
ABSTRACT
Introduction: The revised Ghent nosology presents the classical features of Marfan syndrome. However, behind its familiar face, Marfan syndrome hides less well-known features.
Areas covered: The German Marfan Organization listed unusual symptoms and clinical experts reviewed the literature on clinical features of Marfan syndrome not listed in the Ghent nosology. Thereby we identified the following features: (1) bicuspid aortic valve, mitral valve prolapse, pulmonary valve prolapse, tricuspid valve prolapse, (2) heart failure and cardiomyopathy, (3) supraventricular arrhythmia, ventricular arrhythmia, and abnormal repolarization, (4) spontaneous coronary artery dissection, anomalous coronary arteries, and atherosclerotic coronary artery disease, tortuosity-, aneurysm-, and dissection of large and medium-sized arteries, (5) restrictive lung disease, parenchymal lung disease, and airway disorders, (6) obstructive- and central sleep apnea, (7) liver and kidney cysts, biliary tract disease, diaphragmatic hernia, and adiposity, (8) premature labor, and urinary incontinence, (9) myopathy, reduced bone mineral density, and craniofacial manifestations, (10) atrophic scars, (11) caries, and craniomandibular dysfunction, (12) headache from migraine and spontaneous cerebrospinal fluid leakage, (13) cognitive dysfunction, schizophrenia, depression, fatigue, and pain, (14) and activated fibrinolysis, thrombin, platelets, acquired von Willebrand disease, and platelet dysfunction.
Expert commentary: Future research, nosologies, and guidelines may consider less well-known features of Marfan syndrome.
1. Introduction
The far side of the Moon is the hemisphere of the Moon that always faces away from Earth. People call this side ‘the dark side’, and many artists have let this side stand for the hidden and the hideous. In contrast, planetary scientists explain the phenomenon of a hidden side of the Moon by tidal locking, where tidal forces on Earth slowed down the rotation of the Moon to the point where always the same side of the Moon was facing Earth. Today, thanks to science and discovery, we know that this far side indeed looks different from the near side of the Moon, but that this far side also receives sunlight.
Today, the Marfan syndrome has a near side as well. Soon after the French pediatrician Antoine Bernard-Jean Marfan (1858–1942) described a combination of symptoms in the 5-year-old Gabrielle, the picture of this bright side came into life [Citation1,Citation2]. Marfan himself described dolichocephaly, long limbs, fingers and toes and contractures of the joints. Pieper and Irvine-Jones described the syndrome as a congenital cardiovascular disease [Citation3], Börger [Citation4] and in another paper on the same individual, von Pfaundler [Citation5], added ectopia lentis and high-grade myopia to the list of features. Weve described the autosomal dominant trait of inheritance [Citation6], Jequier added pneumothorax [Citation7], Etter and Glover aortic dissection [Citation8], and soon thereafter other cardiovascular features such as dilatation of the pulmonary artery, and mitral valve prolapse were added (for detailed review see [Citation9]). McKusick postulated a defect of the connective tissue as the cause of the disease [Citation10], which was confirmed more than 30 years later when Lynn Sakai’s group identified fibrillin as the specific fibril that was affected by Marfan disease [Citation11]. Shortly thereafter, Dietz and colleagues reported the first pathogenic variant in the fibrillin1 gene [Citation12]. Various systematic descriptions and nosologies confirmed and further elaborated this picture of Marfan syndrome which today forms a familiar face and well-known side of Marfan syndrome.
However, as in planetary science, medical science moves on to explore the far side of the Marfan syndrome. Today, this far side may indeed have a ‘dark’ face to many patients and physicians. The disease may hide something unknown and poorly described that threatens the affected patients and that takes by surprise the physicians who are not prepared appropriately. For this article, we asked patient representatives about their less usual experiences with the Marfan syndrome, and we sampled a team of well-known experts to take a trip to the dark side of Marfan syndrome. We evaluated huge fields of literature reporting unfamiliar features of Marfan syndrome, which we sampled and mapped carefully to present it in this review. In this way, we sought to encourage future investigations. Further, we made some preliminary conclusions on the refinement of diagnostic routines in the medical care for patients with Marfan syndrome.
2. Methods
In a first step, we asked the German Marfan Organization to compile a list of unusual symptoms and complaints among their members and other people seeking their advice (see ‘the patients’ perspective’ for further description; ). At the same time, by mid-2017, the first and the senior author of this article launched an extensive analysis of Medline with ‘Marfan syndrome’ as key word to search for features or symptoms of Marfan syndrome that were not listed in any of the three international nosologies of Marfan syndrome. This search yielded about 6,200 original articles including case reports. Our search was qualitative in nature, where we intended only to pick up reports of unusual manifestations and symptoms of Marfan syndrome. This search yielded a list of 259 original publications, which we used to derive distinct clusters of manifestations and symptoms. We present the results of our initial Medline search in . In a next step, we set up a list of clinical scientists, whom we considered as leading experts in the respective cluster of clinical features. All experts whom we invited agreed to contribute. We provided our table of literature (), and asked each expert to repeat the literature search on their respective cluster of clinical features and to perform an in depth analysis of the cited literature to present their view on the scope and clinical relevance of distinct organ system features. We asked all experts to provide their opinion on the clinical and diagnostic consequences of these features. In a final step, we compiled a final list of organ features. The first and the senior author used a Likert scale to quantify their evaluative estimation of the scientific and clinical significance of each feature of Marfan syndrome () [Citation13]. In addition, we asked all experts to comment on patients’ reported symptoms in their respective organ chapter.
Figure 1. Evaluative estimation of the clinical significance of suggested features of the Marfan syndrome.
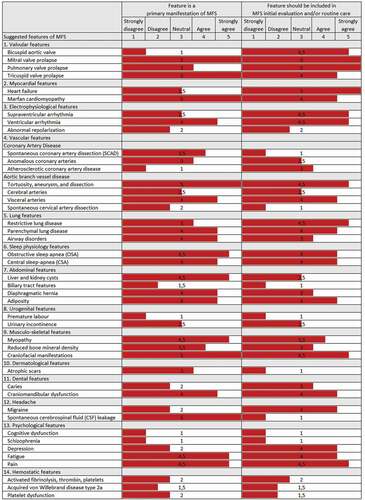
Table 1. Patients‘ symptoms reported by the German Marfan Organization
Table 2. Initial Medline search for features or symptoms of Marfan syndrome
Finally, all potential features of Marfan syndrome may also occur in other genetically caused thoracic aortic syndromes such as Loeys-Dietz syndrome or in vascular Ehlers-Danlos syndrome. Therefore, we provide a separate table, where we list all clinical features along with the first and senior authors’ evaluative opinion on the association of these features with the respective syndrome. To support our evaluation, we screened Medline for the presence of these features, where we combined the search term ‘Loeys-Dietz syndrome’, or ‘Ehlers-Danlos syndrome vascular type’, respectively, with a clinical feature as the other search term, respectively. We display in all original publications including case reports that we found informative on the occurrence of clinical features in Loeys-Dietz or vascular Ehlers-Danlos syndrome ().
Table 3. Association of feature with syndrome
3. The patients’ perspective by Marina Vogler
Authors of this article asked me as a patient representative of the German Marfan organization to report, what patients with Marfan syndrome experience as features on the ‘dark side’ of the Marfan syndrome. We defined as ‘dark side’ what we experience quite often as symptoms and features which we think may belong to the Marfan syndrome, but which do not show up in official descriptions of the syndrome or in official patient guides [Citation14,Citation15], and which are usually not routinely checked for by medical experts of the Marfan syndrome. We present a list of symptoms and complaints, which we derived from three sources: First, from notes that we took over many years during our telephone counseling sessions with members of our organization, or other people from the general German population telling us that they had Marfan syndrome. Second, from information collected from a Marfan-Facebook-group survey that we started from Friday 29th to Sunday, 31st October 2018. Third, from asking our members and visitors with Marfan syndrome on our official ‘23rd German Marfan Day’, held in Fulda, 4 May 2019. We graded reported complaints as ‘major’ whenever these were mentioned by more than 5 independent individuals, and ‘minor’, whenever mentioned less often ().
4. Valvular features by Yskert von Kodolitsch and Evaldas Girdauskas
4.1. Aortic valve disease
Aortic valve disease presents as an integral part of the well-described aortic root disease in Marfan syndrome. Therefore, aortic valve regurgitation, caused by dilatation of aortic valve annulus and the sinotubular junction, in combination with aortic valve cusp prolapse, and frequently aortic valve cusp commissural fenestrations are well-described features of Marfan syndrome that already have their established place on the bright side of clinical attention. However, there is an emerging discussion on the causal relationship of Marfan syndrome and the congenitally bicuspid aortic valve. The classical view sees both entities as differential diagnosis of alternative causes of aneurysmal formation in the proximal aorta [Citation16]. Most advocators of this view emphasize that aneurysms in Marfan syndrome are located primarily in the sinuses of Valsalva, whereas aneurysms in bicuspid aortic valve tend to be located in the ascending aorta beyond the sinotubular junction [Citation17,Citation18]. The opposing view holds, that a congenitally bicuspid aortic valve is an integral part of the Marfan syndrome. Advocators of this view describe an increased frequency of 4.5–4.7% of the bicuspid aortic valve in Marfan syndrome, and even 6.8–8.5% in pediatric Marfan patients [Citation19,Citation20], compared to 0.5–1.5% BAV prevalence in the general population. It is argued that common pathogenic mechanisms are likely to underlie both entities [Citation21,Citation22]. However, in a larger population of Marfan patients, the prevalence of bicuspid aortic valve was 1.8% [Citation23], which compares well to the prevalence in the general population. Irrespective of the causal unity, the coexistence of both entities appears to relate to a more pronounced aortic disease [Citation21–Citation23]. A third view, which we call the differentiated view, identifies a subgroup of bicuspid aortic valve disease with aneurysm located in the sinuses of Valsalva (so-called bicuspid root-phenotype), which mimics the aneurysmal pattern of the Marfan syndrome. This bicuspid aortic valve subtype lacks other clinical features of the Marfan syndrome, but it shares some of its pathogenic and genetic features. Such aortic phenotypes have been described for decades in conjunction with, but also independently of their etiology, as ‘annuloaortic ectasia’ [Citation24], or as ‘forme fruste’ of Marfan syndrome [Citation25], Today, this bicuspid aortic phenotype has been associated with nucleotide sequence variants in the FBN1 gene [Citation26,Citation27], but also with other genes known to cause genetic aortic diseases [Citation27].
