Abstract
Objective. Fetal death can lead to disseminated intravascular coagulation or fetal death syndrome. However, currently it is not clear what are the changes in the coagulation system in patients with a fetal death without the fetal death syndrome. This study was undertaken to determine: (1) whether fetal death in the absence of fetal death syndrome is associated with changes in hemostatic markers in maternal plasma and amniotic fluid; and (2) whether maternal hypertension or placental abruption are associated with further changes in the hemostatic profile of these patients.
Methods. A cross-sectional study included the following: (1) determination of changes in markers of coagulation and platelet activation in patients with a normal pregnancy (n = 71) and patients with fetal demise (FD) without disseminated intravascular coagulation (n = 65); (2) determination of the amniotic fluid (AF)–tissue factor concentration and activity, as well as the concentrations of thrombin–antithrombin III (TAT) complexes in patients with a normal pregnancy (n = 25) and those with a FD (n = 36) who underwent amniocentesis. Plasma and AF concentrations of TAT complexes and TF (an index of thrombin generation), as well as maternal plasma concentrations of sCD40L (a marker of platelet activation), tissue factor pathway inhibitor (TFPI) and prothrombin fragments (PF) 1 + 2 (also an indicator of in vivo thrombin generation) were measured by ELISA. TF and TFPI activity were measured using chromogenic assays.
Results. (1) patients with FD without hypertension had a higher median maternal plasma sCD40L concentration than normal pregnant women (P < 0.001); (2) patients with FD had a higher median maternal plasma TAT III complexes than women with a normal pregnancy (P < 0.001); (3) the median AF–TF concentration and activity were higher in the FD group than in the normal pregnancy group (P < 0.001 for both); (4) patients with preeclampsia and FD had a higher median maternal plasma immunoreactive TF concentration than both normotensive patients with FD and women with normal pregnancies (P < 0.001 and P = 0.001, respectively); (5) the median plasma TF activity was higher in patients with preeclampsia and FD than that of women with normal pregnancies (P = 0.003); (6) among patients with a FD, those with placental abruption had a higher median AF–TAT complexes concentration than those without abruption (P = 0.0004).
Conclusions. Our findings indicate that: (1) mothers with a FD have evidence of increased in vivo thrombin generation and platelet activation than women with normal pregnancies; (2) patients with a FD and hypertension had a higher degree of TF activation than those with fetal death but without hypertension; (3) the AF of women with a FD had a higher median TF concentration and activity than that of normal pregnant women. AF can be a potential source for tissue factor and it participates in the development of fetal death syndrome in patients with a retained dead fetus.
Introduction
The term fetal demise syndrome refers to the association between fetal death and maternal coagulopathy Citation[1] reported in the 1950's among patients with Rh isoimmunization Citation[1-6]. Subsequent observations demonstrated that maternal disseminated intravascular coagulation (DIC) develops only if the dead fetus is retained in utero for more than 5 weeks, suggesting a chronic rather than an acute process as the underlying mechanism of disease Citation[4-6]. However, the precise genesis of this chronic process of the fetal death syndrome remains unclear, largely because no adequate assays for tissue factor were available when the pathophysiology of fetal death syndrome was attributed to an excess of tissue thromboplastin generated from the dead fetus.
The changes in the coagulation system during pregnancy are considered to be adaptive mechanisms for the prevention of bleeding at the time of delivery Citation[7-11]. Indeed, normal pregnancy is associated with a substantial increase in TF concentrations in both the decidua and myometrium Citation[12-15]. Similarly, high TF concentrations have been detected in the fetal membranes (mainly the amnion) and amniotic fluid (AF) Citation[7],Citation[16-19]. In contrast to the changes detected in AF and decidua, the maternal plasma immunoreactive TF concentrations of women with a normal pregnancy do not differ significantly from that of non-pregnant women Citation[11],Citation[20]. However, women in labor at term have significantly higher maternal plasma immunoreactive TF concentrations than that of non-pregnant women Citation[19]. In addition to the changes in TF, normal pregnancy is associated with excessive thrombin generation Citation[11],Citation[21], as determined by increased maternal concentrations of fibrinopeptide A, prothrombin fragments (PF) 1 and 2, and thrombin–antithrombin (TAT) III complexes Citation[7],Citation[22-24]. The concentration of these complexes increases further during and after normal labor Citation[25] and delivery Citation[23],Citation[25], and decreases later during the puerperium Citation[23],Citation[25]. Increased thrombin generation has been implicated as a mechanism of disease in several obstetrical syndromes including: preeclampsia Citation[26-33], small-for-gestational age (SGA) Citation[30],Citation[34],Citation[35], preterm labor Citation[21],Citation[36], and preterm prelabor rupture of membranes (PROM) Citation[21],Citation[37].
This study was undertaken to determine whether: (1) fetal death prior to the development of fetal death syndrome is associated with changes in hemostatic markers in the maternal plasma as well as AF; and (2) whether maternal hypertension or placental abruption are associated with further changes in the hemostatic profile of these patients.
The following analytes were studied in the maternal plasma: (1) TF concentration and activity, as well as markers of platelet activation (sCD40L), all needed for thrombin generation; (2) markers of thrombin generation (TAT III complexes, PF 1 + 2); and (3) the concentration and activity of tissue factor pathway inhibitor (TFPI), the extrinsic pathway inhibitor. The intra-amniotic concentrations of TAT III complexes, TF concentrations, and activity were studied as well.
Material and methods
Study groups and inclusion criteria
A cross-sectional study was conducted in two steps: (1) determination of the maternal plasma concentration of TF, TFPI, PF 1 + 2, TAT III complexes and sCD40L, as well as the activity of TF and TFPI in patients with a normal pregnancy (n = 71) and patients with a fetal demise (FD) without (DIC) (n = 65); (2) determination of the AF–TF concentration and activity as well as the TAT complexes concentrations in patients with normal pregnancies and those with a FD. AF was available for analysis from 36 (55.4%) patients in the FD group and 3 (4.2%) patients in the normal pregnancy group. Therefore, we identified 22 patients with a normal pregnancy outcome who underwent amniocentesis at a similar gestational age as patients with a FD, and included them as normal controls.
Samples and data were retrieved from the Perinatology Research Branch bank of biological samples and clinical databases. Many of these samples have been previously employed to study the biology of inflammation, hemostasis, angiogenesis regulation, and growth factor concentrations in non-pregnant women, normal pregnant women, and those with pregnancy complications. All participating women provided a written informed consent prior to the collection of maternal blood and AF. The Institutional Review Boards of both Wayne State University and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD/NIH/DHHS) approved the collection and utilization of samples for research purposes.
Clinical definitions
Women with normal pregnancies met the following criteria: (1) no medical, obstetrical, or surgical complications at the time of the study; (2) gestational age at blood collection or amniocentesis ranging from 20 to 41 weeks; and (3) delivery of a term neonate, appropriate for gestational age, without complications. Patients with multiple pregnancies or fetuses with congenital and/or chromosomal anomalies were excluded. Fetal death was defined as a FD diagnosed and confirmed by ultrasound ≥ the 19th week of gestation. SGA was diagnosed as a birth weight below the 10th percentile for gestational age Citation[38]. Hypertensive disorders of pregnancy included gestational hypertension and preeclampsia. Gestational hypertension was defined as hypertension (systolic blood pressure ≥140 mmHg or diastolic blood pressure ≥90 mmHg on at least two occasions, 4 h to 1 week apart) that was diagnosed after 20 weeks of gestation without proteinuria. Preeclampsia was defined in the presence of hypertension and proteinuria diagnosed after 20 weeks of gestation (≥300 mg in a 24-h urine collection or at least one dipstick measurement ≥2+) Citation[39]. DIC was defined according to the DIC score of the scientific and standardization committee on disseminated intravascular coagulation of the international society on thrombosis and hemostasis Citation[40],Citation[41]. The parameters included in this score are: platelet count, fibrin related markers, prothrombin time and fibrinogen level, and a score ≥5 is required for diagnosis. Upon admission to the labor and delivery suite (the time when the maternal blood sample was collected), none of the women included in the FD group had an abnormal value in platelet count (<100,000/ml), prothrombin time (>3 s), or fibrinogen level (<1 g/l or 100 mg/dl).
Placental histopathologic examinations
Placental tissue samples were taken by systematic random sampling, subsequently fixed in 10% neutral buffered formalin overnight and embedded in paraffin. Five micrometer-thick paraffin sections, stained with haematoxylin and eosin, were examined using bright-field light microscopy. Three pathologists blinded to the clinical information performed histopathologic examinations, and the placental lesions were classified according to a diagnostic schema proposed by Redline et al. Citation[42]. For the analysis of the association between the different analytes and specific placental lesions, we grouped them into three categories: (1) lesions associated with intra-amniotic infection and/or inflammation; (2) findings associated with maternal under perfusion; and (3) lesions associated with fetal vascular thrombotic occlusive disease.
Sample collection
Maternal blood collection
Blood samples were collected with a vacutainer into 0.109 M trisodium citrate anticoagulant solution (BD; San Jose, CA), centrifuged at 1300 g for 10 min at 4°C, and stored at −70°C until assayed.
