
Abstract
Objective: Biallelic pathogenic variants in TOE1 cause pontocerebellar hypoplasia type 7 (PCH7), a rare neurological condition characterized by psychomotor retardation, spastic paraplegia, seizures, gonadal abnormalities and brain anomalies. Currently, only 14 postnatally diagnosed PCH7 patients have been described. However, the prenatal clinical profile of PCH7 has not yet been reported.
Method: Whole-exome sequencing (WES) was performed to screen for causal variants.
Results: We report the pedigree of a Chinese woman with two eventful pregnancies with fetuses that showed brain anomalies, including microcephaly, cerebral anomalies, enlarged ventricles, corpus callosum thinning, abnormal lateral fissure, underdeveloped insula and pons and brainstem hypoplasia. Interestingly, corpus callosum thinning was observed in fetus 1 but not in fetus 2. An abnormal lateral fissure and an underdeveloped insula were shown in fetus 2 but not fetus 1. Biallelic variants c.716T > C (p.Phe239Ser) and c.955C > T (p.His319Tyr) in TOE1 were identified in both fetuses.
Conclusion: We first describe the prenatal features of a Chinese pedigree with PCH7 caused by biallelic pathogenic variants in TOE1, with phenotypic variability observed even within the same family. Novel phenotypes, an abnormal lateral fissure and an underdeveloped insula were observed in the fetus in our study. These findings will enrich our knowledge of the clinical characteristics, management and genetic counseling of PCH7.
1. Introduction
Pontocerebellar hypoplasia (PCH) is a heterogeneous group of neurodegenerative disorders affecting the function of the brainstem and cerebellum, often leading to severe psychomotor retardation, microcephaly, hypotonia and cognitive impairments [Citation1,Citation2]. The onset of PCH occurs mainly in the prenatal period or at birth, with varying severity from the lethal neonatal subtype to milder forms in which children can survive into adolescence [Citation3]. Currently, at least 23 genes have been recognized as causing 17 subtypes of PCH [Citation3–9].
Pontocerebellar hypoplasia type 7 (PCH7, MIM #614969) is caused by biallelic pathogenic variants in TOE1. PCH7 is characterized by early-onset progressive microcephaly, severe psychomotor delay, hypotonia, breathing abnormalities and hypogonadism. Brain imaging has shown pontocerebellar hypoplasia, cerebral atrophy and thinning of the corpus callosum in patients with PCH7 [Citation10–12]. PCH7 is a very rare autosomal recessive disorder, with only 14 postnatally diagnosed PCH7 patients reported worldwide; 14 variants in TOE1 have been identified to date [Citation10,Citation12]. Chen and coworkers recently reported a Chinese patient with atypical PCH7, suggesting highly variable clinical phenotypes caused by variants in TOE1 [Citation12]. Currently, no prenatal diagnosis of PCH7 caused by pathogenic TOE1 variants has been reported. Here, we report the pedigree of a Chinese woman with two sequential eventful pregnancies with fetuses that showed similar brain anomalies. WES revealed biallelic variants in TOE1 in both fetuses: c.716T > C (p. Phe239Ser) and c.955C > T (p. His319Tyr). Each one of the two variants was separately reported in two unrelated patients with PCH7. We describe the clinical phenotypes of the two fetuses in detail. Our findings provide the first insight into the prenatal clinical characteristics of PCH7 and reconfirm the pathogenic nature of both variants.
2. Materials and methods
2.1. Ethical compliance
This study was granted by the Ethics Committee of Dongguan Maternal and Child Health Care Hospital. Written informed consent was gained from the legal guardians for the publication of any potentially identifiable images or data involved in this study.
2.2. Trio-based whole exome sequencing
Trio-based whole exome sequencing (WES) was performed for the pedigree to screen for causal variants. Sequencing was performed with an Illumina NovaSeq 6000 system (Illumina, San Diego, CA, USA). Suspected variants were confirmed by Sanger sequencing. The pathogenicity of the variants was evaluated according to ACMG/AMP guidelines [Citation13].