In conclusion, we consider bicuspid aortic valve disease as a distinct cause of aortic aneurysm. Bicuspid aortic valve may coincide with Marfan syndrome and thereby aggravates the proximal aortic aneurysmal disease. There is an aortic phenotype of bicuspid aortic valve disease that we call the ‘bicuspid root-phenotype’, which represents most probably a congenital type of bicuspid aortopathy and is associated with mutations in various genes, while some of them are known to cause specific genetic aortic syndromes such as Marfan syndrome.
4.2. Mitral valve disease
In Marfan syndrome, severe myxomatous mitral valve prolapse usually coexists with aortic root aneurysm. However, mitral valve prolapse and mitral valve regurgitation were listed as independent diagnostic features of Marfan syndrome already in the first international nosology of heritable disorders of connective tissue, established in September 1986, in Berlin [Citation28]. Since then, subsequent revisions of this nosology, first in 1996 and then in 2010, both in Ghent, corroborated the independent diagnostic importance of mitral valve prolapse for the diagnosis of Marfan syndrome [Citation16,Citation29]. Therefore, it may appear surprising that we include mitral valve disease into the list of dark side features of the Marfan syndrome. The reasons are the following: First, compared to the studies on aortic disease, the data on mitral valve disease in Marfan syndrome are rather scarce. Second, most available studies had been performed in the last millennium, and were often based on today outdated echocardiographic technology including M-Mode, outdated diagnostic criteria of mitral valve prolapse, and in subjects diagnosed with Marfan syndrome using obsolete diagnostic criteria of the syndrome. Finally, classical studies focused on specific clinical subgroups, so-called neonatal Marfan syndrome, children, adults, or included only surgical patients, and carried thereby substantial patient selection bias [Citation30–Citation38].
Here, we summarize the clinical profile of mitral valve disease in Marfan syndrome based on data that utilized modern diagnostic criteria of mitral valve prolapse, current 2-dimensional echocardiographic technology, and Ghent criteria of Marfan syndrome [Citation29], including demonstration of a pathogenic FBN1 variant, without any age limitations. The first large population-based cohort study using these criteria included 204 patients and reported the following findings [Citation39]:
Mitral valve prolapse is an age-dependent manifestation of Marfan syndrome, where the estimated cumulative distribution of mitral valve prolapse is 8.7% at 1 year of age, 26.7% at 10 years, 36.1% at 20 years, 42.6% at 30 years, 47.7% at 40 years, 51.8% at 50 years, and 60.8% at 80 years of age.
Severe mitral valve regurgitation is also age-dependent, with an estimated cumulative distribution of 0 at 1 year, 2.5% at 20 years, 13.3% at 40 years, 32.7% at 60 years, and 55.8% at 80 years of age.
Infective endocarditis of the mitral valve has an estimated cumulative probability of <1% up to the age of 30 years, but then increases rapidly up to almost 20% at the age of 80 years.
Mitral valve prolapse is associated with dural ectasia, ectopia lentis, and skeletal involvement.
Severe mitral valve regurgitation is related to tricuspid valve prolapse and to the sporadic form of Marfan syndrome.
Another population-based cohort study of 112 patients with Marfan syndrome and the criteria of mitral valve prolapse established clinical predictors of mitral valve complications [Citation40]. The progression of mitral valve regurgitation of ≥1 grade was independently associated with flail mitral leaflet, and increased indexed end-systolic left ventricular diameters. Clinical adverse events of the mitral valve including endocarditis, heart failure, and mitral valve surgery correlated significantly with a flail mitral leaflet and mild or moderate degree of mitral valve regurgitation. Aortic dilatation, dural ectasia, and sporadic mode of inheritance were not associated with outcome [Citation40].
Finally, a cohort study of 116 patients with a causative FBN1 gene mutation and ≤moderate mitral valve regurgitation at baseline examined the impact of FBN1 gene mutation characteristics. This study found only a marginal relationship between FBN1 gene mutations (i.e., located both in a transforming-growth-factor beta-binding protein-like (TGFb-BP) domain and in the calcium-binding epidermal growth factor-like (cbEGF) domain) and the risk of mitral valve surgery [Citation41].
In contrast to mitral valve prolapse alone, moderate mitral valve regurgitation was related to both ventricular premature complexes >10/h, and to ventricular couplets, or non-sustained ventricular arrhythmia in adults with Marfan syndrome, whereas both failed to predict ventricular tachycardia events [Citation42].
Premature calcification of mitral valve ring was present in only one of 138 adults with Marfan syndrome (0.7%) [Citation43]. Such calcification was previously considered as a diagnostic sign of Marfan syndrome [Citation29], but has been eliminated from the list of Marfan signs in the recent nosology [Citation9,Citation16]. Given the prevalence of mitral valve prolapse in 31.7% pediatric Marfan patients, it was considered important for an early diagnosis of Marfan syndrome in children [Citation19]. In some children with severe variants of Marfan syndrome, such as neonatal forms, or de-novo mutations of the FBN1 gene, severe valvular disease may occur with increased frequency.
In conclusion, mitral valve prolapse is an age-dependent manifestation of the Marfan syndrome, with clinical consequences of mitral valve regurgitation and endocarditis that usually progress with an increasing age.
4.3. Pulmonary valve disease
Dilatation of the main pulmonary artery, ‘in the absence of valvular or peripheral pulmonic stenosis, before the age of 40 years’ was a diagnostic criterion of Marfan syndrome in the first Ghent nosology, but it was removed from the list of diagnostic signs [Citation44] in the revised nosology due to the lack of diagnostic specificity [Citation16,Citation29]. The prevalence of pulmonary artery dilatation ranges between 8% and 16% in children [Citation20,Citation45] and 37% and 74% in adults with Marfan syndrome [Citation20,Citation44,Citation46]. In addition, the pulmonary valve was shown to exhibit echocardiographic signs of cusp prolapse in 6% children and in 4% adults with Marfan syndrome, as well as of mild pulmonary regurgitation in 11% children, and 6% adults with Marfan syndrome [Citation47]. Therefore, surgeons tend to assume pulmonary root disease in Marfan syndrome that leads them to dissuade a Ross procedure in Marfan syndrome [Citation48,Citation49]. However, main pulmonary artery aneurysms >6 cm diameter as an isolated indication for elective surgery [Citation44], or more than mild pulmonary regurgitation occurs only exceptionally in Marfan syndrome [Citation20]. In conclusion, the pulmonary artery root may be affected by the same connective tissue defect as the aortic root in Marfan syndrome. However, clinical consequences of pulmonary root disease are rare and may occur only with increased pulmonary artery pressure.
4.4. Tricuspid valve disease
Tricuspid valve disease was almost completely disregarded as a manifestation of the Marfan syndrome. However, tricuspid valve prolapse appears to be quite common in Marfan syndrome, a finding which emerged from some more recent studies:
The overall prevalence of tricuspid valve prolapse was 22% in a cohort of 204 individuals with Marfan syndrome of all ages. With 4% in the first decade of life, 17% in the second, 29% in the third, 32% in the fourth decade, there appeared to be a slight increase of the prevalence of tricuspid valve prolapse in the first half of life. However, the prevalence appeared to be lower in the second half of life, where the prevalence was 24% in the fifth, 15% in the sixth, and 0% beyond the sixth decade of life [Citation39].
Tricuspid valve prolapse presents with echocardiographic features of valvular thickening and prolapse with chordal elongation which involves all 3 leaflets. Histologic examination revealed findings consistent with valvular thickening and myxomatous degeneration [Citation50].
Concurrent involvement of the mitral and the tricuspid valves appears to be common in Marfan syndrome [Citation50].
Progression of the severity from mild to moderate or severe regurgitation occurs only in a small fraction of affected individuals [Citation19].
In addition to tricuspid valve degenerative process, myopathy involving also the right ventricle may at least in theory, be another alternative mechanism which may lead to tricuspid valve regurgitation [Citation51]. In conclusion, tricuspid valve disease although neglected, is common and potentially harmful in Marfan syndrome.
4.5. Expert opinion on patients’ reported symptoms ()
We see two reasons why patients do not report ‘valve-related problems’ (). First, valve dysfunction results in heart failure or heart arrhythmia, and then patients may report these symptoms instead of valve dysfunction. Second, fortunately, both heart failure and arrhythmia do not occur often in Marfan syndrome, and if they occur, they usually will lead the patient to a cardiologist who will identify valve dysfunction and provide adequate therapy. Therefore, symptoms of valvular heart failure do not occur often, and if they occur, these symptoms will be usually treated effectively soon.
4.6. Conclusion
Marfan syndrome is a systemic disease of all heart valves, where the left-sided valves account for the majority of clinical symptoms, in an age-dependent manner. However, the right-sided valves are commonly affected by the same tissue disease, and therefore should be similarly included in the clinical surveillance and decision-making process.
5. Myocardial features by Julie De Backer
5.1. Heart failure
In several (historical) series on death in Marfan syndrome, heart failure is mentioned as one of the leading causes. Figures vary between 5% and 30% which puts heart failure at least at the same level as aortic dissection [Citation52,Citation53]. In addition, there are several reports on end-stage heart failure necessitating heart transplantation in patients with Marfan syndrome. The known problems of both aortic valve regurgitation due to aortic root dilatation and important mitral valve regurgitation in the setting of valve prolapse are often the cause of this heart failure [Citation54].
5.2. Marfan cardiomyopathy
In addition, several reports from independent researchers have been published in recent years indicating a primary intrinsic dysfunction of the myocardium in Marfan syndrome. The reported prevalence of what is now addressed as ‘Marfan cardiomyopathy’ ranges from 3% to 68% across different series [Citation54,Citation55], depending on the definition and population characteristics. Involvement of both left and right ventricles with systolic and diastolic dysfunction has been described [Citation56–Citation62]. In the majority of cases, dysfunction is mild, with subclinical abnormalities that do progress much over time. Although it has not yet been clearly demonstrated, a link between such mild intrinsic cardiomyopathy and an unfavorable course in the event of an additional hemodynamic trigger such as valve dysfunction and/or aortic root replacement is not unlikely.