Amniotic fluid collection
Amniocentesis was performed at the discretion of the treating physician. The procedure was done trans-abdominally under ultrasonographic guidance in order to determine the microbiologic state of the amniotic cavity, fetal karyotype, injection of AF dye to rule out rupture of the fetal membranes, or for assessment of fetal lung maturity. AF was transported to the laboratory in a capped plastic syringe and cultured for aerobic and anaerobic bacteria as well as genital mycoplasmas. White blood cell (WBC) count, glucose concentration and Gram stain for microorganisms were performed in AF shortly after collection. Intra-amniotic infection was defined by the presence of a positive AF culture for microorganisms and inflammation by an AF WBC count ≥100 cells/ml. The results of the AF analyses were used for clinical management.
Immuno and chromogenic assays
Human tissue factor immunoassay
TF concentrations were determined by sensitive and specific immunoassays obtained from American Diagnostica (Greenwich, CT), which recognizes TF-apo, TF, and TF-FVII complexes. The assays were conducted according to the manufacturer's recommendations. The calculated coefficient of variation (CV) in our laboratory was 5.3%, and the sensitivity was 10 pg/ml.
Human tissue factor activity assay
TF activity was determined by a chromogenic assay obtained from American Diagnostica (Greenwich, CT). The assays were conducted according to the manufacturer's recommendations. In our laboratory, the calculated intra-assay CV was 3.77%, whereas the inter-assay CV was 6.25%, and the sensitivity of this assay was 0.53 pM.
Human tissue factor pathway inhibitor immunoassay
The concentrations of TFPI in maternal plasma were determined by sensitive and specific immunoassays obtained from American Diagnostica (Greenwich, CT). The TFPI ELISA employs, as the capture antibody, a murine anti-TFPI monoclonal that is directed against the Kunitz-1 domain of the TFPI molecule; therefore, it detects both TFPI-1 and TFPI-2, while measuring the total TFPI plasma concentrations. The assay was conducted according to the manufacturer's recommendations. The calculated CV in our laboratory was 6.6%, and the sensitivity was approximately 10 ng/ml.
Human tissue factor pathway inhibitor activity assay
TFPI activity was determined by a chromogenic assay obtained from American Diagnostica (Greenwich, CT). The assays were conducted according to the manufacturer's recommendations. In our laboratory, the calculated intra-assay CV was 5.51%, whereas the inter-assay CV was 8.74%, and the sensitivity was 0.017 unit/ml.
Human sCD40L immunoassay
Maternal plasma sCD40L concentrations were determined by sensitive and specific immunoassays obtained from R&D Systems (Minneapolis, MN). The assays were conducted according to the manufacturer's recommendations. In our laboratory, the calculated intra-assay CV was 5%, whereas the inter-assay CV was 6.2%, and the sensitivity of the sCD40L assay was 4.2 pg/ml.
Human thrombin–antithrombin III complexes immunoassay
Maternal plasma concentrations of TAT III complexes were determined by a specific immunoassay. The manufacturer (Dade Behring, Marburg, Germany) validated this assays for citrated plasma samples, and we conducted these immunoassays following to their instructions. In our laboratory, the calculated inter-assay CV was 7.67% whereas the intra-assay CVs was 5.98% and the sensitivity was 0.81 μg/L.
Human prothrombin fragments 1 + 2 immunoassay
Maternal plasma concentration of PF 1 + 2 was determined by specific immunoassay. The manufacturer (Dade Behring, Marburg, Germany) validated this assay for citrated plasma samples and we conducted these immunoassays according to their instructions. In our laboratory, the calculated inter-assay CV in our laboratory was 5.35%, whereas the intra-assay CVs was 5.98% and the sensitivity was for PF1 + 2 assay 5.33 pmol/L.
Statistical analysis
Tissue factor concentrations and activity, TFPI concentration and activity, sCD40L, TAT III complexes and PF 1 + 2 concentrations in the plasma and/or AF were not normally distributed; thus, Kruskal–Wallis test with post-hoc Mann–Whitney U test were used for comparisons of continuous variables. Comparisons of proportions were performed by Chi-square and Fisher's exact tests. A P value < 0.05 was considered statistically significant. Analyses were performed with SPSS, version 12 (SPSS, Chicago, IL).
Results
Demographic and clinical characteristics of the study groups
displays the demographic and clinical characteristics of the study groups. Patients with a FD had a lower median gestational age at delivery and neonatal birthweight than women with a normal pregnancy. There were no significant differences between the study groups in the gestational age at sample collection (normal pregnancy: median 31.6 weeks (range 20–38.3) vs. FD: median 31 weeks (range 19.0–40.0), P = 0.7).
Table I. Demographic and clinical characteristics of patients with normal pregnancies and those with fetal demise.
In the FD group, 23.1% (15/65) of the patients had hypertensive disorders of pregnancy, of which 9.3% (6/65) had gestational hypertension and 13.8% (9/65) had preeclampsia. Because our group has previously reported the association between preeclampsia and elevated TF and TFPI maternal plasma concentrations, we analyzed the changes in the coagulation profile of the maternal plasma in three steps: (1) all patients with a FD were included as one group; (2) in order to study the effect of gestational hypertension, we subdivided the FD group into two subgroups, patients with a FD without hypertension( n = 50) and patients with a FD with hypertension (n = 15), and both groups were compared with the normal pregnancy group; (3) in the third analysis, we studied the effect of preeclampsia, and the FD group was subdivided in two subgroups: patients with a FD without hypertension(n = 50) and patients with a FD and preeclampsia (n = 9). Patients with gestational hypertension were excluded from this analysis and both groups were compared with the normal pregnancy group.
Changes in the coagulation profile of women with normal pregnancies and patients with a fetal demise
In the normal pregnancy group, there was a positive correlation between maternal plasma immunoreactive TF concentrations and activity (r = 0.327, P = 0.006), but not between maternal plasma TFPI concentration and activity (r = 0.22, P = 0.06).
The median maternal plasma immunoreactive TF concentration and activity did not differ significantly between patients with a FD and women with normal pregnancies (P = 0.6 and P = 0.06, respectively; ). In contrast, the median maternal plasma TFPI concentration, but not its activity, was lower in patients with a FD than in women with a normal pregnancy (P < 0.001 and P = 0.093, respectively; ).
Table II. Maternal plasma coagulation parameters of patients with a normal pregnancy and those with a fetal demise.
Patients with a FD had a significantly higher median maternal plasma sCD40L than those with a normal pregnancy (P < 0.001). In addition, the median maternal plasma concentrations of TAT III complexes and PF 1 + 2 were higher in patients with a FD than in normal pregnant women (P < 0.001 for both comparisons; ).
The effect of preeclampsia on the coagulation profile of patients with fetal demise
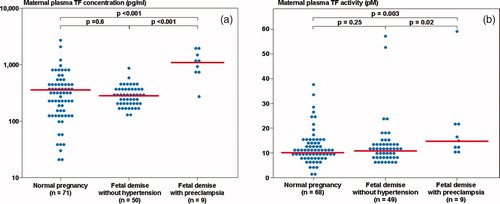
The median maternal plasma TF concentration and activity differed significantly among patients with a FD with and without preeclampsia, as well as women with normal pregnancies (Kruskal–Wallis, P < 0.001 and P = 0.001, respectively). Patients with a FD and preeclampsia had a higher median maternal plasma TF concentration and activity than that of women with normal pregnancies (P = <0.001 and P = 0.003, respectively), and than that of patients with a FD without hypertension (P < 0.001 and P = 0.02, respectively) ((a) and 1(b)).
Figure 1. (a) Maternal plasma tissue factor (TF) concentrations among women with normal pregnancies (median 345.7 pg/ml, range 21.7–2662.2) and patients with a fetal demise with preeclampsia and those without hypertension (with preeclampsia: median 1167.0 pg/ml, range 287.5–1987.0; without hypertension: median 284.2 pg/ml, range 123.6–851.6); (b) Maternal plasma TF activity among women with normal pregnancies (median 9.9 pM, range 0.7–37.6) and patients with a fetal demise with preeclampsia and those without hypertension (with preeclampsia: median 16.2 pM, range 11.6–59.3; without hypertension: median 10.9 pM, range 5.3–57.2).
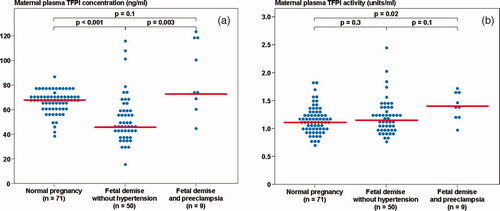
The median maternal plasma TFPI concentration and activity differed significantly among patients with a FD with and without preeclampsia and women with normal pregnancies (Kruskal–Wallis, P < 0.001 and P = 0.021, respectively). Patients with a FD and preeclampsia had a higher median maternal plasma TFPI activity, but not TFPI concentration, than women with normal pregnancies (P = 0.005, P = 0.1, respectively). Among the FD group, patients with preeclampsia had a higher median maternal plasma TFPI concentration than those without hypertension, but the median TFPI activity did not differ significantly between the groups (P = 0.003 and P = 0.1, respectively) ((a) and 2(b)).
