solicited local and general adverse events (AEs) occurring within 4 days postvaccination were recorded by the parent(s) or legally acceptable representative(s) in the participants’ diary cards. Solicited local AEs at the injection site were pain, redness, and swelling, and general AEs were drowsiness, irritability, loss of appetite, fever, vomiting, and diarrhea. Vomiting and diarrhea were solicited due to coadministration of HRV vaccine and were thus only solicited after vaccination with that vaccine at the age of 10 and 14 weeks. The other general AEs were solicited at visits 1, 2, and 3, also for the 2 + 1 group which did not receive PHiD-CV but DTPw-HBV/Hib, HRV, and OPV at visit 2. Fever was defined as axillary temperature ≥37.5°C. Diarrhea was defined as ≥3 looser than normal stools per day. Symptom intensity was graded on a scale of 1 (mild) to 3 (severe). Grade 3 solicited AEs were redness or swelling at the injection site with a diameter >30 mm; for pain, crying when the limb was moved or the limb being spontaneously painful; for irritability, crying that could not be comforted or prevented normal activity; for fever, axillary temperature >39.5°C; for drowsiness, preventing normal activity; for loss of appetite, not eating at all; for diarrhea, ≥6 looser than normal stools per day; and for vomiting, ≥3 episodes of vomiting (≥1 h after feeding) per day. Large swelling reactions after booster vaccination, defined as swelling with a diameter >50 mm, noticeable diffuse swelling or increase of limb circumference, were also recorded.
Unsolicited AEs occurring within 31 days postvaccination were recorded, and serious AEs (SAEs), defined as any medical occurrence that resulted in death, disability, or incapacity, was life-threatening, required hospitalization, or was considered serious by the investigator, were recorded during the entire study.
2.6. Statistical analysis
All study objectives presented here were descriptive and the study had no confirmatory objectives that would drive its sample size. A total of 100 children were planned to be enrolled per group, to achieve 80 evaluable children per group in the according-to-protocol (ATP) cohort for immunogenicity, when assuming an elimination rate of 20%.
Potential trends in group differences were highlighted by nonoverlapping confidence intervals (CIs). No adjustment for multiplicity of analyses was performed; thus, results should be interpreted with caution.
Immunogenicity analyses were performed on the ATP cohort for immunogenicity, comprising vaccinated children who met all eligibility criteria, complied with the protocol-defined procedures, and with assay results available for antibodies against ≥1 study vaccine antigen. Safety analyses were performed on the total vaccinated cohort, including all children with ≥1 administered vaccine dose.
Geometric mean antibody concentrations (GMCs) and geometric mean OPA titers (GMTs) were calculated for each serotype or antigen with 95% CIs by taking the antilog of the mean of the log antibody concentration or titer transformations. Antibody concentrations or OPA titers below assay cutoffs were given an arbitrary value of half the cutoff for GMC and GMT calculations. Percentages of children with serological results above respective thresholds or cutoffs were calculated with exact 95% CIs. Postprimary immunogenicity results for the 3 + 1 and 3 + 0 groups are presented separately as well as pooled.
Statistical analyses were performed using SAS Drug and Development web portal version 3.5, SAS version 9.2, and Proc StatXact-8.1 procedure on SAS.
3. Results
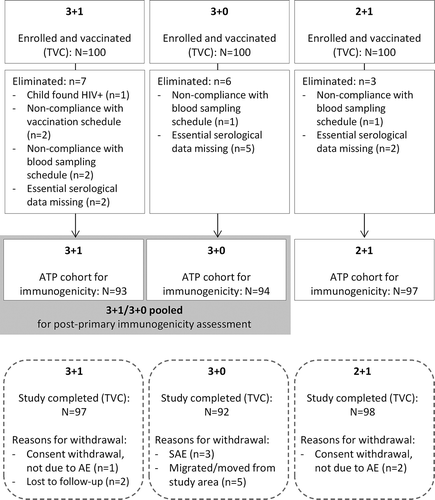
Demographic characteristics were similar between groups (). In each of the groups, 100 children were enrolled; of these, 93 (3 + 1), 94 (3 + 0), and 97 (2 + 1) were included in the ATP cohort for immunogenicity ().
