
ABSTRACT
Introduction: Innate immunity is armed with interferons (IFNs) that link innate immunity to adaptive immunity to generate long-term and protective immune responses against invading pathogens and tumors. However, regulation of IFN production is crucial because chronic IFN responses can have deleterious effects on both antitumor and antimicrobial immunity in addition to provoking autoinflammatory or autoimmune conditions.
Areas covered: Here, we focus on the accumulated evidence on antimicrobial and antitumor activities of type I and II IFNs. We first summarize the intracellular and intercellular mechanisms regulating IFN production and signaling. Then, we discuss the mechanisms modulating the dual nature of IFNs for both antitumor and antimicrobial immune responses. Finally, we review the detrimental role of IFNs for induction of autoinflammation and autoimmunity.
Expert opinion: The current evidence suggests that the dual role of IFNs for antimicrobial and antitumor immunity is dependent not only on the timing, administration route, and dose of IFNs but also on the type of pathogen/tumor. Therefore, we think that combinatorial therapies involving IFN-inducing adjuvants and immune-checkpoint blockers may offer therapeutic potential, especially for cancer, whereas infectious, autoinflammatory or autoimmune diseases require fine adjustment of timing, dose, and route of the administration for candidate IFN-based vaccines or immunotherapies.
1. Introduction
Innate immune sensing of pathogens, tumors, or danger signals via the pattern recognition receptors (PRRs), such as AIM2-like receptors, C-type lectin receptors (CLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), Toll-like receptors (TLRs), or cytosolic DNA/RNA sensors signaling via the adapter molecule stimulator of interferon genes (STING), facilitates efficient priming and activation of adaptive immune cells resulting in the elicitation of robust adaptive immune responses that provide long-term protection through several mechanisms dependent on type I and II interferons (IFNs) produced by innate and adaptive immune cells [Citation1,Citation2]. In particular, type I IFNs and Th1-polarizing cytokine IL-12 released upon PRR signaling from antigen- presenting cells (APCs) acts on CD4 T cells of the adaptive immunity to drive the development of IFN-γ-producing Th1 cells capable of eliciting robust antigen-specific cytotoxic T cell (CTL) responses that can efficiently kill tumors or the infected cells [Citation3–7]. Additionally, upon stimulation by IL-12 and type I IFNs, natural killer (NK) cells of the innate immune system can also produce the tumoricidal and antiviral cytokine IFN-γ to elicit antitumor and antimicrobial immune responses [Citation6,Citation8–10]. Collectively, these responses, defined as type 1 immunity, play crucial roles in defending the host against various threats including tumors and pathogens [Citation6,Citation10]. In contrast, improper activation of the immune responses against the host factors leading to chronic IFN production can result in autoimmune or autoinflammatory conditions (e.g., type I interferonopathies, such as STING-associated vasculopathy and infantile onset (SAVI)) [Citation1,Citation11,Citation12]. Therefore, to prevent collateral tissue damage, IFNs also induce immune-regulatory pathways, such as indoleamine 2,3-dioxygenase (IDO) [Citation1,Citation11,Citation12]. Hence, vaccines or immunotherapies targeting IFN-inducing pathways must be carefully designed.
In this review, we discuss the mechanisms regulating not only the antimicrobial and antitumor effects but also the pro-tumorigenic, immunosuppressive, and inflammatory aspects of types I and II IFNs, aiming to provide further insights into the development of ideal vaccines and immunotherapies relying on IFNs.
2. Type I and II IFNs in cancer and immunity
IFNs are antiviral cytokines that were first defined in the 1950s and named ‘interfer(e)-ons’ because they could ‘interfere’ with viral replication and their production was induced by viral infection [Citation13]. Moreover, IFNs play crucial roles in the clearance of tumors by modulating the immune system, as well as directly acting on tumor cells to induce apoptosis [Citation12,Citation14,Citation15]. According to their structure, function, and source, IFNs are divided into three groups: type I (mainly IFN-α and β), type II (IFN-γ), and type III IFNs (IFN-λ) [Citation12]. Here, we focus on the well-studied types I and II IFNs and explain their role in eliminating tumors and pathogens and in inflammation-induced collateral damage control.
2.1. Type I IFN (IFN-α/β) signaling pathways
The type I IFN family of cytokines comprises IFN-α, IFN-β, and the less well characterized IFNs κ, δ, ε, ζ, τ, and ω. Furthermore, IFN-α has 13 subtypes (IFN-α1, 2, 4, 5, 6, 7, 8, 10, 13, 14, 16, 17, and 21) with similar structures, among which IFN-α2 is the one that has been widely studied and used in preclinical and clinical studies [Citation16,Citation17]. In this review, the term type I IFN applies to IFN-α/β as these are the widely studied types.
Type I IFNs are secreted by various types of cells, including innate immune cells, adaptive immune cells, and nonimmune cells (e.g., fibroblasts and endothelial cells) through activation of the innate immune signaling pathways, such as TLR, CLR, and NLR, by DAMPs and PAMPs derived not only from pathogens and tumors but also from self [Citation12,Citation16]. In particular, nucleic acids in the cytosol can be sensed by STING-dependent (e.g., cGAS) or independent (e.g., RIG-I and AIM2) cytosolic nucleic acid-sensing pathways whereas endosomal TLRs, TLR3, TLR7/8, and TLR9 detect nucleic acids within the endosomes to induce type I IFN production () [Citation1,Citation2]. Conversely, extracellular pathogen-derived factors such as LPS and flagellin can be detected by TLR1/2, TLR2/6, TLR4 (both endosomal and cell surface-bound), and TLR5 positioned on the cell surface to trigger type I IFN production [Citation1,Citation2]. Activation of PRRs, including TLRs or cytosolic nucleic acid sensors, is generally followed by recruitment of various adaptor molecules (e.g., MyD88 and STING) and kinases (e.g., TBK1), leading to phosphorylation, dimerization, and subsequent nuclear translocation of interferon regulatory factors (IRFs), such as IRF3 and IRF7. These factors can further associate with the co-activator CREB-binding protein (CBP) and directly bind to IFN-stimulated response elements (ISREs) of type I IFN promoters, ultimately causing type I IFN production () [Citation1,Citation2].
Figure 1. Nucleic acid sensing-mediated IFN induction and IFN receptor signaling pathways. Pathogenic ssDNA containing unmethylated CpG motifs and ssRNA are detected by endosomal TLR9 and TLR7, respectively, resulting in MyD88-mediated induction of IRF7- and NF-κB-dependent IFNs. In contrast, dsRNA can be detected both by endosomal TLR3 that signals via the adaptor molecule TRIF and by the cytosolic DNA sensors RIG-I and MDA5. While both RNA sensors stimulate the TBK1-IRF3 pathway, cytosolic RNA sensors utilize IPS-1 and TRAF3 adaptor molecules to induce TBK1-dependent IRF3-IRF7 heterodimerization, phosphorylation, nuclear translocation, and subsequent IFN induction. Nevertheless, cytosolic dsDNA can be sensed by various cytosolic nucleic acid sensing pathways, including the AIM2-dependent inflammasome pathway capable of inducing IL-1β production and the STING-dependent nucleic acid sensing pathways (e.g., cGAS, DDX41, and IFI16), which stimulates TBK1-IRF3-dependent IFN production. Once released, type I IFNs bind to their receptors composed of IFNAR1 and IFNAR2 subunits on adjacent cells to activate Jak1 and Tyk2 kinases that aids in phosphorylation and nuclear translocation of STAT1 and STAT2, which binds to IRF9 to form ISGF3 complex culminating in the expression of various ISGs with antitumor and antimicrobial functions via direct binding to ISRE regions of the ISG promoters. Besides, IFNAR signaling also activates STAT3-dependent expression of genes, such as SOCS3, which is a negative regulator of IFN receptor signaling via inhibition of Jak activity. Moreover, secreted IFN-γ binds to the IFNGR composed of IFNGR1 (binding chain) and IFNGR2 (signal-transducing chain) subunits, which signal via Jak1 and Jak2 tyrosine kinases, to induce STAT1 homodimerization, phosphorylation, and nuclear translocation. STAT1 homodimers bind to the GAS regions of the ISG promoters and induce the expression of IFN-γ-regulated genes, including the negative regulatory molecules, such as SOCS1 and SOCS3
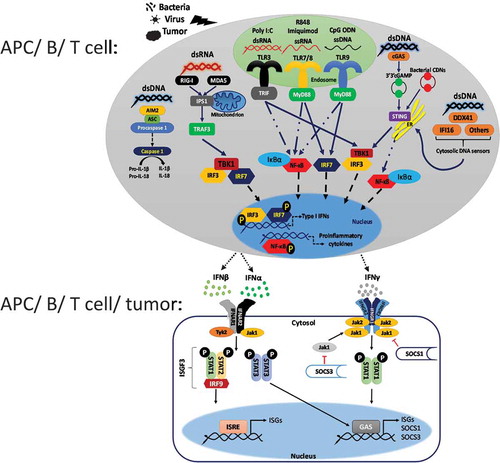
Secreted type I IFNs bind to type I IFN receptor IFNAR, which is composed of IFNAR1 and IFNAR2 subunits that associate with kinases Tyk2 and Jak1, respectively [Citation12]. Binding of type I IFNs to IFNAR triggers clathrin-mediated endocytosis and leads to the activation of Jak1 and Tyk2 kinases, thereby causing phosphorylation and nuclear translocation of signal transducer and activator of transcription 1 (STAT1) and STAT2, which then binds to IRF9 to form the IFN-stimulated gene factor 3 (ISGF3) complex capable of driving the expression of various IFN-stimulated genes (ISGs) with antitumor and antimicrobial functions via direct binding to ISRE regions of the ISG promoters () [Citation12,Citation18]. Moreover, type I IFN receptor signaling provides a positive feedback loop for inducing further type I IFNs via the induction of various ISGs, such as STING [Citation19], in addition to causing the activation of the STAT3, STAT4, STAT5, STAT6, PI3 kinase, and Map kinase pathways [Citation12,Citation20–22]. While type I IFN-induced responses are negatively regulated by STAT3, they are enhanced by STAT5 and STAT6 [Citation22–24]. In contrast, STAT4 induced by type I IFNs is necessary for IFN-γ production in response to viral infection [Citation25].
The mechanisms of action of IFN-α and IFN-β differ from each other owing to several reasons, including the differences in their binding affinity to IFNAR [Citation12,Citation22]. The IFNAR binding affinity of IFN-β is much stronger (50-fold for IFNAR1 and 1000-fold for IFNAR2) than that of IFN-α [Citation22,Citation26]. Indeed, IFNAR1-IFNAR2 complex formation can be achieved by IFN-β but not by IFN-α stimulation [Citation27]. Furthermore, Weerd et al. reported that IFN-β is capable of binding to IFNAR1 even in the absence of the IFNAR2 subunit to activate a distinct set of genes than the ones that are activated by the Jak-STAT pathway [Citation28]. Owing to these differential signaling pathways activated by the IFNAR1 and IFNAR1/IFNAR2 complex, Ifnar1-/- and Ifnar2-/- mice had different responses to inflammatory stimuli; LPS-mediated endotoxin shock was prevented in Ifnar1-/- but not in Ifnar2-/- mice [Citation28]. Moreover, IFNAR is ubiquitously expressed in various cell types including lymphocytes, myeloid cells, stromal cells, and tumors [Citation29]. Although IFNAR expression in immune or stromal cells generally contributes to antimicrobial and antitumor immune responses, the outcome of the IFNAR signaling may vary based on the cell type, level, and duration of IFNAR signaling as comprehensively explained in the current review [Citation30–34].
2.2. Type II IFN (IFNγ) signaling pathways
IFN-γ is mainly produced by NK cells or innate lymphoid cells (ILCs) associated with the innate immunity (e.g., by IL-12, IL-15, or type I IFN stimulation) and later by Th1 type CD4 T cells and CTLs associated with the adaptive immunity [Citation16,Citation35]. In NK cells, IFN-γ that was secreted in a STAT4-dependent manner binds to its heterodimeric receptor composed of IFNGR1 (binding chain) and IFNGR2 (signal-transducing chain) subunits, which also signal via Jak1 and Jak2 tyrosine kinases, culminating in STAT1 homodimerization, phosphorylation, nuclear translocation, binding of STAT1 homodimers to gamma-activated sites (GAS) of ISG promoters, and subsequent induction of IFNγ-regulated genes, as well as some alternative signal transduction pathways such as the Map kinase pathways () [Citation12,Citation36]. Nonetheless, autocrine IFN-γ signaling within the CTLs promotes their cytotoxicity and motility, which are critical for the clearance of infections as well as tumors [Citation37].
Although IFNGR is expressed in various kinds of cells, the level of IFNGR expression may determine the fate of certain immune cells. For instance, certain Th1 cells with a low IFNGR2 expression level can still survive and proliferate in the presence of IFN-γ whereas Th2 cells with a high IFNGR2 expression level undergo apoptosis due to rapid induction of the apoptosis-inducing STAT1-IRF1 pathway [Citation38–40]. Therefore, IFN-γ can also support Th1 type response development via indirectly killing Th2 cells with a high IFNGR2 activity [Citation40]. This phenomenon has also been observed in B and myeloid cells with a high IFNGR2 expression level [Citation40].