Figure 2. (a) Maternal plasma tissue factor pathway inhibitor (TFPI) concentration among women with normal pregnancies (median 66.7 ng/ml, range 37.4–86.5) and patients with a fetal demise with preeclampsia and those without hypertension (with preeclampsia: median 72.4 ng/ml, range 45.3–123.7; without hypertension: median 45.6 ng/ml, range 13.6–115.2); (b) Maternal plasma TFPI activity among women with normal pregnancies (median 1.1 unit/ml, range 0.7–2.7) and patients with a fetal demise with preeclampsia and those without hypertension (with preeclampsia: median 1.4 unit/ml, range 1.0–1.7; without hypertension: median 1.15 unit/ml, range 0.8–2.5).
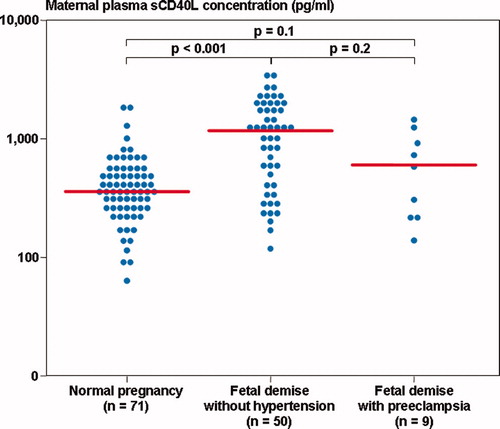
The median maternal plasma sCD40L differs significantly between patients with a FD with and without preeclampsia and women with normal pregnancies (Kruskal–Wallis, P < 0.001). However, in the comparison between the groups, the median maternal plasma sCD40L concentration did not differ significantly between patients with a FD with preeclampsia and women with a normal pregnancy (P = 0.1) or those with a FD without hypertension (P = 0.1) ().
Figure 3. Maternal plasma sCD40L concentration among women with normal pregnancies (median 369.5 pg/ml, range 63.5–1848.7) and patients with a fetal demise with preeclampsia and those without hypertension (with preeclampsia: median 732.0 pg/ml, range 271.3–1714.0; without hypertension: median 1213.3 pg/ml, range 118.0–3818.0).
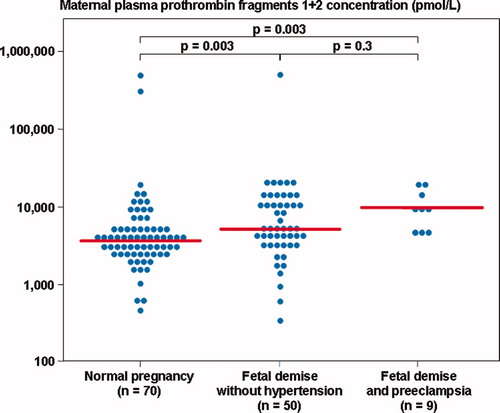
The median plasma TAT III complexes and PF 1 + 2 concentration differed significantly among these groups (Kruskal–Wallis, P < 0.001, for both comparisons). However, patients with preeclampsia and FD had a significantly higher median plasma TAT III complexes and PF 1 + 2 concentration than women with a normal pregnancy (P = 0.003 for both comparisons), but not those with FD without hypertension (P = 0.1 and P = 0.25, respectively) ( and ).
Figure 4. Maternal plasma thrombin–antithrombin (TAT) III complexes concentration among women with normal pregnancies (median 15.7 μg/l, range 5.2–107, 941.0) and patients with a fetal demise with preeclampsia and those without hypertension (with preeclampsia: median 105.8 μg/l, range 11.2–2002.8; without hypertension: median 28.9 μg/l, range 6.6–61, 256.8).
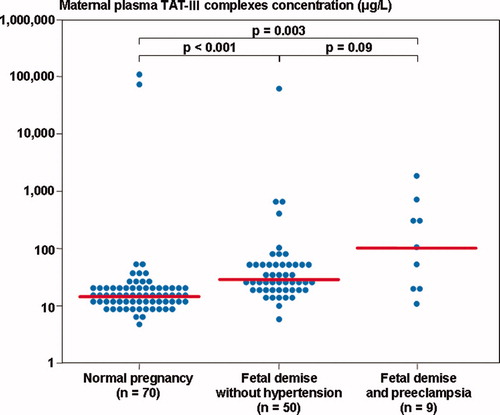
Figure 5. Maternal plasma prothrombin fragments 1 + 2 concentration among women with normal pregnancies (median 3786.0 pmol/l, range 513.4–486, 310.0) and patients with a fetal demise with preeclampsia and those without hypertension (with preeclampsia: median 8715.4 pmol/l, range 4320.4–18, 954.2; without hypertension: median 5205.4 pmol/l, range 343.4–494, 564.0).
The association between placental lesions and changes in markers of coagulation, platelet activation and thrombin generation in patients with a normal pregnancy and those with a fetal demise
Histologic examination of the placenta was performed in 15.5% (11/71) of women with a normal pregnancy and 80% (52/65) of patients with a FD. Within the latter group, 82% (41/50) of patients with a FD without hypertension and 73.3% (11/15) of those with a FD and hypertension had histologic examination of their placenta. Among patients with a FD, those with hypertension had a higher rate of villous infarcts, syncytial knots, intervillous fibrin deposition, and acute atherosis of basal plate arteries or decidual arterioles than patients with FD without hypertension ().
Table III. Placental lesions in patients with a fetal demise according to the presence of hypertensive diseases of pregnancy
Table IV. Demographic and clinical characteristics of patients with normal pregnancies and those with a fetal demise that had amniocentesis
Among patients with a FD and hypertension, women with placental fetal vascular thrombotic occlusive lesions had a significantly higher median maternal plasma sCD40L concentration than those without these placental findings (P = 0.036). In addition, among patients in the FD group without hypertension, those who had placental lesions associated with maternal under perfusion had a significantly higher PF 1 + 2 than those without these lesions (P = 0.025). Of note, among patients with normal pregnancies there were no associations between the median maternal plasma concentration of the different analytes and a specific placental lesion (–).
Supplementary table I. The association between placental lesion and markers for coagulation in women with normal pregnancy.
Supplementary table II. The association between placental lesion and markers for coagulation in women with fetal demise without hypertension.
Supplementary table III. Supplementary The association between placental lesion and markers for coagulation in women with fetal demise with hypertension.
Changes in the median amniotic fluid TF concentrations and activity as well as the concentrations of thrombin–antithrombin III complexes
The demographic and clinical characteristic of the patients are presented in .
The TFPI concentrations, TFPI activity and PF 1 + 2 assays had severe matrix effect in AF and were excluded from the study. Of note, we could not detect immunoreactive sCD40L in the AF.
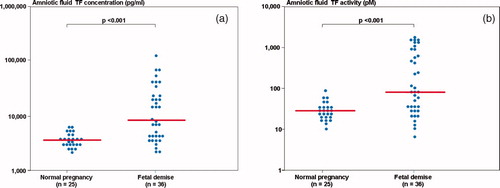
Patients with a FD had higher median AF–TF concentration and activity than those with normal pregnancies (P < 0.001, for both comparisons; (a) and 6(b), respectively). Moreover, among patients with a FD there was a significant correlation between the AF–TF concentrations and activity (r = 0.88, P < 0.0001). The median AF–TAT III complexes concentration did not differ significantly between the groups (normal pregnancy: median: 66.3 μg/l, range 11.4–2265.4 vs. FD: median: 59.3 μg/l, range: 13.6–15,425.3; P = 0.7).
Figure 6. (a) Amniotic fluid tissue factor concentration among women with normal pregnancies (median 3710.4 pg/ml, range 2198.8–6268) and patients with a fetal demise (median 8535.4 pg/ml, range 2208.2–125,990.0); (b) Amniotic fluid tissue factor activity among women with normal pregnancies (median 28.4 pM, range 10.2–84.9) and patients with a fetal demise (median 81.6 pM, range 7.2–1603.4).
The three patients in the FD group with placental abruption had a higher median AF–TAT III complexes concentration than women with normal pregnancies (FD with abruption: median 754.1 μg/l range 398.8–10,573.8; P = 0.015) and patients with FD without abruption (FD without abruption: median 55.5 μg/l range 13.6–15,425.3; P = 0.012).
Discussion
Principal findings of the study
(1) The median maternal plasma sCD40L (an indicator of in vivo platelet activation) concentration was higher in patients with FD than that of normal pregnant women; (2) similarly, patients with a FD had a higher median maternal plasma concentration of TAT III complexes and PF 1 and 2, indices of increase thrombin generation in vivo, than those of women with a normal pregnancy; (3) a higher median AF–TF concentration and activity was detected in patients with a FD than in women with normal pregnancies. However, this was not detected in maternal plasma. Collectively, these results suggest that mothers with a fetal death have a state of platelet activation, and increased thrombin generation, even in the absence of DIC.
Evidence of maternal platelet activation in fetal death
Soluble CD40L, a marker for platelet activation Citation[43-45], participates in platelet aggregation and platelet–leukocyte conjugation Citation[46]. Upon activation, platelets express CD40L on their membrane (from which it is subsequently cleaved by CD40), and the soluble form of CD40L (sCD40L) can be measured in the plasma Citation[47],Citation[48]. Activated platelets are the source of more than 90% of soluble sCD40L in the plasma Citation[47], and this cytokine has been proposed as a measurable marker of platelet activation Citation[43],Citation[44].