Table 1. Demographic characteristics (total vaccinated cohort)
Figure 2. Participant Flow Diagram
3.1. Immunogenicity results
One month postprimary vaccination, for each vaccine serotype, the percentage of infants with antibody concentrations ≥0.2 µg/mL was ≥94.8% and 100% for the 2- and 3-dose primary vaccination courses, respectively, except for serotypes 6B and 23F, with 95% CIs overlapping between the post-dose 2 (2 + 1 group) and post-dose 3 (pooled 3 + 1/3 + 0 groups) for each vaccine and vaccine-related serotype (). Post‐priming antibody levels tended to be lower after 2- than 3-dose priming (), with nonoverlapping 95% CIs between post-dose 2 (2 + 1 group) and post-dose 3 (pooled 3 + 1/3 + 0 groups) for serotypes 4, 5, 7F, 9V, 14, and 19F (). For each vaccine serotype, the percentage of infants with OPA titers ≥8 was ≥80.0% 1 month after 2-dose primary vaccination, and ≥85.8% for the 3-dose primary vaccination course, with overlapping 95% CIs between the 2 + 1 and pooled 3 + 1/3 + 0 groups except for serotype 14 (). OPA GMTs tended to be lower after 2- than 3-dose priming (), with nonoverlapping 95% CIs between the 2 + 1 and pooled 3 + 1/3 + 0 groups for serotypes 4, 7F, 9V, 14, and 19F ().
Table 2. Immunogenicity assessed by GSK 22F ELISA for antipneumococcal antibodies and anti-protein D ELISA (ATP cohort for immunogenicity)
Table 3. Functional immune response assessed by opsonophagocytic activity assay (ATP cohort for immunogenicity)
Figure 3. Kinetics of anti-pneumococcal antibody GMCs (ATP cohort for immunogenicity)
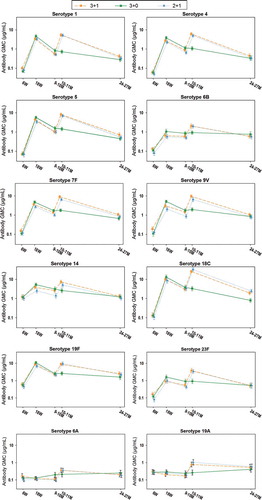
Figure 4. Kinetics of anti-pneumococcal OPA GMTs (ATP cohort for immunogenicity)
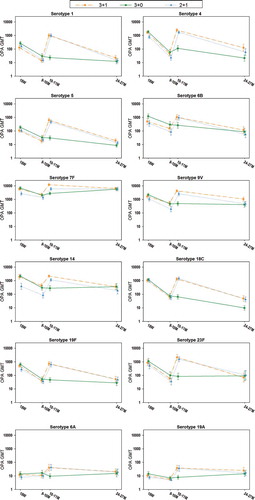
At 9–10 months of age, i.e. 6 months post‐priming, antibody GMCs and OPA GMTs were generally within similar ranges after 2‐ or 3‐dose priming ( and ), although 95% CIs were not overlapping between the 2 + 1 and pooled 3 + 1/3 + 0 groups for antibody GMCs and OPA GMTs for serotypes 4, 6B, 9V, and 14, as well as for the antibody GMCs for serotypes 7F and 23F ( and ). While the percentages of children with antibody concentrations ≥0.2 µg/mL were in similar ranges between the 2 + 1 and pooled 3 + 1/3 + 0 groups for each vaccine serotype, the percentages of children with OPA titers ≥8 were lower in the 2 + 1 group for serotypes 9V and 14 ( and ).
At age 10–11 months, i.e. 1 month post-booster, robust increases in responses for vaccine pneumococcal serotypes were observed compared to pre-booster in the 2 + 1 and 3 + 1 groups ( and , and ); antibody GMCs increased 3.63–9.88-fold and 2.80–9.61-fold, while OPA GMTs increased 4.42–74.68-fold and 4.95–71.97-fold, for the 2 + 1 and 3 + 1 group, respectively. Post-booster antibody GMCs and OPA GMTs were in the same range between the 2 + 1 and 3 + 1 group for all vaccine serotypes except 7F for which antibody GMCs and OPA GMTs were lower in the 2 + 1 group, and serotype 14 for which OPA GMTs were lower in the 2 + 1 group ( and , and ). For all vaccine pneumococcal serotypes, antibody GMCs and OPA GMTs at the age of 10–11 months were lower in children who had not received a booster dose (3 + 0 group) than for the 3 + 1 and 2 + 1 groups ( and , and ).