Studies in mouse models of Marfan syndrome suggest such a relationship, where the application of pressure overload due to partial ligation of the aortic arch leads to clear cardiomyopathy [Citation63]. These models suggest mechanosignaling as a possible cause for cardiomyopathy [Citation64]. The presence of fibrillin-1 in the myocardium has clearly been evidenced in wild-type mice, with indications of more abundant amounts in the atria [Citation65]. It is assumed that the underlying abnormality in the FBN1 gene results in a deficient mechanosignaling function of the fibrillin microfibrils in the extracellular matrix and that mechanical factors such as volume- or pressure overload are inadequately compensated, which in turn leads to myocardial dysfunction. In recent years, the concept of abnormal mechanosignaling has also been introduced in the explanation for aortic pathology in Marfan syndrome [Citation66].
Whether the type of underlying pathogenic FBN1 variant matters in whether or not people are more susceptible to developing cardiomyopathy is still unclear, but in a small study, a difference was observed with patients carrying non-missense variants having more left ventricular dilatation than patients with missense variants [Citation67].
5.3. Expert opinion on patients’ reported symptoms ()
does not list classical symptoms of heart failure including dyspnea, orthopnea, nycturia, and ankle edema, which is not surprising since myocardial dysfunction usually remains mild in Marfan patients. However, according to our experience, patients with Marfan syndrome and clinically significant heart failure will present such symptoms.
5.4. Conclusion
Although more extensive studies in the longer term are certainly necessary to further unravel the clinical relevance of cardiomyopathy, the findings from human studies and mouse models, in any case, indicate that we must be vigilant about this problem. Careful monitoring of myocardial function and potential consequences such as (severe) heart failure and arrhythmia in patients with Marfan syndrome is warranted.
6. Electrophysiological features by Anthony Demolder
Ever since the first report in 1985 by Chen et al., growing evidence has revealed an elevated risk of arrhythmia in patients with Marfan syndrome [Citation68]. In both children and adults, an arrhythmic predisposition for supraventricular as well as ventricular arrhythmias can be noted, sometimes independent from valvular abnormalities [Citation68,Citation69].
6.1. Supraventricular arrhythmia
Among the supraventricular arrhythmias, atrial fibrillation and Wolff-Parkinson-White syndrome have been described. Atrial fibrillation is reported to be present in 8% of patients with Marfan syndrome [Citation69]. In the general population, the prevalence of atrial fibrillation is 1% [Citation70], indicating that Marfan syndrome has an increased prevalence of this arrhythmia. In contrast, Wolff-Parkinson-White syndrome has only been described in case reports of Marfan syndrome suggesting a mere coincidental association [Citation71,Citation72].
6.2. Ventricular arrhythmia
More frequently observed are the ventricular arrhythmias in Marfan syndrome, as evidenced by multiple studies. Significant ventricular ectopy defined as >10 premature ventricular contractions per hour occurs in 20-30%, whereas non-sustained ventricular tachycardia is shown to be present in 10-20% of Marfan syndrome patients [Citation55,Citation69,Citation73]. Three studies report life-threatening arrhythmias in 7-9% of their patients along with sudden cardiac death most likely due to arrhythmia occurring in up to 4% [Citation55,Citation69,Citation74]. These numbers indicate that ventricular arrhythmias are an important cause of death in Marfan syndrome in addition to aortic dissection or -rupture and heart failure. Studies on causes of death after aortic root replacement report fatal arrhythmias in 12-18% of patients with Marfan syndrome, making it the 2nd most frequent cause of death after surgery of the aorta [Citation52,Citation75]. A possible association with underlying intrinsic myocardial dysfunction, as described above, seems plausible and warrants further investigation.
6.3. Abnormal repolarization
In addition to an arrhythmic predisposition, some reports have described associations between Marfan syndrome and abnormal repolarization. One study demonstrated prolonged conduction times and abnormal repolarization, as evidenced by longer PQ-intervals, longer QT-intervals, and more ST-segment depressions compared to controls [Citation73]. In perspective, the PQ-intervals remained within normal boundaries (<200 ms). In line with this, current literature holds only case reports on Marfan syndrome and heart block with no hard evidence regarding an association [Citation76,Citation77].
In contrast, 16–20% of patients with Marfan syndrome were reported to have a prolonged QTc-interval (>440 ms) [Citation55,Citation73] with similar percentages seen in children with Marfan syndrome (9-20%) [Citation68,Citation78]. As opposed to the PQ-interval duration, the QT-interval prolongation was unrelated to the degree of aortic root dilation, the dimension, and function of the left ventricle and the presence of valvular prolapse [Citation73]. Extended prolongation of the QT-interval combined with early after depolarization can trigger arrhythmia with potentially life-threatening complications. Based on current evidence, careful consideration when initiating drugs with pro-arrhythmogenic/QT prolonging potential seems warranted.
The combination of arrhythmic predisposition along with abnormal repolarization in Marfan syndrome strongly suggests the presence of an underlying subclinical aberration in the electrophysiological substrate. To date, the mechanism underlying these cardiac disorders remains unknown. Most studies do show an association between left ventricular dilation, altered repolarization and ventricular arrhythmia [Citation55,Citation69,Citation73,Citation74]. Valvular disease and cardiovascular surgery, both frequently encountered in Marfan syndrome, may be additional contributing factors. In fact, moderate mitral valve regurgitation was associated with ventricular premature complexes >10/h, ventricular couplets and non-sustained ventricular tachycardia in adults with Marfan syndrome [Citation69]. However, both mitral valve prolapse and mitral valve regurgitation failed to predict ventricular tachycardia events [Citation69].
Attempts at risk stratification have described some determinants, including ventricular ectopy, non-sustained ventricular tachycardia, ventricular tachycardia, impaired left ventricular systolic function and elevated serum N-terminal pro-brain natriuretic peptide (NT-proBNP) levels [Citation74]. Of these, serum NT-proBNP level was shown to be the strongest independent predictor of arrhythmogenic events [Citation69,Citation74].
6.4. Expert opinion on patients’ reported symptoms ()
Patient-reported symptoms such as tachycardia or palpitations may be an indicative feature of both supraventricular and ventricular arrhythmia. Considering the arrhythmic predisposition in Marfan syndrome, screening tools such as resting ECG and ambulatory ECG are justified to uncover the presence of arrhythmias, especially in patients reporting suggestive symptoms.
6.5. Conclusion
Since the potential complications are life-threatening arrhythmia and sudden cardiac death, identifying patients at risk is warranted and a low threshold for arrhythmic follow-up investigations (e.g., resting ECG, ambulatory ECG, and echocardiography) is justified.
7. Aortic branch vessels by Shaine Morris and Anji Yetman
Given that the majority of vascular disease in Marfan syndrome is aortic, little attention has been paid to aortic branch vessels. However, tortuosity, aneurysm, and dissection have been reported throughout the arterial system.
7.1. Coronary artery disease
In Marfan syndrome, myocardial ischemia most often occurs secondary to dissection of the aortic root with propagation of the dissection into the coronary vessels [Citation79], or iatrogenic postoperative coronary obstruction. Other potential types of coronary artery disease include: (1) spontaneous coronary artery dissection, (2) anomalous coronary artery origin and (3) atherosclerotic coronary artery disease. The three causes are discussed below.
7.1.1. Spontaneous coronary artery dissection (SCAD)
Although the most common associations with SCAD are coronary atherosclerotic disease, pregnancy/peripartum status, and fibromuscular dysplasia, some cases of Marfan syndrome have been described [Citation80–Citation92]. In a population-based study of SCAD in the United States using administrative records, 10 of 66,360 admissions for SCAD had a co-existing diagnosis of Marfan syndrome [Citation84]. In a study of 114 cases of SCAD retrospectively evaluating for connective tissue disease, 3 were diagnosed with connective tissue disease, including 1 with Marfan syndrome and 2 with vascular Ehlers-Danlos syndrome [Citation82]. Another study of 107 patients with SCAD were prospectively considered for genetic evaluation [Citation93]. Among this group, no patients with Marfan syndrome were detected, but 3 with COL3A1 variants and 1 with a SMAD3 variant were diagnosed. Although several case studies reported pathological findings consistent with cystic medial necrosis in rare cases of SCAD, as far as we know, the first patient with a clinical diagnosis of Marfan syndrome was reported in by Corrado et al. in 1992 [Citation86,Citation94,Citation95]. The youngest individual to date with SCAD in Marfan syndrome was in a 12-year-old girl [Citation91].
Medical treatment rather than intervention is often pursued if possible [Citation90,Citation92]. If intervention is necessary, both percutaneous intervention and bypass surgery have been successfully employed [Citation88,Citation90,Citation91]. Percutaneous intervention can be complicated, and risk of extending the dissection is possible [Citation91].
7.1.2. Anomalous coronary arteries
Multiple case reports [Citation96–Citation101] and one case series [Citation102] note the presence of anomalous coronary artery origin in association with Marfan syndrome. Anomalous right and left coronary arteries, as well as aberrant origin of the circumflex from the right coronary artery have been described.
The frequency of this association, and whether this truly differs from the general population (who may be more apt to go unrecognized) is unknown. Mulder and colleagues identified 3 patients with Marfan syndrome who had anomalous coronary arteries resulting in a prevalence of 5.6%, significantly greater than the 1.6% prevalence quoted for the general population [Citation102].
Anomalous left coronary artery, and to a lesser extent, anomalous right coronary artery are recognized causes of myocardial ischemia and sudden death, particularly in the presence of an intramural or interarterial coronary course [Citation103]. The patient with Marfan syndrome presenting with chest pain or loss of consciousness, invokes concern for aortic dissection or ventricular arrhythmias [Citation55] and the potential for ischemic coronary artery disease may be overlooked particularly in the absence of acquired risk factors. A normal CTA or MRA of the aorta may provide false reassurance regarding a pathologic cause of chest pain. Evaluation for ischemia may be warranted, irrespective of acquired risk factors for coronary artery disease.
