
ABSTRACT
Introduction: For a long time, vitamin K antagonists (VKA) have been the preferred drugs for the anticoagulation management of both atrial fibrillation and venous thromboembolism. Hereby, the major purpose is to attenuate the onset of thrombosis without affecting hemostasis.
Areas covered: Nowadays, non-vitamin K anticoagulants (NOAC), a new class of oral anticoagulants is available for the above-mentioned indications. NOAC are at least as effective and safer with regard to intracranial bleedings compared to VKA, but major bleedings still occur. For this reason, the search for safer anticoagulants is still ongoing.
Expert commentary: There are several unmet needs in NOAC management, including selection of optimal drug and dose, uncertainty on specific conditions and lack of drug persistence. There remains to be an important need for safer anticoagulants; ‘upstream’ anticoagulants including inhibitors of factor XIa may provide additional benefit related to fewer bleeding complications.
1. Introduction
For many decades, the use of anticoagulant agents for the prevention and treatment of thrombotic diseases has been confined to warfarin, heparin, and more recently low-molecular-weight heparins (LMWHs). Roughly since the 1950s, these agents have become crucial in the management of a broad range of patients at risk of (recurrent) thromboembolic diseases. For various reasons, new classes/types of drugs were developed and introduced into the clinical arena over the past two decades. As parental agents, fondaparinux was the first chemically synthesized agent to potentially replace heparin and in part LMWH [Citation1].
Figure 1. a. The figure shows the coagulation cascade and the different anticoagulant drugs presented in this article. On the left side; vitamin K antagonists inhibit vitamin K-related clotting factors II, VII, IX, and X. Further, the NOAC drugs; direct thrombin inhibitors and factor Xa inhibitors are presented in this scheme. Factor X inhibitors inhibit PAR 1 and possibly PAR 2 mediated cell signaling, were direct thrombin inhibitors primarily target PAR1. The other ‘upstream’ targets (factor XI and XII) and the possible inhibitors are also shown here. b. This box shows the possible vascular effects of the different oral anticoagulants. c. This figure shows the possible patient-related factors that determine the individual safety and efficacy of the prescribed drug.
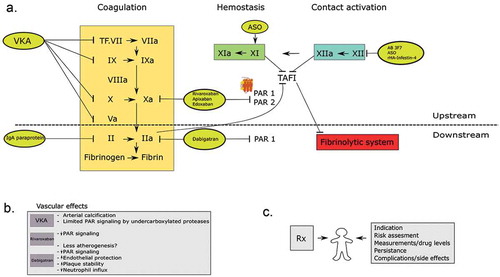
As oral agents, vitamin K antagonists (VKAs) have been the cornerstone of antithrombotic management for decades. While the clinical efficacy of warfarin and other VKA has been hard to beat, both safety (risks of major bleeding) and practical matters have been a consistent challenge for physicians managing the patient on VKA. Well-known limitations are a narrow therapeutic window, drug–drug interactions, interactions with diet, and interpatient variability in dose response [Citation2]. For those reasons, there has been a long-standing desire to obtain safer and easier-to-manage drugs, to eventually replace VKA for most indications. A number of novel synthetic compounds have been produced and clinically introduced for the main indications: prevention of ischemic stroke in patients with atrial fibrillation (AF) and prevention and treatment of venous thromboembolism (VTE). VTE includes two interrelated conditions that are part of the same spectrum, deep venous thrombosis (DVT) and pulmonary embolism (PE). The new class of oral agents was originally referred to as new oral anticoagulants (NOACs), later referred to as non-vitamin K oral anticoagulants (still NOAC, but in order to maintain the abbreviation and favored by cardiology societies; other societies such as the International Society of Thrombosis and Haemostasis [ISTH] favor DOAC, for direct oral anticoagulants). These NOACs directly inhibit thrombin (FIIa) or the upstream coagulation factor Xa (FXa). The US FDA and European Medicines Agency (EMA) approved the first oral FIIa inhibitor dabigatran in 2009. Currently, NOACs cover four registered agents that will be further discussed in the next paragraphs [Citation3–Citation11]. The agents are now globally on the market since several years and are increasingly replacing warfarin (or other VKAs) for the principal indications.
More recent drug developments include agents that tackle even more upstream coagulation proteases, including inhibitors of factor XIa and factor XIIa. Proof-of-principle clinical studies include the inhibition of synthesis of factor XI by a ribonucleic acid (RNA) silencing strategy, which successfully reduced the risk of postoperative VTE in patients undergoing knee surgery [Citation12]. Factor XIIa has also been tackled through several different inhibitors, either synthetic agents or antibodies, and in experimental studies, these agents appear to prevent thrombosis; clinical studies have not yet been initiated [Citation13,Citation14].
In general, anticoagulant drug developers have become interested in these ‘upstream’ (arbitrarily defined as anything proximal from thrombin) targets for several reasons: first, the ‘rediscovery’ of upstream coagulation proteases as relevant contributors to thrombosis (particularly for factors XIa and XIIa); second, the improved opportunities to design and manufacture highly selective (oral) inhibitors; third, a key consideration is the assumption that upstream targeting will have less impact on hemostasis, thus reducing bleeding risks commonly associated with oral anticoagulants.
The aim of the present expert review paper was to discuss various aspects of the different generations of oral anticoagulants, distinguished as ‘upstream’ (proximal to thrombin) and ‘downstream’ targeted oral agents. We will do this from a perspective of an era of VKA and more recently NOACs, and we will concisely summarize some of the major findings and differences among the agents, related to efficacy, safety, and side (unintended) effects. We will focus on recent developments involving the further upstream antagonists of intrinsic and contact coagulation proteins that carry promise as safer anticoagulants. shows a brief summary of the presented topics in this review.
2. The benchmark anticoagulant: warfarin
The principle of VKA, with warfarin as the globally most used representative, is based on the inhibition of the enzyme vitamin K oxidase reductase (VKOR) in hepatocytes. Dose-dependent inhibition of VKOR reduces the ability of the hepatocyte to carboxylate vitamin K-dependent proteins of which factors II, VII, IX, and X are most relevant for procoagulant effects. The net anticoagulant effect of VKA in blood is reflected by a prolongation of the prothrombin time (PT), expressed as international normalized ratio (INR). One ‘side effect’ of VKA treatment is the inhibition of carboxylation of proteins C and S, thus impairing this important anticoagulant pathway [Citation15]. Whether the impaired function of the anticoagulant proteins C and S is clinically or biologically relevant is not fully understood. Clinically, the rare complication of warfarin-associated skin necrosis is attributed to the rapid decline in functional protein C and S by VKA as compared to the slower decay in procoagulant factors [Citation16]. This difference in pharmacodynamics causes a temporary prothrombotic state, particularly in subjects deficient in proteins C or S (in whom VKA should never be started without simultaneous, initial LMWH). Interestingly, impaired functional levels of functional proteins C and S may have impact on cell protective effects mediated by activated protein C (aPC), but clinically, there are no indications that impaired cell defense or altered immune status is a feature of warfarin treatment, although this topic has probably never been studied specifically.
