
ABSTRACT
Introduction: BCG remains the only vaccine against tuberculosis (TB) in use today and despite its impressive global coverage, the nature of BCG protection against the pulmonary forms of TB remains subject to ongoing debate. Because of the limitations of BCG, novel TB vaccine candidates have been developed and several have reached the clinical pipeline. One of these candidates is MTBVAC, the first and only TB vaccine in the clinical pipeline to date based on live-attenuated Mycobacterium tuberculosis that has successfully entered clinical evaluation, a historic milestone in human vaccinology.
Areas covered: This review describes development of MTBVAC from discovery to clinical development in high burden TB-endemic countries. The preclinical experiments where MTBVAC has shown to confer improved safety and efficacy over BCG are presented and the clinical development plans for MTBVAC are revealed. The search of all supportive literature in this manuscript was carried out via Pubmed.
Expert commentary: Small experimental medicine trials in humans and preclinical efficacy studies with a strong immunological component mimicking clinical trial design are considered essential by the scientific community to help identify reliable vaccine-specific correlates of protection in order to support and accelerate community-wide efficacy trials of new TB vaccines.
1. Introduction
Bacille Calmette–Guérin (BCG) vaccine is an attenuated strain of Mycobacterium bovis, the etiologic agent of tuberculosis (TB) in cattle [Citation1]. BCG was introduced for the first time into clinical use almost a hundred years ago, when in 1921 it was given orally to an infant whose mother had died of TB a day after delivery. The infant showed no adverse events to vaccination with BCG and importantly, did not develop TB. At that time, the oral route of BCG administration was considered the natural (gastrointestinal tract) route for acquiring TB in infants and children fed with unpasteurized milk [Citation2].
TB is poverty related with major burden in the poor and developing parts of the world. The incidence of TB is increasing worldwide due to poverty and inequity and is aggravated with the HIV/AIDS pandemic, which greatly increases risk of infection proceeding to active disease [Citation3]. Diabetes, metabolic syndrome, smoking and more recently vitamin deficiencies due to malnutrition and poor socioeconomic conditions are emerging as important risk factors for TB [Citation4,Citation5]. Importantly, how these factors can influence efficacy evaluation of new TB vaccines requires specific attention when defining clinical trial designs that involve study or patient populations with a variety of such risk factors. Because of the rising globalization and emergence of multidrug-resistant (MDR) and extensively drug-resistant TB (XDR) strains, TB is increasingly becoming a serious threat for the entire world. Today TB has reached alarming proportions of 10.4 million incidence cases and 1.8 million deaths attributed to the disease as reported by the latest World Health Organization (WHO) global TB report 2016 [Citation4]. Globally, some 50 million individuals are already latently infected with MDR M. tuberculosis strains creating a remarkable resource for future cases of active TB with insufficient treatment options [Citation3]. Nevertheless, the WHO End TB Strategy has vowed to reduce TB morbidity by 90% and TB mortality by 95% by 2035 and recognizes the urgent need for more accessible diagnostic tools that are rapid and reliable, new less toxic and more efficacious antibiotics to shorten therapy and ultimately new vaccines to prevent pulmonary TB in order to achieve this ambitious goal [Citation4].
2. Search strategies
The search of all supportive literature was carried out via the PubMed service of the US National Library of Medicine and key articles, which we considered representative of the topics reviewed, including BCG history, current state-of-the-art in the field of TB vaccine research and development, were selected.
3. BCG
3.1. The debatable efficacy of BCG
BCG remains the only licensed vaccine against TB. It is inexpensive and requires only one contact with health services at birth. Despite its impressive global coverage, the nature of BCG protection and the risks of BCG vaccination in HIV-infected children remain subject to ongoing debate [Citation6,Citation7]. In infants, BCG is acknowledged to afford protection against disseminated (miliary and meningeal) forms of TB [Citation8,Citation9], whereas in adolescents and adults BCG vaccinated at birth, lack of BCG efficacy against respiratory (pulmonary) TB disease is considered to be a result of waning BCG immunity with time that is gradually lost 10 years after vaccination [Citation10]. In high-burden TB-endemic developing countries, newborns are highly susceptible to infection with M. tuberculosis for which they receive BCG immediately after birth to ensure TB prevention [Citation11]. However, emerging epidemiological data show that BCG protection against miliary TB and TB meningitis is not absolute. In the Western Cape Province of South Africa, where BCG coverage approaches 95%, TB meningitis is currently the most common cause of childhood bacterial meningitis [Citation12]. Earlier studies in the same region show that more than 80% of children with TB meningitis had received BCG [Citation13,Citation14]. Moreover, the recent infant efficacy trial with the subunit candidate MVA85A conducted in the Western Cape Province of South Africa, revealed an alarming TB incidence rate of ~1.5% in infants who had been recently vaccinated with BCG at birth [Citation15], and, what is more, this incidence is twice that of adolescents or young adults in the same geographical area, the age group where BCG-induced immunity at birth is considered to have no effect [Citation11]. Although final conclusions cannot be drawn from a single study, these data raise serious concerns as to whether BCG is really effective in preventing TB even in newborn infants.
There are many and varied hypotheses as to why there is such variation in BCG efficacy; however, none has given the definite answer. It is thought that over-attenuation of BCG due to loss of important immunodominant antigens (e.g. those in the region of difference 1 (RD1) region) in the process of its attenuation by repeated subcultivation passaging in nonstandardized conditions could be one of the reasons [Citation16,Citation17]. Moreover, large number of human clinical trials suggest that revaccination with BCG does not provide additional benefit, which could be due to preexisting heterologous immunity resulting from infection with nontuberculous environmental mycobacteria (NTM), a process known as masking and blocking [Citation18,Citation19].
Although BCG efficacy against TB is debatable, its safety in healthy infants is unquestionable, as no clinical trial to date has shown BCG to have important harmful effects. However, BCG remains one of most reactogenic (local skin reaction) vaccines known today [Citation20,Citation21], which possibly explains in part why it has been removed from the vaccination calendar in low-burden developed countries [Citation4]. Given the limitations of BCG and the WHO declaration in 1993 of TB as a global health emergency, the past 20 years have seen an intensive upsurge in the research and discovery of new TB vaccine candidates. Mainly European driven, the work has been highly fostered by the European Commission Framework Funding Programs resulting in many of these candidates to reach current clinical development.
3.2. Nonspecific benefits of BCG vaccination at birth
An increasing number of observational studies have provided evidence that live BCG may reduce child morbidity and mortality in ways other than TB prevention [Citation22]. For example, trials from West Africa revealed that BCG reduces neonatal mortality by more than 40%, mainly by preventing neonatal sepsis and respiratory infections and fever [Citation23]. Another small study in low-birth-weight children conducted in the USA and the UK deduced that administration of BCG vaccine at first contact might contribute to lower mortality [Citation24,Citation25]. Moreover, neonatal BCG vaccination in clinic has been shown to induce a strong TH1-cell-polarizing cytokine response that can enhance immune responses to other neonatal vaccines [Citation26]. Recently, protection of neonatal BCG vaccination against hospitalization due to respiratory infection and sepsis in low TB endemic countries has been described [Citation27]. Kleinnijenhuis et al. recently provided evidence that BCG may induce nonspecific resistance (T- and B-cell dependent) to pathogens via epigenetic reprogramming of monocytes [Citation28]. Moreover, these effects are shown to be mediated by the functional and epigenetic reprogramming of innate immune cells such as monocytes, macrophages, and NK (natural killer) cells, a process termed ‘trained immunity’ [Citation29,Citation30]. And most recently, Arts et al. [Citation31], showed essential roles for metabolic pathways in BCG induced trained immunity in human monocytes. Moreover, induction of these pathways is regulated by epigenetic mechanisms at the level of chromatin organization, calling for future studies on the potential therapeutic role that the modulation of these pathways may have during vaccination [Citation31]. Finally, most European countries where incidence of TB is now low, have relinquished BCG from their vaccination schedule or restricted it to high-risk groups; however, subsequent studies suggest that this policy may have increased the incidence of a wide variety of conditions, including atopic dermatitis [Citation12].
Considering that BCG is consistently used as the reference gold standard comparator in preclinical and clinical evaluation of new TB vaccines (e.g. it has a well-established safety profile) [Citation32,Citation33], it is important to consider the nonspecific immunity attributed to BCG as potential benefit to be incorporated into the development of new TB vaccines, especially those designed as BCG-replacement strategies.