At 24–27 months of age, i.e. 15 months post-booster or 21 months postprimary vaccination, for each vaccine serotype, antibody GMCs and OPA GMTs were generally within the same range in all three groups, except for serotype 18C ( and , and ).
One month postprimary vaccination, all infants who received three primary doses had anti-protein D antibody concentrations ≥100 EL.U/mL, while two of the infants in the 2 + 1 group did not have measurable anti-protein D antibodies. Anti-protein D antibody GMCs were lower after 2-dose compared to 3-dose priming (). At 10–11 months of age, all infants (including those from the 3 + 0 group who did not receive a booster) except one in the 2 + 1 group had anti-protein D antibody concentrations ≥100 EL.U/mL ().
3.2. Safety results
Postprimary and post-booster vaccination, pain was the most frequently reported solicited local symptom at the PHiD-CV injection site in all groups (). Irritability was the most frequently reported solicited general symptom during the 4-day postvaccination period after each primary dose in all groups. Post-booster, the most common solicited general symptoms were irritability in the 2 + 1 group and drowsiness in the 3 + 1 group (). No large swelling reactions were reported after PHiD-CV booster vaccination.
Figure 5. Solicited local and general symptoms (overall per dose; total vaccinated cohort)
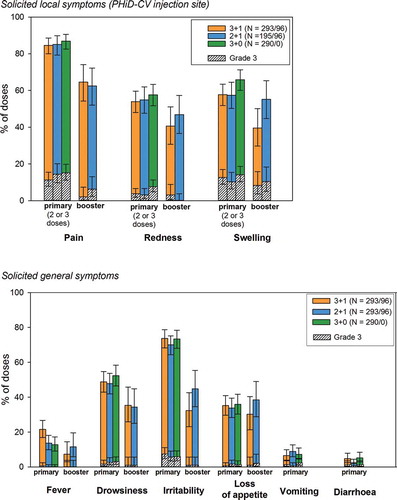
During the 4-day postvaccination period, antipyretic treatments were taken after 48.0%, 35.9%, and 36.5% of all primary doses in the 3 + 1, 3 + 0, and 2 + 1 groups, respectively, and after 17.3% and 19.4% of booster doses in the 3 + 1 and 2 + 1 group, respectively.
Unsolicited AEs were reported after 59.8%, 59.0%, and 62.5% of primary doses in the 3 + 1, 3 + 0, and 2 + 1 group, respectively, and after 51.0% and 44.9% of booster doses in the 3 + 1 and 2 + 1 group, respectively. Cough was the most common unsolicited AE in all groups.
Over the entire study period, at least 1 nonfatal SAE was reported for 20 children each in the 3 + 1 and 2 + 1 groups, and for 12 children in the 3 + 0 group. Of these, three were assessed by the investigator to be causally related to vaccination (two events of febrile convulsions in the 3 + 1 group and one in the 2 + 1 group); all were recovered or resolved by study end. In addition, three fatal SAEs were reported in the 3 + 0 group (sudden death, croup infectious, and convulsion); the case of sudden death occurred 1 day after the first dose and was assessed by the investigator to be causally related to vaccination given the time elapsed from vaccination. The other two fatal SAEs were not assessed as causally related.
4. Discussion
PHiD-CV was immunogenic when administered according to different schedules (3 + 1, 3 + 0, or 2 + 1) to HIV-unexposed-uninfected infants, with the booster dose of the 3 + 1 and 2 + 1 schedules administered at the age of 9–10 months. PHiD-CV vaccination was generally well tolerated and no safety concerns were reported.
Following 2-dose priming, there was a trend toward lower immune responses versus 3-dose priming, but the differences were minor. No obvious differences were observed in percentages of children with serotype-specific IgG levels ≥0.2 µg/mL or with OPA titers ≥8. Strong increases in immune responses were observed from before to after the booster given at 9–10 months of age in the 2 + 1 and 3 + 1 groups, indicative of effective priming with 2 or 3 doses and boostability; antibody GMCs and OPA GMTs at 10–11 months of age were higher than in the 3 + 0 group which did not receive a booster. Persistence of the immune response at 24–27 months of age, as measured by antibody GMCs, appeared to be similar for all three schedules.