From a clinical point of view, bleeding is the most feared and prevalent side effect of VKA. While there is a clear association with INR, most bleedings occur while patients’ INRs are within preferred INR ranges [Citation17]. Thus, other factors contribute to bleeding risk. For VKA, these factors have been characterized and are summarized for patients with AF on VKA in the Hypertension, Abnormal renal/liver function, Stroke, Bleeding history or predisposition, Labile international normalized ratio, Elderly (> 65 years), Drugs/alcohol concomitantly (HASBLED) score [Citation18]. Fixed rather than transient risk factors contribute to bleeding risk in VKA treatment [Citation19]. Genetic factors have not been extensively characterized yet. Biochemically, there is no indication of impaired platelet function in those who bleed versus nonbleeding VKA users [Citation20]. At the same time, impaired whole blood thrombin generation may indicate an underlying hemostatic impairment in VKA users (Bloemen S, de Laat, ten Cate-Hoek et al., unpublished). There are no preferred sites for major bleeding, and any organ tract may be affected; however, gastrointestinal (GI) tract bleeds are most common (1% to 2% annually). Traditionally, GI bleeding on warfarin is considered a signal of underlying disease including cancer (and most likely unrelated to the use of VKA). Most feared is intracranial bleeding occurring at a rate from 0.1% to 0.7%/year, depending on age and also on quality of anticoagulation (time in therapeutic range, TTR). Precipitating factors of intracranial bleeding may be common risk factors such as uncontrolled hypertension but may also be the presence of microbleeds [Citation21]. Currently, considering the lower incidence of intracranial bleeding in NOAC versus VKA-treated patients in the phase 3 trials (fewer and perhaps smaller, see further), a conceptual pathogenesis of VKA-associated intracranial bleeding involves VKA-mediated inhibition of four procoagulant factors including factor VII, which may pose a relatively high burden on tissue factor (TF)-driven hemostasis; the brain is relatively dense in TF [Citation22], and for that matter, inhibition of the factor VII(a)-TF axis may be a particular risk factor for intracranial bleeds while on VKA.
Warfarin-associated arterial calcification has emerged as one of the long-term complications of VKA treatment [Citation23]. Specific cofactors associated with this complication have not been identified, and at the same time, the clinical impact of this type of calcification, related to the inhibition of other vitamin K-dependent proteins in the vessel wall, including matrix Gla protein and calcitonin, has also not been delineated. In contrast to ‘spontaneous’ calcification, a hallmark of atherosclerosis and a biomarker of cardiovascular mortality, similar risk associations have not been shown for VKA-related calcification. The fact that this complication is only detected after many years of VKA treatment suggests that the clinical impact may not be strong and/or dramatic, but it is essentially unknown. Along the line of ‘pleiotropic’ actions of VKA, also influences on the course of atherogenesis and plaque stability should be considered. Experimental studies have shown that except for calcification, atherogenesis may be delayed in onset, particularly in mice with a strong hyperlipidemic background (under revision, Thromb Res 2016), but whether this is associated with detectable anti-inflammatory or other cell stabilizing effects remains to be shown (in contrast to NOAC-related effects, see further). Clinically, an increased prevalence of intraplaque hemorrhages has been reported for patients on warfarin, and this may be linked to reduced plaque stability, but again, prospective studies are lacking [Citation24].
Other than that, side effects and complications of VKA are remarkably limited (except for the known warfarin-induced syndrome in pregnancy). Alopecia is a rare complication, while moderate hair loss is more frequently reported. The mechanism of this side effect is unknown.
3. Downstream inhibitors
3.1. Thrombin inhibition
3.1.1. Dabigatran etexilate
Direct thrombin inhibitors (DTIs) inactivate thrombin through a high-affinity binding with this enzyme. After the failure of ximelagatran (terminated because of liver toxicity concerns), dabigatran was the first DTI approved for clinical use in AF patients in 2009 [Citation3]. Dabigatran is a potent and reversible direct inhibitor of the active site of thrombin, thereby inactivating fibrin-bound and free thrombin in vitro [Citation25]. Inhibition of thrombin prevents the conversion of fibrinogen to fibrin but also the positive feedback activation of other coagulation activation, the cross-linking of fibrin monomers, platelet activation, and inhibition of fibrinolysis [Citation26]. Dabigatran etexilate is an oral prodrug, which is rapidly converted by esterases in blood to its active form dabigatran [Citation26]. The absolute bioavailability is merely 6.5%, so patients ingest relatively high doses to reach an adequate plasma concentration [Citation26]. The absorption of dabigatran etexilate is independent of GI tract acidity or the use of a proton pomp inhibitor, because of the structure with tartaric acid and a coating onto a tartaric acid core [Citation27]. The main pharmacokinetic (PK) data are summarized in . The main outcome data from clinical trials comparing dabigatran to warfarin for prevention of stroke in patients with AF as well as for the treatment of VTE comparing dabigatran plus initial LMWH and warfarin plus LMWH are summarized in and , respectively.
Table 1. Summary pharmacology of the four FDA/EMA-approved NOACs.
Table 2. Overview of the published phase 3 randomized control trails in patients with nonvalvular atrial fibrillation.
Table 3. Overview of the published phase 3 randomized control trails in patients with venous thromboembolism.
Antithrombotic efficacy is comparable (noninferior) to warfarin for both indications, with superiority proven for prevention of ischemic stroke at the 150-mg bid dose of dabigatran in patients with AF [Citation3]. Hence, direct thrombin inhibition with dabigatran provides effective antithrombotic protection as compared to VKA. An increased rate of myocardial infarction (MI) was observed in the RE-LY study [Citation3]; this was also noted for the previous thrombin inhibitor, ximelagatran. Whether the occurrence of this complication is a chance finding or whether it reflects a distinct paradoxical prothrombotic consequence of direct thrombin inhibition in patients at risk of plaque rupture remains unknown. In this light, it should be kept in mind that thrombin has multiple actions, including activation of protein C through its cofactor thrombomodulin. Like for VKA, but through different mechanisms, reduced thrombin availability will have impact on the protein C-activating potential and possibly have untoward effects in patients at risk of thrombosis (vulnerable plaques).