Transcriptional regulation of IFN-γ production within Th1 cells is achieved by the interactions between transcriptional repressor Twist1 and the transcriptional factors STAT4, T-bet, and Runx3 that drive IFN-γ induction. Mechanistically, Twist1 suppresses IFN-γ production through negative regulation of these transcription factors [Citation41]. In contrast, suppressor of cytokine signaling proteins (SOCS), SOCS1, and SOCS3, and Src-homology-2 (SH2)-domain-containing tyrosine kinases, such as SHP1 and SHP2, can suppress both the type I and type II IFN receptor signaling pathways [Citation42,Citation43]. In particular, direct binding of SOCS to JAK via its SH2 domain can suppress the kinase activity of JAK or aid in proteasomal degradation of the IFNAR-protein complexes whereas SHP achieves this suppression via STAT1 de-phosphorylation [Citation42,Citation43]. In addition, micro RNAs miR-155 and miR-146a achieve post-transcriptional regulation of IFN-γ production from T cells [Citation44]. While miR-146a suppresses IFN-γ production from T cells diminishing antitumor T cell responses, miR-155 promotes IFN-γ production through repression of SH2 domain-containing inositol phosphatases-1 (Ship-1) and elicits antitumor immunity [Citation44,Citation45].
Tumor suppressor genes are also capable of regulating IFN-γ signaling via several mechanisms [Citation46,Citation47]. For example, the post-transcriptional regulation of IFN-γ signaling by the tumor suppressor p14ARF (ARF) is executed via mechanisms involving SUMOylation of protein inhibitor of activated STAT1 (PIAS1), resulting in inactivation of PIAS1 and subsequent rescue of PIAS1-mediated suppression of the IFN-γ-mediated signaling pathways [Citation46]. Moreover, another tumor suppressor gene, LNK (SH2B3), also suppresses IFN-γ-induced JAK-STAT1 pathway activation [Citation47]. Indeed, Ding et al. showed that tumors with enhanced RAS-RAF-MEK signaling have a high LNK expression level, which restrains the apoptotic effect of IFN-γ on tumor cells via suppressing IFN-γ-induced JAK-STAT1 pathway activation [Citation47].
2.3. IFN-mediated antitumor immunity
2.3.1. Type I IFN
Type I IFNs aid in boosting immune clearance of the tumors via several mechanisms as described below (summarized in ):
Table 1. Yin and yang of type I IFNs for anti-tumor immunity
2.3.1.1. By directly acting on tumor cells (antiproliferative and proapoptotic action).
Both IFN-α and IFN-β can directly induce apoptosis in several cancer cell lines, such as human breast cancer and neuroblastoma, via upregulation of tumor suppressor genes (e.g., p53) or proapoptotic genes, such as CD95 [Citation48–50]. IFN-α activates the STAT1-p53-CD95 pathway to induce apoptosis in human breast cancer and neuroblastoma cells [Citation48], whereas IFN-β induces STAT1-IRF1-p38 MAPK-caspase-7-dependent apoptotic cell death in Ewing’s sarcoma cells, independent of p53 expression and better than IFN-α [Citation51]. Similarly, by using peripheral blood mononuclear cells (PBMCs) from adult T cell leukemia/lymphoma patients, Dierckx et al. demonstrated that IFN-β is better than IFN-α at inducing the p53-dependent proapoptotic and antiproliferative pathways, confirming the potency of IFN-β over IFN-α as an anticancer agent [Citation52]. Furthermore, the antitumor effect of IFN-β is also observed in human hepatocellular carcinoma (HCC) cells both in vitro and in vivo [Citation53]. Preclinical studies in mice using HCC xenograft models suggested that IFN-β can directly suppress tumor cell proliferation and angiogenesis in addition to boosting the antitumor effect of sorafenib therapy. Sorafenib is the currently available HCC treatment option capable of suppressing tumor proliferation and angiogenesis via inhibiting the activity of multiple kinases, including vascular endothelial growth factor receptor and serine-threonine kinases such as Raf-1 [Citation53]. In contrast, the IFN-β-induced signaling pathways within triple-negative breast cancer (TNBC) cells are capable of inducing potent antitumor immunity via the mechanisms acting directly on tumor cells to suppress cancer stem cell (CSC) properties of the TNBC cells, leading to reduced migration and tumor sphere-forming ability of tumor cells [Citation54]. Indeed, ISGs are repressed in TNBC cells with CSC-like properties, which could be reversed by IFN-β treatment, revealing IFN-β as a promising therapeutic that can potentially prevent metastasis or recurrence of TNBC by directly acting on tumor cells in addition to acting on immune cells in the case of in vivo conditions [Citation54]. On the contrary, the IL-6 family cytokine oncostatin-M can oppose the antitumor effect of IFN-β in TNBC by both directly repressing IFN-β expression at the mRNA levels and inducing CSC phenotype of TNBC cells via upregulating tumor-promoting factors SNAIL and CD44, which can be reversed by IFN-β treatment [Citation55]. In correlation with this data, patients with TNBC having enhanced IFN-β signature exhibit better prognosis, owing to the suppressed CSC phenotype in their tumors, compared to the patients with TNBCs lacking IFN-β signature [Citation55]. Another mechanism by which both types I and II IFNs exert antitumor effect is through suppression of matrix metalloproteinase-9 (MMP-9), which is capable of enhancing invasiveness and angiogenesis of tumors via activation of the IFN-induced STAT1 signaling pathway within astroglioma and fibrosarcoma cells [Citation56].
2.3.1.2. By acting on stromal cells in the tumor microenvironment (TME; anti-angiogenic action):
High-dose intratumor IFN-β is capable of suppressing tumor growth via directly acting on tumor vasculature to suppress angiogenesis [Citation30,Citation57]. Using explanted tumor model with IFN-β-expressing B16 F10 melanoma cells and vascular endothelial cell-specific IFNAR KO mice, Spaapen et al. demonstrated that the IFN-β receptor signaling in the tumor endothelial cells is necessary for IFN-β-mediated rejection of explanted tumors via directly suppressing angiogenesis rather than activating immune cells [Citation57]. In response to this anti-angiogenic effect of type I IFNs, vascular endothelial cells have evolved a defense mechanism, by which angiogenesis-inducing VEGF signaling activates protein kinase D2 (PDK2) recruitment to IFNAR1, leading to the phosphorylation and subsequent degradation of IFNAR1 to inhibit type I IFN signaling and promote angiogenesis, suggesting that PDK2 inhibitors are promising anticancer drugs for shutting down this defense mechanism and inducing type I IFN-dependent suppression of angiogenesis within the tumors [Citation30].
Type I IFNs can directly affect T cells or APCs to promote antigen-specific T cell responses through mechanisms such as enhancement of co-stimulation, cross-priming, or antigen presentation [Citation33,Citation58–61]. For example, Sikora et al. reported that IFN-α exerts potent adjuvant activity to maximize the antitumor effect of melanoma peptide-based cancer vaccine by affecting DCs and CD8 T cells to promote the magnitude and longevity of tumor-specific CD8 T cell responses via the mechanisms depending on IFNAR signaling on T cells and IL-15 receptor signaling on APCs [Citation58]. Further, Zhang et al. have developed a recombinant IFN-α (named super-compound IFN-I (sIFN-I)) with improved IFNAR affinity and pharmacokinetics using a de novo engineering approach based on the structure of clinically available IFN-α-2b and showed that intratumor sIFN-I injection exerts robust antitumor effect through the inhibition of tumor angiogenesis and recruitment of CTLs to the tumors in both immune-competent and immune-deficient mice [Citation62]. Despite the robust antitumor effect of type I IFNs, their therapeutic use is limited owing to toxic side effects, including anemia, leukopenia, nausea, hepatotoxicity, and even depression [Citation31,Citation63]. To avoid these toxicities, Cauwels et al. have developed a novel approach where they combined Clec9A-targeting antibodies with genetically engineered IFN-α (AcTaferon), which has up to 1000-fold more potency than wild type IFN-α2 does, to achieve robust activation of Clec9A-expressing conventional DCs by type I IFNs [Citation31]. Importantly, Clec9A+ DC-targeting IFN-α therapy has reportedly elicited a robust antitumor effect in murine lymphoma, melanoma, and breast carcinoma models without significant toxicity via the mechanisms dependent on CTLs and IFNAR signaling on DCs [Citation31].
Similar to the antitumor effect of IFN-α, IFN-β can boost antitumor immunity via immune cell activation or polarization into an antitumor phenotype [Citation33,Citation59]. Tumor-associated neutrophils (TANs) can be polarized into an antitumor N1 phenotype with upregulated TNF-α expression and elevated neutrophil extracellular trap (NET)-forming ability, consequently promoting tumor cytotoxicity via the direct effect of IFN-β on TANs that are observed both in mice and human melanoma tumors [Citation64]. Particularly, IFNAR signaling in breast cancer TME is one of the key determinants of immune clearance over immune-dormancy [Citation32,Citation55]. In estrogen receptor-negative breast cancer cells, chemotherapy-induced IFN-β activates IFNAR signaling within the TME and induces type I IFN-dependent expansion of both CD4 and CD8 T cells, in addition to suppressing MDSCs to overcome immunosuppression and boost antitumor immunity [Citation32]. Furthermore, epigenetic therapy using inhibitors of histone deacetylase and DNA methyl transferase promotes immune clearance of the epithelial ovarian cancer tumors via mechanisms regulated by type I IFN-induced CTL and NK cell recruitment and activation in the TME [Citation34].
Effect of STING-mediated type I IFN production
Cumulative evidence has suggested that type I IFN production in response to tumor DNA sensing via the cGAS/STING pathway in tumor-associated DCs is crucial for the generation of not only spontaneous but also radiation-induced antitumor immune responses in vivo [Citation65–69]. Specifically, tumor-derived DNA-mediated induction of cGAS-STING-type I IFN axis in DCs aids in CTL priming against tumors via optimally activated DCs to promote immune clearance of the tumors by antigen-specific CTLs [Citation66,Citation67,Citation69]. In addition, STING agonist treatment synergizes with radiation for the induction of antitumor immune responses in vivo [Citation69]. Nevertheless, the antitumor effect of the immunogenic cell death-inducing drug topoisomerase II inhibitor has recently been reported to depend on cGAS-STING-mediated induction of type I IFNs and high-mobility group box 1 from tumor cells activating antitumor CTL responses in vivo [Citation70]. Notably, innate immune detection of cytosolic chromatin fragments from senescent cells by the cGAS-STING pathway can promote cellular senescence, which is one of the important anticancer pathways that can control the proliferation of damaged cells either by arresting the cell cycle or by facilitating their immune clearance through secretion of cytokines and chemokines, and contribute to antitumor immune responses via the mechanisms dependent on STING-induced cytokines including type I IFNs [Citation65]. Furthermore, Liu et al. have analyzed ISG signatures from primary tumors in the Cancer Genome Atlas and found elevated ISG signatures even in tumors without immune cell infiltration, indicating that tumor cells or the stroma itself could be the source of type I IFNs [Citation71]. Further analysis revealed that tumor cells with continuous STING-mediated type I IFN production have elevated RNA sensors such as MDA5 and RIG-I and therefore accumulate double-stranded RNA, rendering the cells susceptible to death mediated by depletion of the RNA editing enzyme ADAR, which is a pro-tumorigenic factor overexpressed in several types of tumors [Citation71,Citation72]. Conversely, preclinical studies of Schadt et al. using murine adenocarcinoma tumor model have demonstrated that cGAS-mediated cyclic guanosine monophosphate–adenosine monophosphate (cGAMP) production occurs within the tumor cells and that it is transferred to tumor-associated DCs, which produce type I IFNs in a STING-dependent manner to prime robust antitumor CTL responses [Citation68]. They also found a correlation between cGAS expression and CTL infiltration in tumors from patients with colorectal adenocarcinoma [Citation68]. Therefore, depending on the tumor type, it is possible that STING signaling within the tumors and/or tumor-associated DCs promotes the antitumor effect of type I IFNs.
Considering the potential of the STING-type I IFN pathway for cancer immunotherapy, several studies have currently been conducted using various STING agonists, such as cyclic dinucleotide derivatives or small chemicals, either as immunotherapeutic agents or cancer vaccine adjuvants [Citation73–84]. Especially, intratumor STING agonist injection has shown robust antitumor effects in mouse models of melanoma, glioma, pancreatic cancer, and lymphoma via the mechanisms regulated by STING-induced type I IFN-dependent priming of antitumor immune responses [Citation75,Citation78,Citation81,Citation82]. Although systemic STING agonist injection has managed to boost the antitumor immunity of murine tumor models of melanoma, breast cancer, ovarian cancer, and colon cancer [Citation73–75,Citation80,Citation83,Citation84], data from the study by Shae et al. indicate that the systemic administration of STING-targeting nanoparticles causes mild toxicity characterized by temporary weight loss without causing liver damage [Citation75]. Moreover, a combination of the STING agonist immunotherapy with an immune checkpoint blockade therapy such as PD-1, CTLA-4 blockade [Citation74–76], radiotherapy [Citation82], chemotherapy [Citation74], or other types I IFN-inducing agents such as TLR9 agonists [Citation78,Citation79], significantly improves or even synergizes with the antitumor effect of the STING agonist-based immunotherapies, suggesting that the STING-based type I IFN-inducing combination therapies offers therapeutic potential for cancer.