Given the need for coronary artery reimplantation at the time of proximal aortic replacement, the takeoff of the coronary arteries may be important in surgical planning. While routine MRA or CTA are typically performed to rule out distal aneurysmal disease prior to surgical aortic root replacement, it has been suggested that coronary artery CT should additionally be performed in order to rule out anomalous coronaries [Citation97,Citation102].
Of the three patients described by Mulder et al. all had a favorable outcome although lack of pre-operative diagnosis was associated with a major perioperative complication [Citation102].
Echocardiographic and angiographic focused imaging of the aortic root may result in the finding of coronary anomalies that might otherwise go unnoticed. Pre-operative coronary imaging may provide useful information for our surgical colleagues. The approach to the patient with an asymptomatic anomalous left coronary artery who does not yet meet surgical criteria for aortic root replacement remains uncertain with regards to whether the timing of surgical intervention for anomalous left coronary artery should be altered to coincide with root replacement, or whether root replacement should be performed at a smaller size to avoid two aortic root surgeries.
7.1.3. Atherosclerotic coronary artery disease
The frequency of atherosclerotic coronary artery disease in Marfan syndrome is unknown. Risk factors for atherosclerosis, including obesity, smoking, hyperlipidemia, and hypertension do not differ from the general population [Citation104]. In addition, carotid intimal thickness, a surrogate measure for coronary artery disease, has been shown not to differ between Marfan syndrome and the general population [Citation105]. Together, this data suggests that patients with Marfan syndrome should carry the same risk of atherosclerotic coronary artery disease as those without. The current literature however is devoid of reports of atherosclerotic coronary artery disease in Marfan syndrome. Whether this relates to a younger age of death due to aortic complications (particularly in those with risk factors for coronary artery disease), underreporting of a less than novel phenomenon, or an inherent protective mechanism remains unknown. Impaired fibrillin-1 function has been associated with increased arterial wall stiffness leading to increased atherosclerotic burden in an ApoE deficient mouse model [Citation106]. Similarly, elastic fiber fragmentation is associated with increased plaque rupture and myocardial infarction in atherogenic mice [Citation107]. While there is an established relationship between increased aortic stiffness and atherosclerotic burden in humans without Marfan syndrome, the same relationship has not been documented in patients with Marfan syndrome. The pleiotropic effects of the cytokine TGFbeta are poorly understood as they relate to atherosclerosis with both protective and deleterious effects documented. This raises the question as to whether TGFbeta may in fact play a beneficial role in patients with Marfan syndrome. Becker and colleagues examined the coronaries in five young adults with Marfan syndrome and death from aortic complications [Citation79]. They noted the extramural coronaries in all five patients to be similarly affected to the aorta with elastic lamella fragmentation in the vascular media. Associated aneurysmal disease of the extramural coronaries was noted in one of the five patients [Citation79]. A larger vessel diameter could, in theory, provide some degree of protection from occlusion from atherosclerotic plaque despite the presence of increased vascular stiffness and elastic fiber fragmentation.
The above-mentioned risk factors for coronary artery disease have also been implicated in aortic aneurysm formation and dissection. While patients with Marfan syndrome have historically been considered to have an asthenic body habitus, the presence of overweight or obesity was documented to be present in 26% of an adult patient cohort, a rate not different than that of the general population [Citation104]. Adipose tissue is known to be metabolically active with the production of several different cytokines and vasoactive substances including angiotensin II and TGF-beta which can adversely affect aortic histology and biomechanics [Citation104].
While the risk of atherosclerotic coronary disease remains undefined in Marfan syndrome, modification of standard risk factors remains essential to limiting aortic disease [Citation104]. Healthy body weight, normal levels of glycated hemoglobin (HgbA1C) and fasting lipid profile should remain a focus of preventative cardiac care. Other established risk factors for coronary artery disease in the general population and aortic disease in the Marfan population include elevated homocysteine [Citation108–Citation110] and impaired endothelial function [Citation111]. Elevated homocysteine levels can occur secondary to genetic variants or nutritional deficiency and have been associated with a significantly greater degree of aortic pathology in patients with Marfan syndrome [Citation109,Citation110]. Treatment with B vitamins may improve outcome [Citation112]. Impaired endothelial relaxation occurs in Marfan syndrome and has been shown to be responsive to combined Losartan and Doxycycline therapy [Citation113] but to neither therapy alone. While coronary artery disease is associated with endothelial dysfunction in the general population, the vasomotor properties of the coronary arteries in Marfan syndrome have not been studied.
Giusti documented that Marfan patients with the most severe vascular changes, including aortic dissection, had significantly higher homocysteine levels when compared to patients with mild changes or normal controls [Citation110]. Therapies shown to be both beneficial in aortic and coronary artery disease, namely statins for hyperlipidemia [Citation114,Citation115] and peroxisome proliferator-activated receptor (PPAR) gamma agonists for diabetes [Citation116] may be considered as adjuvant therapies in at-risk patients. Both classes of drugs have been shown to block TGF-beta activation.
In conclusion, genetic and environmental modifiers of atherosclerotic coronary artery disease have been shown to play a role in aortic pathologic changes in Marfan syndrome. Attention toward the elimination of the risk factors may improve long-term aortic health and reduce coronary artery atherosclerosis.
7.2. Arterial tortuosity
Increasing attention to arterial tortuosity suggests that this is quite common in Marfan syndrome, and increased tortuosity is a sign of more aggressive systemic disease [Citation117]. Tortuosity of the carotid, subclavian, vertebral, and iliac arteries has been described in Marfan syndrome since the late 1980s [Citation117–Citation121]. Once Loeys-Dietz syndrome was described as a separate group of disorders commonly associated with arterial tortuosity, there was discussion as to whether the tortuosity previously described was actually in cases of Loeys-Dietz syndrome [Citation122]. Studies then explored arterial tortuosity using strict diagnostic criteria comparing Loeys-Dietz syndrome and Marfan syndrome [Citation117,Citation120]. In these two studies of vertebral artery tortuosity, vertebral artery tortuosity was common in both Loeys-Dietz syndrome (77-90%) and Marfan syndrome (72-77%), although the degree of tortuosity was less severe in Marfan syndrome. Using quantitative measures, the degree of arterial tortuosity has been shown to be one of the only independent predictors of adverse cardiovascular events in Marfan syndrome apart from aortic dimension. This has been demonstrated using both vertebral arteries and aortic tortuosity indices [Citation117,Citation123,Citation124].
7.3. Aneurysm and dissection of large- and medium-sized arteries
Extra-aortic aneurysms and dissections are most commonly noted as a consequence of aortic dissection, in which the dissection extends to more distal vessels [Citation119,Citation121,Citation125–Citation127]. Less well characterized are aneurysms and dissection of the peripheral vessels independent of aortic dissection. Isolated peripheral aneurysms have been described in Marfan syndrome in the carotid, subclavian, axillary, internal mammary, ulnar, iliac, and superficial femoral arteries [Citation128–Citation138]. A few studies have attempted to characterize the true prevalence of dilation or aneurysms in those without prior intervention. A study of 35 patients with Marfan syndrome without prior aortic intervention were studied with thoracoabdominal MRA [Citation121]. This study demonstrated that 7 (20%) had aneurysmal dilation of the peripheral vessels including the iliac arteries, celiac artery, renal artery, and right subclavian artery. Kono et al. also showed iliac artery aneurysm in 20% of patients with Marfan syndrome who did not have dissection extending to the abdominal aorta [Citation120].
In individuals with prior intervention, aneurysms and dissection are more common [Citation121,Citation125,Citation126]. In the study by Mariucci, of the 29 patients who were status post-intervention (although excluding patients with dissections), 14 (43%) had dilation of an iliac, celiac, renal or subclavian artery [Citation121]. In a study of 100 patients with Marfan syndrome who had undergone arterial interventions for miscellaneous indications, including aortic dissection, almost every patient had dilation of at least 2 non-aortic arteries, and 20% needed additional intervention in non-aortic arteries, most within 6 years of the initial intervention [Citation125]. In this study, 35% of patients who suffered from type A or type B aortic dissection underwent intervention of an extra-aortic arterial segment at some point.
A study by Yetman demonstrated that one-third of individuals in an adult cohort of patients with Marfan syndrome (both with and without intervention/dissection) had peripheral vascular aneurysms, most detected incidentally during CTA or MRA of the thoracoabdominal aorta [Citation126]. Of the 47 peripheral aneurysms noted, 26 (55%) required intervention. Gaertner et al. screened a smaller cohort of 21 patients with Marfan syndrome (both with and without prior intervention) for peripheral aneurysms, and similarly noted that 10 (48% of all, 67% of the adults) had peripheral aneurysms, including in the iliac, vertebral, brachiocephalic trunk, renal, subclavian, femoral, popliteal, and axillary arteries [Citation139]. Treatment of peripheral aneurysms and dissection has been varied depending on location and extent [Citation119,Citation125,Citation126]. Of note, vascular tortuosity may make traditional techniques more challenging [Citation119].
7.3.1. Cerebral arteries
Rare cases of cerebral aneurysm in Marfan syndrome have been documented since the first reported observation in 1971 [Citation118,Citation140–Citation152]. These case reports suggested an increased prevalence of cerebral aneurysms in Marfan syndrome compared to the general population. Followed were a variety of conflicting publications, triggering debate on the necessity of neurovascular screening. Depending on the type of study and strictness of diagnostic criteria for Marfan syndrome, estimates of the prevalence of cerebral aneurysm have ranged from 0% to >20% [Citation143,Citation153–Citation156].
As better understanding has evolved regarding genetic causes of aortopathy and arteriopathy, it is likely that some of the early cases were actually separate genetic syndromes with phenotypic overlap with Marfan syndrome. These include conditions caused by mutations the TGF-ß pathway genes causing Loeys-Dietz syndrome that are known to commonly have neurovascular symptoms. Of note, as far as we know, none of the case studies of cerebral aneurysm in Marfan syndrome reported a known pathogenic FBN1 variant, although the original case by Speciali did report ectopia lentis [Citation140].