Like for warfarin, a clear dose–response relation was shown for dabigatran in a substudy from RE-LY; trough concentrations were independently associated with thromboembolism or bleeding, in case of relatively low versus high trough levels [Citation28]. Given the wide interindividual variation in plasma concentrations, the net biochemical effect may also be variable. In an attempt to document this effect, Dale et al. showed that at therapeutic INR levels, warfarin provided greater suppression of thrombin generation indices as compared to dabigatran, although this study was quite small [Citation29]. A recent study shows that in ‘real-life’ patients, there is a wide range of dabigatran concentrations, but it also indicates that patients that have an initial high concentration tend to remain high on repeated testing [Citation30]. A similar pattern was seen for low concentrations that however tended to increase toward a more ‘therapeutic’ range, but this may have been due to patient selection, where only compliant patients remained in study. Although the authors of the latter study do not suggest that dabigatran doses should be tailored on the basis of plasma concentrations in the individual patient, one may be tempted to do so anyway, initially, to ascertain that trough concentrations are not too far off the observed therapeutic window.
4. Upstream thrombin inhibition
As factor Xa is the enzyme that activates prothrombin, it is another important target for inhibiting thrombin generation. Three oral FXa inhibitors are reviewed here: rivaroxaban, apixaban, and edoxaban, all approved for AF and VTE by the regulatory authorities.
4.1. Rivaroxaban
Rivaroxaban is the first orally active direct factor Xa inhibitor, thereby preventing conversion of fibrinogen to fibrin. Rivaroxaban binds directly to the active site of FXa, thereby blocking the interaction with its substrate. It has been shown to inhibit free FXa, prothrombinase-bound FXa, and clot-bound FXa [Citation31]. Rivaroxaban is an oral agent which is rapidly absorbed and reaches a peak plasma concentration in 2–4 h after administration [Citation32]. Compared to dabigatran, the bioavailability of the 10-mg dose is much higher; 80%. However, the 15-mg and 20-mg dose has a lower bioavailability if taken in the fasted state and consequently should be taken with food [Citation33]. Approximately two-thirds of the dose is metabolized through cytochrome (CYP) enzymes in the liver (CYP3A4 and CYP2C8) and CYP-independent mechanisms [Citation34]. For this reason, rivaroxaban is contraindicated in patients with severe liver disease. Other PK characteristics are summarized in .
4.2. Apixaban
Apixaban is a highly selective inhibitor of FXa. Similar to rivaroxaban, apixaban inhibits FXa, whether it is free, prothrombinase bound, or clot bound [Citation35]. It is designed as a follow-up compound to razaxaban, with superior anti-FXa potency, selectivity, and oral bioavailability [Citation36]. Apixaban is rapidly absorbed from the GI tract reaching peak-plasma levels in about 3 h. The bioavailability of apixaban is approximately 50%. The mean half-time is 12 h [Citation37], and steady-state concentrations are reached within 3 days [Citation38].
4.3. Edoxaban
The fourth FXa inhibitor edoxaban is a direct, selective, synthetic inhibitor of FXa [Citation39], similar to the previously described FXa inhibitors. After oral administration, edoxaban reaches peak concentrations in 1–3 h, and the half-life time is 5.8–10.7 h for single doses 10–150 mg and 8–10 h after multiple doses 90–120 mg daily [Citation40,Citation41]. Edoxaban is primarily absorbed in the upper GI tract, and the oral availability is 62%. It is excreted through renal (35.4%) as well as nonrenal routes such as biliary secretion [Citation40]. Since edoxaban is a substrate for the efflux transporter P-glycoprotein (P-gp), dose adjustment is required in concomitant administration of strong P-gp inhibitors [Citation42]. Given the renal route of elimination, PK changes in patients with renal impairment deserve attention and dose adjustment [Citation42]. Edoxaban should not be used in patient with severe renal impairment (<15 mL/min) [Citation43].
The three factor Xa inhibitors were noninferior to warfarin in the major phase 3 clinical trials, and no obvious differences in efficacy among the products are discernable. Comparable efficacy may also be expected, since the drugs are all based on the same principle of action, direct blockade of the catalytic effect of FXa. Comparative network analyses provide insight in the relative efficacy and safety issues related to these products in comparison with dabigatran and warfarin [Citation44,Citation45]. In terms of bleeding complications, there appear to be some differences in that rivaroxaban and high-dose edoxaban (60 mg) are associated with more frequent GI tract bleeding as compared to warfarin. Like with dabigatran, relatively high concentrations of drug in the GI tract may contribute to a higher risk of GI bleeding with some of the Xa inhibitors, but this remains somewhat speculative. Intracranial bleeding is also less frequent for the factor Xa inhibiting NOAC, probably for similar reasons as outlined earlier for dabigatran and warfarin.
When summarizing some important observations from the NOAC trials, it appears that in patients with AF, there was either noninferiority of NOAC as compared to warfarin, with some exceptions: dabigatran 150 mg showed a lower incidence of ischemic stroke/systemic embolism, while this risk was increased for the lower 30-mg dose of edoxaban 30 mg. NOAC treatment was associated with a consistently lower risk of intracranial bleeding as compared to warfarin. There was a higher incidence of GI bleeding with rivaroxaban and dabigatran 150-mg dosage.
For VTE treatment, the NOAC trials overall did not show major differences with VKA for efficacy or safety outcomes. Considering bleeding, all studies demonstrated relative risks in favor of the NOACs, but the effect size was different. This is probably explained by the different PK properties of the drugs. A caveat in the safety analyses is the fact that the TTR in the warfarin arms of the trials was suboptimal overall, ranging from 57% to 64% [Citation7–Citation11]. Furthermore, the TTR for VKA was similar in all studies: 55–65% [Citation3–Citation11]. While additional analyses ruled out an association of TTR with intracranial hemorrhage, it should be taken into account that in countries with particular well-controlled VKA, the rate of intracranial bleeding on VKA may still be in the range observed for NOACs [Citation46].