4. The current clinical TB vaccine pipeline
Today, there are a total of 13 candidates in the current clinical TB vaccine pipeline and they can be classified into three groups [Citation2]. One encompasses preventive pre-exposure vaccines, aimed for administration prior to first exposure to M. tuberculosis. They can target either BCG-naive newborns, hence priming BCG-replacement strategies, or to boost immunity to BCG-induced immune responses in healthy infants, adolescents and adults vaccinated at birth, hence subunit BCG boosters. The second category includes the preventive post-exposure vaccines, which primarily comprise subunit vaccines that target individuals who have latent TB infection (LTBI) with the aim to reduce the risk of progression to active TB in those who do not receive treatment. The third class of vaccine candidates are aimed as immune-therapeutic strategies in conjunction with antibiotic therapy for active TB. These candidates consist normally of fractioned or killed whole cell mycobacteria pertaining to the M. tuberculosis complex (MTBC) or to other distal mycobacterial species. The most recent detailed revision of these candidates and where they are to date can be found in Kaufmann et al., 2016 [Citation2], the WHO Report 2016 [Citation3], or 2016 TAG Report on TB Prevention by Mike Frick [Citation34], among others.
Although, prevention of infection (primary or secondary) or sterilizing protection would be the optimum target for any of the preventive class of vaccines, most TB vaccines in the clinical and preclinical portfolios today are intended for prevention of disease. Concerning subunit vaccines, they are based on one or more immunodominant antigens specific to BCG and to early and or late phase M. tuberculosis infection, using live viral vectors or adjuvants as delivery systems [Citation2]. In general, these subunit candidates are intended to enhance preexisting immune responses to certain immunodominant antigens induced upon BCG vaccination at birth and or upon M. tuberculosis infection in BCG vaccinated individuals (infants, e.g. MVA85A vaccine trial in adolescents and adults) [Citation35–Citation37].
In the design of new BCG-replacement strategies, it is important to consider the well-acknowledged BCG efficacy against extrapulmonary TB in children [Citation8,Citation9], making it highly desirable that such potential new candidates include this characteristic of BCG. The current pipeline has only two candidates aiming to replace BCG and they are based on live-attenuated mycobacteria. The most advanced candidate in clinical development today is the recombinant BCG (rBCG) VPM1002 which is now in efficacy trials aimed to prevent TB in infants with and without HIV exposure [Citation12]. The other candidate is the live-attenuated M. tuberculosis MTBVAC, which is the first vaccine of its kind to successfully enter human clinical evaluation in the history of human vaccinology. These two candidates could eventually replace BCG, if able to demonstrate improved safety and or greater efficacy than BCG when administered in newborns in high-burden TB-endemic settings.
The main advantage of using rationally attenuated live M. tuberculosis as vaccine is that many genetic regions encoding important immunodominant antigens absent in BCG are still present in attenuated M. tuberculosis, while chromosomal deletions in virulence genes provide assurance for safety and genetic stability. These vaccines are expected to safely induce more specific and longer-lasting immune responses in humans that can provide protection against TB [Citation38]. This is the rationale that has been followed in the development of the live-attenuated MTBVAC.
5. MTBVAC: the unfolding story of the development of the first live-attenuated M. tuberculosis vaccine
MTBVAC was constructed by rational attenuation of the clinical isolate M. tuberculosis Mt103 [Citation39]. This isolate belongs to the modern M. tuberculosis Lineage 4, which together with Lineage 2 (Beijing strains), represents the most geographically widespread lineages of MTBC transmitted by the aerosol route between humans [Citation40]. The primary reasons for using a clinical strain for MTBVAC design and construction were to achieve a representative antigen repertoire of the clinical M. tuberculosis strains transmitted between humans and at the same time to avoid the inherent effects that come with laboratory strains (such as H37Rv) as result of repeated sub-cultivation in vitro [Citation41–Citation44], the same process that lead to BCG attenuation (between 1908 and 1921) and strain diversity afterwards [Citation1]. For example, a single-nucleotide insertion in the whiB6 promoter in M. tuberculosis H37Rv has shifted the role of PhoP from transcriptional activator to transcriptional repressor, resulting in lower levels of ESAT-6 expression and secretion in H37Rv [Citation43]. This polymorphism was shown to be absent in any of the other publicly available MTBC genomes or in 76 clinical M. tuberculosis isolates (including MTBVAC parental strain Mt103) analyzed [Citation43].
6. MTBVAC
6.1. MTBVAC and BCG: phylogenetic similarities and differences
MTBVAC and BCG are both live-attenuated derivatives of the TB pathogens in humans (M. tuberculosis) and in cattle (M. bovis), respectively (). Both M. tuberculosis and M. bovis are related members of the MTBC, which are characterized by more than 99.95% similarity at the nucleotide level, identical 16S rRNA sequences and a strictly clonal population structure without ‘recent’ evidence of ongoing horizontal gene transfer between them [Citation45]. Both M. tuberculosis and M. bovis emerged after an evolutionary bottleneck from a most recent common ancestor (MRCA) [Citation46]. Today, global genome sequencing analyses have shown that M. bovis has smaller chromosomal content relative to the human-adapted pathogen M. tuberculosis, due to numerous genomic deletion events. Specifically, the absence of region of difference 9 (RD9) and other specific RD regions allows branching M. bovis from M. tuberculosis [Citation46] ().
Figure 1. Phylogenetic tree of the M. tuberculosis complex and depiction of M. tuberculosis, BCG and MTBVAC. The tree depicts the regions of difference (RDs) deleted between M. tuberculosis and M. bovis in the process of evolution with their host. BCG has lost additional RDs that have led to attenuation. The number of epitopes of some RD regions is also indicated. MTBVAC is represented to have the antigenic repertoire of M. tuberculosis absent in BCG, expected to confer protection against disease similar to latent M. tuberculosis infection in humans [Citation47], and at the same time presenting attenuation profile that is comparable to BCG
![Figure 1. Phylogenetic tree of the M. tuberculosis complex and depiction of M. tuberculosis, BCG and MTBVAC. The tree depicts the regions of difference (RDs) deleted between M. tuberculosis and M. bovis in the process of evolution with their host. BCG has lost additional RDs that have led to attenuation. The number of epitopes of some RD regions is also indicated. MTBVAC is represented to have the antigenic repertoire of M. tuberculosis absent in BCG, expected to confer protection against disease similar to latent M. tuberculosis infection in humans [Citation47], and at the same time presenting attenuation profile that is comparable to BCG](/cms/asset/336cc44a-616c-43cd-9379-9adaff166c74/ierv_a_1324303_f0001_c.jpg)
During the in vitro process of BCG attenuation between 1908 and 1921, more than 100 genes were lost from BCG relative to M. tuberculosis [Citation1]. Among them, deletion of the RD1 region, which encodes the protein secretion system ESX-1, is considered the most relevant deletion responsible for BCG attenuation [Citation1]. It is generally well assumed that T cell epitopes are evolutionarily conserved in the MTBC [Citation48,Citation49]. Copin et al. [Citation50] compared the sequence diversity of 1530 experimentally verified human T cell epitopes and found that 23% of the known MTBC T cell epitopes are absent in BCG. The majority (82%) of these absent epitopes are contained in 6 antigenic proteins encoded in RD1 and RD2 deleted in BCG strains. These and related studies further suggested that epitope sequence variation in BCG potentially affects human T cell recognition that could result in ineffective priming of the host immune system [Citation16,Citation17,Citation50–Citation52].
6.2. MTBVAC vaccine construction
MTBVAC is a live vaccine based on genetically attenuated M. tuberculosis of the clinical isolate Mt103 [Citation39,Citation53]. MTBVAC attenuation is based on two independent stable genetic deletions, without antibiotic resistance markers, in the genes phoP and fadD26 encoding two major virulence factors [Citation39]. The gene phoP encodes the transcription factor of the two-component virulence system PhoPR and the gene fadD26 is one of the 13 genes responsible for biosynthesis and export of phthiocerol dimycocerosates (PDIM), the main virulence-associated cell-wall lipids of M. tuberculosis [Citation54–Citation56]. PhoP has been shown to regulate more than 2% of M. tuberculosis genome, most of which implicated in virulence [Citation57,Citation58], and this regulation could be mediated through other transcription factors, for example, Whib6 of the ESX-1 system [Citation43] or via noncoding RNAs (ncRNAs), such as the recently described mcr7 and its role in activating the TAT secretion system in MTBVAC leading to over secretion of major antigens such as the Ag85 complex [Citation59]. Other relevant virulence genes regulated by PhoP include lipid metabolism genes (e.g. pks2, pks3) involved in biosynthesis of the polyketide-derived acyltrehaloses (DAT, PAT) and sulfolipids (SL), which are front-line lipid constituents of the cell wall thought to have a role in host immune modulation upon first contact of M. tuberculosis with the human host cells [Citation41,Citation60]. PhoP also regulates genes within ESX-1 (e.g. the espACD locus) implicated in the secretion of the major antigen and virulence factor ESAT-6, so that phoP mutants can produce but are unable to export ESAT-6 [Citation61].