While the WHO recommends a 3 + 0 or 2 + 1 schedule, our findings indicate that the 2 + 1 schedule may be preferred due to the different kinetics in immunogenicity when the 3 doses are given at the age of 6 and 14 weeks and 9–10 months (2 + 1), compared to 6, 10, and 14 weeks (3 + 0), which potentially allows a better duration of protection. This appears to be especially important for serotype 1, as vaccine failures in terms of serotype 1 IPD have been reported after the first year of life in children vaccinated with three primary doses of 9-valent PCV without booster [Citation7]. In many developing countries, serotype 1 is one of the most common pneumococcal serotypes causing IPD [Citation22,Citation23].
Our findings are similar to those from a study in Nepal where PHiD-CV was administered as a 2 + 1 (6, 14 weeks, and 9 months) or 3 + 0 (6, 10, 14 weeks) schedule [Citation18]. At age 18 weeks, the 2 + 1 and 3 + 0 groups did not differ in percentages of children with IgG concentrations ≥0.2 μg/mL or with OPA titers ≥8 for any of the PHiD-CV serotypes. Antibody GMCs and OPA GMTs tended to be lower after 2-dose than after 3-dose priming but increased substantially following the booster dose in the 2 + 1 group. Antibody persistence at 2–4 years was similar in both groups, except for 18C for which IgG levels were lower in the 3 + 0 group, which is in line with the persistence results in our trial. In addition to the study in Nepal and our study in South Africa, one other trial has meanwhile evaluated PHiD-CV booster vaccination at 9–10 months of age but only after 3-dose priming starting at 8–11 weeks of age. In this study, conducted in Burkina Faso in infants with or without sickle cell disease, PHiD-CV induced a robust booster response [Citation24].
Immune responses to 2 + 1 and 3 + 1 PHiD-CV schedules have been compared in healthy children in Sweden and Slovakia who were primed at either 3 and 5; or 3, 4, and 5 months of age and received a booster dose at 11–12 months of age. Higher postprimary and post-booster antibody levels and OPA titers were observed for most serotypes following the 3 + 1 schedule [Citation17]. Children in that study received one additional dose of PHiD-CV at 36–46 months of age to test anamnestic responses; these appeared comparable, suggesting that similar protective efficacy may be achieved with both schedules [Citation25].
Impact of different vaccination schedules has been assessed for other PCVs. In a trial assessing PCV13 schedules, healthy infants were vaccinated at ages 2 and 4 months; 3 and 5 months; 2, 3, and 4 months; or 2, 4, and 6 months, with a booster dose at age 11.5 months. For almost all vaccine serotypes, the four schedules did not differ significantly in antibody levels after the booster dose [Citation26]. A meta-analysis of randomized trials comparing immunogenicity of PCV7, PCV13, or PHiD-CV after two or three doses concluded that while most children were protected by two primary doses of PCV, protection may be reduced for serotype 6B and 23F [Citation27]. Another systematic review on the effect of PCV dosing schedules on immunogenicity concluded that while giving the third dose in the second year of life produces a higher antibody response than when given as part of the primary series in the first 6 months, the lower antibody levels between the 2-dose primary series and booster may result in less disease protection for infants in that interval [Citation28]. This might however be redressed through the indirect effect of PCV immunization which results from reduced circulation of vaccine serotypes in the community [Citation29]. Other systematic reviews assessing the effect of PCV dosing schedules concluded that available data on vaccine-type IPD supported the use of 3 + 1, 3 + 0, and 2 + 1 schedules [Citation30], and that all three schedules reduced clinical and radiologically confirmed pneumonia [Citation31]; finally, indirect benefit concerning vaccine-type IPD, vaccine-type nasopharyngeal carriage, and syndromic pneumonia could be demonstrated for the 3 + 1 schedule, while so far the 3 + 0 and 2 + 1 schedules have demonstrated indirect benefit for vaccine-type IPD only [Citation32].