The finding of a higher median plasma concentration of sCD40L in patients with a FD is novel and can be interpreted as indicating in vivo platelet activation in these patients. There is a solid body of evidence supporting platelet activation in preeclampsia and fetal growth restriction Citation[49-73]. Indeed, elevated maternal plasma and platelet expression of markers for platelet activation, including P-selectin (CD62p) Citation[52-54],Citation[61],Citation[63],Citation[65],Citation[74],Citation[75], Annexin V Citation[53],Citation[57],Citation[76], platelet factor 4 Citation[29],Citation[49],Citation[77-81], and sCD40L concentrations Citation[82-85] were reported in these conditions. Moreover, we have recently reported that the median maternal plasma sCD40L concentration is elevated in patients with preterm labor and intact membranesCitation[86]; of note, all these obstetrical syndromes were associated with elevated median maternal plasma TAT III complexes concentration Citation[21],Citation[30]. There is, however, a paucity of data about the degree and mechanisms responsible for platelet activation in cases of fetal death.
Normotensive patients with a FD had a higher median plasma sCD40L concentration than women with a normal pregnancy as well as than that of patients with preeclampsia and a FD. Thus, patients with a FD have platelet activation that cannot be accounted by hypertensive disorders of pregnancy. Moreover, this finding suggests that in patients with a dead fetus, activated platelets may play a role in the generation of thrombin as reflected by the elevated median plasma concentration of PF 1 + 2 and TAT complexes. However, the association between platelet activation and higher indices of thrombin generation is complex, and each system can be activated by itself, or by the other Citation[87-95].
Several possible mechanisms may contribute to platelet activation in normotensive patients with a FD: (1) platelet activation can be mediated by local/intervillous activation of the coagulation cascade, which may be initiated by the loss of inhibitory mechanisms of TF activation in the trophoblasts. This can lead to an increased thrombin generation, which may activate platelets through the protease-activated receptors (PAR) Citation[96-103], and the glycoprotein Ib receptors Citation[102-105] that also amplify the effect of thrombin on PAR-1 Citation[106]. The activation of maternal platelets leads to an increased median plasma sCD40L concentration and to the release of partially-activated factor V from α granules onto the platelet surface Citation[107], as well as the activation of the intrinsic pathway of coagulation through factors IX and XI Citation[108-111]. (2) Platelet activation can occur as a consequence of maternal inflammation associated with active villous destruction by infiltrating neutrophils. This has been reported in RU486 treated cynomolgus monkeys with a FD Citation[112]. Furthermore, in women with a dead fetus, neutrophils may be activated by the elevated maternal plasma C5a concentrations Citation[113] and subsequently, these neutrophils can activate platelets by secreting cathepsin G Citation[114]. Collectively, the increased platelet activation reported here in patients with a FD may further propagate the increase in indices for thrombin generation associated with this obstetrical syndrome.
Changes in indices for thrombin generation, thrombin–antithrombin complexes and prothrombin fragments 1 + 2, in patients with a fetal demise
The finding of higher median maternal plasma concentrations of TAT III complexes and PF 1 + 2 in patients with a FD without maternal DIC is novel, as they represent indices of increased in vivo thrombin generation. This observation is in keeping with previous reports of an increased thrombin generation in several obstetrical syndromes, including pre-eclampsia Citation[26-32],Citation[115], a small-for-gestational age neonateCitation[30],Citation[34],Citation[35], preterm laborCitation[21],Citation[36], and preterm PROM Citation[21],Citation[37]. Although DIC and hypofibrinogenemia in patients presenting with a retained dead fetus were reported already in the early 1950's Citation[1-6], the maternal plasma thrombin generation in women with a dead fetus without DIC was not studied. This is the first report of elevated median maternal plasma concentrations of TAT III complexes and PF 1 + 2, indices for in vivo thrombin generation, in these patients.
The underlying mechanisms leading to excessive thrombin generation in patients with a fetal death have not been elucidated. Abnormalities of the placenta, specifically vascular placental lesions, may be associated with the above observation. Indeed, among patients with a FD without hypertension, those who had vascular placental lesions consistent with maternal under perfusion had a higher median plasma PF 1 + 2 concentration than those without these lesions. Moreover, the human trophoblast has properties of endothelial cells and can regulate the degree of activation of the coagulation cascade in the intervillous space Citation[116],Citation[117]. Unlike the endothelium of other organs, the trophoblast constantly presents the active placental isoform of TF on its surface Citation[15],Citation[116-120]. This isoform is larger than recombinant TF and has a higher affinity for FVIIa Citation[121],Citation[122], which may lead to increased activation of the coagulation cascade. Consequently, in dead fetuses, the trophoblast may no longer suppress the procoagulant activity of the placental isoform of TF, leading to subclinical activation of the hemostatic system and increased thrombin generation in the maternal plasma, even in the absence of DIC.
Changes in amniotic fluid and maternal plasma tissue factor concentration and activity in patients with a fetal demise
This is the first study to report that patients with a FD have an increased median AF–TF concentration and activity. This is relevant because the release of TF has traditionally been considered responsible for the initiation of the maternal consumptive coagulopathy in cases of FD Citation[1]. High TF concentrations have been previously detected in the fetal membranes (mainly the amnion) and AF of normal pregnant women Citation[16-19]. Moreover, the AF–TF concentrations were higher than that of the maternal plasma Citation[19]. In our study, the median AF–TF concentration in normal pregnant women was 10 fold higher than in maternal plasma. Furthermore, the median AF–TF concentration and activity in women with a FD without DIC, was higher than those in the maternal plasma of normal pregnant women (30 fold and 7–8 fold, respectively).
In contrast to the changes observed in AF, the median maternal plasma TF concentration and activity did not differ between patients with a FD without hypertension and those with a normal pregnancy. This raises a question regarding the role of circulating immunoreactive TF in the pathophysiology of fetal death syndrome.
It is unclear whether the maternal plasma TF concentration reflects activation of the coagulation cascade or systemic maternal inflammation. Indeed, the procoagulant activity of plasma immunoreactive TF (blood-borne TF) of non-pregnant patients Citation[123-128] is controversial. Blood-borne TF has very little or no procoagulant activity, and only the administration of exogenous active TF generates a whole blood and plasma clot after the inhibition of the contact factor (factor XIIa) Citation[128]. Others Citation[124-127] have proposed that blood-borne TF does not initiate the coagulation cascade but rather, propagates clot formation by attaching to activated platelets, further enhancing the coagulation process. However, our finding of a positive correlation between maternal plasma TF concentration and its activity in women with a normal pregnancy suggests that circulating TF has procoagulant properties that may be part of the hypercoagulable state of pregnancy.
The absence of a significant difference in the median maternal plasma immunoreactivity and activity of TF between women with normal pregnancies and normotensive patients with a FD raises a question regarding the validity of the proposed role of tissue thromboplastin in the mechanism leading to DIC in women with fetal death syndrome. When these mechanisms were proposed, there was no direct assay to measure the presence and concentrations of placental TF in the maternal circulation of patients with a FD. However, this view was supported by the experiment reported by Schneider Citation[129] which demonstrated that the intravenous injection of placental extracts into mice leads to the death of the animal through DIC, which can be prevented by the administration of heparin Citation[129]. The author identified thromboplastin as the causative agent through its effect on the clotting time and its chemical properties, and measured its activity by the one-stage prothrombin-time method Citation[129]. Currently, early detection of fetal death by ultrasound and the practice of induction of labor have almost abolished the development of fetal death syndrome in western societies. Thus, the verification of this hypothesis by advanced immunoreactive and functional assays is less feasible. Indeed, we did not have any patient with maternal DIC in our study group. Therefore, we suggest that among patients presenting with a FD without preeclampsia and without DIC, blood-borne TF may have little or no role in the increased maternal plasma indices for thrombin generation detected in these patients, and the role of the increased AF–TF concentration and activity awaits further study.
Preeclampsia and tissue factor concentration and activity
In the FD group, preeclampsia was associated with a higher median maternal plasma TF concentration and activity than that of patients with a normal pregnancy. Moreover, the median TF concentration of patients with preeclampsia was also higher than in patients with a FD without hypertension. These findings are consistent with previous studies Citation[130],Citation[131], suggesting that elevated TF immunoreactivity and activity may be associated with the pathophysiologic process leading to preeclampsia, rather than a consequence of the fetal death.
The hemostatic system in patients having a fetal demise without DIC
FD is syndromic in nature and results from different underlying mechanisms, which can be acute (i.e. placental abruption Citation[132-140], penetrating trauma Citation[141], prolapse of umbilical cord Citation[142],Citation[143]) or chronic (placental infarcts Citation[144-160], infection Citation[145],Citation[146],Citation[161-163], chromosomal or anatomic malformation Citation[145],Citation[146],Citation[161],Citation[163],Citation[164]). The profile of maternal plasma and AF coagulation presented herein may be associated with some of the underlying mechanisms leading to fetal death.
In patients with preeclampsia and a FD, the median TF plasma concentration and activity were higher than those of normal pregnant women, suggesting activation of the coagulation cascade that is probably due to preeclampsia rather than the dead fetus. On the other hand, patients with a FD without hypertension had an elevated median plasma sCD40L concentration, which indicates increased platelet activation.