6.3. MTBVAC characterization process
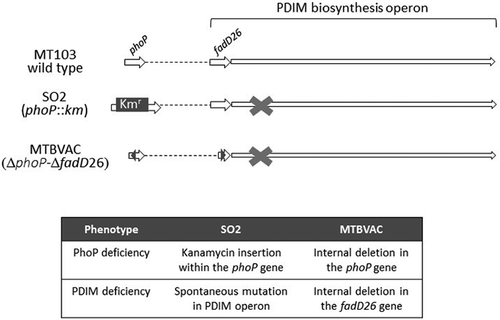
MTBVAC was constructed to contain two independent nonreverting deletion mutations, without antibiotic markers, fulfilling the first Geneva consensus safety requirements for advancing live mycobacterial vaccines to phase I clinical evaluation [Citation33]. MTBVAC was genetically engineered to phenotypically and functionally resemble its prototype SO2. SO2 is a marked Mt103 phoP mutant by the insertion of a kanamycin resistance cassette (kmr) (Mt103phoP::kmr) [Citation62], which in addition to the engineered PhoP-deficient phenotype, SO2 has an acquired spontaneous loss in PDIM biosynthesis [Citation53] (), a process described to be common in M. tuberculosis as result of repeated laboratory subculture and manipulation practices [Citation55,Citation63]. In MTBVAC, the introduction of an unmarked deletion in fadD26 ensures a genetically stable abolishment of PDIM biosynthesis [Citation39]. SO2 has a thorough and complete preclinical history demonstrating robust safety and attenuation profile and promising efficacy compared to BCG in relevant animal models [Citation64–Citation68]. Most of these preclinical studies have been reproduced with MTBVAC to confirm functional profile and biological activity of the double attenuating PhoP-/PDIM-deficient phenotype. Lipid profile analyses have demonstrated that MTBVAC and its prototype SO2 are phenotypically comparable lacking DAT, PAT, and PDIM [Citation39]. To progress to first-in-human phase I clinical evaluation, MTBVAC was developed and characterized as a freeze-dried vaccine product by the Spanish Biopharmaceutical company, Biofabri, in compliance with current Good Manufacturing Practices (cGMP), fulfilling regulatory guidelines for assuring the quality, safety and stability, and efficacy of BCG freeze-dried vaccine [Citation39]. A summary of the preclinical development of MTBVAC is presented in continuation placing special emphasis on those studies where improved efficacy and safety relative to BCG were observed.
Figure 2. MTBVAC and SO2 genetic and phenotypic profile. Figure depicts the marked phoP mutation by insertion of a Kanamycin resistance cassette (Kmr) insertion and spontaneous loss in the virulence-associated phthiocerol dimycocersate (PDIM) lipids in SO2 and the stable unmarked genetic deletions in phoP and fadD26 in MTBVAC. Since MTBVAC was constructed using the SO2 background, MTBVAC also has the spontaneous loss in PDIM present in SO2 and deletion in fadD26 ensuring a genetically stable PDIM deficient phenotype
7. Preclinical characterization of MTBVAC
7.1. Safety and attenuation profile of MTBVAC and its prototype SO2 relative to BCG
Any novel live TB vaccine candidate must demonstrate comparable or better safety and attenuation profile than BCG in recognized preclinical animal models as a safety requirement for entry into first-in-human clinical evaluation [Citation32,Citation33]. The most recognized animal models for evaluation of safety attenuation of new TB vaccine candidates to date include the extremely TB susceptible immunocompromised SCID mice, which lack adaptive immune responses (T and B cells) important for containing TB infection, and guinea pigs, which can develop disease after infection with very few bacilli <5 [Citation69]. In these models, vaccine safety is commonly measured in terms of survival kinetics, viable vaccine bacteria in lung and spleen and signs of disease (pathology) at necropsy.
Robust safety attenuation profile of the doubly attenuated PhoP-/PDIM-deficient prototype SO2 was demonstrated in two SCID mouse models published in Martin et al., 2006 [Citation66]. One study showed that by the aerosol route SO2 is unable to cause disease or adverse events, and mice survived >245 days, when experiment was ended. The other study showed that by intravenous (IV) route, SO2 inoculation renders greater survival to SCID mice than BCG Pasteur [Citation66]. The remaining safety studies were designed in agreement with regulatory and Geneva consensus guidelines exclusively for the characterization of the final vaccine product MTBVAC in mice (SCID and BALB/c) and guinea pigs [Citation39]. Designs mainly followed administration by the subcutaneous (SC) route (the accepted route mimicking clinical intradermal administration of BCG) and a dose equivalent to 50 standard human doses of BCG, approx. 2.5×107 CFU. By this route and dose of administration, all data showed comparable safety and attenuation profile for MTBVAC (and SO2) and BCG SSI, which is the strain used as control comparator in MTBVAC phase I clinical evaluation in adults [Citation70] and in newborns (NCT02013245). Additionally, biodistribution and formal toxicity studies showed that intradermal inoculation of MTBVAC presents lack of toxicity, limited organ biodistribution and persistence, and untraceable excretion in urine and stool up to six months post vaccination, comparable to BCG SSI, as well as appears to be less reactogenic than BCG in mice and guinea pigs [Citation39]. Finally, in newborn mice MTBVAC and BCG SSI presented a comparable safety and lack of toxicity profile, as measured by health appearance, normal growth and development behavior, survived until fixed end point (6 months), and there were no effects on organ development at necropsy [Citation71].
In the context of vaccine safety, horizontal transmission of genetic material is a topic of increasing interest in relationship to TB. In this regard, it should be considered that like BCG, MTBVAC is a rational derivative of the related members of MTBC, characterized by more than 99.95% similarity at the nucleotide level, identical 16S rRNA sequences and a strictly clonal population structure [Citation45]. To our knowledge, with the exception of M. canettii (the more closely related MTBC ancestor), there has been no evidence of horizontal gene transfer between members of the MTBC [Citation72]. This provides strong bases to consider that the probabilities of horizontal gene transfer events and reversion to virulence of MTBVAC and BCG are highly unlikely. Supporting this hypothesis, it should be considered that over the long history of BCG use in humans, which is now more than 96 years of global vaccination coverage in more than 3 billion individuals worldwide (>100,000,000 newborns vaccinated per year), to our knowledge of the existing literature, there have been no reported adverse events due to reversion to partial or full virulence of BCG as result of horizontal gene transfer events with other related or unrelated mycobacterial species of the MTBC. In addition, preclinical studies with MTBVAC and its prototype SO2, have confirmed lack of horizontal gene transfer upon infection with wild-type M. tuberculosis in MTBVAC or SO2 vaccinated animals.
7.2. Protective efficacy and immunogenicity of MTBVAC and prototype SO2 relative to BCG
Protective efficacy in preclinical animal models, together with safety and immunogenicity in phase I and phase IIa clinical trials, are the three main gate criteria used to select the most promising candidates to progress to phase IIb/III efficacy testing [Citation73].
Since 2001, rigorous preclinical efficacy and immunogenicity studies of prototype SO2 and final vaccine construct MTBVAC were conducted in recognized animal models, including immunocompetent BALB/c or C57BL/6 mice and guinea pigs, and nonhuman primates [Citation33,Citation74], by independent (national and international) laboratories. As for all TB vaccine candidates to date, the protective efficacy of MTBVAC and SO2 was measured in terms of improvement of bacterial load (expressed as colony forming units or CFU) in lungs and spleen at a fixed time point post challenge, body weight gain, reduced lung pathology score, and when possible, long-term survival relative to gold standard BCG [Citation73]. In the recognized small animal models (mice and guinea pigs), BCG is considered to confer a relatively good protection against M. tuberculosis challenge, as measured by 1–3 log reduction in CFU counts (variable between models), or a significant degree of improvement in survival or pathology score [Citation69,Citation73–Citation76]. With this consideration, the minimum acceptable efficacy criteria set by the Geneva Consensus requirements for advancing new TB vaccines to clinical development include a significant higher reduction in lung bacterial burden of >0.5 logs CFU in comparison to BCG [Citation32,Citation33], which was followed in the MTBVAC preclinical development program. Regarding evaluation of vaccine-induced immunogenicity, various parameters can be measured in mice, nonhuman primates (NHP) and humans, however, in the face of the current lack of immune correlates of protection against TB, the immunogenicity readouts to date have predominantly allowed to deduce vaccine take.