2.3.2. Type II IFN
The critical role of IFN-γ in antitumor immunity has been reported extensively in preclinical and clinical studies [Citation50]. Here, we review the antitumor effect of IFN-γ by dividing it into three main mechanisms, which usually occur simultaneously in vivo (summarized in ):
Table 2. Yin and yang of type II IFNs for anti-tumor immunity
2.3.2.1. By directly acting on tumor cells (antiproliferative and proapoptotic action)
IFN-γ treatment can cause cell cycle arrest and induce apoptosis of human pancreatic cancer cells via triggering IRF1- and caspase-1-dependent apoptosis in vitro [Citation85]. In addition, IFN-γ activates the STAT1-IRF1-dependent or STAT1-independent CD95 upregulation pathways for promoting apoptosis in human breast cancer and neuroblastoma cells [Citation48]. Similarly, Wang et al. have reported that IFN-γ can activate cytosolic phospholipase A2 (cPLA2), which induces mitochondrial reactive oxygen species (ROS) generation and subsequent autophagy-associated apoptosis in colorectal cancer cells in vitro [Citation15]. Furthermore, IFN-γ-induced cancer cell apoptosis has also been observed by El Jamal et al. in head and neck squamous cell carcinoma cells. Notably, they also demonstrated that IFN-γ induces IDO activation that suppresses the expression of antioxidant molecule heme oxygenase-1(HO-1) and thereby causes the accumulation of ROS and the activation of proapoptotic Noxa protein promoters, ultimately leading to apoptosis mediated by ER stress as well as mitochondrial dysregulation [Citation14]. Moreover, IFN-γ suppresses the metastatic capacity of several cancer cells, such as osteosarcoma, colon carcinoma, and breast cancer cells, by indirectly repressing the transcription of the metastasis-inducing protein S100A4 via induction of class II transactivator, which can sequester the transcriptional coactivator CBP/p300 away from the S100A4 promoters to suppress promoter activity of S100A4 [Citation86,Citation87].
2.3.2.2. By acting on stromal cells in the TME (anti-angiogenic action)
Using a genetically modified fibrosarcoma cell line, in which IFN-γ production can be induced by doxycycline treatment, and a conditional IFN-γ-GFP transgenic mouse model, Kammertoens et al. demonstrated that the antitumor effect of IFN-γ is dependent on the direct effect of IFN-γ on tumor endothelial cells but not on immune cells. Specifically, IFN-γ produced by the tumors causes regression of tumor blood vessels, leading to decreased blood flow to tumors and ischemia-associated tumor remission unlike the necrotic collapse of the tumors induced by TNF-α [Citation88].
2.3.2.3. By acting on immune cells such as CTLs for effective priming, activation. and tumor clearance
One of the mechanisms by which IFN-γ exerts antitumor effect is by directing M1 macrophage polarization rather than M2 polarization [Citation50,Citation89]. Indeed, Redente et al. showed that urethane-induced lung tumors grow larger in IFN-γR KO mice than in IL-4 RαKO mice owing to polarization of the tumor-associated macrophages (TAMs) into the M2 phenotype in IFN-γR KO mice rather than the M1 phenotype, which was observed in mice deficient for IL-4R signaling [Citation89]. In addition, the antitumor activity of the macrophages is synergistically promoted by IFN-γ and the calcineurin B subunit of the serine/threonine phosphatase calcineurin via the mechanisms regulated by the co-ordinate action of the IFN-γ-triggered p38 MAPK/PKC-δ-Jak2-STAT1 and the calcineurin B-integrin αM-p38-STAT1 signaling pathways that stimulate IRF1- and IRF9-induced antitumor cytokine production from the M1-polarized macrophages [Citation90]. Nonetheless, IFN-γ can also act on CTLs to facilitate tumor clearance [Citation37,Citation91]. For instance, Gerber et al. demonstrated that radiation-induced suppression of implanted colon tumors is mediated by IFN-γ mainly produced by CTLs, and that it acts not on tumor cells but on CTLs within the TME as the diminished antitumor effect of radiation was observed in IFN-γ KO mice or CD8 T cell-depleted mice [Citation91].
To escape from elimination by the immune response, tumors have developed several immune escape mechanisms, such as maintenance of an immunosuppressive TME through suppressive cytokines (e.g., IL-10) and upregulation of suppressive molecules such as PD-L1 and IDO against antitumor cytokines such as IFN-γ [Citation92]. Indeed, preclinical studies have indicated that despite the expression of considerable amounts of Ifng mRNA within the TILs of melanoma-bearing mice, posttranscriptional mechanisms suppress IFN-γ production at the protein levels via destabilization of Ifng mRNA, thereby impeding TIL-dependent antitumor immune responses, which could be reversed by CD28 co-stimulation that promotes mRNA stabilization and subsequent IFN-γ-dependent tumor clearance [Citation93]. Another mechanism by which tumors maintain a suppressive microenvironment is through the secretion of galectin-3, which can bind glycosylated proteins including IFN-γ within the TME, into the tumor matrix [Citation94]. As galectin-3 captures IFN-γ and prevents IFN-γ-mediated chemokine (CXCL9/10) gradient formation to attract T cells to the tumors, antitumor immunity is impaired due to impaired T cell infiltration into the tumors [Citation94]. Nonetheless, many reports have suggested that immune checkpoint blockade therapy-promoted antitumor immunity requires induction of IFN-γ-dependent mechanisms [Citation95–98]. When Garris et al. investigated the mode of action of anti-PD-1-mediated antitumor immunity using a combinatorial approach involving single-cell RNA sequencing and intravital real-time imaging, they discovered that PD-1 blockade first activates CD8 T cells for IFN-γ production, which then activates DCs for noncanonical NF-Κb-dependent IL-12 production; this, in turn, licenses T cells for tumor clearance via the paracrine IL-12 receptor signaling [Citation98]. Furthermore, Tian et al. invented a novel therapeutic cancer vaccine that can produce granulocyte-macrophage colony-stimulating factor (GM-CSF) and anti-PD-1 antibody by using B16-F10 melanoma and CT26 colon cancer cells and showed that the potent antitumor effect of the cancer vaccine in tumor-bearing mice is dependent on IFN-γ-producing Th1 CD4 cells induced via recruitment of activated DC and macrophages, as well as enhancement of effector T cell to regulatory T cell ratio (Teff/Treg) within the TME rather than that of innate NK cell responses [Citation96]. Besides, Overacre-Delgoffe et al. introduced a new concept of Treg fragility, which is regulated by Treg intrinsic neuropilin-1 (Nrp1) and hypoxia-inducible factor 1 alpha (Hif1α) expression, required for the optimal antitumor effect of anti-PD-1 immune checkpoint blockade therapy [Citation99]. According to their findings, owing to the hypoxic TME and consequently elevated Hif1α levels, Tregs lacking Nrp1 within the tumors produce IFN-γ, which in turn converts the surrounding Tregs into fragile Tregs and culminates in antitumor immunity-boosting effects [Citation99]. Consistent with these preclinical studies, biopsy analysis of tissue samples from patients with melanoma, non-small cell lung cancer (NSCLC), and urothelial cancer treated with immune checkpoint inhibitors (anti-PD-1/PD-L1) suggested that IFN-γ expression in the tumor is a good predictive marker for responsiveness to treatment and found that patients with high intratumor IFN-γ levels benefit more from immune checkpoint blockade therapy than those with low intratumor IFN-γ levels [Citation95,Citation97].
Myeloid-derived suppressor cells (MDSC) are one of the factors possessing a suppressive TME to achieve immune escape [Citation100]. However, IFN-γ can eliminate MDSC-mediated immune escape of the tumors by directly activating the IFNγ-R signaling pathway in MDSCs, leading to elevated levels of phosphorylated STAT1 capable of binding to antiapoptotic Bcl2a1 promoters and resulting in decreased survival and suppressor functions of MDSCs [Citation101].
2.4. IFN-mediated antimicrobial immunity
2.4.1. Type I IFNs
Type I IFNs generate antimicrobial immunity via various ways (summarized in ). First, they directly act on pathogens to suppress viral replication [Citation13] or kill bacteria, such as Staphylococcus aureus [Citation102]. The antibacterial effect is due to the cationic and amphipathic properties of a part of IFN-β (called helix 4) that can directly bind to the bacterial cell wall and disrupt its integrity, a mechanism commonly used also by antimicrobial peptides [Citation102]. The second and the most common mode of action of type I IFNs for induction of antimicrobial state is through their direct effect on innate immune cells, such as induction of antimicrobial ISGs in innate immune and stromal cells, recruitment of inflammatory cells to the site of infection, and enhancement of antigen presentation to adaptive immune cells for generating robust pathogen-specific cellular and humoral immune responses [Citation103,Citation104]. Indeed, various reports have indicated that mice deficient in IFNAR signaling are unable to efficiently clear viruses, such as herpes simplex virus (HSV) [Citation105–108]. Three independent studies demonstrated that HSV-induced type I IFN production is required to elicit protective antiviral responses through the mechanisms dependent on type I IFN-induced CCL2-mediated recruitment of inflammatory monocytes [Citation104,Citation107,Citation108]. While Iijima et al. showed the importance of antiviral Th1 responses, Uyangaa et al. further demonstrated that CCL2 produced at earlier time points by various cells at the infected sites, including monocytes and DCs, together with type I IFN-induced CCL3 produced by infected epithelial cells later cooperatively recruit NK cells that play key roles in anti-HSV immunity [Citation107]. In addition, upon type I IFN stimulation, inflammatory monocytes recruited to the site of HSV infection produce IL-18, which in turn activates NK cells for IFN-γ production via IL-18 receptor signaling on NK cells [Citation108]. In contrast, the direct effect of type I IFNs on NK cells to enhance their effector function and activation is required for NK cell-mediated clearance of vaccinia virus infection [Citation109]. Besides, the study by Li et al. indicated that the antiviral effect of type I IFNs, particularly IFN-α, for hepatitis B virus and adenovirus infection is mediated by the cell-to-cell transfer of IFN-α-stimulated exosomes equipped with antiviral molecules, suggesting that exosomal delivery of antiviral molecules is a better treatment option for viral infections than systemic type I IFN treatment having toxic side effects [Citation110].
Table 3. Dual role of IFNs for anti-microbial immunity against various pathogens
Influenza virus is an RNA virus that can be sensed by innate RNA sensors, such as RIG-I, capable of inducing IRF3-dependent type I IFN production, which is important for immune clearance of the viral infection [Citation1,Citation111,Citation112]. Nonetheless, comprehensive analysis of two non-adjuvanted influenza vaccines from the clinic revealed that the trivalent vaccine capable of activating monocyte-derived DCs (moDCs), cDCs, and pDCs for type I IFN production has greater efficacy than the monovalent vaccine that can activate pDCs but not moDCs, or cDCs for type I IFN production [Citation111]. Indeed, comparison of transcriptional profiles of the vaccinated individuals also showed that the trivalent vaccine induced a temporary type I IFN signature 24–48 h upon immunization, which was not observed in the monovalent vaccination [Citation111]. More recently, Pernet et al. demonstrated the regulation of type I IFN-mediated antiviral immune responses by the lipid mediator leukotriene B4 (LTB4)-BLT1 receptor signaling in murine influenza infection model [Citation112]. Mechanistically, influenza-induced LTB4 activated BLT1 receptor signaling to potentiate IFNAR signaling in interstitial macrophages for elevated IFN-α production and suppressed the detrimental inflammatory monocyte-derived macrophage accumulation in the lungs thereby eliciting protective antiviral immunity without collateral lung damage [Citation112]. Similarly, cholesterol metabolism pathways, specifically the 7-dehydrocholesterol reductase (DHCR7) enzyme that catalyzes the conversion of 7-dehydrocholesterol (7-DHC) to cholesterol can also regulate type I IFN-mediated antiviral immunity [Citation113]. Xiao et al. demonstrated that macrophage expression of DHCR7 is downregulated by RNA/DNA virus infections, which leads to accumulation of 7-DHC, which in turn can activate the PI3K-AKT3 pathway and induce phosphorylation of IRF3, resulting in the promotion of IRF3-dependent type I IFN production, as well as boosting antiviral immunity [Citation113]. Importantly, in vivo DHCR7 inhibitor treatment protected from viruses including H1N1 and vesicular stomatitis virus (VSV), suggesting the therapeutic potential of cholesterol metabolism targeting for the treatment of viral infections [Citation113].
Conversely, distinct mechanisms regulate antiviral responses against enteric viruses in the mucosal compartments. Owing to the differential expression of IFNAR among the subset cells of the gut, type I IFNs produced by leukocytes and type III IFNs (IFN-λ) produced by intestinal epithelial cells can cooperatively induce antiviral responses against enteric viruses [Citation114]. Furthermore, by using a murine norovirus infection model, Neil et al. demonstrated that virus-induced type I IFN production in the colon leads to recruitment of both CCR2-dependent macrophages, which help to control infection in the gut, and IL-22-producing ILCs, which protect intestinal tissues from collateral damage induced by chemicals or chronic type I IFN signaling via IL-22-STAT3-dependent pathways [Citation115]. Moreover, intranasal IFN-α treatment in neonatal mice prior to Respiratory syncytial virus (RSV) injection could promote the expression of B cell activation factor (BAFF) and subsequent IgA production from B cells in the nasal mucosa that are crucial for RSV clearance from the mucosal sites [Citation116]. In addition, two different groups also showed that plasmacytoid DC (pDC)-derived type I IFNs that are induced via the TLR7-MyD88 pathway play key roles in controlling RSV infection in mice [Citation117,Citation118]. Mechanistically, Kim et al. found that pDCs activated via the TLR7 pathway produce type I IFNs and prime robust antiviral CTL responses [Citation118].