All the patients included in our study did not have DIC, suggesting that the increased platelet activation and elevated indices of thrombin generation reflect subclinical activation of the hemostatic system. Evidence in support of this view is derived from the study by Brummel et al. Citation[165], who calculated that at least 26 ± 6 nM of TAT III complexes are needed in order to reach the propagation phase of thrombin generation (909 μg/l of TAT III complexes). In the current study, translation of the median TAT III complexes concentration into molar quantity revealed that women with normal pregnancies had median maternal plasma of TAT complexes 0.17 nM, which increased to 0.31 nM in the case of patients with a FD, and to 0.55 nM among those with a FD and hypertension. Moreover, the highest concentration was observed in the AF of patients with placental abruption, reaching 7.96 nM of TAT III complexes.
Consequently, these findings suggest that although women with a FD have higher thrombin generation than women with normal pregnancies, they may still be at the initiation phase of thrombin generation. Thus, in the absence of an acute cause of fetal death, like abruption, that will lift the dam and unleash the waterfall of the coagulation cascade, leading to uncontrolled thrombin generation, maternal consumption coagulopathy and DIC; the activation of the hemostatic system is subclinical and well tolerated by patients with a dead fetus at least for a limited time period.
Summary
We report the maternal plasma and AF elements of the initiation and propagation steps of the coagulation cascade, as well as indices of thrombin generation, which is the outcome of the activity of the formers and the inhibitor of initiation step. The question regarding the role of TF in the development of maternal DIC remains unanswered and can not be addressed by our study, because none of the patients had DIC and the current medical practice of induction of labor near to the diagnosis of a dead fetus seems to prevent this maternal complication. On the other hand, we report here for the first time that patients with a FD have increased platelet activation and indices of thrombin generation even before the development of DIC, suggesting subclinical activation of the hemostatic system.
Acknowledgement
This research was supported (in part) by the Perinatology Research Branch, Division of Intramural Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH, DHHS.
References
- Romero R, Copel J A, Hobbins J C. Intrauterine fetal demise and hemostatic failure: the fetal death syndrome. Clin Obstet Gynecol 1985; 28: 24–31
- Weiner A E, Reid D E, Roby C C, Diamond L K. Coagulation defects with intrauterine death from Rh isosensitization. Am J Obstet Gynecol 1950; 60: 1015–1022
- Reid D E, Weiner A E, Roby C C, Diamond L K. Maternal afibrinogenemia associated with long-standing intrauterine fetal death. Am J Obstet Gynecol 1953; 66: 500–506
- Pritchard J A, Ratnoff O D. Studies of fibrinogen and other hemostatic factors in women with intrauterine death and delayed delivery. Surg Gynecol Obstet 1955; 101: 467–477
- Tricomi V, Kohl S G. [Fetal death in utero.]. Am J Obstet Gynecol 1957; 74: 1092–1097
- Pritchard J A. Fetal death in utero. Obstet Gynecol 1959; 14: 573–580
- de Boer K, ten Cate J W, Sturk A, Borm J J, Treffers P E. Enhanced thrombin generation in normal and hypertensive pregnancy. Am J Obstet Gynecol 1989; 160: 95–100
- Yuen P M, Yin J A, Lao T T. Fibrinopeptide A levels in maternal and newborn plasma. Eur J Obstet Gynecol Reprod Biol 1989; 30: 239–244
- Sorensen J D, Secher N J, Jespersen J. Perturbed (procoagulant) endothelium and deviations within the fibrinolytic system during the third trimester of normal pregnancy. A possible link to placental function. Acta Obstet Gynecol Scand 1995; 74: 257–261
- Walker M C, Garner P R, Keely E J, Rock G A, Reis M D. Changes in activated protein C resistance during normal pregnancy. Am J Obstet Gynecol 1997; 177: 162–169
- Bellart J, Gilabert R, Miralles R M, Monasterio J, Cabero L. Endothelial cell markers and fibrinopeptide A to D-dimer ratio as a measure of coagulation and fibrinolysis balance in normal pregnancy. Gynecol Obstet Invest 1998; 46: 17–21
- Lockwood C J, Krikun G, Schatz F. The decidua regulates hemostasis in human endometrium. Semin Reprod Endocrinol 1999; 17: 45–51
- Lockwood C J, Krikun G, Schatz F. Decidual cell-expressed tissue factor maintains hemostasis in human endometrium. Ann N.Y Acad Sci 2001; 943: 77–88
- Kuczynski J, Uszynski W, Zekanowska E, Soszka T, Uszynski M. Tissue factor (TF) and tissue factor pathway inhibitor (TFPI) in the placenta and myometrium. Eur J Obstet Gynecol Reprod Biol 2002; 105: 15–19
- Erlich J, Parry G C, Fearns C, Muller M, Carmeliet P, Luther T, Mackman N. Tissue factor is required for uterine hemostasis and maintenance of the placental labyrinth during gestation. Proc Natl Acad Sci U.S.A 1999; 96: 8138–8143
- Creter D. Amnioplastin: new reagent for coagulation tests. Lancet 1977; 2: 251
- Omsjo I H, Oian P, Maltau J M, Osterud B. Thromboplastin activity in amniotic fluid. Gynecol Obstet Invest 1985; 19: 1–5
- Lockwood C J, Bach R, Guha A, Zhou X D, Miller W A, Nemerson Y. Amniotic fluid contains tissue factor, a potent initiator of coagulation. Am J Obstet Gynecol 1991; 165: 1335–1341
- Uszynski M, Zekanowska E, Uszynski W, Kuczynski J. Tissue factor (TF) and tissue factor pathway inhibitor (TFPI) in amniotic fluid and blood plasma: implications for the mechanism of amniotic fluid embolism. Eur J Obstet Gynecol Reprod Biol 2001; 95: 163–166
- Holmes V A, Wallace J M. Haemostasis in normal pregnancy: a balancing act?. Biochem Soc Trans 2005; 33: 428–432
- Chaiworapongsa T, Espinoza J, Yoshimatsu J, Kim Y M, Bujold E, Edwin S, Yoon B H, Romero R. Activation of coagulation system in preterm labor and preterm premature rupture of membranes. J Matern Fetal Neonatal Med 2002; 11: 368–373
- Reinthaller A, Mursch-Edlmayr G, Tatra G. Thrombin-antithrombin III complex levels in normal pregnancy with hypertensive disorders and after delivery. Br J Obstet Gynaecol 1990; 97: 506–510
- Uszynski M. Generation of thrombin in blood plasma of non-pregnant and pregnant women studied through concentration of thrombin-antithrombin III complexes. Eur J Obstet Gynecol Reprod Biol 1997; 75: 127–131
- Reber G, Amiral J, de M P. Modified antithrombin III levels during normal pregnancy and relationship with prothrombin fragment F1 + 2 and thrombin-antithrombin complexes. Thromb Res 1998; 91: 45–47
- Andersson T, Lorentzen B, Hogdahl H, Clausen T, Mowinckel M C, Abildgaard U. Thrombin-inhibitor complexes in the blood during and after delivery. Thromb Res 1996; 82: 109–117
- VanWijk M J, Boer K, Berckmans R J, Meijers J C, van der Post J A, Sturk A, Vanbavel E, Nieuwland R. Enhanced coagulation activation in preeclampsia: the role of APC resistance, microparticles and other plasma constituents. Thromb Haemost 2002; 88: 415–420
- Kobayashi T, Sumimoto K, Tokunaga N, Sugimura M, Nishiguchi T, Kanayama N, Terao T. Coagulation index to distinguish severe preeclampsia from normal pregnancy. Semin Thromb Hemost 2002; 28: 495–500
- Hayashi M, Numaguchi M, Ohkubo N, Yaoi Y. Blood macrophage colony-stimulating factor and thrombin-antithrombin III complex concentrations in pregnancy and preeclampsia. Am J Med Sci 1998; 315: 251–257
- Hayashi M, Inoue T, Hoshimoto K, Negishi H, Ohkura T, Inaba N. Characterization of five marker levels of the hemostatic system and endothelial status in normotensive pregnancy and pre-eclampsia. Eur J Haematol 2002; 69: 297–302
- Chaiworapongsa T, Yoshimatsu J, Espinoza J, Kim Y M, Berman S, Edwin S, Yoon B H, Romero R. Evidence of in vivo generation of thrombin in patients with small-for-gestational-age fetuses and pre-eclampsia. J Matern Fetal Neonatal Med 2002; 11: 362–367
- Kobayashi T, Terao T. Preeclampsia as chronic disseminated intravascular coagulation. Study of two parameters: thrombin-antithrombin III complex and D-dimers. Gynecol Obstet Invest 1987; 24: 170–178