Considering the limitations of the current animal models, there has not been an experiment to date (published or unpublished) where protective efficacy, immunogenicity or safety of MTBVAC (and SO2) has not been comparable or superior to standard BCG vaccination. Most MTBVAC and SO2 protection and immunogenicity studies (which follow standard design of low-dose challenge and efficacy evaluation at short term (8–12 weeks) post challenge) have shown comparability to gold standard BCG. However, there are few exceptions where superior efficacy and immunogenicity were observed, and these studies varied in using high challenge dose and evaluation of long-term efficacy, including survival. One of these exceptional studies was the high-dose challenge long-term survival experiment in guinea pigs conducted at Public Health England (PHE), where MTBVAC prototype SO2 conferred 100 % survival (6/6 guinea pigs) relative to 33% survival in the BCG SSI group (2/6 guinea pigs), as well as significantly greater reduction in severity of disease and bacterial burden in lungs and spleen [Citation66,Citation75]. Improved protective efficacy of SO2 relative to BCG SSI was also observed in the nonhuman primate rhesus macaque model at the National Primate Research Centre, where, despite ability of both vaccines to confer efficacy compared to unvaccinated controls (e.g. by increase in total body weight, significantly reduced gross lung lesions, wasting and systematic inflammation), SO2 showed an average of reduction in lung bacillary load of 1.2 log CFU as compared to 0.43 log CFU conferred by BCG relative to the unvaccinated control group [Citation68]. Moreover, SO2 but not BCG vaccination revealed significant body weight gain and marked reduction in lung pathology when compared to unvaccinated controls. Serum C-reactive protein assessment indicated that vaccination with prototype SO2 or BCG Danish leads to significant reduction in systemic inflammatory response after challenge when compared to unvaccinated controls [Citation68]. In adult and newborn C57BL/6 mice MTBVAC confers significantly greater protection than BCG SSI as measured by significant CFU reduction (>0.5 log) in lung bacterial burden [Citation39,Citation71]. Of note, in the well-established guinea pig model at PHE, BCG is given at a lower 5×104 CFU dose, and in those studies MTBVAC and SO2 administration normally followed the same vaccination regimen as that of BCG to allow for data comparison. Importantly, preclinical efficacy data in mice and guinea pigs have demonstrated that efficacy of BCG and MTBVAC vaccines is dose independent by the SC route, at least in short-term protection experiments [Citation39,Citation77].
Some of the most abundant antigenic proteins in M. tuberculosis described include Ag85B and Ag85A. These antigens are constituents of some of the most clinically advanced subunit vaccines in the pipeline to date [Citation2,Citation3], designed to boost BCG-induced immunity at birth mainly in adolescents and adults [Citation78]. Ag85B was expressed in the recombinant BCG, rBCG30, demonstrating improved efficacy in guinea pigs over BCG [Citation79]. This led us to study expression and immunogenicity of Ag85B in prototype SO2 and MTBVAC in more detail. In a model of adult C57BL/6 transgenic mice expressing a T cell receptor (TCR) specific for a peptide of Ag85B, immunogenicity studies at University of Sydney showed that vaccination with SO2 induced a marked expansion of Ag85B-specific CD4 + T cell responses [Citation67]. These Ag85B-specific T cells presented an increased frequency of persisting at extended time points displaying a central memory phenotype, a property not shared by BCG. Moreover, upon M. tuberculosis challenge, SO2 group showed significantly greater recall of vaccine-induced memory cells to secondary infection which was of increased magnitude compared to that of the BCG group [Citation67]. In newborn C57BL/6 mice, MTBVAC vaccination induced differential immune response to Ag85B compared to BCG vaccination, as measured by IFNγ secretion in splenocytes [Citation71]. A plausible explanation for these differential immune responses to Ag85B between MTBVAC (or SO2) and BCG could be due to an acquired point mutation in the fbpB gene in all BCG sub-strains, which results in an unstable protein [Citation50]. Interestingly, in addition to the rich antigen-specific repertoire inherent of MTBVAC, recent data by our group observed that the PhoP-dependent ncRNA mcr7 leads to an increased activity of the Tat-C secretion system resulting in greater secretion of Ag85 complex proteins in MTBVAC [Citation44,Citation59]. Intriguingly, MTBVAC vaccination in newborn C57BL/6 mice revealed greater immune response to stimulation with purified protein derivative (PPD) of secreted M. tuberculosis antigens, relative to BCG, whereas adult C57BL/6 mice showed comparable PPD responses for both vaccine groups [Citation39]. This makes us suspect that in clinical trials, the immune response induced by MTBVAC vaccination in naïve newborns could be more robust or distinguished than that induced by neonatal BCG vaccination.
8. MTBVAC GMP and clinical development
8.1. GMP development of MTBVAC to support entry into first-in-human clinical trials
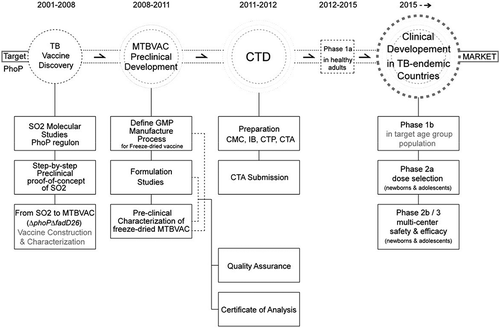
The success of the preclinical and GMP development of MTBVAC to support phase I clinical evaluation is result of the close collaborative partnership between University of Zaragoza (Unizar), Biofabri, and the European non-for-profit TuBerculosis Vaccine Initiative (TBVI) [Citation80]. Moreover, the MTBVAC project has counted with a robust preclinical, regulatory and clinical expertise from the TBVI Product Development and Clinical Development Teams (TBVI PDT and CDT), gratefully acknowledged in [Citation39] and [Citation70]. In Oct 2012, MTBVAC was granted approval to enter a first-in-human phase Ia clinical trial at Lausanne University Hospital (CHUV) in Switzerland, led by the Principal Investigator (PI) Prof. François Spertini. The study primary objective was safety and local tolerance of MTBVAC in comparison with BCG SSI with minimum secondary immunogenicity in healthy adult volunteers without any history of BCG vaccination, or exposure to TB (PPD-negative) and or HIV. Successful completion demonstrated that vaccination with MTBVAC was at least as safe as BCG, and did not induce serious adverse events [Citation70]. And although no statistical significance could be achieved due to the small group size of the trial, the immunogenicity data showed that MTBVAC was at least as immunogenic as BCG, and at the same dose level as BCG, MTBVAC group showed greater frequency of polyfunctional CD4+ central memory T cells. This first phase I trial marks a historic milestone in human vaccinology, as for the first time a live-attenuated M. tuberculosis vaccine has entered clinical trials showing excellent safety profile and differential immunogenicity profile compared to BCG supporting clinical development in high-burden TB endemic countries ().