Cerebral artery dissection in Marfan syndrome appears to be uncommon, but of patients presenting with cerebral dissection, a diagnosis of Marfan syndrome should be considered. In a 44-person cohort from China with intracranial vertebral–basilar artery dissection, whole-exome sequencing demonstrated that one patient had a FBN1 pathogenic variant and clinical features consistent with Marfan syndrome. In a series by Kim et al., 2 dissections were noted in the 59 patients with Marfan syndrome who had neurovascular imaging [Citation156]. In conclusion, given the rarity of the condition in those with clear Marfan syndrome, neurovascular screening is not currently recommended, but there should be a low threshold for imaging if symptoms suggest cerebrovascular involvement.
7.3.2. Spontaneous cervical artery dissection
Spontaneous dissection of the carotid and vertebral arteries, a major cause of ischemic stroke in the young, is most often idiopathic [Citation157]. However, Marfan syndrome is often considered in the differential diagnosis. Both ultrastructural aberration of the arterial wall and arterial tortuosity have been noted to be common in carotid artery dissection, invoking a possible etiology as connective tissue disease [Citation158,Citation159]. However, cases to date of confirmed Marfan syndrome and carotid or vertebral artery dissection are almost always in the presence of prior aortic dissection or surgery [Citation160,Citation161]. In two studies systemically evaluating 84 and 43 patients, respectively, with spontaneous cervical artery dissection, none had Marfan syndrome [Citation162,Citation163]. In a large study of 1,934 consecutive patients with cervical artery dissection specifically evaluating for inherited connective tissue disease, only one had clinical Marfan syndrome; FBN1 testing was not performed [Citation157]. This prevalence is similar to that of Marfan syndrome in the general population [Citation164]. Given literature to date, there does not appear to be a significant association between Marfan syndrome and spontaneous cervical artery dissection without a prior inciting event.
7.3.3. Visceral arteries
In Marfan syndrome, the visceral arteries are most commonly involved after descending aortic dissection or graft repair, when the visceral arteries are at risk of dissection and patch aneurysm [Citation127]. Rare cases of visceral artery aneurysms unrelated to dissection have also been reported in Marfan syndrome including in the gastric, celiac artery, splenic, and hepatic arteries, almost all necessitating intervention [Citation120,Citation121,Citation125,Citation137,Citation165–Citation167]. One study demonstrated that Marfan syndrome was present in 3% of cases of visceral artery aneurysms [Citation165].
7.4. Expert opinion on patients’ reported symptoms ()
Hypotension and syncope can both be indirectly related to imbalances in blood pressure regulation which may be impaired in patients with Marfan syndrome. Indeed, both orthostatic hypotension and autonomic dysfunction are more commonly occurring in patients with Marfan syndrome. Treatment with beta-blockers, or angiotensin receptor blockers, to slow aortic root growth, could be a reinforcing factor of both. Ceasing beta-blockers did not normalize the abnormal blood pressure response in one study [Citation168].
7.5. Conclusion
In conclusion, extra-aortic vascular disease may occur in Marfan syndrome. Both spontaneous coronary artery dissection and anomalous coronary artery origins have been described, although it is unclear if the prevalence of these conditions is higher than in the healthy population. Minimal data exist about the atherosclerotic disease in Marfan syndrome, but clinical risk factors appear to be similar to the rest of the population. The most common extra-aortic vascular manifestations of Marfan syndrome are aneurysms of the medium and large vessels and the visceral arteries. These aneurysms are most common in patients who have had a prior aortic dissection or prior cardiovascular surgery. Intermittent serial screening by MR and CT angiography from neck to pelvis in this subpopulation of patients with Marfan syndrome should be considered. Literature to date does not support a significantly increased risk of spontaneous artery dissection or cerebral artery aneurysms in Marfan syndrome.
8. Lung features by Enid Neptune
The lung manifestations of Marfan syndrome are burdensome and complex [Citation169]. More importantly, they can lead to substantial disability and reduced quality of life. Scoliosis, pectus deformities and respiratory muscle weakness contribute to restrictive lung disease. Parenchymal lung disease, often developmental in origin, leads to upper lobe blebs, pneumothoraces and, especially in the setting of neonatal Marfan syndrome, overt emphysema. Both chest wall deformities and airway wall defects can manifest disorders such as asthma and bronchiectasis. Finally, soft tissue laxity and a predilection for upper airway obstruction confer the high prevalence of sleep-disordered breathing. This spectrum of lung disorders is challenging for care providers who often have to evaluate dyspnea, chest pain and reduced exercise capacity in the context of the known risks of highly morbid and fatal cardiac and aortic disease. Unfortunately, no reliable contemporary (post-Ghent 2010) assessment of lung disease prevalence is available given that screening for pulmonary impairment is inconsistent across centers and mostly symptom- or finding-triggered [Citation170]. However, even small, single-center retrospective studies confirm a high burden of respiratory disease in Marfan syndrome [Citation171].
8.1. Restrictive lung disease
Chest wall deformities such as scoliosis and pectus deformities are common with Marfan Syndrome often resulting in restrictive lung disease [Citation172]. With severe restriction, thoracic insufficiency syndrome (TIS) develops punctuated by impaired ventilation and gas exchange [Citation173]. In pediatric Marfan syndrome, lung growth is further compromised by these disorders directly impacting peak lung function attainment and accelerating the onset of TIS. Scoliosis is the most common spinal deformity in Marfan syndrome afflicting more than 60% of patients [Citation174]. Up to one-half of affected persons require surgery for progression and/or associated respiratory compromise [Citation175]. Pectus deformities, namely pectus excavatum and to a lesser degree pectus carinatum, are common congenital disorders that are highly represented in Marfan syndrome [Citation176]. Most pectus deformities are mild and do not produce marked alterations in lung function. However, more severe pectus defects, especially when coupled to scoliosis, can cause clinically consequential restrictive disease [Citation177]. Less invasive and more customizable surgical options (e.g., vertical expandable prosthetic titanium ribs (VEPTR) magnetic rods, or Nuss bars) for both scoliosis and pectus deformities are now employed early in disease development leading to fewer complications and lung function stabilization in selected patients [Citation178–Citation182]. As yet, no long-term studies documenting stability or improvement in lung function with these newer interventions in the Marfan population are available.
8.2. Parenchymal lung disease
Long before the genetic cause of Marfan syndrome was discovered, multiple case reports described spontaneous pneumothorax as a feature [Citation183]. Even now, spontaneous pneumothorax, a minor systemic criterion in the Ghent algorithm for Marfan syndrome, can be the initial clinical context that ultimately leads to the Marfan syndrome diagnosis; nonetheless, the true prevalence among all Marfan syndrome patients is unknown [Citation170,Citation184,Citation185]. Small studies indicate a much higher prevalence in Marfan syndrome compared with the general population [Citation171,Citation186,Citation187]. Upper lobe blebs, pulmonary cysts, and overt emphysema, sometimes revealed as incidental findings on imaging, all predispose to pneumothorax. Because of this, the true prevalence of the airspace findings in Marfan syndrome is unknown, but is likely much higher than the incidence of pneumothorax. Recurrent and bilateral pneumothoraces are also more common in Marfan syndrome and despite surgical resection, blebs can reform quickly. Accordingly, prophylactic surgical interventions are not supported. Moderate or severe pneumothoraces, especially if tension physiology is demonstrated, merit mechanical pleurodesis and local bleb resection. Chemical pleurodesis can be hazardous if future aortic surgeries are anticipated. Other approaches such as blood patches or bronchoscopic airway valves (especially if a bronchopleural fistula is present) may have special utility in the Marfan population but are understudied [Citation188,Citation189].
Mouse models of Marfan syndrome consistently show airspace enlargement suggesting that airspace dysmorphology is a primary and early feature of the syndrome. The likely mechanism is a developmental disturbance in airspace formation triggered by the lack of fibrillin-1 during critical stages of lung development. This pathogenetic scheme is supported by the complex constellation of neonatal Marfan syndrome which includes pulmonary emphysema as well as severe cardiovascular impairments [Citation190,Citation191]. Further, a histologic survey in early-onset acquired emphysema showed reduced levels of fibrillin-1 expression in the airspace compartment [Citation192]. Fortunately, therapeutic considerations are emerging. While Losartan shows efficacy for emphysema reversal in mouse models of Marfan syndrome, no clinical trials have thus far included pulmonary outcomes [Citation193].
8.3. Airway disorders
Airways obstruction on pulmonary function testing and/or recurrent wheezing have been reported in Marfan syndrome [Citation194]. The anatomic basis for this manifestation is unclear and may reflect airway smooth muscle dysfunction as a consequence of altered matrix composition or direct airway compression from chest wall deformities. Additionally, primary alterations in the immune milieu might contribute similarly to the allergic airways disease observed in Loeys-Dietz Syndrome [Citation195]. A few case reports detail bronchiectasis in Marfan Syndrome which could also reflect primary large airway wall pathology [Citation196,Citation197]. For all of these disorders, the use of aerosolized β-adrenergic agonists is discouraged given potential cardiovascular side effects as well as antagonistic interactions with β-blockers. Inhaled anticholinergic and inhaled corticosteroids are the preferred treatments.
8.4. Expert opinion on patients’ reported symptoms ()
does not include very common symptoms that Marfan patients, and their physicians, often attribute to cardiac disease or deconditioning such as dyspnea with exertion, reduced exercise capacity or chest pain. These symptoms may reflect lung disorders that are underdiagnosed in persons with Marfan syndrome such as asthma, bullous lung disease, or restrictive lung disease. Early involvement of lung specialists to evaluate these symptoms would serve the interests of the Marfan population.
8.5. Conclusions
In conclusion, a variety of lung features occur with Marfan syndrome that can lead to reduced quality of life and progressive disability. Awareness of these complications with early diagnosis and appropriate interventions is crucial. Close monitoring of lung function, standardized to sitting rather than standing height, and exercise capacity by pulmonary specialists should be incorporated into a coherent multidisciplinary Marfan care plan [Citation198].
9. Sleep physiology features by Laura Muiño-Mosquera
9.1. Obstructive sleep apnea (OSA)
The first reference to sleep apnea in Marfan syndrome dates from almost 30 years ago. In 1991 Cistulli and Sullivan first described six patients with Marfan syndrome (2 females, age range 15-43yrs), all of whom presented obstructive sleep apnea syndrome [Citation199]. Further research confirmed that the prevalence of sleep apnea in patients with Marfan was relatively high, ranging from 31% to 64% [Citation200–Citation202] depending on the studied cohort and the method used to test sleep apnea. In fact, two studies found that in comparison to sex- and age-matched controls, the prevalence of sleep apnea in Marfan patients was significantly higher [Citation200,Citation201].