- Kobayashi T, Tokunaga N, Sugimura M, Suzuki K, Kanayama N, Nishiguchi T, Terao T. Coagulation/fibrinolysis disorder in patients with severe preeclampsia. Semin Thromb Hemost 1999; 25: 451–454
- Schjetlein R, Abdelnoor M, Haugen G, Husby H, Sandset P M, Wisloff F. Hemostatic variables as independent predictors for fetal growth retardation in preeclampsia. Acta Obstet Gynecol Scand 1999; 78: 191–197
- Hayashi M, Ohkura T. Elevated levels of serum macrophage colony-stimulating factor in normotensive pregnancies complicated by intrauterine fetal growth restriction. Exp Hematol 2002; 30: 388–393
- Ballard H S, Marcus A J. Primary and secondary platelet aggregation in uraemia. Scand J Haematol 1972; 9: 198–203
- Elovitz M A, Baron J, Phillippe M. The role of thrombin in preterm parturition. Am J Obstet Gynecol 2001; 185: 1059–1063
- Rosen T, Kuczynski E, O'Neill L M, Funai E F, Lockwood C J. Plasma levels of thrombin-antithrombin complexes predict preterm premature rupture of the fetal membranes. J Matern Fetal Med 2001; 10: 297–300
- Gonzalez R P, Gomez R M, Castro R S, Nien J K, Merino P O, Etchegaray A B, Carstens M R, Medina L H, Viviani P G, Rojas I T. [A national birth weight distribution curve according to gestational age in chile from 1993 to 2000]. Rev Med Chil 2004; 132: 1155–1165
- ACOG practice bulletin. Diagnosis and management of preeclampsia and eclampsia. Number 33, January 2002. Obstet Gynecol 2002; 99: 159–167
- Taylor F B, Jr, Toh C H, Hoots W K, Wada H, Levi M. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost 2001; 86: 1327–1330
- Toh C H, Hoots W K. The scoring system of the Scientific and Standardisation Committee on Disseminated Intravascular Coagulation of the International Society on Thrombosis and Haemostasis: a 5-year overview. J Thromb Haemost 2007; 5: 604–606
- Redline R W, Heller D, Keating S, Kingdom J. Placental diagnostic criteria and clinical correlation–a workshop report. Placenta 2005; 26(Suppl A)S114–S117
- Patel J V, Lim H S, Nadar S, Tayebjee M, Hughes E A, Lip G Y. Abnormal soluble CD40 ligand and C-reactive protein concentrations in hypertension: relationship to indices of angiogenesis. J Hypertens 2006; 24: 117–121
- Lip G Y, Patel J V, Hughes E, Hart R G. High-sensitivity C-reactive protein and soluble CD40 ligand as indices of inflammation and platelet activation in 880 patients with nonvalvular atrial fibrillation: relationship to stroke risk factors, stroke risk stratification schema, and prognosis. Stroke 2007; 38: 1229–1237
- Hermann A, Rauch B H, Braun M, Schror K, Weber A A. Platelet CD40 ligand (CD40L)–subcellular localization, regulation of expression, and inhibition by clopidogrel. Platelets 2001; 12: 74–82
- Chakrabarti S, Varghese S, Vitseva O, Tanriverdi K, Freedman J E. CD40 ligand influences platelet release of reactive oxygen intermediates. Arterioscler Thromb Vasc Biol 2005; 25: 2428–2434
- Henn V, Slupsky J R, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek R A. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998; 391: 591–594
- Henn V, Steinbach S, Buchner K, Presek P, Kroczek R A. The inflammatory action of CD40 ligand (CD154) expressed on activated human platelets is temporally limited by coexpressed CD40. Blood 2001; 98: 1047–1054
- Saleh A A, Bottoms S F, Farag A M, Dombrowski M P, Welch R A, Norman G, Mammen E F. Markers for endothelial injury, clotting and platelet activation in preeclampsia. Arch Gynecol Obstet 1992; 251: 105–110
- Leiberman J R, Hagay Z J, Mazor M, Aharon M, Nathan I, Dvilansky A. Plasma and urine beta-thromboglobulin in severe preeclampsia. Arch Gynecol Obstet 1988; 243: 165–168
- Holthe M R, Lyberg T, Staff A C, Berge L N. Leukocyte-platelet interaction in pregnancies complicated with preeclampsia. Platelets 2005; 16: 91–97
- Lok C A, Nieuwland R, Sturk A, Hau C M, Boer K, Vanbavel E, Vanderpost J A. Microparticle-associated P-selectin reflects platelet activation in preeclampsia. Platelets 2007; 18: 68–72
- Tomer A. Platelet activation as a marker for in vivo prothrombotic activity: detection by flow cytometry. J Biol Regul Homeost Agents 2004; 18: 172–177
- Yoneyama Y, Suzuki S, Sawa R, Kiyokawa Y, Power G G, Araki T. Plasma adenosine levels and P-selectin expression on platelets in preeclampsia. Obstet Gynecol 2001; 97: 366–370
- Bar J, Zosmer A, Hod M, Elder M G, Sullivan M H. The regulation of platelet aggregation in vitro by interleukin-1beta and tumor necrosis factor-alpha: changes in pregnancy and in pre-eclampsia. Thromb Haemost 1997; 78: 1255–1261
- Bagamery K, Landau R, Kvell K, Graham J. Different platelet activation levels in non-pregnant, normotensive pregnant, pregnancy-induced hypertensive and pre-eclamptic women. A pilot study of flow cytometric analysis. Eur J Obstet Gynecol Reprod Biol 2005; 121: 117–118
- Harlow F H, Brown M A, Brighton T A, Smith S L, Trickett A E, Kwan Y L, Davis G K. Platelet activation in the hypertensive disorders of pregnancy. Am J Obstet Gynecol 2002; 187: 688–695
- Missfelder-Lobos H, Teran E, Lees C, Albaiges G, Nicolaides K H. Platelet changes and subsequent development of pre-eclampsia and fetal growth restriction in women with abnormal uterine artery Doppler screening. Ultrasound Obstet Gynecol 2002; 19: 443–448
- Felfernig-Boehm D, Salat A, Vogl S E, Murabito M, Felfernig M, Schmidt D, Mittlboeck M, Husslein P, Mueller M R. Early detection of preeclampsia by determination of platelet aggregability. Thromb Res 2000; 98: 139–146
- Myatt L, Miodovnik M. Prediction of preeclampsia. Semin Perinatol 1999; 23: 45–57
- Konijnenberg A, van der Post J A, Mol B W, Schaap M C, Lazarov R, Bleker O P, Boer K, Sturk A. Can flow cytometric detection of platelet activation early in pregnancy predict the occurrence of preeclampsia? A prospective study. Am J Obstet Gynecol 1997; 177: 434–442
- Bodis J, Torok A, Tinneberg H R. Hypothesis of preeclampsia requires inclusion of the role of platelets. Am J Obstet Gynecol 1997; 177: 243–244
- Konijnenberg A, Stokkers E W, van der Post J A, Schaap M C, Boer K, Bleker O P, Sturk A. Extensive platelet activation in preeclampsia compared with normal pregnancy: enhanced expression of cell adhesion molecules. Am J Obstet Gynecol 1997; 176: 461–469
- Ahlawat S, Pati H P, Bhatla N, Fatima L, Mittal S. Plasma platelet aggregating factor and platelet aggregation studies in pre-eclampsia. Acta Obstet Gynecol Scand 1996; 75: 428–431
- Halim A, Kanayama N, el M E, Nakashima A, Bhuiyan A B, Khatun S, Terao T. Plasma P selectin (GMP-140) and glycocalicin are elevated in preeclampsia and eclampsia: their significances. Am J Obstet Gynecol 1996; 174: 272–277
- Janes S L, Kyle P M, Redman C, Goodall A H. Flow cytometric detection of activated platelets in pregnant women prior to the development of pre-eclampsia. Thromb Haemost 1995; 74: 1059–1063
- Janes S L, Goodall A H. Flow cytometric detection of circulating activated platelets and platelet hyper-responsiveness in pre-eclampsia and pregnancy. Clin Sci (Lond) 1994; 86: 731–739
- Hutt R, Ogunniyi S O, Sullivan M H, Elder M G. Increased platelet volume and aggregation precede the onset of preeclampsia. Obstet Gynecol 1994; 83: 146–149
- Norris L A, Gleeson N, Sheppard B L, Bonnar J. Whole blood platelet aggregation in moderate and severe pre-eclampsia. Br J Obstet Gynaecol 1993; 100: 684–688
- Louden K A, Broughton P F, Heptinstall S, Fox S C, Mitchell J R, Symonds E M. Platelet reactivity and serum thromboxane B2 production in whole blood in gestational hypertension and pre-eclampsia. Br J Obstet Gynaecol 1991; 98: 1239–1244
- Pekonen F, Rasi V, Ammala M, Viinikka L, Ylikorkala O. Platelet function and coagulation in normal and preeclamptic pregnancy. Thromb Res 1986; 43: 553–560
- Romero R, Snyder E, Scott D, Oyarzun E, Hobbins J C, Duffy T P. Beta-thromboglobulin during normal pregnancy, labor, and puerperium. Am J Perinatol 1988; 5: 109–112
- Hagay Z J, Leiberman J R, Mazor M, Aharon M, Dvilansky A. [Plasma and urinary beta-thromboglobulin as indicators of platelet activation in pre-eclampsia]. Harefuah 1988; 114: 427–429