3. Results
3.1. Clinical presentation
In the first pregnancy of a 26-year-old primiparous woman, the prenatal ultrasonography at 23 weeks detected microcephaly (187.4 mm; −2.9 SD), cerebellar hypoplasia (22.9 mm; −1.0 SD), dilated bilateral lateral ventricles (>10 mm), no cavum septum pellucidum and corpus callosum thinning in the female fetus (). A precise midsagittal T2-weighted brain MRI sequence determined that the diameter of the pontine was 5.69 mm (reference: 8.48 mm) at 24 weeks of pregnancy, implying pons and brainstem hypoplasia (). The karyotype and chromosomal microarray of the amniocytes were normal, confirming a female karyotype (46,XX). The woman decided to terminate the pregnancy at 27 weeks of gestation considering a possible poor prognosis caused by brain structure anomalies.
Figure 1. Radiographic findings for the two fetuses. An ultrasound image showing microcephaly (187.4 mm; -2.9 SD) (a), cerebellar hypoplasia (22.9 mm; –1.0 SD) (b), dilated bilateral lateral ventricles (>10 mm) (c), and corpus callosum thinning (d, e). A precise midsagittal T2-weighted brain MRI sequence determined that the diameter of the pontine was 5.69 mm (reference: 8.48 mm) at 24 weeks of pregnancy, implying pons and brainstem hypoplasia in the first fetus (f). B Ultrasound images showing the second fetus at 25 weeks of gestation with recurrent microcephalia (216.2 mm; -2.4 SD) (a), cerebellar hypoplasia (24.9 mm; -2SD) (b), an abnormal lateral fissure and an underdeveloped insula (6.5 mm) (c). The precise midsagittal T2-weighted brain MRI sequence showed that the diameter of the pontine was 7.14 mm (reference: 8.96 mm) at 26 weeks of pregnancy, and the shape was not full, indicating pons and brainstem hypoplasia (d). Ultrasonic scans at 29 weeks showed persistent microcephaly (255.2 mm; –2.3 SD) (e), cerebellar hypoplasia (29.7 mm; -2.6 SD) (f), an underdeveloped insula (9.0 mm) (g), an abnormal lateral fissure and bilateral lateral ventricles with a diameter of 9.6 mm (h).
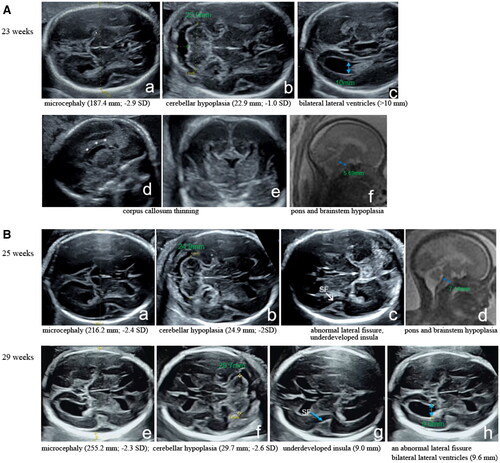
The woman had another planned pregnancy after two years. Unfortunately, the anomaly scans at 25 weeks of gestation showed recurrent microcephalia (216.2 mm; −2.4 SD), cerebellar hypoplasia (24.9 mm; –2SD), an abnormal lateral fissure, an underdeveloped insula (6.5 mm) and fetal growth retardation in the male fetus. The precise midsagittal T2-weighted brain MRI sequence showed that the diameter of the pontine was 7.14 mm (reference: 8.96 mm) at 26 weeks of pregnancy, and the shape was not full, indicating pons and brainstem hypoplasia. Ultrasonic scans at 29 weeks showed persistent microcephaly (255.2 mm; −2.3 SD), cerebellar hypoplasia (29.7 mm; −2.6 SD), an underdeveloped insula (9.0 mm), an abnormal lateral fissure, bilateral lateral ventricles with a diameter of 9.6 mm and fetal growth retardation (). Ambiguous genitalia were not observed. The karyotype and chromosomal microarray of the amniocytes were normal, confirming a male karyotype (46,XY). Then the woman again decided to terminate the pregnancy and undergo WES. In this study, fetal growth estimation was calculated using Hadlock’s formula.
3.2. Genetic analysis
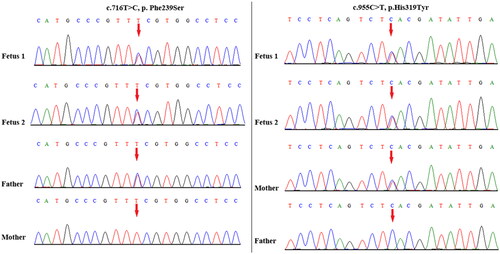
Trio-based WES revealed the biallelic variants c.716T > C (p. Phe239Ser) and c.955C > T (p. His319Tyr) in TOE1, which were inherited from the father and mother, respectively (). Both variants have been previously reported in two unrelated PCH7 patients diagnosed postnatally and were confirmed to be pathogenic by functional experiments in the literature [Citation10]. Next, we analyzed the DNA sample from the first fetus, which had been retained in our laboratory, and it revealed the same TOE1 biallelic variants as expected.