Incidences of unsolicited AEs were in similar ranges as previously reported in Mali [Citation33,Citation34]. At least one SAE was reported over a total follow-up period of approximately 22 months for 15–20% of children per group. These incidences are in a similar range as those reported for PHiD-CV recipients in the COMPAS study (21.5%), which also had a long follow-up period (more than 2 years) [Citation14]. Considering the child mortality rates in the study area [Citation35], the occurrence of three fatal SAEs in HIV-unexposed-uninfected children did not raise safety concerns. One case of sudden death in the 3 + 0 group occurred one day following administration of the first dose of PHiD-CV, DTPw-HBV/Hib, and OPV vaccines. Given the short time elapsed from vaccination, the investigator considered that a causal association with the administered vaccines could not be excluded.
The main limitation of our study is that we did not evaluate efficacy of the different schedules against pneumococcal disease; thus, the clinical relevance of immunological findings remains unknown, especially for mucosal disease. Also, a formal correlate of protection for PCVs has not yet been established, and assessments for PCV13 indicate that correlates of protection against IPD may differ between the various vaccine serotypes [Citation36]. Nevertheless, effectiveness of PHiD-CV against several disease end points has been shown in the Finnish invasive pneumococcal disease trial, both for a 3 + 1 schedule (age: approximately 3, 4, 5, and 11–12 months) and a 2 + 1 schedule (age: approximately 3, 5, and 11–12 months). Both schedules have shown effectiveness against culture-confirmed IPD [Citation12], clinically suspected IPD [Citation13], hospital-diagnosed pneumonia [Citation15,Citation16], antimicrobial purchases [Citation37], and a trend toward reduction in the frequency of tympanostomy tube placements [Citation38]. In a nested study in Finland, reductions in vaccine-type carriage, as well as a trend toward a reduction in acute otitis media, were observed with both schedules [Citation39]. Effectiveness of PCVs administered as a 2 + 1 schedule has also been demonstrated in post-marketing studies [Citation40–Citation42]. The clinical relevance of the observed differences in antibody kinetics after 2 + 1 or 3 + 0 vaccination is however unknown as there are no clinical outcome data from randomized trials that directly compare a 2 + 1 and 3 + 0 schedule with marketed higher valent PCVs in developing countries.
Other study limitations include the small sample size, the fact that no confirmatory objectives were defined and no non-inferiority assessment between the different schedules was performed, therefore potential group differences should be interpreted with caution and assessed for their clinical relevance, as well as the variability observed in OPA and IgG responses (noticeable when comparing post-priming immune responses in the 3 + 1 and 3 + 0 groups). In addition, the open study design could have caused a biased reactogenicity assessment.
5. Conclusions
Overall, our study is the first performed in Africa to assess a 2 + 1 vaccination schedule of PHiD-CV with a booster dose administered at the age of 9–10 months, as well as the first study to assess the three different schedules of a higher valent PCV in a developing country. Post‐priming antibody levels tended to be lower in the 2 + 1 group, but by 6 months post-priming, antibody levels were in similar ranges in all groups. The robust response observed post-booster supports administration of three infant PCV doses in a 2 + 1 rather than a 3 + 0 vaccination schedule. Regardless of whether two or three primary doses are administered, the booster dose can be given from 9 months of age, which fits the Expanded Program on Immunization of routine childhood vaccines and is thereby likely to increase compliance of receiving the PCV booster dose.
Key issues
The World Health Organization (WHO) recommends pneumococcal conjugate vaccines (PCVs) to be included in infant immunization programs worldwide, according to either a 3 + 0 or a 2 + 1 schedule.
However, limited clinical data are currently available to assess differences between various infant PCV schedules.
We evaluated immunogenicity of the 10-valent pneumococcal non-typeable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV) administered using 3 different immunization schedules.
PHiD-CV was administered according to the following schedules: 3 + 1 (6, 10, 14 weeks of age with booster at 9–10 months of age); 3 + 0 (6, 10, 14 weeks); or 2 + 1 (6, 14 weeks, with booster at 9–10 months).
PHiD‐CV was immunogenic when administered in the different schedules, with minor differences in post-priming antibody levels.