5. Nonhemostatic functions of FIIa and FXa inhibitors
With regard to vascular effects, FIIa inhibitor dabigatran may well have impact on atherogenesis as well as plaque stability in the long run. In analogy with VKA where calcification turned out to be an unintended consequence of local inhibition of vessel wall proteins, dabigatran (and possibly other NOACs) may show to have local effects in the vessel wall that may be clinically relevant. NOACs are very small molecules that will easily pass the endothelium and interact with coagulation proteases in the vessel wall. Indeed, a number of experimental studies have shown that administration of dabigatran inhibits atherogenesis in mice with an apoE null background, prone to develop atherosclerosis [Citation47–Citation49]. These studies also indicated several protective mechanisms resulting from thrombin inhibition, including endothelial cell protection, reduced inflammation by diminished neutrophil influx, and increased plaque stability. Many of these effects are thought to be due to inhibition of thrombin’s protease-activated receptor (PAR) mediated cell signaling [Citation50] (Posma J, et al., J Thromb Haemost 2016 under revision). If one would like to extrapolate these results to clinical implications, it should be taken into account that all the results were obtained in mouse models. There are differences between the mouse models and humans in the cellular composition of the arterial intima, and mouse models do not experience spontaneous plaque rupture or thrombosis. As a consequence, these animals do not suffer from MI or stroke [Citation51].
The effects of dabigatran are also quite pronounced as compared to rivaroxaban, while data on other NOAC have not yet been published. This may point to an advantage of direct thrombin inhibition over factor Xa inhibition, although published rivaroxaban effects on atherogenesis may have been studied at too low drug levels (Posma J et al., unpublished data). Many of the effects of dabigatran on atherosclerosis may be explained by antagonizing thrombin in its ability to activate PAR-1. However, also other interactions may be relevant as observed in the clinical setting where dabigatran was shown to be associated with lower apolipoprotein B (apoB) levels in a subanalysis from RE-LY [Citation52]. Given that the size of the effect was comparable to statins, an indirect lipid-modifying effect may also have consequences in patients treated with dabigatran.
Regarding pleiotropic effects of factor Xa inhibition, it is interesting to speculate that targeting factor Xa may have different effects from direct thrombin inhibition. Theoretically, factor Xa inhibition attenuates all downstream effects including PAR-1-mediated and maybe PAR-2-mediated cell signaling by FXa and thrombin, while direct thrombin inhibition would primarily target PAR-1 effects. However, since thrombin feedback activates its own formation (including factor Xa generation), it is unlikely that inhibition of factor Xa would yield essentially different outcomes vis-a-vis PAR activation [Citation53]. In comparison, inhibition of vitamin K-dependent proteins by warfarin may still allow limited PAR signaling by undercarboxylated proteases [Citation54]. However, much of the cellular receptor-mediated effects will also critically depend on local enzyme and drug concentrations (Posma et al., JTH submitted).
5.1. Inhibiting factor XI (a)
Factor XI is a coagulation protein that hinges between the contact system factors (factor XII linked) and the intrinsic coagulation cascade [Citation55]. Its role in hemostasis is evident from the hemophilia-like phenotype associated with severe factor XI deficiency; however, its impact is moderate as compared to the classic hemophilia’s A and B and predominantly confined to bleeding in association with trauma and in specific organ systems such as bladder and mucous membranes [Citation56]. It is only since a few decades that a physiologic route of factor XI activation has been characterized, showing that thrombin feedback activates this protein [Citation57]. Activated factor XI drives factor IX conversion and subsequent downstream intrinsic coagulation. Excess factor XI (activity) is a risk factor for VTE, and absence of factor XI may provide some protection against ischemic stroke [Citation58]. For all these reasons, targeting factor XI may provide a reasonably effective antithrombotic therapy, perhaps with less bleeding tendency as compared to more centrally acting anticoagulants.
Experimental studies in small and larger animals provide substantial evidence of antithrombotic activity. A proof-of-concept clinical study in patients undergoing orthopedic surgery of the knee provides evidence of antithrombotic protection as compared to LMWH prophylaxis [Citation12]. In this study, factor XI antigen levels were lowered prior to surgery with RNA interference in factor XI protein synthesis. This strategy resulted in a dose-dependent reduction of factor XI levels to about 20% on average at the highest RNAi dose. While antithrombotic efficacy was excellent in this trial, bleeding side effects were less as compared to LMWH. This study may pave the way to development of factor XIa inhibitors particularly for indications where bleeding risks limit the use of downstream acting anticoagulants. Two aspects merit discussion. First, the authors noted that thrombi, when these developed in the FXI intervention arm, appeared to be smaller in size, which was interpreted as a sign of efficacy. However, it is quite possible that an effect on fibrinolysis explains the smaller clots, where factor XIa is an important contributor in (in part thrombin mediated) fibrinolysis impairment through activation of thrombin activatable fibrinolysis inhibitor (TAFI) [Citation59–Citation61]. A second issue is the postulated safety. Bleeding was rare in this study [Citation12], but it remains a question how the outcome will be in other types of surgery. Organs with high local fibrinolytic activity such as the bladder may be prone to bleeding upon factor XI inhibition. Future studies should take these points into careful consideration.
5.2. Inhibiting factor XIIa
The well-known but poorly understood contact coagulation system appears to go through a revival following experimental studies from Renne and Nieswandt showing that factor XII is an important contributor to arterial thrombosis, at least in animal models [Citation13,Citation62,Citation63]. Clinically, the impact of factor XII is enigmatic; its absence does not inflict a bleeding tendency in man, and variations in factor XII levels are variably associated with thrombosis risk; there is no association with venous thrombosis, and there are possible associations with manifestations of arterial thrombosis, MI, and ischemic stroke [Citation64].
One intriguing and poorly understood aspect is the position of factor XII in the contact system. On one side, factor XII triggers coagulation through activating factor XI, but the source of factor XII activation has not yet been convincingly demonstrated. A number of candidates have been proposed including polyphosphates, collagen/platelets, and deoxyribonucleic acid (DNA). Artificial surfaces with a negative charge are traditionally known activators of the contact system, but whether these play any role in vivo is questionable [Citation65].
On the other hand, factor XII hinges with inflammatory cascades through the kininogen/kallikrein/bradykinin system and complement activation. Experimental studies showed that depending on the trigger, the direction of factor XIIa effects is either in a proinflammatory or procoagulant direction [Citation66]. Also, the stages of factor XII activation are important (alpha or beta forms of XIIa) as well as the localization of the activation process at a phospholipid surface. Endothelial cells have been shown to provide such a surface, involving uPAR, cytokeratin-1, and gC1qR as cofactors [Citation67]. Pathologic conditions involving activated platelets and neutrophils may also provide surface and cofactors to start up contact activation in one or the other direction. This pathway selectivity is intriguing in that for instance in typical inflammatory syndromes such as angioedema unimpaired contact activation is pathognomonic [Citation68], however, hardly ever associated with thrombosis.