In murine models of peritonitis and pneumonia, IFN-β contributes to the resolution of bacterial inflammation by inducing the apoptosis of inflammatory neutrophils, as well as promoting the removal of dying cells by macrophages, also known as efferocytosis, through IFNAR-STAT3 signaling-mediated re-programming of macrophages into a pro-resolving phenotype [Citation119]. Conversely, clearance of intracellular bacterial infections also relies on type I IFNs. Specifically, apoptosis of macrophages infected with Listeria monocytogenes requires type I IFN-mediated boosting of macrophage death for efficient clearance of Listeria infection [Citation120]. In addition to regulating macrophage functions, L. monocytogenes-induced type I IFNs can also induce IL-15 production from DCs and cause recruitment of NK cells to lymph nodes, where IL-15-dependent priming and activation of NK cells occur to boost the robust antibacterial immunity [Citation121].
Effect of STING-mediated type I IFN production
Cumulative evidence suggests that the detection of viral/bacterial nucleic acids via the STING-dependent cytosolic nucleic acid sensing pathways (e.g., cGAS-STING pathway) to induce robust type I IFN production is one of the key mechanisms of host defense against several pathogens, including human immunodeficiency virus (HIV), cytomegalovirus (CMV), HSV, Legionella pneumophila, L. monocytogenes, and Mycobacterium tuberculosis (Mtb) [Citation122–127]. Indeed, cGAS-mediated sensing of cDNA from HIV-2, which is capable of replicating both in T cells and DCs unlike more pathogenic HIV-1 that can replicate in T cells only, within the DCs can activate the STING pathway to induce a strong type I IFN response allowing immune clearance of the infection [Citation128]. Nevertheless, in the murine CMV (MCMV) infection model, Tegtmeyer et al. demonstrated that STING is necessary for the induction of early IFN-β responses in the Kupffer cells of the liver and restrict the dissemination of the virus by myeloid cells although STING deficiency did not affect the survival of the MCMV-infected mice owing to the compensation of other TLR and RLR signaling pathways in innate immune cells other than Kupffer cells at later time points to control MCMV infection in a type I IFN-dependent manner [Citation122]. In addition to this study, Baranek et al. found that MCMV-induced type I IFN production exerts antiviral effects via the mechanisms that are differentially regulated in NK cells and DCs [Citation129]. While the IFNAR-ISG pathway is robustly activated in DCs, NK cells are indirectly activated by IL-15 provided by type I IFN-stimulated DCs, owing to STAT1 downregulation; consequently, IFNAR signaling is reduced in NK cells [Citation129]. In contrast, innate immune sensing of L. monocytogenes and Mtb is achieved by binding of bacterial cyclic dinucleotide, cyclic dimeric adenosine monophosphate (c-di-AMP), to STING activating type I IFN-dependent antibacterial immune responses [Citation123,Citation125]. Current evidence also suggests that cGAS-mediated antiviral immunity is negatively regulated via inflammasome-triggered cleavage of cGAS by caspases. Indeed, mice deficient for inflammasome activation showed elevated type I IFN-dependent antiviral immune responses [Citation130].
Based on the data above and the dependency of DNA vaccines on the STING-type I IFN pathway to induce robust adaptive immunity, utilization of the adjuvants targeting the STING pathway could be a promising strategy to develop vaccines with greater efficacy for infectious diseases [Citation126,Citation131,Citation132]. Particularly, cyclic dinucleotides, such as cyclic dimeric guanosine monophosphate (c-di-GMP), c-di-AMP, and cGAMP, show potent mucosal adjuvant activities to elicit type I IFN-dependent protective humoral (both IgG and IgA) and cellular (a mixed Th1/2/17 response) immune responses against pathogens such as Streptococcus pneumoniae [Citation133,Citation134].
2.4.2. Type II IFNs
IFN-γ produced by NK, NK T, ILCs, or T cells elicit antimicrobial immune responses against several pathogens, including but not limited to HSV, VSV, and Mtb, in various ways (summarized in ) [Citation36,Citation103,Citation105,Citation135]. First, IFN-γ can stimulate nitric oxide (NO) production at the infection site in macrophages or stromal cells resulting in direct inhibition of viral replication by NO [Citation136]. Second, IFN-γ supports the polarization of macrophages into the antimicrobial/pro-inflammatory M1 phenotype rather than the immunosuppressive M2 phenotype by preventing binding of enhancers to M2-like genes, as well as enhancing pro-inflammatory activity and the viability of M1 macrophages via metabolic reprogramming [Citation137,Citation138]. Third, via regulating translation and mTORC1 pathways, IFN-γ can modulate macrophage intracellular metabolism and boost autophagy, as well as pro-inflammatory cytokine production to facilitate the killing of the microbes [Citation139]. Moreover, IFN-γ can enhance the phagocytic ability of macrophages to exert antibacterial effect via upregulation of complement receptor 3, which belongs to the non-opsonizing phagocytosis pathway that occurs independently of Fc receptors [Citation140].
IFN-γ induces production of the Th1-inducing cytokine IL-12 and enhances co-stimulatory molecule upregulation on APCs, thereby facilitating the generation of antimicrobial type 1 immunity, involving IFN-γ producing Th1 cells and IgG2c producing B cells [Citation141,Citation142]. Moreover, IFN-γ is an inducer of B cell IgG2a (Th1 type antibody response) class switching in a T-independent manner [Citation143,Citation144]. In addition, by using the LPS-matured DC injection model in mice, it was shown that CXCR3-dependent recruitment of NK cells to lymph nodes is required for efficient priming of T cells for Th1 polarization through mechanisms depending on the action of NK cell-derived IFN-γ on T cells [Citation145].
Primary dengue virus infection with one of the four serotypes of the virus elicits antiviral cellular and humoral immune responses that can control the infection successfully [Citation146,Citation147]. However, in the case of secondary infection with a different dengue serotype, preexisting cross-reactive antibodies bind to viral particles and facilitate invasion of the myeloid cells by the virus, rather than helping viral clearance, in an Fc receptor-dependent manner, a phenomenon causing severe dengue fever called antibody-dependent enhancement (ADE) [Citation146,Citation147]. Indeed, Sun et al. showed that NK cell-derived IFN-γ production prevents ADE development in human PBMCs, thereby aiding in the immune control of dengue infection [Citation148]. In particular, the presence of antibodies that can induce antibody-dependent cell cytotoxicity, a process where antibody-bound infected cells cross-link CD16 receptors on NK cells to cause their degranulation and cytokine production, is also required for this anti-ADE effect of IFN-γ [Citation148].
The control of mycobacterial infections, including Mtb and nontuberculosis mycobacteria, requires IFN-γR signaling [Citation149–151]. In fact, mutations rendering the IFN-γR signaling pathway inactive are associated with mycobacterial disease syndrome (MSMD), a term used to describe elevated susceptibility to nontuberculosis mycobacterial infections [Citation151]. One of the first MSMD-related mutations identified is a missense mutation (T168N) in the IFNGR2 gene [Citation150]. Later, Blouin et al. revealed that T168N mutation is a gain-of-glycosylation mutation adding a neo-glycan to IFN-γR2 leading to excessive galectin binding and IFN-γR partitioning in actin instead of lipid nanodomains, thereby interfering with the IFN-γR conformational changes required for functional JAK/STAT signaling [Citation152]. Conversely, IFN-γ promotes the elimination of Mtb inside the infected macrophages by inducing antimicrobial peptides and expression of inducible nitric oxide synthase (iNOS) to boost antibacterial NO production and by inducing the expression of p47 GTPase Lrg-47 (also known as Irgm1) that can facilitate Mtb-incorporated phagosome maturation and elimination of Mtb infection by autophagy in a vitamin D-dependent manner [Citation149,Citation153].
2.5 The dark side of IFNs
2.5.1. Pro-tumorigenic effects
2.5.1.1. Type I IFNs
Despite the potent antitumor effect of type I IFNs explained in this review, type I IFNs might paradoxically show pro-tumorigenic properties in certain conditions mostly due to chronic type I IFN signaling-mediated induction of inhibitory molecules, such as PD-1, PD-L1, CTLA-4, and IDO that can halt the generation of antitumor immunity (summarized in ) [Citation154–158]. For instance, IFN-α stimulation can enhance the expression of the inhibitory molecule PD-1 on mouse T cells in an IRF9-dependent manner and interfere with the elicitation of antitumor T cell responses both in vitro and in vivo [Citation158]. Indeed, a combinatorial treatment approach using IFN-α and anti-PD-1 antibodies boosted potent antitumor immune responses unlike singular treatments [Citation158]. Similarly, Jacquelot et al. discovered that IFNAR signaling plays a deleterious role in antitumor immunity by supporting the development of resistance to anti-PD1 checkpoint blockade therapy in murine tumor models [Citation154]. Mechanistically, sustained type I IFN production within the TME of anti-PD-1 blockade resistant tumors leads to IFNAR signaling-induced elevation of iNOS in both tumor and immune cells resulting in recruitment of suppressive Tregs, as well as MDSCs to TME and consequently decreased antitumor CTL responses [Citation154]. Nevertheless, IFN-β treatment enhances melanoma tumor dormancy, which helps tumors avoid immune clearance and metastasize, through activation of the IDO pathway that converts tryptophan to kynurenine, which in turn binds to aryl hydrocarbon receptor (AhR) to induce the expression of the cell cycle inhibitor gene p27 in tumor cells [Citation157]. Therefore, type I IFN-mediated tumor dormancy can successfully be broken by combination therapies involving AhR or IDO inhibitors as AhR blockade redirected IFNAR signaling to the STAT3-p53 pathway and induced dormant tumor cell death due to p53-dependent ROS accumulation in the cells [Citation157].
Chronic inflammation is another well-known driver of tumorigenesis [Citation159]. Ahn et al. demonstrated that STING KO mice were resistant to the development of carcinogen-induced skin tumors in mice, suggesting that the STING-type I IFN pathway activated by chronic cytosolic DNA leakage is the driver of chronic inflammation-mediated tumorigenesis [Citation155]. Conversely, activation of the STING-type I IFN pathway within the TME of tumors with low but not high antigenicity drives tumorigenesis of these tumors in an IDO-dependent manner as observed in a mouse model of Lewis lung cancer (LLC) [Citation156].
2.5.1.2. Type II IFNs
IFN-γ is also capable of upregulating inhibitory molecule expression to induce tolerogenic/immunosuppressive responses and interfere with the generation of the antitumor immunity via the IFN-γR-JAK1/2-STAT1 signaling pathway (summarized in ) [Citation160–164]. In particular, IFN-γ exposure of tumors, such as hematopoietic tumors, melanoma, and gastric cancer, causes PD-L1 upregulation on the tumors leading to the generation of poor CTL- and NK cell-mediated antitumor immune responses [Citation163–165]. Moreover, IFN-γ decreased the efficacy of a therapeutic melanoma peptide vaccine by upregulating noncognate major histocompatibility complex (MHC) class I and PD-L1 molecules on tumor cells to interfere with proper CTL responses [Citation165]. Furthermore, Mo et al. showed that human melanocytes and melanoma cell lines upregulate the co-inhibitory molecule CTLA-4 in response to IFN-γ through binding of STAT1 to GAS sites on the CTLA-4 promoter, thereby preventing co-stimulatory signals on the T cells to halt antitumor immunity [Citation162]. In contrast, IFN-γ-induced IDO upregulation on both tumor cells and DCs can diminish antitumor immunity [Citation160,Citation161]. While IDO-kynurenine-AhR-mediated p27 upregulation in tumor cells prevents STAT1-mediated tumor cell apoptosis and drives tumor cell dormancy, IDO-kynurenine-AhR-mediated IDO upregulation within tolerogenic DCs is necessary to maintain the immunosuppressive phenotype of tolerogenic DCs [Citation160,Citation161]. Nevertheless, IFN-γ-induced upregulation of SOCS2 in tumor-infiltrating DCs is responsible for the generation of limited antimelanoma immunity due to lack of efficient T cell priming by those tolerogenic DCs [Citation166].
IFN-γ-mediated immune escape of the tumors may occur via downregulation of the tumor antigens on the tumors that are exposed to IFN-γ produced by tumor-infiltrating CTLs as shown by Beatty et al. in a mouse colon carcinoma model [Citation167]. Nonetheless, by using an NSCLC tumor model in mice, Song et al. clearly showed that intratumoral dose of IFN-γ is the determinant of antitumor vs. pro-tumorigenic effects of IFN-γ [Citation168]. While lower doses of IFN-γ activate intercellular adhesion molecule-1 (ICAM-1)-PI3K-Akt-Notch1 signaling pathway to drive tumor stemness and proliferation, higher doses of IFN-γ activates JAK1-STAT1-caspase-3/7-mediated apoptosis of tumor cells [Citation168]. Besides, the suppressive effect of TLR2 agonist immunotherapy on antitumor immunity is also linked to IFN-γ produced by tumor-infiltrating CTLs as shown in OVA-expressing LLC and EG-7 tumor models [Citation169]. Mechanistically, the TLR2 agonist supports the survival and differentiation of monocytic MDSCs into macrophages, which can perform antigen presentation to CTLs to induce transient IFN-γ production, which promotes iNOS upregulation and NO production in macrophages culminating in NO-mediated suppression of antitumor CTL proliferation [Citation169].