Post‐booster responses suggest effective immunological priming with both 2- and 3-dose primary series and boostability.
While the WHO recommends a 3 + 0 or 2 + 1 schedule, our findings indicate that the 2 + 1 schedule may be preferred due to the different kinetics in immunogenicity, which potentially allows a better duration of protection for the 2 + 1 schedule.
Administration of the booster dose at 9–10 months of age corresponds to the Expanded Program on Immunization of routine childhood vaccines and is thereby likely to increase compliance of receiving the PCV booster dose.
Declaration of interest
S.A Madhi received grants from GSK, Pfizer, Novartis and Sanofi for the conduct of clinical trials and consulting fees for advisory boards and/or speaker’s bureaus from GSK, Pfizer and Sanofi. M Moreira is an employee of the GSK group of companies, owns shares of the GSK group of companies. N François is an employee of the GSK group of companies. J Ruiz-Guiñazu is an employee of the GSK group of companies, owns shares of the GSK group of companies. J.P Yarzabal is an employee of the GSK group of companies, owns shares of the GSK group of companies. D Borys is an employee of the GSK group of companies, owns shares of the GSK group of companies. L Schuerman is an employee of the GSK group of companies, owns shares of the GSK group of companies. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Trademarks
Synflorix, Tritanrix-HepB/Hib, and Rotarix are trademarks of the GSK group of companies.
Acknowledgments
The authors wish to thank investigator Dr Peter Adrian and the clinical and serological laboratory teams of the GSK group of companies for their contribution to this study, in particular Sudheer Ravula (GSK) for statistical analysis, Mireille Venken (GSK), Janice Beck, Kristel Vercauteren, Domenica Majorino (XPE Pharma & Science c/o GSK) and Ann Dhoest (freelance for GSK) for protocol and clinical report writing; Charlotte de Buck van Overstraeten, Catena Lauria (GSK) and Katleen Van Hoefs (Keyrus Biopharma c/o GSK) for global study management. The authors also thank Joke Vandewalle (XPE Pharma & Science c/o GSK) for drafting the manuscript; and Stéphanie Deroo (XPE Pharma & Science c/o GSK) for manuscript coordination.
Additional information
Funding
References
- Pneumonia: the forgotten killer of children [Internet]. UNICEF/WHO; [ cited February 2017]. Available from: http://www.childinfo.org/files/Pneumonia_The_Forgotten_Killer_of_Children.pdf
- Despite steady progress, pneumonia remains one of the single largest killers of young children worldwide [Internet]. UNICEF; [ cited February 2017]. Available from: http://data.unicef.org/child-health/pneumonia.html
- O’Brien KL, Wolfson LJ, Watt JP, et al. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet. 2009;374(9693):893–902.
- Summary of WHO Position Papers - Recommended Routine Immunizations for Children [Internet]. WHO; [ cited February 2017]. Available from: http://www.who.int/immunization/policy/Immunization_routine_table2.pdf
- WHO. Pneumococcal vaccines WHO position paper – 2012. Weekly Epidemiol Rec. 2012;87(14):129–144. Available from: http://www.who.int/wer/2012/wer8714.pdf
- Madhi SA, Cohen C, von Gottberg A. Introduction of pneumococcal conjugate vaccine into the public immunization program in South Africa: translating research into policy. Vaccine. 2012;30(Suppl 3):C21–27.
- Klugman KP, Madhi SA, Adegbola RA, et al. Timing of serotype 1 pneumococcal disease suggests the need for evaluation of a booster dose. Vaccine. 2011;29(18):3372–3373.
- Schoub BD, Ngcobo NJ, Madhi S. The National Advisory Group on Immunization (NAGI) of the Republic of South Africa. Vaccine. 2010;28(Suppl 1):A31–34.
- WHO vaccine-preventable diseases: monitoring system. 2016 Global summary. Coverage time series for South Africa (ZAF) [Internet]. WHO; [ cited February 2017]. Available from: http://apps.who.int/immunization_monitoring/globalsummary/coverages?c=ZAF
- Summary of product characteristics [Internet]. EMA; [ cited February 2017]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000973/WC500054346.pdf
- Prymula R, Schuerman L. 10-valent pneumococcal nontypeable Haemophilus influenzae PD conjugate vaccine: synflorix. Expert Rev Vaccines. 2009;8(11):1479–1500.