9.2. Central sleep apnea (CSA)
Three different types of sleep apnea can be distinguished: central, obstructive and mixed. Most commonly, patients with Marfan syndrome present obstructive episodes, but one study also found high prevalence of central sleep apnea [Citation202]. The obstructive episodes can be explained by the characteristic facial features (retrognathia, malar hypoplasia, dolichocephaly) in combination with higher airway collapsibility [Citation203,Citation204]. Furthermore, patients with a high body mass index seem to have higher prevalence of obstructive sleep apnea [Citation201,Citation202,Citation205]. Why patients with Marfan syndrome present central sleep apnea is not clear, but it may be related to impaired left ventricular function in some patients, and autonomic system dysregulation in others [Citation202,Citation205]. Detection of sleep apnea through a questionnaire is cumbersome because there is a poor correlation between the score and the presence of sleep apnea. In our experience, the STOP-bang questionnaire seems to have the best positive predictive value [Citation205].
9.3. Clinical consequences of sleep apnea
Individuals with sleep apnea that is unrelated to Marfan syndrome appear to have a higher cardiovascular risk, showing a higher prevalence of hypertension, stroke, and arrhythmia [Citation206–Citation210]. Higher systolic blood pressure and higher prevalence of ventricular arrhythmia were mildly associated with sleep apnea in Marfan syndrome in one study [Citation205] and with higher incidence of atrial fibrillation in another [Citation202]. In the general population, the relationship between sleep apnea and aortic diameters is less-well studied and the findings are inconsistent [Citation211–Citation214]. Similarly, in Marfan syndrome patients the relation between sleep apnea and aortic root growth is debated: while some studies find an association [Citation201,Citation215], others fail to do so [Citation202,Citation205]. The influence of sleep apnea in other organ systems and in the quality of life has not been studied in Marfan syndrome but could be very relevant. In our own experience, treatment of sleep apnea in patients with Marfan syndrome is successful and improves fatigue and quality of life.
Treatment strategy is the same as in the general population and consists of weight reduction (in those patients with overweight) and nasal continuous positive airway pressure (CPAP).
9.4. Expert opinion on patients’ reported symptoms ()
Interestingly, patients do not report symptoms of sleep-disordered breathing (). Patients will typically not report sleep apnea as a symptom but will often complain of fatigue which is an inherent consequence. Also, interrogating the patient’s bed partner may reveal relevant information regarding snoring and arousals.
9.5. Conclusion
In conclusion, sleep apnea is highly prevalent in patients with Marfan syndrome. Although the typical Marfan physiognomy and higher airway collapsibility are, in part, responsible for this higher prevalence, overweight, especially in older ages, is an important and modifiable risk factor to be taken into consideration. Although sleep apnea does not clearly increase cardiovascular risk in patients with Marfan syndrome, further research in this field might be necessary to elucidate whether patients with Marfan syndrome might benefit from an earlier and more aggressive treatment.
10. Abdominal features by reed Pyeritz
No abdominal features are currently included in the nosology of Marfan syndrome, although several features with variable relevance have been reported.
10.1. Liver and kidneys cysts
A manifestation that clearly is of increased prevalence is cyst formation in the liver and kidneys [Citation216]. These first were recognized incidentally on imaging of the abdomen performed for cardiovascular indications. Their prevalence is clearly greater than in age-matched controls (59% vs. 30% for renal and 35% vs. 17% for hepatic cysts). Some cysts can become quite large, but other than a mass effect on surrounding tissue, the cysts are benign and harmless. Their age of first appearance and growth in both number and size over time have not been examined. Some confusion with adult polycystic kidney disease exists in the older literature.
10.2. Biliary tract features
Cholelithiasis may be more common in Marfan syndrome (in the only study of unselected, asymptomatic patients, 18% vs. 2% of controls) [Citation216]. Biliary duct ectasia, likely contributing to bile stasis has been reported [Citation217].
10.3. Diaphragmatic hernia
In individual patients, especially those with severe, neonatal Marfan syndrome, a dozen or so reports of diaphragmatic hernias of various types occur. In fact, the presence of a diaphragmatic hernia in children is one predictor of reduced life-expectancy [Citation218]. Congenital hernias may not be detected until later in life [Citation219]. A reporting bias in older patients undoubtedly exists, certainly when an abdominal complication has occurred in association with a hernia. Variants in FBN1, as well as other genes, contribute to familial congenital diaphragmatic hernia independent of Marfan syndrome [Citation220].
10.4. Other abdominal features
The same reporting bias pertains to intestinal issues, such an intussusception, volvulus, diverticulosis, and rupture. In one retrospective examination of adults admitted for diverticulosis, a slight association with Marfan syndrome (odds ratio = 2.4) was found [Citation221].
10.5. Adiposity
Most people with Marfan syndrome accrue adiposity as they age [Citation104]. In Marfan syndrome, this is most pronounced in the abdomen, and the appearance is accentuated by the persistent slimness of the extremities. Increased visceral fat predisposes to insulin resistance and type 2 diabetes mellitus, and may predispose to aortic complications [Citation104]. The adiposity is composed of white fat, in distinction to brown fat that is subcutaneous. Fibrillin-1 plays both a structural and hormonal role in the differentiation of adipocytes, so pathogenic variants in FBN1 may likely contribute to the deposition of fat in the abdomen. Since the BMP pathway is involved in adipocyte differentiation, it would be interesting to determine if the TGF-β pathway is involved and if medication, such as angiotensin receptor blockers, might decrease visceral fat accumulation. This hypothesis might be pursued with additional data [Citation222–Citation224].
10.6. Expert opinion on patients’ reported symptoms ()
The patients’ complaints of abdominal symptoms do not appear to relate to any of the Marfan-specific features discussed in this chapter or to any other potential features of Marfan syndrome that I am aware of.
10.7. Conclusion
All of these potential abdominal issues, as well as cardiovascular ones, should be considered in any person with Marfan syndrome presenting with abdominal features, particularly in older patients.
11. Urogenital features by Thy Thy Vanem
11.1. Kidney cysts
An increased prevalence of small renal cysts was already mentioned above. Although small renal cysts usually are benign, it has been assumed that cystic kidney disease is associated with aneurysm progression in Marfan syndrome and that such cysts should be considered as markers for aortic aneurysm development [Citation225,Citation226]. It is not known if the formation of renal cysts in Marfan syndrome is influenced by TGFβ1 or if it is due to a structural failing of microfibrils [Citation216]. A case report of Marfan syndrome mouse with renal cystic disease, extreme aortic dilatation and increased vascular inflammation suggests that cystic renal disease may be causally involved in the pathogenesis of aortic aneurysm formation by promoting aortic inflammation [Citation226].
Linking renal cysts with Marfan syndrome is also suggested from the opposite way: patients with autosomal dominant polycystic kidney disease (ADPKD) clearly show an increased prevalence of aortic root aneurysms [Citation227]. In patients with ADPKD, some systemic features reminiscent of Marfan syndrome have been reported in ADPKD patients [Citation228–Citation230]. The process of aortic aneurysm formation in ADPKD is incompletely understood. The proteins encoded by the genes involved in ADPKD (PKD1 and PKD2) encode components of the polycystin complex which is regarded as an important mechanosensor in the kidney – a role in the aortic wall is not established [Citation231]. Dysregulated TGF-β signaling has also been suggested to play a role in the development of aortic aneurysms based on studies in mouse models. Double heterozygous mice with targeted mutations in Pkd1 and Fbn1 displayed a more severe aortic phenotype, which is according to further experiments related to further upregulation of TGF-ß signaling caused by Pkd1 haploinsufficiency.
11.2. Other renal features
Several other renal features have been described in case reports in Marfan syndrome patients, such as nephrotic syndrome due to focal segmental glomerulosclerosis, medullary sponge kidney, recurrent nephrolithiasis, glomerular basement alterations, renal vascular anomaly and renovascular hypertension [Citation232–Citation239]. The prevalence of kidney disease in Marfan syndrome is not known and there is no established evidence of the association between kidney diseases and Marfan syndrome. However, one paper suggests that microfibrillar disarrangement can be the cause of glomerular basement membrane alterations in Marfan syndrome [Citation232].
11.3. Premature labor
Cardiovascular complications are known risks in pregnant Marfan syndrome women, as well as increased risk of obstetric and fetal/neonatal complications compared to healthy controls [Citation240]. There are several studies on pregnancy outcome in Marfan syndrome women, but few studies describing specifically premature labor in Marfan syndrome women, although premature labor is one of the most common reasons for hospitalization of pregnant women in the general population. One paper reports a case of a Marfan syndrome woman who experienced premature labor in five out of six pregnancies, but with no evidence of cervical incompetence [Citation241].
Several studies have reported similar rates of preterm births in Marfan syndrome groups compared to the general population [Citation242–Citation245]. However, one retrospective study from 2006 found an increased percentage of preterm deliveries due to the preterm premature rupture of membranes and cervical incompetence in Marfan syndrome women [Citation246]. In a study from 2014, 29 pregnancies in 21 Marfan syndrome women were compared to 116 controls [Citation240]. Only pregnancies beyond 24 weeks of gestational age and only singleton pregnancies were included in this study. Two babies (7%) in the Marfan syndrome group were delivered preterm iatrogenically due to maternal reasons, none due to spontaneous premature labor.
11.4. Urinary incontinence
It has been assumed that connective tissue abnormality can contribute to urinary incontinence [Citation247,Citation248], and a high prevalence of urinary incontinence has been reported in women with Marfan syndrome [Citation247–Citation249]. However, urinary incontinence is common in women in general and the estimated prevalence varies depending on several factors, among them the population studied. In adult women in the general population, an estimated prevalence of nearly 50% has been reported, but few patients seek help for the condition [Citation250–Citation252]. Lower prevalence of 10-17% has been reported in non-pregnant women age 20 years and above [Citation253,Citation254]. In a study by Jabs, a significantly higher prevalence was found in female Marfan syndrome patients compared to the general population [Citation255]. Eighty-eight patients reported a history of urinary incontinence and 72% had experienced episodes of stress incontinence. Of these, 52% considered the problems as significant. In this Marfan syndrome population, stress incontinence was not associated with parity. In a study by Chan et al. patients with Marfan syndrome had significantly higher incidence of urinary symptoms, stress incontinence and urge incontinence compared to controls, despite lower parity in the Marfan group [Citation247]. None of the studies have found correlations between joint hypermobility and stress incontinence.