2.5.2. Suppressive effects on antiviral/antimicrobial immunity
2.5.2.1. Type I IFNs
Despite the data showing beneficial effects of type I IFNs for anti-L. monocytogenes immunity development [Citation120,Citation121], Auerbuch et al. found that mice deficient for IFNAR signaling are indeed more resistant to infection with high dose L. monocytogenes revealing a paradoxically deleterious role of type I IFNs in antibacterial immunity [Citation170]. In the absence of IFNAR signaling, elevated levels of IL-12p70 and TNF-α-producing CD11b+ macrophages 72 h postinfection correlated with enhanced resistance to L. monocytogenes infection [Citation170]. In contrast, Pontiroli et al. demonstrated that exogenous type I IFN administration at earlier time points of L. monocytogenes infection aids in the immune clearance of the bacterial infection via potentiating NK cell activation [Citation171]. Therefore, duration, magnitude, and perhaps the source of type I IFNs are crucial in antimicrobial immunity. This hypothesis is also supported by the study by Teijaro et al. showing that chronic type I IFN signaling observed with persistent viral infections (e.g., lymphocytic choriomeningitis virus (LCMV)) can abolish antiviral immunity by enhancing the suppressive factors PD-L1 and IL-10 and disrupting the lymphoid architectures in lymphoid organs, such as the spleen [Citation172]. In fact, type I IFN signaling blockade reversed these deleterious roles of type I IFNs and provided CD4 T cell-dependent clearance of the persistent virus infection [Citation172]. In addition, studies using mouse models of LCMV and measles virus (MV) infection further revealed that virus-induced type I IFNs diminish antiviral immunity through the suppression of DC differentiation and proliferation in a STAT2-dependent but STAT1-independent manner [Citation173]. Consistent with these LCMV studies, Sandler et al. also demonstrated that although type I IFN treatment of the rhesus macaques infected with simian immunodeficiency virus-induced beneficial antiviral immune responses to limit systemic infections, prolonged type I IFN treatment conversely suppressed antiviral immunity by suppressing antiviral gene expression and enhancement of SIV-mediated CD4 T cell loss [Citation174].
Clearance of the fungal infections with Cryptococcus neoformans in the lungs was also negatively affected by type I IFNs [Citation175]. In particular, IFNAR deficient mice were able to clear fungal infections better than wild type mice due to the presence of enhanced Th1, Th2, and IL-4-mediated mucin production by bronchiolar epithelial cells in the absence of type I IFN signaling [Citation175]. Moreover, type I IFNs were reported to exacerbate lung damage during influenza infection and decrease survival of the infected mice [Citation176]. Mechanistically, excessive type I IFN production by pDCs in the lungs causes lung epithelial cell death via TNF-related apoptosis-inducing ligand (TRAIL)-dependent apoptotic pathways as mice with hyperactive pDCs producing excessive amounts of type I IFNs were more susceptible to influenza [Citation176]. In contrast, comprehensive analysis of self-healing and disseminated forms of human leprosy (caused by the intracellular bacteria Mycobacterium leprae) samples revealed that type I IFNs can halt IFN-γ-induced vitamin D-dependent anti-mycobacterial immune responses via the mechanisms regulated by type I IFN-induced IL-10 production [Citation177]. While self-healing lesions had a Th1-weighted cytokine response, such as IFN-γ, lesions from disseminated forms of leprosy showed a Th2-weighted immune response in addition to elevated levels of IL-10 [Citation177]. Similarly, by using the Mtb infection model and tuberculosis patient samples, it was shown that the crosstalk between type I IFN and interleukin-1 (IL-1) regulates anti-mycobacterial immune responses via modulation of eicosanoid metabolism [Citation178]. Specifically, an elevated type I IFN response can suppress IL-1 responses causing eicosanoid imbalance to decrease prostaglandin E2 levels (PGE2) and aggravate disease progression [Citation178].
2.5.2.2. Type II IFNs
Although vast majority of the evidence suggests that IFN-γ derived from innate (e.g., NK and ILC1) or adaptive (e.g., Th1 cells and CTLs) type 1 immune cells plays a protective role against several pathogens, some reports have demonstrated that IFN-γ can diminish antimicrobial immunity through induction of suppressive immune-regulatory molecules, such as PD-L1 (summarized in ) [Citation103,Citation179]. For instance, in the mouse model of endotoxemia, IFN-γ causes PD-L1 upregulation on splenic neutrophils, which then gain the ability to suppress antigen-specific T cell proliferation [Citation179]. Importantly, these IFN-γ-induced PD-L1-expressing suppressive neutrophils are also observed in experimental human endotoxemia samples [Citation179].
Type 1 immunity plays important roles in clearance of intracellular infections or cancers, whereas type 2 immunity is beneficial for clearance of extracellular pathogens, such as helminths, as well as aiding in resolution of inflammation [Citation180]. Therefore, suppression of type 2 immunity by the type 1 cytokine IFN-γ can be harmful to the host in the case of infections with extracellular pathogens or exacerbate collateral tissue damage due to chronic type 1 inflammation, which is normally controlled by type 2 cytokines [Citation180–182]. IFN-γ-induced STAT1 signaling is capable of suppressing proliferation, as well as effector functions of ILC2 that produce type 2 cytokines, which provide anti-helminth immunity and protect the host from tissue damage [Citation182,Citation183]. STAT1-deficient mice exhibited decreased ILC1 and elevated ILC2 and ILC3 responses that worsened RSV-mediated lung pathology. In contrast, Califano et al. demonstrated that mice lacking IFN-γ is less susceptible to lethal influenza infection due to the inability of IFN-γ to limit the protective ILC2 responses, especially IL-5 and amphiregulin, in the lungs of the infected mice, revealing the protective role of IFN-γ in influenza-mediated lung pathology unlike RSV-mediated lung pathology [Citation181]. Thus, these data prove that IFN-γ plays a dual role in the pathogenesis of infectious diseases similar to type I IFNs.
2.5.3. IFN-mediated autoimmunity and autoinflammation
2.5.3.1. Type I IFNs
Chronic type I IFN production due to improper detection of host-derived nucleic acids via cytosolic (e.g., cGAS) or endosomal (e.g., TLR7 and TLR9) nucleic acid sensors have been proven to exacerbate the pathology of autoimmune or autoinflammatory conditions in several clinical and preclinical studies [Citation11]. Among the cytosolic nucleic acid sensing pathways, the cGAS-STING-type I IFN pathway has attracted a considerable amount of attention as a significant driver of various autoimmune or autoinflammatory conditions, such as systemic lupus erythematosus (SLE), Aicardi-Goutieres Syndrome (AGS), and SAVI [Citation184–186]. In particular, defects or deficiencies in DNases, such as DNase II and III (DNase III is also known as three prime exonuclease 1 (TREX1)), result in the development of severe type I IFN-mediated autoinflammation due to the sustained activation of the cGAS-STING pathway by the phagocytosed DNA, which is normally cleared by DNases, derived from apoptotic cells [Citation184,Citation186]. Indeed, several TREX1 mutations have been discovered in patients with severe SLE and AGS who are susceptible to chronic inflammation-mediated premature death [Citation187]. In addition, the mice lacking DNase II or III exhibited either embryonic lethality (for DNase II) or premature death (for Dnase III), which was rescued by crossing them with STING or cGAS deficient mice [Citation184,Citation187]. Moreover, more direct evidence linking the STING-type I IFN axis to autoinflammation is the disease called SAVI, which is caused by mutations in STING revealing it is constitutively active, thereby causing continuous type I IFN production, which causes pathologies generally affecting the lungs and skin [Citation185]. Importantly, the JAK1/2 inhibitor, baricitinib, improved disease symptoms, such as elevated IFN signature, in autoinflammatory interferonopathies including SAVI and chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperatures (CANDLE) in the clinic [Citation188].
In addition to cytosolic nucleic acid sensors, improper activation of endosomal TLR7 and TLR9 capable of detecting RNA and DNA, respectively, have also been associated with autoimmune conditions, such as lupus [Citation11,Citation189–193]. In fact, several chemically-induced or genetic mouse models of SLE are dependent on TLR7-mediated type I IFN production, which was reported to be the driver of myeloid cell expansion in TLR7-overexpressing mice that spontaneously develop SLE [Citation192,Citation193]. Recently, Negishi et al. identified U11snRNA as the pathogenic TLR7 ligand responsible for the pathogenesis of TLR7-dependent autoimmunity [Citation189]. In addition, they have developed a novel antagonist targeting U11snRNA and successfully prevented TLR7-dependent autoimmunity development [Citation189]. Nevertheless, in a TLR7-dependent murine lupus model, a CXCR4-targeting small molecule managed to suppress TLR7-mediated IFN-α production from pDCs to ameliorate disease progression [Citation190]. Furthermore, despite the contradictory reports about the pathogenic role of TLR9 signaling in autoimmunity, Barrat et al. demonstrated that an inhibitor targeting both TLR7 and TLR9 can improve the clinical manifestations of lupus-prone (NZB x NZW) F1 mice in vivo [Citation191].
2.5.3.2 Type II IFNs
Chronic inflammation followed by the generation of pathogenic T or B cells specific for self-antigens leads to autoimmune diseases, such as multiple sclerosis (MS), lupus nephritis, and rheumatoid arthritis, in which Th1 and Th17 type pathogenic T cells are predominant [Citation194]. Since the Th1 cytokine IFN-γ can drive M1 macrophage generation and support type 1 immunity, it was thought that IFN-γ would play a pathogenic role in Th1 dominant autoimmune diseases. Indeed, IFN-γ was shown to aggravate pathogenesis in collagen-induced arthritis (CIA) and proteoglycan-induced arthritis models of mice [Citation195,Citation196]. Nevertheless, IFN-γ treatment aggravated MS progression [Citation197]. Mechanistically, two independent studies have demonstrated that IFN-γ promotes disease development in mouse models of arthritis and patients with human RA via activating inflammatory macrophages [Citation198,Citation199].
Despite the evidence above showing the pathogenic role of IFN-γ in autoimmunity, several reports have indicated that IFN-γ could play a paradoxically protective role depending on the timing, route of administration, as well as disease type [Citation200–202]. While the administration of IFN-γ at the early phases promoted inflammation, IFN-γ exhibited a self-limiting effect to ameliorate Th1-mediated autoinflammation when administered at later time points of the disease in a murine Th1-mediated delayed type hypersensitivity (DTH) model [Citation201]. Similarly, anti-IFN-γ treatment starting from the early time points of CIA mice ameliorated disease progression while later anti-IFN-γ treatment had neutral or disease-promoting effects in CIA mice [Citation202]. In contrast, a recent study showed that a genetically engineered mouse, in which TLR9 activation is not limited to endosomal compartments-only, developed a lethal autoinflammatory disease that was dependent not on type I IFNs but on type II IFNs unlike TLR7-mediated autoinflammatory conditions [Citation203]. In particular, IFN-γ production from NK cells was responsible for this TLR9-mediated lethal autoinflammation [Citation203].
3. Conclusion
Here, we reviewed the dual role of type I and II IFNs in antimicrobial and antitumor immunity based on the evidence obtained from both preclinical and clinical studies. As complex mechanisms modulate the antitumor or antimicrobial effects of IFNs, careful consideration of the timing, dose, and route of administration is required for each type of tumor or pathogen when designing IFN-based therapies, including immunotherapies or vaccines. Improper induction of type I and II IFNs against self-factors, such as nucleic acids, can lead to autoinflammatory or autoimmune conditions. Yet, the data regarding the pathogenic role of IFNs are also contradictory and dependent on timing, dose, route, and the type of disease model as demonstrated by the protective effect of late but not early type II IFNs in DTH and CIA disease models in mice [Citation201,Citation202]. Therefore, IFNs are double-edged swords that could both potentiate and diminish protective antimicrobial and antitumor immune responses.
4. Expert opinion
IFNs were initially discovered as potent antiviral and antitumor agents. However, a comprehensive research of their mode of action revealed the yin and yang of IFNs for both antimicrobial (summarized in ) and antitumor immunity (summarized in ). Among the IFN-inducing pathways, the STING-dependent cytosolic nucleic acid sensing pathways play a key role in the generation of immune responses against various pathogens, as well as tumors. Indeed, spontaneous antitumor immunity is induced in a STING-dependent manner. Particularly, data from cancer studies suggested that tumor-targeting IFN-inducing immunotherapies, involving TLR or STING agonists (e.g., CpG ODN+cGAMP), exert synergistic effects when used in combination or concomitantly with immune-checkpoint blocking agents [Citation31,Citation74,Citation78,Citation79,Citation204]. Indeed, the pro-tumorigenic or suppressive effect of IFNs on antimicrobial immunity is generally due to chronic IFN production to induce suppressive immune-regulatory pathways, such as PD-L1 and IDO.