- Palmu AA, Jokinen J, Borys D, et al. Effectiveness of the ten-valent pneumococcal Haemophilus influenzae protein D conjugate vaccine (PHiD-CV10) against invasive pneumococcal disease: a cluster randomised trial. Lancet. 2013;381(9862):214–222.
- Palmu AA, Jokinen J, Nieminen H, et al. Vaccine effectiveness of the pneumococcal Haemophilus influenzae protein D conjugate vaccine (PHiD-CV10) against clinically suspected invasive pneumococcal disease: a cluster-randomised trial. Lancet Respir Med. 2014;2(9):717–727.
- Tregnaghi MW, Saez-Llorens X, Lopez P, et al. Efficacy of pneumococcal nontypable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV) in young Latin American children: A double-blind randomized controlled trial. Plos Med. 2014;11(6):e1001657.
- Kilpi T, Palmu A, Puumalainen T et al. Effectiveness of the 10-valent pneumococcal Haemophilus influenzae protein D conjugate vaccine (PHiD-CV10) against hospital-diagnosed pneumonia in infants - FinIP trial. Paper presented at the 31st Annual Meeting of the European Society for Paediatric Infectious Diseases. 2013 May 28-Jun 1; Milan, Italy.
- Palmu AA, Kilpi T, Rinta-Kokko H et al. Impact of 10-valent pneumococcal conjugate vaccine (PCV10) against hospital-diagnosed pneumonia among vaccine-eligible children in Finland. Paper presented at the 54th Interscience Conference on Antimicrobial Agents and Chemotherapy; 2014 Sep 5-9; Washington, DC, USA.
- Silfverdal SA, Hogh B, Bergsaker MR, et al. Immunogenicity of a 2-dose priming and booster vaccination with the 10-valent pneumococcal nontypeable Haemophilus influenzae protein D conjugate vaccine. Pediatr Infect Dis J. 2009;28(10):e276–282.
- Hamaluba M, Kandasamy R, Upreti SR, et al. Comparison of two-dose priming plus 9-month booster with a standard three-dose priming schedule for a ten-valent pneumococcal conjugate vaccine in Nepalese infants: a randomised, controlled, open-label, non-inferiority trial. Lancet Infect Dis. 2015;15(4):405–414.
- Madhi SA, Koen A, Jose L, et al. Vaccination with 10-valent pneumococcal conjugate vaccine in infants according to HIV status. Medicine (Baltimore). 2017;96(2):e5881.
- Poolman JT, Frasch CE, Kayhty H, et al. Evaluation of pneumococcal polysaccharide immunoassays using a 22F adsorption step with serum samples from infants vaccinated with conjugate vaccines. Clin Vaccine Immunol. 2010;17(1):134–142.
- Henckaerts I, Durant N, De Grave D, et al. Validation of a routine opsonophagocytosis assay to predict invasive pneumococcal disease efficacy of conjugate vaccine in children. Vaccine. 2007;25(13):2518–2527.
- Hausdorff WP, Feikin DR, Klugman KP. Epidemiological differences among pneumococcal serotypes. Lancet Infect Dis. 2005;5(2):83–93.
- Johnson HL, Deloria-Knoll M, Levine OS, et al. Systematic evaluation of serotypes causing invasive pneumococcal disease among children under five: the pneumococcal global serotype project. Plos Med. 2010;7(10):e1000348.
- Sirima SB, Tiono A, Gansane Z, et al. Immunogenicity and safety of 10-valent pneumococcal nontypeable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV) administered to children with sickle cell disease between 8 weeks and 2 years of age: a phase III, open, controlled study. Pediatr Infect Dis. 2016;36(5):e136–e150.
- Silfverdal SA, Skerlikova H, Zanova M, et al. Anamnestic immune response in 3- to 4-year-old children previously immunized with 10-valent pneumococcal nontypeable Haemophilus influenzae protein D conjugate vaccine as 2-dose or 3-dose priming and a booster dose in the first year of life. Pediatr Infect Dis J. 2011;30(9):e155–163.