11.5. Expert opinion on patients’ reported symptoms ()
Patients reported bladder problems, urinary tract infection and premature labor as major complaints, and urinary incontinence, maldescensus testis, ovarian cysts, and kidney stones as minor/rare complaints (). As far as not addressed above, we will try to comment according to what is known from the literature:
Urinary disorders are not frequent in Marfan syndrome, and apart from urinary incontinence, no studies describe other chronic urinary tract symptoms or increased urinary tract infection in patients with Marfan syndrome. One paper describes four patients with urinary problems secondary to vascular or neurological complications related to Marfan syndrome [Citation256].
We have found one paper describing a case of seminoma in Marfan syndrome [Citation257]. Apart from this, we have not found studies describing associations between specific urogenital features and Marfan syndrome, such as maldescensus testis, which is the most common congenital anomaly in the genitourinary tract [Citation258].
Marfan patients have reported ovarian cysts and kidney stones as minor complaints. However, we have not found any studies to confirm any relations between these findings and Marfan syndrome. Two papers describe cases where a pelvic mass was initially misdiagnosed as an ovarian cyst, but where further investigations revealed an anterior sacral meningocele, which is a severe form of dural ectasia, a known feature of Marfan syndrome [Citation259,Citation260].
11.6. Conclusion
There is no evidence of associations between kidney diseases and Marfan syndrome. One study indicates an increased percentage of preterm deliveries in women with Marfan syndrome. There are few studies on premature labor in women with Marfan syndrome. Urinary disorders are not common in Marfan syndrome, but urinary incontinence has been suggested to be added to the list of clinical features in Marfan syndrome women.
12. Musculoskeletal features by Nina Riise
12.1. Myopathy
Skeletal muscle abnormalities including variation in fiber size, overall reduced fiber size, fatty infiltration, excessive matrix between fibers and a reduced number of proliferating satellite cells have been described in ‘large subsets’ [Citation261] or ‘many individuals’ [Citation262] with Marfan syndrome [Citation263] but the clinical evidence is sparse. However, decreasing muscle mass over time also has been described [Citation264].
TGF-beta1 has a known role in signaling in skeletal muscle. Increased TGF-beta1 activity leads to failed muscle regeneration [Citation262] and fibrosis [Citation265]. It may lead to the formation of scar tissues, weakened muscle function, pain and increased risk of reinjury [Citation266].
Marfan syndrome myopathy may cause muscle fatigue and weakness [Citation267], myalgia, cramps [Citation268], inability to increase muscle mass despite physical exercise, hypotonia [Citation261,Citation262], and reduced muscle mass [Citation264,Citation268]. It may be associated with reduced bone mineral density [Citation264] and respiratory failure [Citation269].
There is no current therapy. Losartan and retinoic acid have shown a beneficial effect on muscles in experimental models [Citation262,Citation270].
12.2. Reduced bone mineral density
Several studies reported reduced average bone mineral density in Marfan syndrome [Citation264,Citation271–Citation277]. However, other studies do not corroborate a reduced bone mineral density [Citation278–Citation280]. The prevalence of osteoporosis in Marfan syndrome is unknown. The studies that are available have several methodological weaknesses, and different diagnostic criteria are used. Some studies point out that bone mineral density decreases over time [Citation264,Citation277]. Defective fibrillin-1 leads to improper over activation of latent TGF-beta, enhanced osteoblast maturation and osteoblast-supported osteoclast activity, which could account for reduced bone mineral density [Citation281–Citation283].
In conclusion, it is unclear whether patients with Marfan syndrome have reduced bone mineral density, and if so, whether this translates into an increased risk for bone fractures [Citation271,Citation275]. However, clinicians should be aware of this possibility, and screening with dual-energy X-ray absorptiometry (DEXA) should be considered in individuals with clinical risk factors or symptoms. There is no evidence on the prevention or treatment of osteopenia or osteoporosis in Marfan syndrome. General guidelines should be followed. A few studies have shown conflicting results on the effect of losartan on bone mineral density [Citation277,Citation284,Citation285]. There are no studies on bone mineral density in groups of older people with Marfan syndrome; hence, the long-term prognosis is unknown.
12.3. Craniofacial features
Information about facial dysmorphism in Marfan syndrome may be useful for early recognition of the disease [Citation286,Citation287]. A recent study using 3-dimensional skeletal imaging reported greater facial divergence and a lower facial height index as a uniform feature in adults with Marfan syndrome [Citation286]. Another study [Citation288] described a retrognathic maxilla in 81% and retrognathic mandible in 89%. The mandibular ramus is short [Citation286,Citation288]. Large frontal sinuses [Citation288] and high nasal airway restriction associated with maxillary restriction have been described [Citation289].
The facial features listed in the revised Ghent criteria [Citation170] comprise dolichocephaly, malar hypoplasia, enophthalmus, retrognathia, and downslanting palpebral fissures. These have been found to yield a diagnostic sensitivity of 28–54% and a specificity of 91–99% for Marfan syndrome [Citation287,Citation290]. The craniofacial features may increase the risk of obstructive sleep apnea [Citation289] and cause dental crowding and overjet [Citation288] necessitating orthodontic treatment in some cases [Citation288].
The cause of the craniofacial abnormalities in Marfan syndrome is not clear. Fibrillin insufficiency in the periosteum and inserting muscles, improper response to deficient tension in periosteum and sutures leading to ‘long face’ growth or oral breathing are suggested explanations [Citation291].
12.4. Expert opinion on patients’ reported symptoms ()
Deviated nasal septum and sinusitis have been described as common, and allegedly large tonsils and adenoids can contribute to otitis in Marfan syndrome [Citation292]. However, there are no data to confirm this. Vertebral body hemangioma and ingrown toenails are not described as features of Marfan syndrome. Manifest osteoporosis may be more frequent than in the general populations, as some studies describe an increased prevalence of decreased bone mineral density. Marked abnormalities in the pelvic values compared with those found in the unaffected population, with increased retroversion of the pelvis, in particular, were found in one study [Citation293].
12.5. Conclusion
People with Marfan syndrome may have myopathy as part of the syndrome, but the prevalence, severity, and mechanisms are poorly described. Decreased bone mineral density has been described in people with Marfan syndrome. However, the evidence is conflicting, and the clinical relevance is uncertain.
13. Dermatological features by Leema Robert
Skin striae are one of the systemic features that is scored in the revised nosology for Marfan syndrome. One of the first studies to document skin and joint hypermobility in patients with Marfan syndrome was undertaken by Grahame and Pyeritz in 1995 [Citation294], clearly documenting striae, atrophic scars, and skin hyperextensibility, all increasing with age. Striae atrophicae were the most common finding and were documented in two-thirds of patients in this study. Papyrous scars were present but not as large as those seen in Ehlers-Danlos syndromes. A clear and significant association of skin features in and presence of hyperextensible joints was also demonstrated in patients with Marfan syndrome. It deserves to be documented that patients ascertained for this study were from a single clinic in Maryland, USA. Their diagnosis was established on the presence of unequivocal clinical features of Marfan syndrome.
Another key paper that documents skin features is the study undertaken by Ledoux et al. [Citation295], which is a systematic review of skin and cutaneous features in 61 consecutive patients with age, sex and height matched controls. Sixty-six percent of individuals with Marfan syndrome had striae when compared to 16% of controls. Similar to the Grahame and Pyeritz paper, large hypertrophic or atrophic scars were also documented in higher frequency in Marfan syndrome vs controls (46% vs 21%). Hyperpigmented or hypopigmented scars were also noted. Other features documented include hernia (5 in Marfan syndrome vs 4 in controls), fibroma (1 vs 1), and purpura (1 vs 1). In contrast to the Grahame and Pyeritz paper, the 49 Marfan syndrome patients in this cohort had undergone genetic testing with a proven pathogenic variant in FBN1 in 34 patients and a TGFBR2 variant in one patient. The incidence of striae however is similar to the previous publication and two-thirds of patients were noted to have this dermal manifestation. The clear distinction however is the presence of striae in uncommon areas of the body in Marfan syndrome patients. Adult patients with Marfan syndrome demonstrated numerous striae on shoulders, breast, flanks, hips, and buttocks. The authors therefore conclude that striae could be a used a good diagnostic criterion for Marfan syndrome, particularly if they located in atypical sites. The authors therefore conclude that striae could be used as a good diagnostic criterion for Marfan syndrome, particularly if they located in atypical sites.
Several single case reports of various skins and cutaneous involvement have been published. It is debatable whether these are just coincidental findings as the larger studies described do not validate these findings. These case reports and case series on skin and cutaneous features reported in Marfan syndrome are briefly outlined below.
Alopecia universalis: Ziv et al. describes non-scarring alopecia of the whole body (alopecia universalis) in a case with clinical features of Marfan syndrome [Citation296].
White forelock: Herman et al. describe an unusual association of white forelock in a case with clinical features of Marfan syndrome [Citation297].
Vermiculate atrophoderma: Harper et al. describe a 14-year-old boy with clear clinical features of Marfan syndrome with vermiculate atrophoderma [Citation298].
Lichen planus: Nallegowda et al. report on pterygium of both upper and lower limb nails in a patient with clear clinical features of Marfan syndrome [Citation299]. This was concluded as Lichen planus where variable involvement of nails are well documented.
Lipodystrophy: A case with de novo pathogenic variant in exon 64 of the FBN1 gene (c.8155_8156del2) was described with severe generalized lipodystrophy and progeroid features. Other lipodystrophy conditions were considered and excluded. Graul- Neumann et al. considered dual pathology in their patient and possibility of a mutation-specific phenotype was raised [Citation300].