Based on the current evidence, since treatment of patients with cancer or infections using type I or II IFNs directly could potentially lead to undesired effects, such as autoimmune or autoinflammatory reactions, we think that utilization of IFN-inducing therapies or vaccines adjuvanted with IFN-inducing agents, rather than systemic IFN treatment, would be a safer option. Local injections of IFNs at the vaccination site would be another advantageous and safer option to achieve efficient activation of APCs taking up vaccine antigens compared to systemic IFN treatment. Similarly, immunization of patients with stage IV melanoma with an IFN-α-adjuvanted peptide vaccine has expanded the effector memory CD8 T cells, as well as activating monocyte-derived APCs [Citation205]. However, improper or continuous activation of nucleic acid-sensing pathways, such as the cGAS-STING pathway, may lead to severe autoinflammatory and autoimmune conditions including SAVI, SLE, and AGS. Notably, besides the general pathological role of IFNs (especially TLR7/9-dependent type I IFN production) in autoimmune and autoinflammatory diseases, IFNs can also play a dual role in such diseases based on the type of the disease and the time of administration. For instance, despite the pathological role of IFN-γ in MS, type I IFN is one of the therapeutic agents used for the treatment of not only MS but also Mediterranean fever and Behcet’s disease [Citation206–208]. In the latter cases, the anti-inflammatory effect of type I IFNs is attributed to type I IFN-mediated IL-10 induction and suppression of the inflammasome-dependent IL-1 production, which contributes to disease pathology [Citation209].
While IFNs protect against various pathogens, excessive IFN production can aggravate bacterial or viral infections by causing collateral tissue damage and weakening the host (e.g., Type I IFNs induce TRAIL-dependent apoptosis in influenza-infected lung epithelial cells [Citation176]). Thus, optimization of the dose, route, and schedule of IFNs or IFN-inducing agents is crucial to get the optimal immune response capable of clearing invading pathogens without causing significant adverse events. A recent study by Palacio et al. clearly showed that transient blockade of early-type I IFNs via systemic administration of anti-IFN-I antibodies during acute viral infection with several murine viruses including arenavirus, coronavirus, flavivirus, and rhabdovirus can improve antiviral immune responses against secondary viral infections. More importantly, early-type I IFN blockade also improved experimental viral vaccine efficacy in mice. Mechanistically, early type I IFN blockade allowed rapid upregulation of viral antigens without increasing the circulating virus levels due to reversion of IFN response after transient IFN blockade and therefore, enhanced antigen capture and presentation by the APCs to elicit more robust antiviral cellular and humoral immune responses [Citation210]. In contrast, early-type I IFN treatment efficiently induced anti-Middle East respiratory syndrome–coronavirus (MERS-CoV) immunity while delayed type I IFN treatment failed to protect mice from lethal infection [Citation211]. Considering the data reviewed in this review and the conflicting data about the effect of IFNs in COVID-19 pathology [Citation212], it is difficult to generalize the effect of IFNs in different types of microbial infections as the outcome will be different in each case.
In conclusion, we believe that therapies based on inducers as well as suppressors of IFNs may offer therapeutic potential for not only infectious diseases and cancer but also autoinflammatory and autoimmune conditions.
Article highlights
IFNs link innate and adaptive immunity and contribute to the development of long-term and effective antimicrobial and antitumor immune responses not only by modulating the innate and adaptive immune cells but also by directly acting on the stromal/tumor cells, as well as pathogens.
Improper/constitutive activation of IFN-inducing pathways, especially by the self-derived factors such as host DNA, leads to pathological conditions involving dampening of antimicrobial/antitumor immunity as well as the development of autoimmune/autoinflammatory conditions.
Despite the robust antitumor effect of IFNs, in certain cases, such as chronic inflammation, IFNs can lead to immunotolerance and support tumorigenesis due to IFN-mediated upregulation of immunoregulatory molecules, such as PD-L1, CTLA-4, and IDO that can dampen antitumor immunity.
Collateral tissue damage caused by an improper activation of IFN-inducing pathways can be overcome either by modulating the dose, route, and timing of the treatment or by using combination therapies involving immunomodulators, such as checkpoint blockade agents.
Immunotherapies/vaccines based on STING-dependent and independent nucleic acid sensing pathways that induce IFNs pose great therapeutic potential for both cancer and infectious diseases.
Considering the potential IFN treatment-related adverse events, such as autoimmunity/autoinflammation, utilization of IFN-inducing agents, including vaccines adjuvanted with IFN-inducing adjuvants could be a safer option than the direct use of IFNs as immunotherapeutics.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384.
- Sun L, Wu J, Du F, et al. Cyclic GMP-AMP Synthase Is a cytosolic DNA sensor that activates type I IFN pathway. Science. 2013;339:786–791.
- Paganin C, Frank I, Trinchieri G. Priming for high interferon-γ production induced by interleukin-12 in both CD4+ and CD8+ T cell clones from HIV-infected patients. J Clin Invest. 1995;96:1677–1682.
- Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity. 2003;19:641–644.
- Iwasaki A, Medzhitov R. Regulation of adaptative immunity bye the innate immune system. Science. 2010;327:291–295.
- Vesely MD, Kershaw MH, Schreiber RD, et al. Natural Innate and Adaptive Immunity to Cancer. Annu Rev Immunol. 2011;29:235–271.
- Starbeck-Miller GR, Xue -H-H, Harty JT. IL-12 and type I interferon prolong the division of activated CD8 T cells by maintaining high-affinity IL-2 signaling in vivo. J Exp Med. 2014;211:105–120.
- Chesler DA, Shoshkes C. The role of IFNg in immune responses to viral infections of the central nervous system. Cytokine Growth Factor Rev. 2002;13:441–454.
- Nguyen KB, Salazar-Mather TP, Dalod MY, et al. Coordinated and Distinct Roles for IFN-αβ, IL-12, and IL-15 Regulation of NK Cell Responses to Viral Infection. J Immunol. 2002;169:4279–4287.
- Wakita D, Chamoto K, Ohkuri T, et al. IFN-γ-dependent type 1 immunity is crucial for immunosurveillance against squamous cell carcinoma in a novel mouse carcinogenesis model. Carcinogenesis. 2009;30:1408–1415.
- Lee-Kirsch MA. The Type I Interferonopathies. Annu Rev Med. 2017;68:297–315.
- Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386.
- Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc B-Biol Sci. 1957;147:258–267.
- El Jamal SM, Taylor EB, Abd Elmageed ZY, et al. Interferon gamma-induced apoptosis of head and neck squamous cell carcinoma is connected to indoleamine-2,3-dioxygenase via mitochondrial and ER stress-associated pathways. Cell Div. 2016;11:1–14.
- Wang QS, Shen SQ, Sun HW, et al. Interferon-gamma induces autophagy-associated apoptosis through induction of cPLA2-dependent mitochondrial ROS generation in colorectal cancer cells. Biochem Biophys Res Commun. 2018;498:1058–1065.
- Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32.
- Belardelli F, Ferrantini M, Proietti E, et al. Interferon-alpha in tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002;13:119–134.
- Marchetti M, Monier MN, Fradagrada A, et al. Stat-mediated signaling induced by type I and type II interferons (IFNs) is differentially controlled through lipid microdomain association and clathrin-dependent endocytosis of IFN receptors. Mol Biol Cell. 2006;17(7):2896–2909.
- Ma F, Li B, Yu Y, et al. Positive feedback regulation of type I interferon by the interferon-stimulated gene STING. EMBO Rep. 2015;16:202–212.
- Uddin S, Majchrzak B, Woodson J, et al. Activation of the p38 mitogen-activated protein kinase by type I interferons. J Biol Chem. 1999;274:30127–30131.
- Uddin S, Yenush L, Sun XJ, et al. Interferon-α engages the insulin receptor substrate-1 to associate with the phosphatidylinositol 3ʹ-kinase. J Biol Chem. 1995;270:15938–15941.
- Schreiber G. The molecular basis for differential type I interferon signaling. J Biol Chem. 2017;292:7285–7294.
- Fish EN, Uddin S, Korkmaz M, et al. Activation of a CrkL-Stat5 signaling complex by type I interferons. J Biol Chem. 1999;274:571–573.
- Uddin S, Lekmine F, Sassano A, et al. Role of Stat5 in Type I interferon-signaling and transcriptional regulation. Biochem Biophys Res Commun. 2003;308:325–330.
- Nguyen KB, Watford WT, Salomon R, et al. Critical role for STAT4 activation by type 1 interferons in the interferon-γ response to viral infection. Science. 2002;297:2063–2066.
- Lavoie TB, Kalie E, Crisafulli-Cabatu S, et al. Binding and activity of all human alpha interferon subtypes. Cytokine. 2011;56:282–289.
- Platanias LC, Uddin S, Domanski P, et al. Differences in Interferon α and β Signaling. J Biol Chem. 1996;271:23630–23633.
- De Weerd NA, Vivian JP, Nguyen TK, et al. Structural basis of a unique interferon-β signaling axis mediated via the receptor IFNAR1. Nat Immunol. 2013;14:901–907.
- De Weerd NA, Nguyen T. The interferons and their receptors-distribution and regulation. Immunol Cell Biol. 2012;90:483–491.
- Zheng H, Qian J, Carbone CJ, et al. Vascular endothelial growth factor – induced elimination of the type 1 interferon receptor is required for efficient angiogenesis. Blood. 2015;118:4003–4007.
- Cauwels A, Van Lint S, Paul F, et al. Delivering type I interferon to dendritic cells empowers tumor eradication and immune combination treatments. Cancer Res. 2018;78:463–474.
- Lan Q, Peyvandi S, Duffey N, et al. Type I interferon/IRF7 axis instigates chemotherapy-induced immunological dormancy in breast cancer. Oncogene. 2019;38:2814–2829.
- Le Bon A, Durand V, Kamphuis E, et al. Direct stimulation of T cells by type I IFN enhances the CD8 + T cell response during cross-priming. J Immunol. 2006;176:4682–4689.
- Stone ML, Chiappinelli KB, Li H, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. PNAS. 2017;114:10981–10990.
- Pegram HJ, Andrews DM, Smyth MJ, et al. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol. 2011;89:216–224.
- Mack EA, Kallal LE, Demers DA, et al. Type 1 interferon induction of natural killer cell gamma interferon production for defense during lymphocytic choriomeningitis virus infection. MBio. 2011;2:13–15.
- Bhat P, Leggatt G, Waterhouse N, et al. Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. 2017;8:2836–2846.
- Maggi E, Parronchi P, Manetti R, et al. Reciprocal regulatory effects of IFN-γ and IL-4 on the in vitro development of human Th1 and Th2 clones. J Immunol. 1992;148:2142–2147.
- Pernis A, Gupta S, Gollob KJ, et al. Lack of interferon γ receptor β chain and the prevention of interferon γ signaling in TH1 Cells. Science. 1995;269:245–247.
- Bernabei P, Coccia EM, Rigamonti L, et al. Interferon-gamma receptor 2 expression as the deciding factor in human T, B, and myeloid cell proliferation or death. J Leukoc Biol. 2001;70:950–960.
- Pham D, Vincentz JW, Firulli AB, et al. Twist1 regulates Ifng expression in Th1 cells by interfering with Runx3 function. J Immunol. 2012;189:832–840.
- Song MM, Shuai K. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon-mediated antiviral and antiproliferative activities. J Biol Chem. 1998;273:35056–35062.
- Rakesh K, Agrawal DK. Controlling cytokine signaling by constitutive inhibitors. Biochem Pharmacol. 2005;70:649–657.
- Huffaker TB, Hu R, Runtsch MC, et al. Epistasis between MicroRNAs 155 and 146a during T cell-mediated antitumor immunity. Cell Rep. 2012;2:1697–1709.
- Huffaker TB, Lee SH, Tang WW, et al. Antitumor immunity is defective in T cell–specific microRNA-155– deficient mice and is rescued by immune checkpoint blockade. J Biol Chem. 2017;292:18530–18541.
- Alagu J, Itahana Y, Sim F, et al. Tumor suppressor p14ARF enhances IFN-γ–activated immune response by inhibiting PIAS1 via SUMOylation. J Immunol. 2018;201:451–464.
- Ding LW, Sun QY, Edwards JJ, et al. LNK suppresses interferon signaling in melanoma. Nat Commun. 2019;10:1–13.
- Porta C, Hadj-Slimane R, Nejmeddine M, et al. Interferons α and γ induce p53-dependent and p53-independent apoptosis, respectively. Oncogene. 2005;24:605–615.
- Kotredes KP, Gamero AM. Interferons as inducers of apoptosis in malignant cells. J Interf Cytokine Res. 2013;33:162–170.
- Castro F, Cardoso AP, Gonçalves RM, et al. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol. 2018;9:1–19.
- Sancéau J, Hiscott J, Delattre O, et al. IFN-β induces serine phosphorylation of Stat-1 in Ewing’s sarcoma cells and mediates apoptosis via induction of IRF-1 and activation of caspase-7. Oncogene. 2000;19:3372–3383.
- Dierckx T, Khouri R, Menezes SM, et al. IFN-β induces greater antiproliferative and proapoptotic effects and increased p53 signaling compared with IFN-α in PBMCs of adult T-cell leukemia/lymphoma patients. Blood Cancer J. 2017;7:10–13.
- Enomoto H, Tao L, Eguchi R, et al. The in vivo antitumor effects of type I-interferon against hepatocellular carcinoma: the suppression of tumor cell growth and angiogenesis. Sci Rep. 2017;7:1–11.
- Doherty MR, Cheon HJ, Junk DJ, et al. Interferon-beta represses cancer stem cell properties in triple-negative breast cancer. PNAS. 2017;114:13792–13797.
- Doherty MR, Parvani JG, Tamagno I, et al. The opposing effects of interferon-beta and oncostatin-M as regulators of cancer stem cell plasticity in triple-negative breast cancer. Breast Cancer Res. 2019;21:1–12.