Figure 3. Development of MTBVAC from discovery to clinical development. The figure represents the chronological order of the steps in the discovery of phoP, construction and characterization of SO2 and MTBVAC, and the GMP, preclinical and clinical development of final product MTBVAC freeze-dried vaccine. Meaning of abbreviations: CTP – Clinical Trial Protocol; CMC – Chemistry, Manufacturing, and Controls; IB – Investigator’s Brochure; CTP – Clinical Trial Protocol; CTA – Clinical Trial Application
8.2. Clinical development plans of MTBVAC
Today, Biofabri is officially the industrial manufacturer and exclusive licensee of MTBVAC (also Sponsor of the Clinical Trials of MTBVAC) and together with its faithful partner and MTBVAC intellectual owner Unizar they have set two independent clinical development plans (CDPs) for MTBVAC under the umbrella of TBVI. One CDP is also in collaboration with US-founded nonprofit TB vaccine R&D organization Aeras [Citation81] and focuses on the development of MTBVAC as a preventive vaccine in adolescents and adults (BCG-vaccinated at birth) living in high-burden countries with or without prior infection with M. tuberculosis (). In support of this strategy are the recently published results of the protection study in the well-established guinea-pig model of aerosol TB infection at PHE (UK), comparing BCG and MTBVAC vaccination schedules in prime-boost combination or alone (Clark et al. JID in press https://doi.org/10.1093/infdis/jix030). The efficacy data of this study suggested that MTBVAC immunity is longer lasting than BCG when given as a single dose and that long and short intervals between BCG prime and MTBVAC boost resulted in improved efficacy in lungs compared to BCG alone. Plans to support a phase IIa dose-defining study (NCT 02933281) before progressing to a multicenter prevention-of-disease phase IIb/III trial are underway. The second CDP of MTBVAC is focused on the development of MTBVAC as a preventive vaccine for newborns, which could replace BCG if improved safety and or efficacy is demonstrated (). Currently, a phase Ib trial of MTBVAC in healthy newborns is being conducted by the South African Tuberculosis Vaccine Initiative (SATVI) in the high-burden TB-endemic region of Worcester, South Africa, with PI Dr. Michele Tameris (NCT02729571). The study design is set to evaluate the safety and immunogenicity of MTBVAC in comparison with BCG SSI in newborns living in a TB endemic region. As a safety step prior to starting vaccination of newborns, a safety arm in healthy M. tuberculosis-unexposed adults was successfully completed in Dec 2015 with no reported vaccine-related adverse events. Newborn vaccination stage has already ended and final data safety and immunogenicity reports are expected 2nd quarter 2017. This newborns phase Ib trial is conducted with the aim to advance to a phase IIa dose-defining study to allow progress to community wide efficacy evaluation.
8.3. The Quantiferon TB-diagnostic test and implications for MTBVAC clinical trials
Being a live-attenuated M. tuberculosis derivative, MTBVAC can express the M. tuberculosis antigen repertoire, which also includes the highly immunodominant ESAT-6 and CFP-10 antigens, albeit impaired secretion of ESAT-6 due to the deletion in phoP. This makes one reason that at some point during vaccine clearance from the organism, the immune system could detect these antigens. This calls for significant considerations for the potential implications of the current Quantiferon assay for in vitro diagnosis of TB infection (based on ESAT-6, CFP-10 and TB7.7) in MTBVAC clinical trials in TB-endemic regions. Relevant to this, in the MTBVAC phase Ia trial in adults in Lausanne, ELISpot conversion data to PPD, and to separate antigens ESAT-6 and CFP-10 showed responses to ESAT-6 that stayed below threshold for a positive test readout of infection with M. tuberculosis, while there was a transient increase in CFP-10-specific IFNγ response over the limit of positivity in two of nine subjects from the intermediate MTBVAC dose group (5×104 CFU) and in one subject of MTBVAC high-dose group (5x105 CFU) at day 28, which at day 270 (end of study) was below threshold for M. tuberculosis infection [Citation70]. These results are supported by recent data in goats where vaccination with SO2 induced a transient ESAT-6/CFP-10 positivity in 50% of the animals [Citation82]. Although the group numbers of the phase Ia are too small to achieve statistical significance for these data, detailed clinical characterization of MTBVAC-induced responses to these and other antigens are needed. Most importantly, there may be a need for adapting the current Quantiferon/ELISpot TB diagnostic tests to reliably distinguish between MTBVAC vaccination and M. tuberculosis infection in clinical trials.
Relevant to these observations, recent studies have shown that most of the experimentally confirmed human T cell epitopes of M. tuberculosis are concentrated in ESAT-6 and CFP-10, and up to 80% of humans and nonhuman primates show reactogenicity to both of them [Citation83–Citation86]. Moreover, these human T cell epitopes were shown to be evolutionary conserved in the related members of the MTBC [Citation48]. Andrews et al., showed that individuals with latent TB infection (LTBI) had up to 79% lower risk of progressive TB disease after reinfection than uninfected individuals [Citation47], powerfully denoting that vaccines that replicate this level of risk reduction would likewise be important in controlling TB. These studies support the idea that the immune response in LTBI, and therefore, selection of immunodominant antigens could be of significant benefit to achieve protection against TB disease [Citation83]. Taken together, these observations call for more in-depth characterization of the ability of MTBVAC to induce immune reactivity to the immunodominant antigens ESAT-6 and CFP-10 in humans. Moreover, it would be important to investigate whether preexisting immune sensitization to BCG, NTM, and or M. tuberculosis in adolescents and adults, which could result in potential masking and blocking effects to vaccination, would affect MTBVAC-induced immunity in presensitized individuals relative to the naïve immune system of newborns. And ultimately, deciphering whether MTBVAC-induced specific immunity to ESAT-6, CFP-10 and or other immune-dominant M. tuberculosis antigens could play a role in improved TB protection in humans may help in the identification of reliable antigen-specific correlates of protection. Elaborate preclinical efficacy studies in macaque models may be a highly valuable opportunity for such studies because of the ability of this species to recognize the majority of the experimentally identified human T cell antigens [Citation83,Citation84].
9. Expert commentary
Priming immunity with live vaccines is considered to induce specific long-lasting memory immune responses, which is not observed with subunit vaccines, and this effect could be related to restricted persistence or replication in vivo as observed for other human live vaccines (e.g. polio, measles, yellow fever) [Citation38]. Although debatable in clinic, live BCG is typically the most protective vaccine against M. tuberculosis in experimental animal models [Citation38], which makes one consider that human vaccination with live-attenuated M. tuberculosis (e.g. MTBVAC) would induce specific long-term immune responses against certain T cell epitopes that are present in M. tuberculosis and absent in BCG and which may be important for protection [Citation38,Citation83]. Moreover, neonatal vaccination with live-attenuated M. tuberculosis vaccines could represent by far the most effective strategy for conferring high-quality protective immunity against TB, as healthy newborns represent the most sensitive and immunologically naïve population without preexisting mycobacterial immunity, which in older age groups can lead to potential masking and blocking effects to vaccination [Citation18,Citation87]. Additionally, considering the similar biological nature of novel live-attenuated M. tuberculosis vaccines and BCG makes one consider that these novel candidates may also possess the inherent nonspecific benefits attributed to BCG and as such, it would be important to consider evaluating them in advanced clinical development trials.
Despite inherent risks with preexisting mycobacterial sensitization, in 2014, the BMGF revised its TB vaccine strategy shifting entirely supporting discovery and development of novel preventive TB vaccines in adolescents and adults, where the greatest impact on TB transmission is expected to be observed [Citation88]. This important shift, which was possibly driven by the unsuccessful outcome of MVA85A phase IIb infant efficacy trial [Citation15], has left infant TB prevention strategies with little to no priority in the current BMGF TB vaccine agenda [Citation34]. Moreover, a strong emphasis on the need for diversifying the vaccine concepts in the clinical pipeline was also placed by the revised TB vaccine strategy, as the majority of candidates are subunit approaches of which six of the eight subunits contain an Ag85 antigen (either Ag85A or Ag85B), most designed to provoke cell-mediated immunity driven by CD4 and CD8 T cells, resulting in vaccines with little immunologic diversity [Citation34]. Driven by this new shift in agenda, in July 2015 BMGF launched the Collaboration for TB Vaccine Discovery (CTVD), which comprises an international network of selected scientists and experts dedicated to innovation and collaboration in basic discovery, preclinical development, and early phase I and smaller experimental medicine trials of new TB vaccines [Citation89]. Likewise, the new TB vaccine strategy has influenced vaccine developers of new live mycobacterial vaccines, like MTBVAC, to consider expanding the target population to encompass M. tuberculosis unexposed and exposed adolescents and adults.
The revised BMGF TB vaccine strategy has also set new gating decisions for clinical evaluation of new TB vaccines, which are based on NHP models and small experimental clinical trials in humans with two outcomes: immune signatures, that is, detailed immune responses measured, and protection following challenge with TB [Citation89]. A product representing a distinct concept/hypothesis that vows promise in these studies will be selected for clinical development [Citation89]. However, there are important limitations with the current NHP models requiring consideration, especially if efficacy in different animal models that could include macaques will be used by BMGF to decide on the fate of a candidate. For example, in the current animal models, including macaques, BCG is considered to work so well that it leaves little room for improvement for new candidates implying the need for large group numbers to achieve statistical significance in improved efficacy, which could make such studies practically and ethically unfeasible, and prohibitively expensive. Moreover, depending on the macaque species, different disease outcomes may be observed that could affect interpretation of study results. Cynomolgus macaques are known to be prone to developing latent infection, so that about 50 % of non-vaccinated controls in a study will not show clinical signs of active disease [Citation90], which may have some resemblance to TB outcome in humans, but it makes data analysis difficult. Rhesus macaques on the other hand are considered more sensitive to TB infection and disease; however, this model calls for or a design that includes larger number of animals per group to achieve statistical significance when comparing new vaccine candidates to BCG, thus highly increasing cost of experiment. Nowadays PET CT scanning is used to evaluate pathology in lungs, and this method is considered more reliable than CFU measure [Citation91]. Despite these important limitations to the nonhuman primate models, a promising ultra-low-dose aerosol challenge (<20 CFU) model in nonhuman primates has been established at the PHE and can use survival end points, as reported for another candidate in the pipeline [Citation92].