Figure 2. Variant confirmation by Sanger sequencing. Compound heterozygous variants c.716T > C and c.955C > T in TOE1 were detected in both fetuses and their asymptomatic parents. The red arrow indicates the variant site.
4. Discussion
TOE1 is a target of the EGR1 transcription factor and encodes an Asp-Glu-Asp-Asp (DEDD) deadenylase that localizes to Cajal bodies in the nucleus, where it acts as a 3-exonuclease to promote the maturation of small nuclear RNAs (snRNAs) instead of mRNA [Citation14,Citation15]. TOE1 plays a crucial role in maintaining genome stability by mediating the maturation of the RNA template used by telomerase for telomere replication [Citation16]. TOE1 is involved in cell cycle regulation through upregulation of p21. TOE1 also binds p53 to modulate its transactivation potential [Citation17]. Morpholino-knockdown of toe1 in zebrafish simulates the human PCH7 phenotype. Thus, TOE1 has been determined to be the causative gene of PCH7 [Citation10]. To date, only 14 postnatally diagnosed PCH7 patients have been reported, and 14 variants have been identified in TOE1, including 10 missense variants, two splicing variants, one nonsense variant and one frameshift variant. The main clinical manifestations include early-onset progressive microcephaly (10/12), developmental delay (14/14), intellectual disability (10/10), hypotonia (8/11), hypertonia (9/11), seizures (7/10), cerebellar anomalies (13/13), pons hypoplasia (12/13), brainstem hypoplasia (10/11), corpus callosum thinning (10/12), cerebral anomalies (9/12), white matter anomalies (8/10), enlarged ventricles (7/12) and ambiguous genitalia in males (10/14) [Citation10,Citation12]. Highly variable clinical phenotypes caused by TOE1 variants have been recently reported [Citation12].
To date, the prenatal clinical picture of PCH7 caused by pathogenic TOE1 variants has never been described. Here, we revealed the following compound heterozygous variants in TOE1 in the pedigree of a Chinese woman with two eventful pregnancies: c.716T > C (p. Phe239Ser) and c.955C > T (p. His319Tyr). We described the clinical phenotypes of the two fetuses in detail. Both fetuses showed microcephaly and cerebellar anomalies. The previous fetus had thinning of corpus callosum, which was not observed in the current fetus. The current fetus showed an abnormal lateral fissure and an underdeveloped insula, whereas the previous fetus did not show these features. Pons and brainstem hypoplasia are the most common features for postnatally diagnosed PCH7 patients. Pons and brainstem hypoplasia was also shown in both fetuses. Ambiguous genitalia are often observed in male PCH7 patients; however, this feature was not seen in the male fetus. Our study indicates that the TOE1 variant can cause phenotypic variability even within the same family. Furthermore, the current fetus displayed an obvious abnormal lateral fissure and an underdeveloped insula, which have not been depicted in previously reported PCH7 patients. Thus, the present case provides the first insight into the prenatal clinical characteristics of PCH7 and enriches the clinical spectrum of PCH7. Moreover, both variants have only been previously reported in two unrelated PCH7 patients diagnosed postnatally [Citation10]. Here, both fetuses with biallelic variants in TOE1 presented similar brain anomalies, which reconfirmed the pathogenic nature of the variants.
5. Conclusions
We first described the prenatal clinical features of a Chinese pedigree with PCH7 caused by compound heterozygous variants in TOE1, with phenotypic variability observed even within the same family. We also reconfirmed the pathogenic nature of the variants. Novel phenotypes, an abnormal lateral fissure and an underdeveloped insula were observed in the fetus in our study. These findings will enrich our knowledge of the clinical characteristics, clinical management and genetic counseling of PCH7.
Ethics approval
This study was approved by the Ethics Committee of Dongguan Maternal and Child Health Care Hospital.
Authors’ contributions
HMG and ZXD drafted the first versions of the manuscript. HMY was responsible for the design of the project, data analysis, and revising the manuscript. QHX, ZW and XMZ collected clinical information on the fetuses in detail. XCH coordinated the clinical evaluation. QMW and YS performed the experiments and data entry. All authors read and approved the final manuscript.