- Ma Z, Qin H, Benveniste EN. Transcriptional suppression of matrix metalloproteinase-9 gene expression by IFN-γ and IFN-β: critical role of STAT-1α. J Immunol. 2001;167:5150–5159.
- Spaapen RM, Leung MYK, Fuertes MB, et al. Therapeutic activity of high-dose intratumoral IFN-β requires direct effect on the tumor vasculature. J Immunol. 2014;193:4254–4260.
- Sikora AG, Jaffarzad N, Hailemichael Y, et al. IFN-α enhances peptide vaccine-induced CD8 + T cell numbers, effector function, and antitumor activity. J Immunol. 2009;182:7398–7407.
- Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249–1255.
- Honda K, Sakaguchi S, Nakajima C, et al. Selective contribution of IFN-α/β signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc Natl Acad Sci U S A. 2003;100:10872–10877.
- Cho HJ, Hayashi T, Datta SK, et al. IFN-αβ promote priming of antigen-specific CD8 + and CD4 + T lymphocytes by immunostimulatory DNA-based vaccines. J Immunol. 2002;168:4907–4913.
- Zhang KJ, Yin XF, Yang YQ, et al. A potent in Vivo antitumor efficacy of novel recombinant type I interferon. Clin Cancer Res. 2017;23:2038–2049.
- Kirkwood JM, Bender C, Agarwala S, et al. Mechanisms and management of toxicities associated with high-dose interferon alfa-2b therapy. J Clin Oncol. 2002;20:3703–3718.
- Andzinski L, Kasnitz N, Stahnke S, et al. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int J Cancer. 2016;138:1982–1993.
- Glück S, Guey B, Gulen MF, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19:1061–1070.
- Andzinski L, Spanier J, Kasnitz N, et al. Growing tumors induce a local STING dependent Type I IFN response in dendritic cells. Int J Cancer. 2016;139:1350–1357.
- Woo S, Fuertes MB, Corrales L, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2015;41:830–842.
- Schadt L, Sparano C, Schweiger NA, et al. Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep. 2019;29:1236–1248.
- Deng L, Liang H, Xu M, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type i interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41:843–852.
- Wang Z, Xia L, Xia X. cGAS/STING axis mediates a topoisomerase II inhibitor-induced tumor immunogenicity. JCI. 2019;129:4850–4862.
- Liu H, Golji J, Brodeur LK, et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat Med. 2019;25:95–102.
- Di XL, Öhman M. ADAR1 editing and its role in cancer. Genes (Basel). 2019;10:1–12.
- Ramanjulu JM, Pesiridis GS, Yang J, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. 2018;564:439–443.
- Ghaffari A, Peterson N, Khalaj K, et al. Sting agonist therapy in combination with pd-1 immune checkpoint blockade enhances response to carboplatin chemotherapy in high-grade serous ovarian cancer. Br J Cancer. 2018;119:440–449.
- Shae D, Becker KW, Christov P, et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat Nanotechnol. 2019;14:269–278.
- Sallets A, Robinson S, Kardosh A, et al. Enhancing immunotherapy of STING agonist for lymphoma in preclinical models. Blood Adv. 2018;2:2230–2241.
- Jing W, McAllister D, Vonderhaar EP, et al. STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models. J Immunother Cancer. 2019;7:1–18.
- Temizoz B, Kuroda E, Ohata K, et al. TLR9 and STING agonists synergistically induce innate and adaptive type-II IFN. Eur J Immunol. 2015;45:1159–1169.
- Yildiz S, Alpdundar E, Gungor B, et al. Enhanced immunostimulatory activity of cyclic dinucleotides on mouse cells when complexed with a cell-penetrating peptide or combined with CpG. Eur J Immunol. 2015;45:1170–1179.
- Li T, Cheng H, Yuan H, et al. Antitumor activity of cGAMP via stimulation of cGAS-cGAMP- STING-IRF3 mediated innate immune response. Sci Rep. 2016;6:1–14.
- Ohkuri T, Ghosh A, Kosaka A, et al. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol Res. 2014;2:1199–1209.
- Baird JR, Friedman D, Cottam B, et al. Radiotherapy combined with novel STING- targeting oligonucleotides results in regression of established tumors. Cancer Res. 2016;76:50–61.
- Nakamura T, Miyabe H, Hyodo M, et al. Liposomes loaded with a STING pathway ligand, cyclic di-GMP, enhance cancer immunotherapy against metastatic melanoma. J Control Release. 2015;216:149–157.
- Chandra D, Quispe-tintaya W, Jahangir A, et al. STING Ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol Res. 2014;2:901–910.
- Detjen KM, Farwig K, Welzel M, et al. Interferon γ inhibits growth of human pancreatic carcinoma cells via caspase-1 dependent induction of apoptosis. Gut. 2001;49:251–262.
- Andersen K, Smith-Sørensen B, Pedersen KB, et al. Interferon-γ suppresses S100A4 transcription independently of apoptosis or cell cycle arrest. Br J Cancer. 2003;88:1995–2001.
- Boye K, Andersen K, Tveito S, et al. Interferon-γ-induced suppression of S100A4 transcription is mediated by the class II transactivator. Tumor Biol. 2006;28:27–35.
- Kammertoens T, Friese C, Arina A, et al. Tumour ischaemia by interferon-γ resembles physiological blood vessel regression. Nature. 2017;545:98–102.
- Redente EF, Dwyer-Nield LD, Barrett BS, et al. Lung tumor growth is stimulated in IFN-γ-/- mice and inhibited in IL,-4Rα-/- mice. Anticancer Res. 2009;29:5095–5101.
- Su Z, Yang R, Zhang W, et al. The synergistic interaction between the calcineurin B subunit and IFN-γ enhances macrophage antitumor activity. Cell Death Dis. 2015;6:1–13.
- Gerber SA, Sedlacek AL, Cron KR, et al. IFN-γ mediates the antitumor effects of radiation therapy in a murine colon tumor. Am J Pathol. 2013;182:2345–2354.
- Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21:687–692.
- Salerno F, Guislain A, Freen-Van Heeren JJ, et al. Critical role of post-transcriptional regulation for IFN-γ in tumor-infiltrating T cells. Oncoimmunology. 2019;8:1–12.
- Gordon-Alonso M, Hirsch T, Wildmann C, et al. Galectin-3 captures interferon-gamma in the tumor matrix reducing chemokine gradient production and T-cell tumor infiltration. Nat Commun. 2017;8:1–15.
- Higgs BW, Morehouse CA, Streicher K, et al. Interferon gamma messenger RNA Signature in tumor biopsies predicts outcomes in patients with non–small cell lung carcinoma or urothelial cancer treated with durvalumab. Clin Cancer Res. 2018;24:3857–3866.
- Tian H, Shi G, Wang Q, et al. A novel cancer vaccine with the ability to simultaneously produce anti-PD-1 antibody and GM-CSF in cancer cells and enhance Th1-biased antitumor immunity. Signal Transduct Target Ther. 2016;1:1–10.
- Karachaliou N, Gonzalez-Cao M, Crespo G, et al. Interferon gamma, an important marker of response to immune checkpoint blockade in non-small cell lung cancer and melanoma patients. Ther Adv Med Oncol. 2018;10:1–23.
- Garris CS, Arlauckas SP, Kohler RH, et al. Successful anti-PD-1 cancer immunotherapy requires t cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity. 2018;49:1148–1161.
- Overacre-Delgoffe AE, Chikina M, Dadey RE, et al. Interferon-γ drives treg fragility to promote anti-tumor immunity. Cell. 2017;169:1130–1141.
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174.
- Medina-Echeverz J, Haile LA, Zhao F, et al. IFN-γ regulates survival and function of tumor-induced CD11b+Gr-1high myeloid derived suppressor cells by modulating the anti-apoptotic molecule Bcl2a1. Eur J Immunol. 2014;44:2457–2467.
- Kaplan A, Lee MW, Wolf AJ, et al. Direct Antimicrobial Activity of IFN-β. J Immunol. 2017;198:4036–4045.
- Lee AJ, Ashkar AA. The dual nature of type I and type II interferons. Front Immunol. 2018;9:1–10.
- Iijima N, Mattei LM, Iwasaki A. Recruited inflammatory monocytes stimulate antiviral Th1 immunity in infected tissue. Proc Natl Acad Sci U S A. 2011;108:284–289.
- Müller U, Steinhoff U, Reis LFL, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921.
- Van Den BROEKMF, Muller U, Huang S, et al. Immune defence in mice lacking type i and/or type ii interferon receptors. Immunol Rev. 1995;148:5–18.
- Uyangaa E, Kim JH, Patil AM, et al. Distinct upstream role of type I IFN signaling in hematopoietic stem cell-derived and epithelial resident cells for concerted recruitment of Ly-6Chi monocytes and NK cells via CCL2-CCL3 cascade. PLoS Pathog. 2015;11:1–30.
- Lee AJ, Chen B, Chew MV, et al. Inflammatory monocytes require type I interferon receptor signaling to activate NK cells via IL-18 during a mucosal viral infection. J Exp Med. 2017;214:1153–1167.
- Martinez J, Huang X, Yang Y. Direct action of type I IFN on NK cells is required for their activation in response to vaccinia viral infection in vivo. J Immunol. 2008;180:1592–1597.
- Li J, Liu K, Liu Y, et al. Exosomes mediate the cell-to-cell transmission of IFN-α-induced antiviral activity. Nat Immunol. 2013;14:793–803.
- Athale S, Banchereau R, Thompson-Snipes L, et al. Influenza vaccines differentially regulate the interferon response in human dendritic cell subsets. Sci Transl Med. 2017;9:1–21.
- Pernet E, Downey J, Vinh DC, et al. Leukotriene B4–type I interferon axis regulates macrophage-mediated disease tolerance to influenza infection. Nat Microbiol. 2019;4:1389–1400.
- Xiao J, Li W, Zheng X, et al. Targeting 7-dehydrocholesterol reductase integrates cholesterol metabolism and IRF3 activation to eliminate infection. Immunity. 2020;52:109–122.
- Mahlakõiv T, Hernandez P, Gronke K, et al. Leukocyte-derived IFN-α/β and epithelial IFN-λ constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS Pathog. 2015;11:1–19.
- Neil JA, Matsuzawa-Ishimoto Y, Kernbauer-Hölzl E, et al. IFN-I and IL-22 mediate protective effects of intestinal viral infection. Nat Microbiol. 2019;4:1737–1749.
- Hijano DR, Siefker DT, Shrestha B, et al. Type I interferon potentiates iga immunity to respiratory syncytial virus infection during infancy. Sci Rep. 2018;8:1–12.
- Wang H, Peters N, Schwarze J. Plasmacytoid dendritic cells limit viral replication, pulmonary inflammation, and airway hyperresponsiveness in respiratory syncytial virus infection. J Immunol. 2006;177:6263–6270.
- Kim TH, Oh DS, Jung HE, et al. Plasmacytoid dendritic cells contribute to the production of ifn-β via tlr7-myd88-dependent pathway and ctl priming during respiratory syncytial virus infection. Viruses. 2019;11:1–11.
- Kumaran Satyanarayanan S, El Kebir D, Soboh S, et al. IFN-β is a macrophage-derived effector cytokine facilitating the resolution of bacterial inflammation. Nat Commun. 2019;10:1–16.
- Stockinger S, Materna T, Stoiber D, et al. Production of type I IFN sensitizes macrophages to cell death induced by listeria monocytogenes. J Immunol. 2002;169:6522–6529.
- Lucas M, Schachterle W, Oberle K, et al. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity. 2007;26:503–517.
- Tegtmeyer PK, Spanier J, Borst K, et al. STING induces early IFN-β in the liver and constrains myeloid cell-mediated dissemination of murine cytomegalovirus. Nat Commun. 2019;10:1–12.
- Dey B, Dey RJ, Cheung LS, et al. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat Med. 2015;21:401–408.
- Lippmann J, Müller HC, Naujoks J, et al. Dissection of a type I interferon pathway in controlling bacterial intracellular infection in mice. Cell Microbiol. 2011;13:1668–1682.
- Sauer J, Sotelo-troha K, Von MJ, et al. The N -ethyl- N -nitrosourea-induced goldenticket mouse mutant reveals an essential function of sting in the in vivo interferon response to listeria monocytogenes and cyclic dinucleotides. Infect Immun. 2011;79:688–694.
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792.
- Hurbain I, Lahaye X, Satoh T, et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. 2013;39:1132–1142.
- Lahaye X, Satoh T, Gentili M, et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. 2013;39:1132–1142.
- Baranek T, Manh TPV, Alexandre Y, et al. Differential responses of immune cells to type i interferon contribute to host resistance to viral infection. Cell Host Microbe. 2012;12:571–584.
- Wang Y, Ning X, Gao P, et al. Inflammasome activation triggers caspase-1-mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity. 2017;46:393–404.
- Li XD, Wu J, Gao D, et al. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 2013;341:1390–1394.
- Ishii KJ, Kawagoe T, Koyama S, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451:725–729.
- Blaauboer SM, Mansouri S, Tucker HR, et al. The mucosal adjuvant cyclic di-GMP enhances antigen uptake and selectively activates pinocytosis-efficient cells in vivo. Elife. 2015;4:1–25.
- Skrnjug I, Guzma CA, Ruecker C. Cyclic GMP-AMP displays mucosal adjuvant activity in mice. PLoS One. 2014;9:1–7.