Finally, if animal models including nonhuman primate models are to be employed in the decision-making process for selecting a candidate to advance to community-wide efficacy trials, considerations for incorporating elements of the planned clinical program (e.g. phase II dose-defining study) are necessary to allow closer comparability of nonhuman primate and clinical data. These elements include target age (e.g. adolescents or young adults), previous mycobacterial sensitization (e.g. to BCG vaccination at birth), exact clinical route, and regimen of vaccination, and the clinical immunological assays used or planned for use for vaccine characterization and dose definition. For example, the whole-blood intracellular staining (WB-ICS) assay, originally developed by Prof. Willem Hanekom (former SATVI director, current deputy director TB vaccines at BMGF) is now widely used as one of the standard immunological characterization tests in TB vaccine clinical trials, as in the phase Ia of MTBVAC [Citation70] or in other trials at SATVI [Citation37,Citation93–Citation96]. Moreover, considerations for investigating ESAT-6 and CFP-10 conversion kinetics and how they can relate to vaccine induced protection will be made. Also, other possibilities for reliable differentiation between MTBVAC vaccination and M. tuberculosis infection should also be investigated.
10. Five-year view
Altogether, the next five years are filled with many challenges and anticipations for MTBVAC, as well as for other vaccines in the TB vaccine development community. Being the only live M. tuberculosis vaccine in the pipeline of clinical development and considering the inherent rich antigen-specific repertoire which is differential to that of BCG, MTBVAC represents the most unique opportunity to study and understand what is needed in terms of an effective immunity against M. tuberculosis infection and disease establishment. Immunological data from the planned dose-defining MTBVAC clinical trials in newborns and adolescents will allow establishing qualitative and quantitative differences between MTBVAC- and BCG-triggered responses. Moreover, parallel protection experiments in NHP, faithfully mirroring the planned immunological tests in phase IIa will permit comparison of immunological and protective efficacy data in the quest to identify reliable correlates of protection for use in community-wide phase IIb efficacy trials. In this context, collaborations on a global level with wide range of expertise in the field of TB and vaccine development would be enthusiastically embraced.
Key issues
MTBVAC is the first live attenuated M. tuberculosis-based vaccine to enter clinical development, a historic milestone in human vaccinology.
MTBVAC has shown improved or comparable safety and efficacy to BCG in relevant preclinical animal models.
The first Phase 1a trial with MTBVAC showed that the vaccine is as at least as safe as BCG SSI.
MTBVAC is currently being evaluated in a Phase 1b trial in newborns, South Africa.
MTBVAC Clinical Development Plans target TB prevention in newborns, as a BCG replacement strategy, and in adolescents and adults.
According to the new TB vaccine strategy of the major funder BMGF, in addition to demonstrated safety and immunogenicity in early human clinical trials, convincing NHP efficacy and experimental medicine studies, are needed to support progress to community-wide Phase 2b efficacy evaluation.
Declaration of interest
C Martin and J Gonzalo-Asensio are co-inventors of the patent ‘tuberculosis vaccine’ filed by the University of Zaragoza, Spain. C Martin and N Aguilo are co-inventors of the patent ‘Inactivated tuberculosis vaccine’ filed by the university of Zaragoza, Spain. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Acknowledgments
The authors gratefully acknowledge TBVI Preclinical and Clinical Development Teams for their dedication, expertise and continuing support in the preclinical, GMP and clinical development of MTBVAC. We thank our industrial partner Biofabri and in particular the team dedicated to the industrial development and production of MTBVAC, including Dr. Eugenia Puentes, Dr. Conchita Fernandez, Dr. Alberto Parra, Dr. Juana Doce, Dr. Esteban Rodriguez, Oswaldo Alvarez and Ingrid Murillo.
Additional information
Funding
References
- Brosch R, Gordon SV, Garnier T, et al. Genome plasticity of BCG and impact on vaccine efficacy. Proc Natl Acad Sci USA. 2007;104(13):5596–5601.
- Kaufmann SH, Weiner J, von Reyn CF. Novel approaches to TB vaccine development. Int J Infect Dis. 2017;56:263–267.
- World Health Organization (WHO). Global tuberculosis report 2016. Geneva, Switzerland: WHO Press; 2016.
- Ai JW, Ruan QL, Liu QH, et al. Updates on the risk factors for latent tuberculosis reactivation and their managements. Emerg Microbes Infect. 2016;5:e10.
- Sadikot RT. Identifying patients at high risk of tuberculosis recurrence. Int J Mycobacteriol. 2016;5(Suppl 1):S66.
- WHO. BCG vaccine. WHO position paper. Wkly Epidemiol Rec. 2004;79(4):27–38.
- Zwerling A, Behr MA, Verma A, et al. The BCG world Atlas: a database of global BCG vaccination policies and practices. PLoS Med. 2011;8(3):e1001012.
- Colditz GA, Berkey CS, Mosteller F, et al. The efficacy of bacillus Calmette-Guerin vaccination of newborns and infants in the prevention of tuberculosis: meta-analyses of the published literature. Pediatrics. 1995;96(1 Pt 1):29–35.
- Trunz BB, Fine P, Dye C. Effect of BCG vaccination on childhood tuberculous meningitis and miliary tuberculosis worldwide: a meta-analysis and assessment of cost-effectiveness. Lancet. 2006;367(9517):1173–1180.
- Abubakar I, Pimpin L, Ariti C, et al. Systematic review and meta-analysis of the current evidence on the duration of protection by bacillus Calmette-Guerin vaccination against tuberculosis. Health Technol Assess. 2013;17(37):1–372, v–vi.
- Kaufmann SH, Evans TG, Hanekom WA. Tuberculosis vaccines: time for a global strategy. Sci Transl Med. 2015;7(276):276fs278.
- Kaufmann SH, Cotton MF, Eisele B, et al. The BCG replacement vaccine VPM1002: from drawing board to clinical trial. Expert Rev Vaccines. 2014;13(5):619–630.
- Donald PR, van Zyl LE, de Villiers J. BCG vaccination status of children with tuberculous meningitis and the use of unsupervised isoniazid prophylaxis. S Afr Med J. 1995;85(3):167–170.
- Wolzak NK, Cooke ML, Orth H, et al. The changing profile of pediatric meningitis at a referral centre in Cape Town, South Africa. J Trop Pediatr. 2012;58(6):491–495.
- Tameris MD, Hatherill M, Landry BS, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet. 2013;381(9871):1021–1028.
- Zhang L, Ru HW, Chen FZ, et al. Variable virulence and efficacy of BCG vaccine strains in mice and correlation with genome polymorphisms. Mol Ther. 2016;24(2):398–405.
- Zhang W, Zhang Y, Zheng H, et al. Genome sequencing and analysis of BCG vaccine strains. PLoS One. 2013;8(8):e71243.
- Barreto ML, Pilger D, Pereira SM, et al. Causes of variation in BCG vaccine efficacy: examining evidence from the BCG REVAC cluster randomized trial to explore the masking and the blocking hypotheses. Vaccine. 2014;32(30):3759–3764.
- Rodrigues LC, Pereira SM, Cunha SS, et al. Effect of BCG revaccination on incidence of tuberculosis in school-aged children in Brazil: the BCG-REVAC cluster-randomised trial. Lancet. 2005;366(9493):1290–1295.
- WHO. World Health Organization. Information sheet observed rate of vaccine reactions Bacille-Calmette Guérin (BCG) vaccine. 2012 Apr . Global Vaccine Safety, Immunization Vaccines and Biologicals 20, avenue Appia, Ch-1211 Geneva 27. Available from: http://www.who.int/vaccine_safety/initiative/tools/BCG_Vaccine_rates_information_sheet.pdf. (Ed.^(Eds).