- Karalis I, Nadar S K, Al Y E, Blann A D, Lip G Y. Platelet activation in pregnancy-induced hypertension. Thromb Res 2005; 116: 377–383
- Yoneyama Y, Suzuki S, Sawa R, Miura A, Doi D, Otsubo Y, Araki T. Plasma nitric oxide levels and the expression of P-selectin on platelets in preeclampsia. Am J Obstet Gynecol 2002; 187: 676–680
- Erez O, Hallak M, Luber A, Maymon E, Mazor M, Tomer A. Platelets Activation Marker as Predictor of Severe Preeclampsia in Patients with History of Preeclampsia. Am J Obstet Gynecol 2001; 185: S175
- Ballegeer V C, Spitz B, De Baene L A, Van Assche A F, Hidajat M, Criel A M. Platelet activation and vascular damage in gestational hypertension. Am J Obstet Gynecol 1992; 166: 629–633
- Ayhan A, Akkok E, Urman B, Yarali H, Dundar S, Kirazli S. Beta-thromboglobulin and platelet factor 4 levels in pregnancy and preeclampsia. Gynecol Obstet Invest 1990; 30: 12–14
- Csaicsich P, Deutinger J, Tatra G. Platelet specific proteins (beta-thromboglobulin and platelet factor 4) in normal pregnancy and in pregnancy complicated by preeclampsia. Arch Gynecol Obstet 1989; 244: 91–95
- Socol M L, Weiner C P, Louis G, Rehnberg K, Rossi E C. Platelet activation in preeclampsia. Am J Obstet Gynecol 1985; 151: 494–497
- Arocha-Pinango C L, Ojeda A, Lopez G, Garcia L, Linares J. beta-thromboglobulin (beta-TG) and platelet factor 4 (PF4) in obstetrical cases. Acta Obstet Gynecol Scand 1985; 64: 115–120
- Alacacioglu I, Ozcan M A, Piskin O, Yuksel F, Alacacioglu A, Demirkan F, Ozsan H G, Polat M, Ozgenc Y, Undar B. Increased concentration of soluble CD40 ligand in preeclampsia. Clin Appl Thromb Hemost 2007; 13: 201–205
- Laskowska M, Laskowska K, Leszczynska-Gorzelak B, Oleszczuk J. sCD40 ligand determined in maternal and umbilical cord blood in pregnancies complicated by pre-eclampsia with and without intrauterine growth retardation. Gynecol Obstet Invest 2007; 64: 8–13
- Mellembakken J R, Solum N O, Ueland T, Videm V, Aukrust P. Increased concentrations of soluble CD40 ligand, RANTES and GRO-alpha in preeclampsia–possible role of platelet activation. Thromb Haemost 2001; 86: 1272–1276
- Oron G, Ben-Haroush A, Hod M, Orvieto R, Bar J. Serum-soluble CD40 ligand in normal pregnancy and in preeclampsia. Obstet Gynecol 2006; 107: 896–900
- Erez O, Romero R, Hoppensteadt D, Fareed J, Chaiworapongsa T, Kusanovic J P, Mazaki-Tovi S, Gotsch F, Than N G, Vaisbuch E, et al. Premature labor: a state of platelet activation?. J Perinat Med 2008; 36: 377–387
- Holvoet P, Collen D. Thrombosis and atherosclerosis. Curr Opin Lipidol 1997; 8: 320–328
- Dimitrow P P, Undas A, Bober M, Tracz W, Dubiel J S. Obstructive hypertrophic cardiomyopathy is associated with enhanced thrombin generation and platelet activation. Heart 2008; 94: e21
- Maroney S A, Haberichter S L, Friese P, Collins M L, Ferrel J P, Dale G L, Mast A E. Active tissue factor pathway inhibitor is expressed on the surface of coated platelets. Blood 2007; 109: 1931–1937
- Sobocka M B, Sobocki T, Babinska A, Hartwig J H, Li M, Ehrlich Y H, Kornecki E. Signaling pathways of the F11 receptor (F11R; a.k.a. JAM-1, JAM-A) in human platelets: F11R dimerization, phosphorylation and complex formation with the integrin GPIIIa. J Recept Signal Transduct Res 2004; 24: 85–105
- Weiss H J, Lages B. Platelet prothrombinase activity and intracellular calcium responses in patients with storage pool deficiency, glycoprotein IIb-IIIa deficiency, or impaired platelet coagulant activity–a comparison with Scott syndrome. Blood 1997; 89: 1599–1611
- Banno Y, Nakashima S, Ohzawa M, Nozawa Y. Differential translocation of phospholipase C isozymes to integrin-mediated cytoskeletal complexes in thrombin-stimulated human platelets. J Biol Chem 1996; 271: 14989–14994
- Miller G J. Lipoproteins and the haemostatic system in atherothrombotic disorders. Baillieres Clin Haematol 1994; 7: 713–732
- Esumi N, Todo S, Imashuku S. Platelet aggregating activity mediated by thrombin generation in the NCG human neuroblastoma cell line. Cancer Res 1987; 47: 2129–2135
- Yoshida K, Kimura H. Presence of calmodulin in human platelet cytoskeletons and its concentration change upon activation of platelets. Biochim Biophys Acta 1984; 801: 290–297
- Sood R, Zogg M, Westrick R J, Guo Y H, Kerschen E J, Girardi G, Salmon J E, Coughlin S R, Weiler H. Fetal gene defects precipitate platelet-mediated pregnancy failure in factor V Leiden mothers. J Exp Med 2007; 204: 1049–1056
- Hoffman M, Monroe D M, III. A cell-based model of hemostasis. Thromb Haemost 2001; 85: 958–965
- Hung D T, Vu T K, Wheaton V I, Ishii K, Coughlin S R. Cloned platelet thrombin receptor is necessary for thrombin-induced platelet activation. J Clin Invest 1992; 89: 1350–1353
- Wu C C, Teng C M. Comparison of the effects of PAR1 antagonists, PAR4 antagonists, and their combinations on thrombin-induced human platelet activation. Eur J Pharmacol 2006; 546: 142–147
- Lisman T, Weeterings C, de Groot P G. Platelet aggregation: involvement of thrombin and fibrin(ogen). Front Biosci 2005; 10: 2504–2517
- Keuren J F, Wielders S J, Ulrichts H, Hackeng T, Heemskerk J W, Deckmyn H, Bevers E M, Lindhout T. Synergistic effect of thrombin on collagen-induced platelet procoagulant activity is mediated through protease-activated receptor-1. Arterioscler Thromb Vasc Biol 2005; 25: 1499–1505
- Lova P, Campus F, Lombardi R, Cattaneo M, Sinigaglia F, Balduini C, Torti M. Contribution of protease-activated receptors 1 and 4 and glycoprotein Ib-IX-V in the G(i)-independent activation of platelet Rap1B by thrombin. J Biol Chem 2004; 279: 25299–25306
- Dubois C, Steiner B, Meyer Reigner S C. Contribution of PAR-1, PAR-4 and GPIbalpha in intracellular signaling leading to the cleavage of the beta3 cytoplasmic domain during thrombin-induced platelet aggregation. Thromb Haemost 2004; 91: 733–742
- Soslau G, Class R, Morgan D A, Foster C, Lord S T, Marchese P, Ruggeri Z M. Unique pathway of thrombin-induced platelet aggregation mediated by glycoprotein Ib. J Biol Chem 2001; 276: 21173–21183
- Dicker I B, Pedicord D L, Seiffert D A, Jamieson G A, Greco N J. Both the high affinity thrombin receptor (GPIb-IX-V) and GPIIb/IIIa are implicated in expression of thrombin-induced platelet procoagulant activity. Thromb Haemost 2001; 86: 1065–1069
- De Candia E, Hall S W, Rutella S, Landolfi R, Andrews R K, De C R. Binding of thrombin to glycoprotein Ib accelerates the hydrolysis of Par-1 on intact platelets. J Biol Chem 2001; 276: 4692–4698
- Monkovic D D, Tracy P B. Functional characterization of human platelet-released factor V and its activation by factor Xa and thrombin. J Biol Chem 1990; 265: 17132–17140
- Ahmad S S, Rawala-Sheikh R, Walsh P N. Comparative interactions of factor IX and factor IXa with human platelets. J Biol Chem 1989; 264: 3244–3251
- Ahmad S S, Rawala-Sheikh R, Ashby B, Walsh P N. Platelet receptor-mediated factor X activation by factor IXa. High-affinity factor IXa receptors induced by factor VIII are deficient on platelets in Scott syndrome. J Clin Invest 1989; 84: 824–828
- Ahmad S S, Rawala-Sheikh R, Walsh P N. Platelet receptor occupancy with factor IXa promotes factor X activation. J Biol Chem 1989; 264: 20012–20016
- Rawala-Sheikh R, Ahmad S S, Ashby B, Walsh P N. Kinetics of coagulation factor X activation by platelet-bound factor IXa. Biochemistry 1990; 29: 2606–2611
- Sinosich M J, Wolf J P, Williams R F, Hodgen G D. RU 486 mediated leukocytic inflammatory reaction at the utero-placental interface. Asia Oceania J Obstet Gynaecol 1989; 15: 375–381
- Richani K, Romero R, Soto E, Espinoza J, Nien J K, Chaiworapongsa T, Refuerzo J, Blackwell S, Edwin S S, Santolaya-Forgas J, et al. Unexplained intrauterine fetal death is accompanied by activation of complement. J Perinat Med 2005; 33: 296–305
- Ferrer-Lopez P, Renesto P, Schattner M, Bassot S, Laurent P, Chignard M. Activation of human platelets by C5a-stimulated neutrophils: a role for cathepsin G. Am J Physiol 1990; 258: C1100–C1107