- Ashkar AA, Rosenthal KL. Interleukin-15 and natural killer and NKT cells play a critical role in innate protection against genital herpes simplex virus type 2 infection. J Virol. 2003;77:10168–10171.
- Karupiah G, Xie QW, Buller RML, et al. Inhibition of viral replication by interferon-γ-induced nitric oxide synthase. Science. 1993;261:1445–1448.
- Wang F, Zhang S, Jeon R, et al. Interferon gamma induces reversible metabolic reprogramming of M1 macrophages to sustain cell viability and pro-inflammatory activity. EBioMedicine. 2018;30:303–316.
- Kang K, Park SH, Chen J, et al. Interferon-γ represses M2 gene expression in human macrophages by disassembling enhancers bound by the transcription factor MAF. Immunity. 2017;47:235–250.
- Su X, Yu Y, Zhong Y, et al. Interferon-γ regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat Immunol. 2015;16:838–849.
- Drevets DA, Leenen PJM, Campbell PA. Complement receptor type 3 mediates phagocytosis and killing of Listeria monocytogenes by a TNF-α- and IFN-γ-stimulated macrophage precursor hybrid. Cell Immunol. 1996;169:1–6.
- Yokozeki H, Katayama I, Ohki O, et al. Interferon-gamma differentially regulates CD80 (B7-1) and CD86 (B7-2/B70) expression on human Langerhans cells. Br J Dermatol. 1997;136:831–837.
- Ma BX, Chow JM, Gri G, et al. The interleukin 12 p40 gene promoter is primed by interferon gamma in monocytic cells. J Exp Med. 1996;183:144–157.
- Snapper CM, Paul WE. Interferon-γ and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science. 1987;236:944–947.
- Gerth AJ, Lin L, Peng SL. T-bet regulates T-independent IgG2a class switching. Int Immunol. 2003;15:937–944.
- Martín-Fontecha A, Thomsen LL, Brett S, et al. Induced recruitment of NK cells to lymph nodes provides IFN-γ for TH1 priming. Nat Immunol. 2004;5:1260–1265.
- Balsitis SJ, Williams KL, Lachica R, et al. Lethal antibody enhancement of dengue disease in mice is prevented by Fc modification. PLoS Pathog. 2010;6:1–13.
- Guzmán MG, Kourí G, Valdés L, et al. Enhanced severity of secondary dengue-2 infections: death rates in 1981 and 1997 Cuban outbreaks. Rev Panam Salud Publica/Pan Am J Public Heal. 2002;11:223–227.
- Sun P, Williams M, Nagabhushana N, et al. NK cells activated through antibody-dependent cell cytotoxicity and armed with degranulation/IFN-γ production suppress antibody-dependent enhancement of dengue viral infection. Sci Rep. 2019;9:1–12.
- MacMicking JD, Taylor GA, McKinney JD. Immune Control of Tuberculosis by IFN-γ-inducible LRG-47. Science. 2003;302:654–659.
- Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon-γ receptor and susceptibility to mycobacterial infection. J Clin Invest. 1998;101:2364–2369.
- Bustamante J, Boisson-Dupuis S, Abel L, et al. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol. 2014;26:454–470.
- Blouin CM, Hamon Y, Gonnord P, et al. Glycosylation-dependent IFN-γR partitioning in lipid and actin nanodomains is critical for JAK activation. Cell. 2016;166:920–934.
- Fabri M, Stenger S, Shin DM, et al. Vitamin D is required for IFN-γ-mediated antimicrobial activity of human macrophages. Sci Transl Med. 2011;3:1–11.
- Jacquelot N, Yamazaki T, Roberti MP, et al. Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 2019;29:846–861.
- Ahn J, Xia T, Konno H, et al. Inflammation-driven carcinogenesis is mediated through STING. Nat Commun. 2014;5:5166–5175.
- Lemos H, Mohamed E, Huang L, et al. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res. 2016;76:2076–2081.
- Liu Y, Qin FX, Huang B, et al. STAT3/p53 pathway activation disrupts IFN- b – induced dormancy in tumor-repopulating cells. The Journal of Clinical Investigation. 2018;128(3):1057–1073.
- Terawaki S, Chikuma S, Shibayama S, et al. IFN-α directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. 2011;186:2772–2779.
- Gonda TA, Tu S, Wang TC. Chronic inflammation, the tumor microenvironment and carcinogenesis. Cell Cycle. 2009;8:2005–2013.
- Liu Y, Liang X, Yin X, et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-γ-induced immunologic dormancy of tumor-repopulating cells. Nat Commun. 2017;8:1–15.
- Li Q, Harden JL, Anderson CD, et al. Tolerogenic phenotype of IFN-γ–induced IDO + dendritic cells is maintained via an autocrine IDO–Kynurenine/AhR–IDO Loop. J Immunol. 2016;197:962–970.
- Mo X, Zhang H, Preston S, et al. Interferon-γ signaling in melanocytes and melanoma cells regulates expression of CTLA-4. Cancer Res. 2018;78:436–450.
- Mimura K, Teh JL, Okayama H, et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 2018;109:43–53.
- Bellucci R, Martin A, Bommarito D, et al. Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology. 2015;4:1–10.
- Il CH, Lee YR, Celis E. Interferon γ limits the effectiveness of melanoma peptide vaccines. Blood. 2011;117:135–144.
- Nirschl CJ, Suárez-Fariñas M, Izar B, et al. IFNγ-dependent tissue-immune homeostasis is co-opted in the tumor microenvironment. Cell. 2017;170:127–141.
- Beatty GL, Paterson Y. IFN-γ can promote tumor evasion of the immune system in vivo by down-regulating cellular levels of an endogenous tumor antigen. J Immunol. 2000;165:5502–5508.
- Song M, Ping Y, Zhang K, et al. Low-dose IFNg induces tumor cell stemness in tumor microenvironment of non–small cell lung cancer. Cancer Res. 2019;79:3737–3748.
- Shime H, Maruyama A, Yoshida S, et al. Toll-like receptor 2 ligand and interferon-γ suppress anti-tumor T cell responses by enhancing the immunosuppressive activity of monocytic myeloid-derived suppressor cells. Oncoimmunology. 2017;7:1–13.
- Auerbuch V, Brockstedt DG, Meyer-Morse N, et al. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med. 2004;200:527–533.
- Pontiroli F, Dussurget O, Zanoni I, et al. The timing of IFNβ production affects early innate responses to listeria monocytogenes and determines the overall outcome of lethal infection. PLoS One. 2012;7:1–13.
- Teijaro JR, Ng C, Lee AM, et al. Interferon signaling. Science (New York, N.Y.). 2013;340(6128):27–29.
- Hahm B, Trifilo MJ, Zuniga EI, et al. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity. 2005;22:247–257.
- Sandler NG, Bosinger SE, Estes JD, et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature. 2014;511:601–605.
- Sato K, Yamamoto H, Nomura T, et al. Cryptococcus neoformans infection in mice lacking type i interferon signaling leads to increased fungal clearance and IL-4-dependent mucin production in the lungs. PLoS One. 2015;10:1–19.
- Davidson S, Crotta S, McCabe TM, et al. Pathogenic potential of interferon αβ in acute influenza infection. Nat Commun. 2014;5:1–15.
- Teles RMB, Graeber TG, Krutzik SR, et al. Type I interferon suppresses type ii interferon–triggered human anti-mycobacterial responses. Science (New York, N.Y.). 2013;339(6126):1448–1454.
- Mayer-Barber KD, Andrade BB, Oland SD, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type i interferon crosstalk. Nature. 2014;511:99–103.
- Langereis JD, Pickkers P, De Kleijn S, et al. Spleen-derived IFN-γ induces generation of PD-L1 + -suppressive neutrophils during endotoxemia. J Leukoc Biol. 2017;102:1401–1409.
- Spellberg B, Edwards JE. Type 1/Type 2 immunity in infectious diseases. Clin Infect Dis. 2001;32:76–102.
- Califano D, Furuya Y, Roberts S, et al. IFN-γ increases susceptibility to influenza A infection through suppression of group II innate lymphoid cells. Mucosal Immunol. 2018;11:209–219.
- Moro K, Kabata H, Tanabe M, et al. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat Immunol. 2016;17:76–86.
- Stier MT, Goleniewska K, Cephus JY, et al. STAT1 represses cytokine-producing group 2 and group 3 innate lymphoid cells during viral infection. J Immunol. 2017;199:510–519.
- Ahn J, Gutman D, Saijo S, et al. STING manifests self DNA-dependent inflammatory disease. PNAS. 2012;109:19386–19391.
- Liu Y, Jesus AA, Marrero B, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371:507–518.
- Rice GI, Rodero MP, Crow YJ. Human disease phenotypes associated with mutations in TREX1. J Clin Immunol. 2015;35:235–243.
- Gao D, Li T, Li X, et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. PNAS. 2015;112:5699–5705.
- Montealegre Sanchez GA, Reinhardt A, Ramsey S, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest. 2018;128:3041–3052.
- Negishi H, Endo N, Nakajima Y, et al. Identification of U11snRNA as an endogenous agonist of TLR7-mediated immune pathogenesis. PNAS. 2019;116:23653–23661.
- Smith N, Rodero MP, Bekaddour N, et al. Control of TLR7-mediated type I IFN signaling in pDCs through CXCR4 engagement-A new target for lupus treatment. Sci Adv. 2019;5:1–14.
- Barrat FJ, Meeker T, Chan JH, et al. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol. 2007;37:3582–3586.
- Buechler MB, Teal TH, Elkon KB, et al. cutting edge: type I IFN drives emergency myelopoiesis and peripheral myeloid expansion during chronic TLR7 signaling. J Immunol. 2013;190:886–891.
- Lee PY, Kumagai Y, Li Y, et al. TLR7-dependent and FcγR-independent production of type I interferon in experimental mouse lupus. J Exp Med. 2008;205:2995–3006.
- Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-γ: implications for immune responses and autoimmune diseases. Immunity. 2009;31:539–550.
- Finnegan A, Grusby MJ, Kaplan CD, et al. IL-4 and IL-12 regulate proteoglycan-induced arthritis through stat-dependent mechanisms. J Immunol. 2002;169:3345–3352.
- Matthys P, Vermeire K, Mitera T, et al. Enhanced autoimmune arthritis in IFN-gamma receptor-deficient mice is conditioned by mycobacteria in Freund’s adjuvant and by increased expansion of Mac-1+ myeloid cells. J Immunol. 1999;163:3503–3510.
- Panitch HS, Haley AS, Hirsch RL, et al. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet. 1987;329:893–895.
- Hu X, Herrero C, Li WP, et al. Sensitization of IFN-γ Jak-STAT signaling during macrophage activation. Nat Immunol. 2002;3:859–866.
- Wang L, Tassiulas I, Park-Min KH, et al. “Tuning” of type I interferon-induced Jak-STAT1 signaling by calcium-dependent kinases in macrophages. Nat Immunol. 2008;9:186–193.
- Rosloniec EF, Latham K, Guedez YB. Paradoxical roles of IFN-γ in models in Th1-mediated autoimmunity. Arthritis Res. 2002;4:333–336.
- Feuerer M, Eulenburg K, Loddenkemper C, et al. Self-limitation of Th1-mediated inflammation by IFN-γ. J Immunol. 2006;176:2857–2863.
- Boissier MC, Chiocchia G, Bessis N, et al. Biphasic effect of interferon‐γ in murine collagen‐induced arthritis. Eur J Immunol. 1995;25:1184–1190.
- Stanbery AG, Newman ZR, Barton GM. Dysregulation of TLR9 in neonates leads to fatal inflammatory disease driven by IFN-γ. PNAS. 2020;117:3074–3182.
- Temizoz B, Kuroda E, Ishii KJ. Combination and inducible adjuvants targeting nucleic acid sensors. Curr Opin Pharmacol. 2018;41:104–113.
- Di Pucchio T, Pilla L, Capone I, et al. Immunization of stage IV melanoma patients with Melan-A/MART-1 and gp100 peptides plus IFN-α results in the activation of specific CD8+ T cells and monocyte/dendritic cell precursors. Cancer Res. 2006;66:4943–4951.
- Weinstock-Guttman B, Ransohoff RM, Kinkel RP, et al. The interferons: biological effects, mechanisms of action, and use in multiple sclerosis. Ann Neurol. 1995;37:7–15.
- Tweezer-Zaks N, Rabinovich E, Lidar M, et al. Interferon-α as a treatment modality for colchicine-resistant familial Mediterranean fever. J Rheumatol. 2008;35:1362–1365.
- Kötter I, Günaydin I, Zierhut M, et al. The use of interferon α in behçet disease: review of the literature. Semin Arthritis Rheum. 2004;33:320–335.
- Guarda G, Braun M, Staehli F, et al. Type I Interferon Inhibits Interleukin-1 Production and Inflammasome Activation. Immunity. 2011;34:213–223.
- Palacio N, Dangi T, Chung YR, et al. Early type I IFN blockade improves the efficacy of viral vaccines. J Exp Med. 2020;217:1–14.
- Channappanavar R, Meyerholz DK, Perlman S, et al. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest. 2019;129:3625–3639.
- Haji Abdolvahab M, Moradi-kalbolandi S, Zarei M, et al. Potential role of interferons in treating COVID-19 patients. Int Immunopharmacol. 2021;90:1–15.