- WHO. The immunological basis for immunization series module 5: tuberculosis. 2011. Available from: http://whqlibdoc.who.int/publications/2011/9789241502412_eng.pdf. (Ed.^(Eds) (World Health Organization, Deparment of Immunizzation, Vaccines and Biologicals, 2011).
- Shann F. Nonspecific effects of vaccines and the reduction of mortality in children. Clin Ther. 2013;35(2):109–114.
- Aaby P, Roth A, Ravn H, et al. Randomized trial of BCG vaccination at birth to low-birth-weight children: beneficial nonspecific effects in the neonatal period? J Infect Dis. 2011;204(2):245–252.
- Aaby P, Roth A, Biering-Sorensen S, et al. No evidence of bias in trial showing BCG reduces neonatal mortality. J Infect Dis. 2012;205(3):515–517.
- Biering-Sorensen S, Aaby P, Napirna BM, et al. Small randomized trial among low-birth-weight children receiving bacillus Calmette-Guerin vaccination at first health center contact. Pediatr Infect Dis J. 2012;31(3):306–308.
- Ota MO, Vekemans J, Schlegel-Haueter SE, et al. Influence of Mycobacterium bovis bacillus Calmette-Guerin on antibody and cytokine responses to human neonatal vaccination. J Immunology. 2002;168(2):919–925.
- de Castro MJ, Pardo-Seco J, Martinon-Torres F. Nonspecific (Heterologous) protection of neonatal BCG vaccination against hospitalization due to respiratory infection and sepsis. Clin Infect Dis. 2015;60(11):1611–1619.
- Kleinnijenhuis J, Quintin J, Preijers F, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA. 2012;109(43):17537–17542.
- Netea MG, Joosten LA, Latz E, et al. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352(6284):aaf1098.
- Netea MG, Quintin J, van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host & Microbe. 2011;9(5):355–361.
- Arts RJ, Carvalho A, La Rocca C, et al. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. 2016;17(10):2562–2571.
- Walker KB, Brennan MJ, Ho MM, et al. The second Geneva consensus: recommendations for novel live TB vaccines. Vaccine. 2010;28(11):2259–2270.
- Kamath AT, Fruth U, Brennan MJ, et al. New live mycobacterial vaccines: the Geneva consensus on essential steps towards clinical development. Vaccine. 2005;23(29):3753–3761.
- The tuberculosis prevention pipeline. 2016 Jul 15. by Mike Frick. Available from: http://pipelinereport.org/2016/tb-prevention
- Hawkridge T, Scriba TJ, Gelderbloem S, et al. Safety and immunogenicity of a new tuberculosis vaccine, MVA85A, in healthy adults in South Africa. J Infect Dis. 2008;198(4):544–552.
- Sander CR, Pathan AA, Beveridge NE, et al. Safety and immunogenicity of a new tuberculosis vaccine, MVA85A, in Mycobacterium tuberculosis-infected individuals. Am J Respir Crit Care Med. 2009;179(8):724–733.
- Scriba TJ, Tameris M, Smit E, et al. A phase IIa trial of the new tuberculosis vaccine, MVA85A, in HIV- and/or Mycobacterium tuberculosis-infected adults. Am J Respir Crit Care Med. 2012;185(7):769–778.
- Scriba TJ, Kaufmann SH, Henri Lambert P, et al. Vaccination against tuberculosis with whole-cell mycobacterial vaccines. J Infect Dis. 2016;214(5):659–664.
- Arbues A, Aguilo JI, Gonzalo-Asensio J, et al. Construction, characterization and preclinical evaluation of MTBVAC, the first live-attenuated M. tuberculosis-based vaccine to enter clinical trials. Vaccine. 2013;31(42):4867–4873.
- Stucki D, Brites D, Jeljeli L, et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat Genet. 2016;48(12):1535–1543.
- Chesne-Seck ML, Barilone N, Boudou F, et al. A point mutation in the two-component regulator PhoP-PhoR accounts for the absence of polyketide-derived acyltrehaloses but not that of phthiocerol dimycocerosates in Mycobacterium tuberculosis H37Ra. J Bacteriol. 2008;190(4):1329–1334.
- Gonzalo-Asensio J, Soto CY, Arbues A, et al. The Mycobacterium tuberculosis phoPR operon is positively autoregulated in the virulent strain H37Rv. J Bacteriol. 2008;190(21):7068–7078.
- Solans L, Aguilo N, Samper S, et al. A specific polymorphism in Mycobacterium tuberculosis H37Rv causes differential ESAT-6 expression and identifies WhiB6 as a novel ESX-1 component. Infect Immun. 2014;82(8):3446–3456.
- Broset E, Martin C, Gonzalo-Asensio J. Evolutionary landscape of the Mycobacterium tuberculosis complex from the viewpoint of PhoPR: implications for virulence regulation and application to vaccine development. MBio. 2015;6(5):e01289–01215.
- Brosch R, Gordon SV, Marmiesse M, et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci USA. 2002;99(6):3684–3689.
- Boritsch EC, Brosch R. Evolution of mycobacterium tuberculosis: new insights into pathogenicity and drug resistance. Microbiol Spectr. 2016;4:5.
- Andrews JR, Noubary F, Walensky RP, et al. Risk of progression to active tuberculosis following reinfection with Mycobacterium tuberculosis. Clinical Infect Dis. 2012;54(6):784–791.
- Comas I, Chakravartti J, Small PM, et al. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet. 2010;42(6):498–503.
- Coscolla M, Copin R, Sutherland J, et al. M. tuberculosis T cell epitope analysis reveals paucity of antigenic variation and identifies rare variable TB antigens. Cell Host & Microbe. 2015;18(5):538–548.
- Copin R, Coscolla M, Efstathiadis E, et al. Impact of in vitro evolution on antigenic diversity of mycobacterium bovis bacillus Calmette-Guerin (BCG). Vaccine. 2014;32(45):5998–6004.
- Copin R, Wang X, Louie E, et al. Within host evolution selects for a dominant genotype of mycobacterium tuberculosis while T cells increase pathogen genetic diversity. PLoS Pathog. 2016;12(12):e1006111.
- Yruela I, Contreras-Moreira B, Magalhaes C, et al. Mycobacterium tuberculosis complex exhibits lineage-specific variations affecting protein ductility and epitope recognition. Genome Biol Evol. 2017.
- Arbués A. Construction and characterization of a new generation of phoP-based vaccines against tuberculosis [Doctorate Thesis; Thesis Director Prof. Carlos Martin Montañes]. Grupo de Genetica de Micobacterias. Available form: http://genmico.unizar.es/. Departamento de Microbioligía, Medicina Preventiva y Salud Publica (Ed.^(Eds) (Universidad de Zaragoza, Facultad de Medicina, Ed. A, 2010).
- Cox JS, Chen B, McNeil M, et al. Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. 1999;402(6757):79–83.
- Kirksey MA, Tischler AD, Simeone R, et al. Spontaneous phthiocerol dimycocerosate-deficient variants of Mycobacterium tuberculosis are susceptible to gamma interferon-mediated immunity. Infect Immun. 2011;79(7):2829–2838.
- Trivedi OA, Arora P, Vats A, et al. Dissecting the mechanism and assembly of a complex virulence mycobacterial lipid. Mol Cell. 2005;17(5):631–643.
- Gonzalo-Asensio J, Mostowy S, Harders-Westerveen J, et al. PhoP: a missing piece in the intricate puzzle of Mycobacterium tuberculosis virulence. PLoS One. 2008;3(10):e3496.
- Gonzalo-Asensio JÁ. Deciphering the role of PhoP in Mycobacterium tuberculosis virulence [Doctorate Thesis; Director Prof. Carlos Martin]. Grupo de Genetica de Micobacterias. Available from: http://genmico.unizar.es/. Departamento de Microbioligía, Medicina Preventiva y Salud Publica (Ed.^(Eds) (Universidad de Zaragoza, Facultad de Medicina, Ed. A, 2006)
- Solans L, Gonzalo-Asensio J, Sala C, et al. The PhoP-dependent ncRNA Mcr7 modulates the TAT secretion system in Mycobacterium tuberculosis. PLoS Pathog. 2014;10(5):e1004183.
- Gonzalo Asensio J, Maia C, Ferrer NL, et al. The virulence-associated two-component PhoP-PhoR system controls the biosynthesis of polyketide-derived lipids in Mycobacterium tuberculosis. J Biol Chem. 2006;281(3):1313–1316.