Several strategies to target factor XII each with a different mechanism of action have been established. First, antibodies against factor XII, which bind factor XII and block its activation. For example, animal studies with antibody 3F7 provided thromboprotection as efficiently as heparin, but 3F7 treatment did not impair the hemostatic function and did not increase bleeding in rabbits [Citation69]. This suggests that it provides anticoagulation without an increase in bleeding tendency. Another strategy is the use of antisense oligonucleotides (ASO), which reduce the hepatic synthesis of factor XII. Furthermore, knockdown of factor XII with ASO in a rabbit model of catheter thrombosis prolonged the time to catheter occlusion by twofold in rabbits [Citation70]. Recently, it was demonstrated that the protein rHA-Infestin-4, a strong FXII inhibitor, decreased occlusion rates in both arterial and venous thrombosis model in mice [Citation62]. Against this background, factor XIIa inhibitors may have antithrombotic potential, but it remains to be shown at what price this would come in vivo. Animal experiments are promising in demonstrating antithrombotic activity without apparent inflammatory side effects, but this may also have been ignored to some extend in experiments aimed at thrombosis (and bleeding). On top of this, fibrinolysis is also regulated by factor XII, possibly linked to factor XI-related effects on TAFI activation (see above), such that factor XIIa inhibitors such as Infestin-4 should be evaluated for fibrinolytic effects.
5.3. Fibrin polymerization
Baglin et al. described an interesting finding leading toward a new antithrombotic approach. They found an IgA paraprotein with antithrombin activity in a patient identified during screening for coagulation abnormalities [Citation71]. Further characterization of this antibody showed that it specifically binds to thrombin exosite I and is not associated with a severe bleeding phenotype. Experimental studies suggest that this antibody may be potentially useful as an antithrombotic tool, apparently without overt bleeding enhancing tendency. It remains to be shown whether this methodology can be further exploited toward clinical testing.
6. Where are we now and what are the unmet needs of currently available therapies?
Although one might argue that we have come a long way in satisfying the need for safer and easier-to-use anticoagulant drugs, there are still some concerns that need to be addressed. The fixed dosing principle might not be as adequate as presented, at least for several indications the NOAC cannot be applied (e.g. mechanical valves, acute coronary syndrome [ACS], antiphospholipid syndrome [APS], pregnancy and breast feeding, children, and malignancy); what are the options here? Finally, it is not only the properties of the drugs that are important, but also patient-related factors that determine the individual safety and efficacy of drugs.
Optimal drug and doses? One could be concerned about the fixed-dose NOAC policy for several reasons. Although warfarin has been used as the benchmark drug in the clinical trials evaluating the safety and efficacy of NOACs, some critical remarks should be made. One is that NOACs were shown to be globally noninferior to warfarin, the quality of which was suboptimal (TTR 55–65%) [Citation3,Citation5,Citation6]. There may therefore be room for further improvement of NOAC therapy. This could involve optimizing drug selection and NOAC dose optimization. Although NOAC doses are registered, leaving little room for dose fine-tuning on individual basis, the choice among anticoagulants creates room for selecting the best agent for a given patient. Knowing the approximate therapeutic ranges, one can test trough levels upon starting an NOAC and determine whether these are within the therapeutic window; if not and without room for tailoring, one could switch to another anticoagulant. This policy may not require new trials, as this type of selection is on rationale grounds and does not involve any experimentation with new doses. This way improved and more personal therapy may be the consequence.
Uncertain indications? There are populations in whom the application of NOAC has not yet been successful. The RE-ALIGN study was developed to test dabigatran in the setting of mechanical heart valves (MHV). However, this study had to be stopped because of increased rates of thromboembolic and bleeding complications compared with warfarin [Citation72]. No other NOAC trials have been undertaken in these patients, and the use of NOAC is contraindicated after implantation of mechanical heart valves. The explanation for the failure of dabigatran in patients with MHV remains equivocal. One study showed that key components of MHV are prothrombotic and promote thrombin generation through the intrinsic pathway of coagulation. VKA suppressed this thrombin generation at INR values of 1.5 or higher. In contrast, dabigatran concentrations >200 ng/mL were required to suppress thrombin generation to the same extent as warfarin at an INR of 2.0–3.5. These dabigatran concentrations were above those achieved in the RE-ALIGN study and induce an increased bleeding risk. In conclusion, a possible explanation that dabigatran failed to prevent thromboembolic complications is because the target trough dabigatran concentration was too low [Citation73]. Alternatively, the intraindividual variation in peak and trough may be too large as compared to the more stable anticoagulant intensity achieved with VKA. It also remains questionable whether a single target approach will be effective for this indication, but given the negative dabigatran data, other NOAC studies are unlikely to occur.
The role of NOACs in patients with acute coronary syndrome (ACS) has also been explored. The Acute Coronary Syndrome – Thrombolysis In Myocardial Infarction 51 (ATLAS ACS 2 – TIMI 51) trial tested the addition of rivaroxaban to aspirin and clopidogrel following ACS. Rivaroxaban reduced cardiovascular death, MI, stroke, and all-cause mortality. However, this was associated with a threefold increase in major bleeding and intracranial hemorrhage [Citation74]. The Apixaban for Prevention of Acute Ischemic and Safety Events (APPRAISE-2) trial failed to find the benefits of adding a high dose of apixaban to single or dual antiplatelet therapy in ACS patients [Citation75]. Dabigatran also showed no added efficacy in triple therapy but, on the other hand, an increased risk of major bleeding [Citation76]. The role of novel anticoagulants in combination with dual antiplatelet therapy in ACS remains under discussion, and currently ongoing trials will refine our knowledge for the use of NOACs in patients with coexisting AF and coronary heart disease (CHD). Another area where NOACs have not been convincing is in prevention of VTE in medical patients, and bleeding complications still offset efficacy benefits.
Conditions with high bleeding risk. This is a particular area where there may be a place for drugs targeted at upstream factors FXIIa or FXIa, or for selected downstream agents such as fibrin formation inhibitors. Essentially, any agent with truly reduced impact on hemostasis may find a place in the management of patients at risk of thrombosis, but with (relative) contraindications because of bleeding risk. Examples are (1) anticoagulation in the patient with acute ischemic stroke having received thrombolytic therapy; (2) adjunct therapy for patients with other types of ischemia–reperfusion injury such as MI where primary and secondary prevention now consists of dual platelet inhibition; combination with anticoagulants is challenging because of the high bleeding risk; (3) any indication for anticoagulation in patients with high HASBLED scores or with renal insufficiency; (4) indications for anticoagulation in the setting of manifest bleeding (trauma, intracranial bleeding and concurrent thrombosis, disseminated intravascular coagulation [DIC] etc.); (5) anticoagulation in patients with cancer.