Patient consent
Written informed consent was obtained from the legal guardians for the publication of any potentially identifiable images or data included in this article.
Consent for publication
Consent to publish has been obtained from the parents of the pedigree.
Acknowledgments
We would like to express our sincere gratitude to the families for their cooperation.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Additional information
Funding
References
- Cassandrini D, Biancheri R, Tessa A, et al. Pontocerebellar hypoplasia clinical, pathologic, and genetic studies. Neurology. 2010;75(16):1459–1464. doi:10.1212/WNL.0b013e3181f88173.
- Kasinathan A, Sankhyan N, Dijk TV, et al. Clinico-radiological profile of children with pontocerebellar hypoplasia. J Pediatr Neurosci. 2020;15(2):94–98. doi:10.4103/jpn.JPN_6_19.
- Namavar Y, Barth PG, Kasher PR, et al. Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain. 2011;134(Pt 1):143–156. doi:10.1093/brain/awq287.
- van Dijk T, Baas F, Barth PG, et al. What’s new in pontocerebellar hypoplasia? An update on genes and subtypes. Orphanet J Rare Dis. 2018a;13(1):92. doi:10.1186/s13023-018-0826-2.
- van Dijk T, Ferdinandusse S, Ruiter JPN, et al. Biallelic loss of function variants in COASY cause prenatal onset pontocerebellar hypoplasia, microcephaly, and arthrogryposis. Eur J Hum Genet. 2018b;26(12):1752–1758. doi:10.1038/s41431-018-0233-0.
- Uwineza A, Caberg JH, Hitayezu J, et al. VPS51 biallelic variants cause microcephaly with brain malformations: a confirmatory report. Eur J Med Genet. 2019;62(8):103704. doi:10.1016/j.ejmg.2019.103704.
- Chai G, Webb A, Li C, et al. Mutations in spliceosomal genes PPIL1 and PRP17 cause neurodegenerative pontocerebellar hypoplasia with microcephaly. Neuron. 2021;109(2):241–256.e9. doi:10.1016/j.neuron.2020.10.035.
- Ucuncu E, Rajamani K, Wilson MSC, et al. MINPP1 prevents intracellular accumulation of the chelator inositol hexakisphosphate and is mutated in pontocerebellar hypoplasia. Nat Commun. 2020;11(1):6087. doi:10.1038/s41467-020-19919-y.
- Coolen M, Altin N, Rajamani K, et al. Recessive PRDM13 mutations cause fatal perinatal brainstem dysfunction with cerebellar hypoplasia and disrupt Purkinje cell differentiation. Am J Hum Genet. 2022;109(5):909–927. doi:10.1016/j.ajhg.2022.03.010.
- Lardelli RM, Schaffer AE, Eggens VR, et al. Biallelic mutations in the 3' exonuclease TOE1 cause pontocerebellar hypoplasia and uncover a role in snRNA processing. Nat Genet. 2017;49(3):457–464. doi:10.1038/ng.3762.
- Anderson C, Davies JH, Lamont L, et al. Early pontocerebellar hypoplasia with vanishing testes: a new syndrome? Am J Med Genet A. 2011;155A(4):667–672. doi:10.1002/ajmg.a.33897.
- Chen H, Li N, Xu Y, et al. Novel compound heterozygous variant of TOE1 results in a mild type of pontocerebellar hypoplasia type 7: an expansion of the clinical phenotype. Neurogenetics. 2022;23(1):11–17. doi:10.1007/s10048-021-00675-0.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30.
- Son A, Park JE, Kim VN. PARN and TOE1 constitute a 3' end maturation module for nuclear non-coding RNAs. Cell Rep. 2018;23(3):888–898. doi:10.1016/j.celrep.2018.03.089.
- Lardelli RM, Lykke-Andersen J. Competition between maturation and degradation drives human snRNA 3' end quality control. Genes Dev. 2020;34(13-14):989–1001. doi:10.1101/gad.336891.120.
- Deng T, Huang Y, Weng K, et al. TOE1 acts as a 3' exonuclease for telomerase RNA and regulates telomere maintenance. Nucleic Acids Res. 2019;47(1):391–405. doi:10.1093/nar/gky1019.
- Sperandio S, Tardito S, Surzycki A, et al. TOE1 interacts with p53 to modulate its transactivation potential. FEBS Lett. 2009;583(13):2165–2170. doi:10.1016/j.febslet.2009.06.004.