- Frigui W, Bottai D, Majlessi L, et al. Control of M. tuberculosis ESAT-6 secretion and specific T cell recognition by PhoP. PLoS Pathog. 2008;4(2):e33.
- Perez E, Samper S, Bordas Y, et al. An essential role for phoP in Mycobacterium tuberculosis virulence. Mol Microbiol. 2001;41(1):179–187.
- Domenech P, Reed MB. Rapid and spontaneous loss of phthiocerol dimycocerosate (PDIM) from Mycobacterium tuberculosis grown in vitro: implications for virulence studies. Microbiology. 2009;155(Pt 11):3532–3543.
- Aguilar D, Infante E, Martin C, et al. Immunological responses and protective immunity against tuberculosis conferred by vaccination of Balb/C mice with the attenuated Mycobacterium tuberculosis (phoP) SO2 strain. Clin Exp Immunol. 2007;147(2):330–338.
- Cardona PJ, Asensio JG, Arbues A, et al. Extended safety studies of the attenuated live tuberculosis vaccine SO2 based on phoP mutant. Vaccine. 2009;27(18):2499–2505.
- Martin C, Williams A, Hernandez-Pando R, et al. The live Mycobacterium tuberculosis phoP mutant strain is more attenuated than BCG and confers protective immunity against tuberculosis in mice and guinea pigs. Vaccine. 2006;24(17):3408–3419.
- Nambiar JK, Pinto R, Aguilo JI, et al. Protective immunity afforded by attenuated, PhoP-deficient Mycobacterium tuberculosis is associated with sustained generation of CD4+ T-cell memory. Eur J Immunol. 2012;42(2):385–392.
- Verreck FA, Vervenne RA, Kondova I, et al. MVA.85A boosting of BCG and an attenuated, phoP deficient M. tuberculosis vaccine both show protective efficacy against tuberculosis in rhesus macaques. Plos One. 2009;4(4):e5264.
- Williams A, Hall Y, Orme IM. Evaluation of new vaccines for tuberculosis in the guinea pig model. Tuberculosis (Edinb). 2009;89(6):389–397.
- Spertini F, Audran R, Chakour R, et al. Safety of human immunisation with a live-attenuated Mycobacterium tuberculosis vaccine: a randomised, double-blind, controlled phase I trial. Lancet Respir Med. 2015;3(12):953–962.
- Aguilo N, Uranga S, Marinova D, et al. MTBVAC vaccine is safe, immunogenic and confers protective efficacy against Mycobacterium tuberculosis in newborn mice. Tuberculosis (Edinb). 2016;96:71–74.
- Boritsch EC, Khanna V, Pawlik A, et al. Key experimental evidence of chromosomal DNA transfer among selected tuberculosis-causing mycobacteria. Proc Natl Acad Sci USA. 2016;113(35):9876–9881.
- McShane H, Williams A. A review of preclinical animal models utilised for TB vaccine evaluation in the context of recent human efficacy data. Tuberculosis (Edinb). 2014;94(2):105–110.
- Orme IM. The use of animal models to guide rational vaccine design. Microbes Infect. 2005;7(5–6):905–910.
- Williams A, Hatch GJ, Clark SO, et al. Evaluation of vaccines in the EU TB vaccine cluster using a guinea pig aerosol infection model of tuberculosis. Tuberculosis (Edinb). 2005;85(1–2):29–38.
- Clark S, Hall Y, Williams A. Animal models of tuberculosis: Guinea pigs. Cold Spring Harb Perspect Med. 2015;5(5):a018572.
- Aguilo N, Toledo AM, Lopez-Roman EM, et al. Pulmonary Mycobacterium bovis BCG vaccination confers dose-dependent superior protection compared to that of subcutaneous vaccination. Clin Vaccine Immunol. 2014;21(4):594–597.
- Moliva JI, Turner J, Torrelles JB. Prospects in Mycobacterium bovis Bacille Calmette et Guerin (BCG) vaccine diversity and delivery: why does BCG fail to protect against tuberculosis? Vaccine. 2015;33(39):5035–5041.
- Horwitz MA, Harth G, Dillon BJ, et al. Recombinant bacillus calmette-guerin (BCG) vaccines expressing the Mycobacterium tuberculosis 30-kDa major secretory protein induce greater protective immunity against tuberculosis than conventional BCG vaccines in a highly susceptible animal model. Proc Natl Acad Sci USA. 2000;97(25):13853–13858.
- The TuBerculosis Vaccine Initiative (TBVI) is a non-profit foundation that facilitates the discovery and development of new, safe and effective TB vaccines that are accessible and affordable for all people. Available from: www.tbvi.eu. TBVI provide independent expertis through their Product and Clinical Development Team (TBVI PDT/CDT), which has played an imporant role in MTBVAC development to date.
- Aeras is a nonprofit biotechnology organization developing new, effective TB vaccines through partnerships in both the public and private sectors, with individuals, research organizations, academic institutions, funders, policymakers and others around the world to advance TB vaccine science and development. Available from: www.aeras.org
- Bezos J, Casal C, Puentes E, et al. Evaluation of the immunogenicity and diagnostic interference caused by M. tuberculosis SO2 vaccination against tuberculosis in goats. Res Vet Sci. 2015;103:73–79.
- Carpenter C, Sidney J, Kolla R, et al. A side-by-side comparison of T cell reactivity to fifty-nine Mycobacterium tuberculosis antigens in diverse populations from five continents. Tuberculosis. 2015;95(6):713–721.
- Mothe BR, Lindestam Arlehamn CS, Dow C, et al. The TB-specific CD4(+) T cell immune repertoire in both cynomolgus and rhesus macaques largely overlap with humans. Tuberculosis. 2015;95(6):722–735.
- Lindestam Arlehamn CS, McKinney DM, Carpenter C, et al. A quantitative analysis of complexity of human pathogen-specific CD4 T cell responses in healthy M. tuberculosis infected South Africans. PLoS Pathog. 2016;12(7):e1005760.
- Millington KA, Fortune SM, Low J, et al. Rv3615c is a highly immunodominant RD1 (Region of Difference 1)-dependent secreted antigen specific for Mycobacterium tuberculosis infection. Proc Natl Acad Sci USA. 2011;108(14):5730–5735.
- Arregui S, Sanz J, Marinova D, et al. On the impact of masking and blocking hypotheses for measuring the efficacy of new tuberculosis vaccines. PeerJ. 2016;4:e1513.
- Knight GM, Griffiths UK, Sumner T, et al. Impact and cost-effectiveness of new tuberculosis vaccines in low- and middle-income countries. Proc Natl Acad Sci USA. 2014;111(43):15520–15525.
- The Collaboration for TB Vaccine Discovery (CTVD) was lounched by BMGF in July 2015, as an international network of scientists and experts dedicated to fostering innovation, cooperation, and collaboration in the up-stream TB vaccine discovery space. Available from: www.ctvd.co
- Lin PL, Rodgers M, Smith L, et al. Quantitative comparison of active and latent tuberculosis in the cynomolgus macaque model. Infect Immun. 2009;77(10):4631–4642.
- Coleman MT, Chen RY, Lee M, et al. PET/CT imaging reveals a therapeutic response to oxazolidinones in macaques and humans with tuberculosis. Sci Transl Med. 2014;6(265):265ra167.
- Sharpe SA, McShane H, Dennis MJ, et al. Establishment of an aerosol challenge model of tuberculosis in rhesus macaques and an evaluation of endpoints for vaccine testing. Clinical Vaccine Immunol. 2010;17(8):1170–1182.
- Kagina BM, Abel B, Scriba TJ, et al. Specific T cell frequency and cytokine expression profile do not correlate with protection against tuberculosis after bacillus Calmette-Guerin vaccination of newborns. Am J Respir Crit Care Med. 2010;182(8):1073–1079.
- Kagina BM, Mansoor N, Kpamegan EP, et al. Qualification of a whole blood intracellular cytokine staining assay to measure mycobacteria-specific CD4 and CD8 T cell immunity by flow cytometry. J Immunol Methods. 2015;417:22–33.
- Nemes E, Kagina BM, Smit E, et al. Differential leukocyte counting and immunophenotyping in cryopreserved ex vivo whole blood. Cytometry A. 2015;87(2):157–165.
- Soares AP, Scriba TJ, Joseph S, et al. Bacillus Calmette-Guerin vaccination of human newborns induces T cells with complex cytokine and phenotypic profiles. J Immunol. 2008;180(5):3569–3577.