Drug persistence? The development of the class of NOAC has no doubt changed the anticoagulant landscape. Based on more stable PK, these agents can be administered in fixed doses with limited necessity for routine laboratory testing, although renal and liver functions should be checked at reasonable intervals. Uncertainties remain with regard to proper intake of unsupervised medication, which is notoriously poor. It is known that the nonadherence rate in chronic patients varies between 25% and 55% in chronic conditions such as AF [Citation77]. Yet, there is limited knowledge about the impact of nonadherence on safety and effectiveness outcomes in NOAC treatment. A recent study in AF 7265 patients in Germany showed better NOAC persistence than VKA therapy after 180/360 days. Persistence was significantly higher in rivaroxaban users compared to both VKA (66% vs. 58%) and dabigatran users (66% vs. 60% after 180 days) with a similar trend after 360 days. On the other hand, other data showed lower persistence rates after 24 months of follow-up. About 28% of the newly NOAC patients versus 17% of the newly warfarin patients discontinued their medication [Citation78]. Major causes for discontinuation of NOAC were drug adverse events, worsening kidney functions, and patients’ desire [Citation78]. Further studies are needed to identify other factors that may be responsible for nonadherence and evaluate their impact on clinical outcomes [Citation79].
7. Expert commentary
The unmet needs outlined earlier indicate the actions that should be undertaken. Leaving an era of laboratory-managed anticoagulation (INR-managed VKA) for fixed-dose NOAC administration in major indications, the long-term management of anticoagulation poses new questions. Drug persistence is challenging, and about 30% of patients on NOAC tend to stop taking medication adequately within several months. Thus, programs that focus on optimizing management of patients on NOAC (and also VKA) merit full attention. Recommendations such as from ESC on the follow-up of patients on NOAC need to be implemented and tested on their merits. For instance, in situations of intercurrent disease (infection, dehydration, diarrhea, etc.) where INR values are known to fluctuate substantially: how is NOAC’s pharmacokinetics affected, and how should the physician deal with this? How often do renal and other critical organs functions need to be assessed in elderly subjects? Obviously, these issues are generic and not different for upstream or downstream agents, but because of the importance for health care, we like to emphasize them here too. Optimizing drug and dose policy in any patient on oral anticoagulant remains a challenge, even more so in the NOAC era. As discussed, careful drug selection based on clinical characteristics and taking CHA2DS2-VASC and HASBLED criteria into account should be a starting point. Assessment of individual PK in a limited and practically feasible manner merits attention. This could provide an individual patient’s benchmark for an NOAC trough activity level, to decide on appropriate NOAC selection and useful for incident comorbidity assessment (in case of bleeding, drug activity within the benchmark range for the given patient?).
Selecting new and sometimes challenging indications for studying agents with potentially greater safety–efficacy ratio is an actual theme of interest. Now that factor XI reducing therapy has been shown to provide effective antithrombotic therapy, more agents aimed at this target are being developed. Although it may still take a number of years to obtain practical oral factor XIa inhibitors, in the meantime, one should carefully consider the available evidence from literature and practice. In which settings is factor XIa thought to play a relevant role? Which patients will benefit most from this upstream inhibition? Basically, the same questions are at stake for factor XIIa interventions, although there the concern of interfering in the other contact system driven directions (inflammation) should first be carefully explored in animal experiments and healthy subjects.
8. Five-year view
Many studies with NOAC are undertaken to cover part of the remaining indications such as ACS and management of patients with thrombosis and cancer. Hopefully, management of patients on NOAC (and other anticoagulants) will be optimized aiming at personalized drug selection and patient guidance in the setting of complications, interactions, interventions, and other daily life issues that challenge optimal drug intake and tolerance. Any long-term effects of NOAC on the vasculature (inhibition of atherogenesis) and other vital organs should be carefully looked for, also based on lessons drawn from the VKA era, showing calcification as a long-term unintended side effect. Other areas such as mechanical heart valves will not easily see a solution in terms of simpler or safer drugs. Some studies will be initiated to explore the potential of safer anticoagulants aimed at factors XII, XI, or downstream at the level of fibrin crosslinking. Factor XIa inhibition may be the most straightforward antithrombotic target, in the sense that a priori no major side effects other than (trauma related) bleeding are anticipated. However, care should be taken not to overestimate the safety of factor XIa inhibition, since major bleeds do occur in severe factor XI deficiency, but the pattern and severity are poorly predictable. Development of other inhibitors against factor XIIa or fibrin crosslinking will depend on the absence of unexpected side effects or in case of fibrin, unexpected bleeding complications.
Key issues
NOACs are currently available as an alternative to VKA for anticoagulation in AF and VTE.
NOACs showed noninferiority to warfarin in reducing strokes and embolic events. Although most of these drugs had higher incidence of GI bleeding, major bleeding and intracranial bleeding rates were lower.
Although NOACs are safe and effective for stroke prevention, there remains a need for safer anticoagulants due to the bleeding risk.
Research now is focused on identifying targets ‘upstream’ thrombin, particularly the contact pathway coagulation factors XI and XII.
Declaration of interest
H ten Cate is chair of the Dutch Federation of Anticoagulation Clinics; is a consultant for Stago; received fees for speaking and/or research from Bayer, Boehringer Ingelheim, Pfizer, Stago and Leo Farma; is a fellow of the Gutenberg Research College, Gutenberg University and Center for Thrombosis and Haemostasis, Mainz, Germany. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Additional information
Funding
References
- Turpie AG, Gallus AS, Hoek JA. A synthetic pentasaccharide for the prevention of deep-vein thrombosis after total hip replacement. N Engl J Med. 2001;344(9):619–625.
- Ageno W, Gallus AS, Wittkowsky A, et al. Oral anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141(2 Suppl):e44S–88S.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med. 2009;361(12):1139–1151.
- Giugliano RP, Ruff CT, Braunwald E, et al. Edoxaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2013;369(22):2093–2104.
- Granger CB, Alexander JH, McMurray JJV, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365(11):981–992.
- Patel MR, Mahaffey KW, Garg J, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365(10):883–891.
- Agnelli G, Buller HR, Cohen A, et al. Oral apixaban for the treatment of acute venous thromboembolism. N Engl J Med. 2013;369(9):799–808.
- Buller HR, Büller HR, Décousus H, et al. Edoxaban versus warfarin for the treatment of symptomatic venous thromboembolism. N Engl J Med. 2013;369(15):1406–1415.
- Investigators E, Bauersachs R, Berkowitz SD, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363(26):2499–2510.
- Investigators E-P, Büller HR, Prins MH, et al. Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N Engl J Med. 2012;366(14):1287–1297.
- Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009;361(24):2342–2352.
- Büller HR, Bethune C, Bhanot S, et al. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med. 2015;372(3):232–240.
- Kenne E, Nickel KF, Long AT, et al. Factor XII: a novel target for safe prevention of thrombosis and inflammation. J Intern Med. 2015;278(6):571–585.
- Worm, M., Köhler EC, Panda R, et al. The factor XIIa blocking antibody 3F7: a safe anticoagulant with anti-inflammatory activities. Ann Transl Med. 2015;3(17):247.
- Weiss P, Soff GA, Halkin H, et al. Decline of proteins C and S and factors II, VII, IX and X during the initiation of warfarin therapy. Thromb Res. 1987;45(6):783–790.
- Chan YC, Valenti D, Mansfield AO, et al. Warfarin induced skin necrosis. Br J Surg. 2000;87(3):266–272.
- Witt DM, Delate T, Hylek EM, et al. Effect of warfarin on intracranial hemorrhage incidence and fatal outcomes. Thromb Res. 2013;132(6):770–775.
- Pisters R, Lane DA, Nieuwlaat R, et al. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138(5):1093–1100.
- Van Rein N, Le Cessie S, Van Vliet IP, et al. Increased risk of major bleeding after a minor bleed during treatment with vitamin K antagonists is determined by fixed common risk factors. J Thromb Haemost. 2016;14(5):948–952.
- Van Der Meijden PEJ, Bouman AC, Feijge MAH, et al. Platelet dysfunction in thrombosis patients treated with vitamin K antagonists and recurrent bleeding. PLoS One. 2013;8(5):e64112.
- Lee SH, Ryu WS, Roh JK. Cerebral microbleeds are a risk factor for warfarin-related intracerebral hemorrhage. Neurology. 2009;72(2):171–176.
- Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7(1):41–53.
- Han KH, O’Neill WC. Increased peripheral arterial calcification in patients receiving warfarin. J Am Heart Assoc. 2016;5(1):e002665.
- Derksen WJM, Peeters W, Tersteeg C, et al. Age and coumarin-type anticoagulation are associated with the occurrence of intraplaque hemorrhage, while statins are associated less with intraplaque hemorrhage: a large histopathological study in carotid and femoral plaques. Atherosclerosis. 2011;214(1):139–143.
- Van Ryn J, Goss A, Hauel N, et al. The discovery of dabigatran etexilate. Front Pharmacol. 2013;4:12.
- Hankey GJ, Eikelboom JW. Dabigatran etexilate: a new oral thrombin inhibitor. Circulation. 2011;123(13):1436–1450.
- Stangier J, Stähle H, Rathgen K, et al. Pharmacokinetics and pharmacodynamics of the direct oral thrombin inhibitor dabigatran in healthy elderly subjects. Clin Pharmacokinet. 2008;47(1):47–59.
- Reilly PA, Lehr T, Haertter S, et al. The effect of dabigatran plasma concentrations and patient characteristics on the frequency of ischemic stroke and major bleeding in atrial fibrillation patients: the RE-LY Trial (Randomized Evaluation of Long-Term Anticoagulation Therapy). J Am Coll Cardiol. 2014;63(4):321–328.
- Dale B, Eikelboom JW, Weitz JI, et al. Dabigatran attenuates thrombin generation to a lesser extent than warfarin: could this explain their differential effects on intracranial hemorrhage and myocardial infarction? J Thromb Thrombolysis. 2013;35(2):295–301.
- Chan NC, Coppens M, Hirsh J, et al. Real-world variability in dabigatran levels in patients with atrial fibrillation. J Thromb Haemost. 2015;13(3):353–359.
- Perzborn E, Strassburger J, Wilmen A, et al. In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939 – an oral, direct factor Xa inhibitor. J Thromb Haemost. 2005;3(3):514–521.
- Kubitza D, Becka M, Wensing G, et al. Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939–an oral, direct factor Xa inhibitor–after multiple dosing in healthy male subjects. Eur J Clin Pharmacol. 2005;61(12):873–880.
- Kubitza D, Becka M, Voith B, et al. Safety, pharmacodynamics, and pharmacokinetics of single doses of BAY 59-7939, an oral, direct factor Xa inhibitor. Clin Pharmacol Ther. 2005;78(4):412–421.
- Lang D, Freudenberger C, Weinz C. In vitro metabolism of rivaroxaban, an oral, direct factor Xa inhibitor, in liver microsomes and hepatocytes of rats, dogs, and humans. Drug Metab Dispos. 2009;37(5):1046–1055.
- He K, Luettgen JM, Zhang D, et al. Preclinical pharmacokinetics and pharmacodynamics of apixaban, a potent and selective factor Xa inhibitor. Eur J Drug Metab Pharmacokinet. 2011;36(3):129–139.
- Carreiro J, Ansell J. Apixaban, an oral direct factor Xa inhibitor: awaiting the verdict. Expert Opin Investig Drugs. 2008;17(12):1937–1945.
- Pfizer BMS. Apixaban, summary of the product characteristics; 2011 [2016 Apr 26]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002148/WC500107728.pdf
- Raghavan N, Frost CE, Yu Z, et al. Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab Dispos. 2009;37(1):74–81.
- Furugohri T, Isobe K, Honda Y, et al. DU-176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles. J Thromb Haemost. 2008;6(9):1542–1549.
- Bathala MS, Masumoto H, Oguma T, et al. Pharmacokinetics, biotransformation, and mass balance of edoxaban, a selective, direct factor Xa inhibitor, in humans. Drug Metab Dispos. 2012;40(12):2250–2255.
- Ogata K, Mendell-Harary J, Tachibana M, et al. Clinical safety, tolerability, pharmacokinetics, and pharmacodynamics of the novel factor Xa inhibitor edoxaban in healthy volunteers. J Clin Pharmacol. 2010;50(7):743–753.
- Lip GYH, Agnelli G. Edoxaban: a focused review of its clinical pharmacology. Eur Heart J. 2014;35(28):1844–1855.
- Savaysa. Prescribing information. Parsippany (NJ): Daiichi Sankyo, Inc; 2015.
- Morimoto T, Crawford B, Wada K, et al. Comparative efficacy and safety of novel oral anticoagulants in patients with atrial fibrillation: a network meta-analysis with the adjustment for the possible bias from open label studies. J Cardiol. 2015;66(6):466–474.
- Tereshchenko LG, Henrikson CA, Cigarroa J, et al. comparative effectiveness of interventions for stroke prevention in atrial fibrillation: a network meta-analysis. J Am Heart Assoc. 2016;5(5):e003206.