- Spijkerman J, Veenhoven RH, Wijmenga-Monsuur AJ, et al. Immunogenicity of 13-valent pneumococcal conjugate vaccine administered according to 4 different primary immunization schedules in infants: a randomized clinical trial. JAMA. 2013;310(9):930–937.
- Ruckinger S, Dagan R, Albers L, et al. Immunogenicity of pneumococcal conjugate vaccines in infants after two or three primary vaccinations: a systematic review and meta-analysis. Vaccine. 2011;29(52):9600–9606.
- Deloria Knoll M, Park DE, Johnson TS, et al. Systematic review of the effect of pneumococcal conjugate vaccine dosing schedules on immunogenicity. Pediatr Infect Dis J. 2014;33(Suppl 2):S119–129.
- Madhi SA, Nunes MC. The potential impact of pneumococcal conjugate vaccine in Africa: considerations and early lessons learned from the South African experience. Hum Vaccin Immunother. 2016;12(2):314–325.
- Conklin L, Loo JD, Kirk J, et al. Systematic review of the effect of pneumococcal conjugate vaccine dosing schedules on vaccine-type invasive pneumococcal disease among young children. Pediatr Infect Dis J. 2014;33(Suppl 2):S109–118.
- Loo JD, Conklin L, Fleming-Dutra KE, et al. Systematic review of the effect of pneumococcal conjugate vaccine dosing schedules on prevention of pneumonia. Pediatr Infect Dis J. 2014;33(Suppl 2):S140–151.
- Loo JD, Conklin L, Fleming-Dutra KE, et al. Systematic review of the indirect effect of pneumococcal conjugate vaccine dosing schedules on pneumococcal disease and colonization. Pediatr Infect Dis J. 2014;33(Suppl 2):S161–171.
- Dicko A, Odusanya OO, Diallo AI, et al. Primary vaccination with the 10-valent pneumococcal non-typeable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV) in infants in Mali and Nigeria: a randomized controlled trial. BMC Public Health. 2011;11:882.
- Dicko A, Santara G, Mahamar A, et al. Safety, reactogenicity and immunogenicity of a booster dose of the 10-valent pneumococcal non-typeable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV) in Malian children. Hum Vaccin Immunother. 2013;9(2):382–388.
- Health and Related Indicators [Internet]. South Africa: health systems trust; [ cited February 2017]. Available from: http://www.hst.org.za/uploads/files/chap16_08.pdf
- Andrews NJ, Waight PA, Burbidge P, et al. Serotype-specific effectiveness and correlates of protection for the 13-valent pneumococcal conjugate vaccine: a postlicensure indirect cohort study. Lancet Infect Dis. 2014;14(9):839–846.
- Palmu AA, Jokinen J, Nieminen H, et al. Effect of pneumococcal Haemophilus influenzae protein D conjugate vaccine (PHiD-CV10) on outpatient antimicrobial purchases: a double-blind, cluster randomised phase 3-4 trial. Lancet Infect Dis. 2014;14(3):205–212.
- Palmu AA, Jokinen J, Nieminen H, et al. Effectiveness of the ten-valent pneumococcal conjugate vaccine against tympanostomy tube placements in a cluster-randomized trial. Pediatr Infect Dis J. 2015;34(11):1230–1235.
- Vesikari T, Forsten A, Seppa I, et al. Effectiveness of the 10-Valent Pneumococcal Nontypeable Haemophilus influenzae Protein D-Conjugated Vaccine (PHiD-CV) against carriage and acute otitis media-a double-blind randomized clinical trial in Finland. J Pediatric Infect Dis Soc. 2016;5(3):237–248.
- De Wals P, Lefebvre B, Markowski F, et al. Impact of 2+1 pneumococcal conjugate vaccine program in the province of Quebec, Canada. Vaccine. 2014;32(13):1501–1506.
- Sigurdsson S, Kristinsson KG, Erlendsdottir H, et al. Decreased incidence of respiratory infections in children after vaccination with ten-valent pneumococcal vaccine. Pediatr Infect Dis J. 2015;34(12):1385–1390.
- Jokinen J, Rinta-Kokko H, Siira L, et al. Impact of ten-valent pneumococcal conjugate vaccination on invasive pneumococcal disease in Finnish children–a population-based study. Plos One. 2015;10(3):e0120290.