- Schjetlein R, Haugen G, Wisloff F. Markers of intravascular coagulation and fibrinolysis in preeclampsia: association with intrauterine growth retardation. Acta Obstet Gynecol Scand 1997; 76: 541–546
- Sood R, Sholl L, Isermann B, Zogg M, Coughlin S R, Weiler H. Maternal Par4 and platelets contribute to defective placenta formation in mouse embryos lacking thrombomodulin. Blood 2008; 112: 585–591
- Sood R, Kalloway S, Mast A E, Hillard C J, Weiler H. Fetomaternal cross talk in the placental vascular bed: control of coagulation by trophoblast cells. Blood 2006; 107: 3173–3180
- Aharon A, Brenner B, Katz T, Miyagi Y, Lanir N. Tissue factor and tissue factor pathway inhibitor levels in trophoblast cells: implications for placental hemostasis. Thromb Haemost 2004; 92: 776–786
- Isermann B, Sood R, Pawlinski R, Zogg M, Kalloway S, Degen J L, Mackman N, Weiler H. The thrombomodulin-protein C system is essential for the maintenance of pregnancy. Nat Med 2003; 9: 331–337
- Lanir N, Aharon A, Brenner B. Procoagulant and anticoagulant mechanisms in human placenta. Semin Thromb Hemost 2003; 29: 175–184
- Butenas S, Brummel-Ziedins K E, Mann K G. Real human tissue factor. FASEB J 2006; 20: A47
- Butenas S, Orfeo T, Brummel-Ziedins K E, Mann K G. Tissue factor in thrombosis and hemorrhage. Surgery 2007; 142: S2–14
- Osterud B. Cellular interactions in tissue factor expression by blood monocytes. Blood Coagul Fibrinolysis 1995; 6(Suppl 1)S20–S25
- Kobayashi M, Wada H, Wakita Y, Shimura M, Nakase T, Hiyoyama K, Nagaya S, Minami N, Nakano T, Shiku H. Decreased plasma tissue factor pathway inhibitor levels in patients with thrombotic thrombocytopenic purpura. Thromb Haemost 1995; 73: 10–14
- Balasubramanian V, Grabowski E, Bini A, Nemerson Y. Platelets, circulating tissue factor, and fibrin colocalize in ex vivo thrombi: real-time fluorescence images of thrombus formation and propagation under defined flow conditions. Blood 2002; 100: 2787–2792
- Balasubramanian V, Vele O, Nemerson Y. Local shear conditions and platelet aggregates regulate the incorporation and activity of circulating tissue factor in ex-vivo thrombi. Thromb Haemost 2002; 88: 822–826
- Chou J, Mackman N, Merrill-Skoloff G, Pedersen B, Furie B C, Furie B. Hematopoietic cell-derived microparticle tissue factor contributes to fibrin formation during thrombus propagation. Blood 2004; 104: 3190–3197
- Butenas S, Bouchard B A, Brummel-Ziedins K E, Parhami-Seren B, Mann K G. Tissue factor activity in whole blood. Blood 2005; 105: 2764–2770
- Schneider C. The active principle of placental toxin:thromboplastin its inactivator in blood: antithromboplastin. Am J Physiol 1947; 149: 123
- Erez O, Hoppensteadt D, Than N G, Fareed J, Mazaki-Tovi S, Espinoza J, Chaiworapongsa T, Yoon B H, Hassan S S, Gotsch F, et al. Tissue Factor and its Natural Inhibitor in Preeclampsia and SGA. J Matern Fetal Neonatal Med 2008; 12: 855–869
- Bellart J, Gilabert R, Angles A, Piera V, Miralles R M, Monasterio J, Cabero L. Tissue factor levels and high ratio of fibrinopeptide A:D-dimer as a measure of endothelial procoagulant disorder in pre-eclampsia. Br J Obstet Gynaecol 1999; 106: 594–597
- HOWIE B O. Intra-uterine foetal death with defective maternal blood-clotting mechanism. Proc R Soc Med 1956; 49: 93–96
- DeValera E. Abruptio placentae. Am J Obstet Gynecol 1968; 100: 599–606
- Sher G, Statland B E. Abruptio placentae with coagulopathy: a rational basis for management. Clin Obstet Gynecol 1985; 28: 15–23
- Oyelese Y, Ananth C V. Placental abruption. Obstet Gynecol 2006; 108: 1005–1016
- Beller F K, Uszynski M. Disseminated intravascular coagulation in pregnancy. Clin Obstet Gynecol 1974; 17: 250–278
- Boulton F E, Letsky E. Obstetric haemorrhage: causes and management. Clin Haematol 1985; 14: 683–728
- Finley B E. Acute coagulopathy in pregnancy. Med Clin North Am 1989; 73: 723–743
- Parasnis H, Raje B, Hinduja I N. Relevance of plasma fibrinogen estimation in obstetric complications. J Postgrad Med 1992; 38: 183–185
- Thomson A J, Greer I A. Non-haemorrhagic obstetric shock. Baillieres Best Pract Res Clin Obstet Gynaecol 2000; 14: 19–41
- Awwad J T, Azar G B, Seoud M A, Mroueh A M, Karam K S. High-velocity penetrating wounds of the gravid uterus: review of 16 years of civil war. Obstet Gynecol 1994; 83: 259–264
- Enakpene C A, Omigbodun A O, Arowojolu A O. Perinatal mortality following umbilical cord prolapse. Int J Gynaecol Obstet 2006; 95: 44–45
- Aagaard-Tillery K M, Holmgren C, Lacoursiere D Y, Houssain S, Bloebaum L, Satterfield R, Branch D W, Varner M W. Factors associated with nonanomalous stillbirths: the Utah Stillbirth Database 1992–2002. Am J Obstet Gynecol 2006; 194: 849–854
- Korteweg F J, Gordijn S J, Timmer A, Holm J P, Ravise J M, Erwich J J. A placental cause of intra-uterine fetal death depends on the perinatal mortality classification system used. Placenta 2008; 29: 71–80
- Copper R L, Goldenberg R L, DuBard M B, Davis R O. Risk factors for fetal death in white, black, and Hispanic women. Collaborative Group on Preterm Birth Prevention. Obstet Gynecol 1994; 84: 490–495
- Lammer E J, Brown L E, Anderka M T, Guyer B. Classification and analysis of fetal deaths in Massachusetts. JAMA 1989; 261: 1757–1762
- Menghrajani P, Osterheld M C. Significance of hemorrhagic endovasculitis in placentae from stillbirths. Pathol Res Pract 2008; 204: 389–394
- ACOG Committee Opinion No. 383. Evaluation of stillbirths and neonatal deaths. Obstet Gynecol 2007; 110: 963–966
- Corabian P, Scott N A, Lane C, Guyon G. Guidelines for investigating stillbirths: an update of a systematic review. J Obstet Gynaecol Can 2007; 29: 560–567
- Marchetti D, Belviso M, Marino M, Gaudio R. Evaluation of the placenta in a stillborn fetus to estimate the time of death. Am J Forensic Med Pathol 2007; 28: 38–43
- Pasupathy D, Smith G C. The analysis of factors predicting antepartum stillbirth. Minerva Ginecol 2005; 57: 397–410
- Sander C M, Gilliland D, Richardson A, Foley K M, Fredericks J. Stillbirths with placental hemorrhagic endovasculitis: a morphologic assessment with clinical implications. Arch Pathol Lab Med 2005; 129: 632–638
- Pierce B T, Martin L S, Hume R F, Jr., Calhoun B C, Muir-Padilla J, Salafia C M. Relationship between the extent of histologic villous mineralization and stillbirth in aneuploid and euploid fetuses. J Soc Gynecol Investig 2002; 9: 290–293
- Magee J F. Investigation of stillbirth. Pediatr Dev Pathol 2001; 4: 1–22
- Kraus F T, Acheen V I. Fetal thrombotic vasculopathy in the placenta: cerebral thrombi and infarcts, coagulopathies, and cerebral palsy. Hum Pathol 1999; 30: 759–769
- Incerpi M H, Miller D A, Samadi R, Settlage R H, Goodwin T M. Stillbirth evaluation: what tests are needed?. Am J Obstet Gynecol 1998; 178: 1121–1125
- Adelson P, Spurrett B, Trudinger B, Frommer M. A New South Wales population-based study of stillbirths weighing 2,500 g or more. Aust N.Z J Obstet Gynaecol 1993; 33: 166–173
- Gruenberger W, Gerstner G J. The causes of antepartum fetal death: a clinico-pathological study. Clin Exp Obstet Gynecol 1980; 7: 210–214
- Bendon R W. Review of some causes of stillbirth. Pediatr Dev Pathol 2001; 4: 517–531
- Arias F, Romero R, Joist H, Kraus F T. Thrombophilia: a mechanism of disease in women with adverse pregnancy outcome and thrombotic lesions in the placenta. J Matern Fetal Med 1998; 7: 277–286
- Cartlidge P H, Dawson A T, Stewart J H, Vujanic G M. Value and quality of perinatal and infant postmortem examinations: cohort analysis of 400 consecutive deaths. BMJ 1995; 310: 155–158
- Fretts R C, Boyd M E, Usher R H, Usher H A. The changing pattern of fetal death, 1961–1988. Obstet Gynecol 1992; 79: 35–39
- Pauli R M, Reiser C A. Wisconsin Stillbirth Service Program: II. Analysis of diagnoses and diagnostic categories in the first 1,000 referrals. Am J Med Genet 1994; 50: 135–153
- Faye-Petersen O M, Guinn D A, Wenstrom K D. Value of perinatal autopsy. Obstet Gynecol 1999; 94: 915–920
- Brummel K E, Paradis S G, Butenas S, Mann K G. Thrombin functions during tissue factor-induced blood coagulation. Blood 2002; 100: 148–152