- Sjögren V, Grzymala-Lubanski B, Renlund H, et al. Safety and efficacy of well managed warfarin. A report from the Swedish quality register Auricula. Thromb Haemost. 2015;113(6):1370–1377.
- Borissoff JI, Otten JJT, Heeneman S, et al. Genetic and pharmacological modifications of thrombin formation in apolipoprotein e-deficient mice determine atherosclerosis severity and atherothrombosis onset in a neutrophil-dependent manner. PLoS One. 2013;8(2):e55784.
- Kadoglou NPE, Moustardas P, Katsimpoulas M, et al. The beneficial effects of a direct thrombin inhibitor, dabigatran etexilate, on the development and stability of atherosclerotic lesions in apolipoprotein E-deficient mice: dabigatran etexilate and atherosclerosis. Cardiovasc Drugs Ther. 2012;26(5):367–374.
- Lee I-O, Kratz MT, Schirmer SH, et al. The effects of direct thrombin inhibition with dabigatran on plaque formation and endothelial function in apolipoprotein E-deficient mice. J Pharmacol Exp Ther. 2012;343(2):253–257.
- Borissoff JI, Spronk HMH, Ten Cate H. The hemostatic system as a modulator of atherosclerosis. N Engl J Med. 2011;364(18):1746–1760.
- Bentzon JF, Falk E. Atherosclerotic lesions in mouse and man: is it the same disease? Curr Opin Lipidol. 2010;21(5):434–440.
- Joseph P, Pare G, Wallentin L, et al. Dabigatran etexilate and reduction in serum apolipoprotein B. Heart. 2016;102(1):57–62.
- Schurgers LJ, Spronk HMH. Differential cellular effects of old and new oral anticoagulants: consequences to the genesis and progression of atherosclerosis. Thromb Haemost. 2014;112(5):909–917.
- Spronk HMH, De Jong AM, Crijns HJ, et al. Pleiotropic effects of factor Xa and thrombin: what to expect from novel anticoagulants. Cardiovasc Res. 2014;101(3):344–351.
- Gailani D, Bane CE, Gruber A. Factor XI and contact activation as targets for antithrombotic therapy. J Thromb Haemost. 2015;13(8):1383–1395.
- Wheeler AP, Gailani D. Why factor XI deficiency is a clinical concern. Expert Rev Hematol. 2016;9(7):629–637.
- Gailani D, Broze GJ. Factor XI activation in a revised model of blood coagulation. Science. 1991;253(5022):909–912.
- Kamphuisen PW, Ten Cate H. Cardiovascular risk in patients with hemophilia. Blood. 2014;123(9):1297–1301.
- Bouma BN, Meijers JC. Role of blood coagulation factor XI in downregulation of fibrinolysis. Curr Opin Hematol. 2000;7(5):266–272.
- Colucci M, Incampo F, Cannavò A, et al. Reduced fibrinolytic resistance in patients with factor XI deficiency. Evidence of a thrombin-independent impairment of the thrombin-activatable fibrinolysis inhibitor pathway. J Thromb Haemost. 2016;14(8):1603–1614.
- Dai L, Mitchell M, Savidge G, et al. The profibrinolytic effect of plasma thrombomodulin in factor XI deficiency and its implications in hemostasis. J Thromb Haemost. 2004;2(12):2200–2204.
- May F, Krupka J, Fries M, et al. FXIIa inhibitor rHA-Infestin-4: safe thromboprotection in experimental venous, arterial and foreign surface-induced thrombosis. Br J Haematol. 2016;173(5):769–778.
- Renné T, Pozgajová M, Grüner S, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202(2):271–281.
- Key NS. Epidemiologic and clinical data linking factors XI and XII to thrombosis. Hematol Am Soc Hematol Educ Program. 2014;2014(1):66–70.
- Björkqvist J, Nickel KF, Stavrou E, et al. In vivo activation and functions of the protease factor XII. Thromb Haemost. 2014;112(5):868–875.
- De Maat S, Van Dooremalen S, De Groot PG, et al. A nanobody-based method for tracking factor XII activation in plasma. Thromb Haemost. 2013;110(3):458–468.
- De Maat S, De Groot PG, Maas C. Contact system activation on endothelial cells. Semin Thromb Hemost. 2014;40(8):887–894.
- Konings J, Cugno M, Suffritti C, et al. Ongoing contact activation in patients with hereditary angioedema. PLoS One. 2013;8(8):e74043.
- Larsson M, Rayzman V, Nolte MW, et al. A factor XIIa inhibitory antibody provides thromboprotection in extracorporeal circulation without increasing bleeding risk. Sci Transl Med. 2014;6(222):222ra17.
- Yau JW, Liao P, Fredenburgh JC, et al. Selective depletion of factor XI or factor XII with antisense oligonucleotides attenuates catheter thrombosis in rabbits. Blood. 2014;123(13):2102–2107.
- Baglin TP, Langdown J, Frasson R, et al. Discovery and characterization of an antibody directed against exosite I of thrombin. J Thromb Haemost. 2016;14(1):137–142.
- Eikelboom JW, Connolly SJ, Brueckmann M, et al. Dabigatran versus warfarin in patients with mechanical heart valves. N Engl J Med. 2013;369(13):1206–1214.
- Jaffer IH, Stafford AR, Fredenburgh JC, et al. Dabigatran is less effective than warfarin at attenuating mechanical heart valve-induced thrombin generation. J Am Heart Assoc. 2015;4(8):e002322.
- Mega JL, Braunwald E, Wiviott SD, et al. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med. 2012;366(1):9–19.
- Alexander JH, Lopes RD, James S, et al. Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med. 2011;365(8):699–708.
- Oldgren J, Budaj A, Granger CB, et al. Dabigatran vs. placebo in patients with acute coronary syndromes on dual antiplatelet therapy: a randomized, double-blind, phase II trial. Eur Heart J. 2011;32(22):2781–2789.
- Fischer MA, Stedman MR, Lii J, et al. Primary medication non-adherence: analysis of 195,930 electronic prescriptions. J Gen Intern Med. 2010;25(4):284–290.
- Shiga T, Naganuma M, Nagao T, et al. Persistence of non-vitamin K antagonist oral anticoagulant use in Japanese patients with atrial fibrillation: a single-center observational study. J Arrhythm. 2015;31(6):339–344.
- Beyer-Westendorf J, Ehlken B, Evers T. Real-world persistence and adherence to oral anticoagulation for stroke risk reduction in patients with atrial fibrillation. Europace. 2016;18:1150–1157.