Atrophic skin patches: Bergman et al. report atrophic skin patches with abnormal elastic fibers in a 50 years old patient with a MASS phenotype. Genetic testing identified a missense mutation in exon 27 of the FBN1 gene (c.3422C>T; p.Pro1141Leu) [Citation295].
13.1. Expert opinion on patients’ reported symptoms ()
Psoriasis: This is very likely a coexisting condition rather than a skin manifestation of Marfan syndrome. As outlined by the British skin foundation, psoriasis is a common immune-mediated skin condition that affects 1 in 50 people. This often manifests as dry scaly skin. Common-affected areas include scalp, elbows knees, hands and feet, and skin folds.
Plantar soft tissue tumors: These are not described in association with Marfan syndrome. Piezogenic papules are small soft tissue herniations seen in weight-bearing areas of the foot. These are described in mild connective tissue disorders. Careful evaluation of soft tissue tumors by clinicians would be recommended but a specific association with Marfan syndrome currently remains undetermined.
Dry Scaly Skin: The most common causes of dry, scaly skin are eczema and psoriasis. Although eczema and immune dysregulation are noted in other connective tissue disorders such as Loeys-Dietz syndrome, the association in Marfan syndrome remains undetermined.
13.2. Conclusion
The key skin features noted in Marfan syndrome are striae and atrophic scars. As outlined above systematic documentation of skin and cutaneous manifestations by Grahame and Pyeritz [Citation294] and Ledoux et al. [Citation295] provide data on the prevalence of cutaneous manifestations in contrast to single case reports. Although association with other dermal features has been noted, further systematic reviews are needed to study the prevalence of these features in Marfan syndrome versus controls.
14. Dental features by alexander Rahman and Ingmar Staufenbiel
Only few studies investigated dental aspects of Marfan syndrome. Oral manifestations can affect the dental hard and soft tissues and the periodontium. Marfan syndrome can lead to orthodontic changes as well as degeneration of the dental pulp. It may also increase the prevalence of craniomandibular dysfunction.
14.1. Caries
In 2002 De Coster et al. investigated structural defects of enamel and dentine [Citation301]. They found that hypoplastic enamel areas were more often found in patients with Marfan syndrome. Therefore, they concluded that patients with Marfan syndrome may have a higher risk of caries. In contrast, Rahman et al. investigated the prevalence of dental caries in 31 children with Marfan syndrome. No increased prevalence of caries in Marfan syndrome patients was documented compared to healthy children [Citation302].
Staufenbiel et al. examined the periodontal health of 51 patients with Marfan syndrome as compared to 31 control subjects. We used a complete periodontal status including probing pocket depth, clinical attachment level, and bleeding on probing. The mean values of the clinical attachment level were almost identical in both groups (2.62 ± 0.56 mm in the Marfan syndrome group compared to 2.70 ± 0.61 mm in the control group), indicating that patients with Marfan syndrome did not have a higher risk for periodontal disease [Citation303].
14.2. Orthodontic manifestations
From an orthodontic point of view, a narrow upper jaw with a high palate, a skeletal class II with retrognathy of the lower jaw and crowding of teeth is frequently observed in patients with Marfan syndrome [Citation291].
Additionally, Bauss et al. described a significantly higher prevalence of pulp stones and pulp calcifications in patients with Marfan syndrome [Citation304]. These alterations are not considered to be a disease but they may cause higher complication rates during root canal treatments.
Based on clinical and radiological findings, a significantly higher prevalence of craniomandibular dysfunction was detected in patients with Marfan syndrome [Citation305].
14.3. Expert opinion on patients’ reported symptoms ()
The major complaints described by the patients could be understood as crowded teeth described in several studies. As a result of the crowded teeth the oral hygiene procedures are compromised and could explain the increased gum bleeding. The tendency to more inflammations signs as well as structural defects of enamel is symptoms which can be found in the scientific literature.
14.4. Conclusion
Especially because of the elevated signs of inflammation we recommend more frequent dental maintenance in patients with Marfan syndrome. This should include professional oral hygiene appointments to reduce the bacterial load in the oral cavity. As endocarditis is frequently caused by oral bacteria, oral health care can be regarded as endocarditis prophylaxis.
15. Headache as neurological symptom by Anji Yetman
Headaches are common in Marfan syndrome and may occur secondary to migraines, intracranial hypotension, and carotid artery dissections [Citation306]. The first two are discussed below, the last one is discussed as vascular manifestation.
15.1. Migraine
Numerous case-control studies have documented a higher incidence of migraines both with and without aura in patients with Marfan syndrome [Citation306,Citation307]. In the largest series to date, 40% of patients with Marfan syndrome compared with 28% of age- and gender-matched controls met diagnostic criteria for migraine headache [Citation306]. Treatment for migraines in patients with Marfan syndrome may differ from the general population. Therapies commonly used for the reduction of aortic aneurysmal disease, namely angiotensin receptor blockers, ACE inhibitors, and propranolol have all been used successfully for the reduction of migraine burden [Citation306,Citation308].
15.2. Spontaneous cerebrospinal fluid (CSF) leakage
Much less common than migraine headaches, headaches secondary to intracranial hypotension may occur in Marfan syndrome [Citation306,Citation309–Citation315]. Intracranial hypotension occurs as a result of low pressures of CSF within the subarachnoid space secondary to spontaneous leaks of CSF most commonly from the lumbar or sacral dura at the site of dural ectasia [Citation306]. While dural ectasia has been documented in 40% of children and 63-95% of adults with Marfan syndrome [Citation314], only a small fraction of these patients are diagnosed with intracranial hypotension. The phenomenon has been seen across the age spectrum including children as young as 5 years [Citation306,Citation316].
In contrast to migraines, patients with intracranial hypotension typically experience headaches that worsen with standing and improve with recumbency [Citation306]. Other symptoms are often nonspecific and include nausea, dizziness, neck stiffness, decreased hearing, or blurred vision [Citation315]. Headaches may be non-positional and may be associated with exertion. In some cases, they may be paradoxic, worsening with lying down. Minor trauma, or an intense Valsalva maneuver as may occur with coughing, bowel movements or labor [Citation310,Citation313] have been identified as causative factors in the dural leaks responsible for this phenomenon. Because of the marked clinical variability that may be present with intracranial hypotension, diagnostic criteria for spontaneous intracranial hypotension have been established and include at least one of the following: A. the demonstration of extrathecal CSF on spinal imaging which may include CT myelography, MR imaging or MR myelography, or B. cranial MR imaging findings of spontaneous intracranial hypotension (subdural fluid collections, enhancement of the pachymeninges, sagging of the brain) with at least one of the following: 1. Low opening CSF pressure; 2. Spinal meningeal diverticulum; 3. Improvement of symptoms after epidural blood patch, or in the absence of A or B, C. Presence of 1, 2 and 3 above [Citation317]. Complications may stem from failure to recognize the etiology of the headache thereby not providing adequate therapy. Patients with intracranial hypotension may present with a picture of dementia, quadriplegia or coma [Citation317].
Therapy for intracranial hypotension may include conservative measures such as fluids, bed rest and caffeine [Citation316] however the most commonly used initial treatment is epidural blood patching [Citation317]. The improvement in symptoms after an epidural blood patch may be temporary and the procedure can be repeated.
15.3. Expert opinion on patients’ reported symptoms ()
Patients with extreme height have a higher incidence of orthostatic hypotension manifesting as postural dizziness or syncope. The source of the hypotension in such individuals is said to be cerebral hypotension [Citation318]. Given almost universal tall stature in patients with Marfan syndrome, it is not surprising that postural dizziness is a frequent occurrence. Mulder et al. found both postural dizziness and fatigue to be more frequent in patients with Marfan syndrome than in the general population and noted that the two symptoms were strongly correlated and occurred independent of the use of beta-blocker therapy [Citation168]. Use of physical counter pressure manoeuvers such as knee-bending, or leg crossing may be helpful.
15.4. Conclusion
Headaches in Marfan syndrome are frequent and can be disabling. Accurate diagnosis of the underlying etiology is important to direct effective therapies. Cerebral and spinal imaging may provide important clues as to the underlying etiology. Intracranial hypotension should be considered in patients with refractory, positional or complicated headaches that do not respond to conservative management.
16. Psychological features by Svend Rand-Hendriksen
The literature about psychosocial features in individuals with Marfan syndrome is sparse. In most studies published, the clinical information given does not secure the Marfan syndrome diagnosis in accordance with recent diagnostic criteria (Ghent-1; 1996 or Ghent-2; 2010) [Citation170,Citation319]; DNA investigations are only performed in a few more recent studies. Consequently, the described cases and families may have one of the related heritable thoracic aortic diseases that have been discovered more recently. Whether psychosocial features are inherently part of Marfan syndrome, a consequence of Marfan syndrome, or incidentally co-occur with Marfan syndrome is largely unknown.
16.1. Cognitive dysfunction
A higher prevalence of cognitive dysfunction in children and young adults with Marfan syndrome has been suggested in two papers, based on clinical questioning about the presence of learning disabilities, attention deficit hyperactivity disorder (ADHD), neuro-maturational immaturity and/or performance IQ score was different from their verbal score by more than 20 points: Hofman (1988) reports 50% [Citation320], while Handisides (2019) reports 27% [Citation321]. No prevalence studies have been published. Two studies have published data from neuropsychological testing, both with small samples: Lannoo (1996) found very small differences in neuropsychological functioning in comparison to a group of healthy volunteers among 13 patients aged 14 to 56 years [Citation322], whereas our own group (2007) found cognitive abilities within normal range among 16 young adults [Citation323].
Inborn cognitive dysfunction, reported as prevalent in the primary description of Loeys-Dietz syndrome (2005) [Citation122], does not seem prevalent among persons with Marfan syndrome. However, for the single individual, early diagnostics and relevant stimulation and support are of high importance.
In conclusion, the prevalence of cognitive dysfunction among people with Marfan syndrome is probably comparable to the general population. To find the true prevalence of cognitive dysfunction in Marfan syndrome, larger studies in persons with clinically and genetically confirmed Marfan syndrome must be done.
16.2. Schizophrenia
Case reports reported on individuals and their families with coexistence of Marfan syndrome and schizophrenia [Citation324–Citation334]. Some early papers suggested that symptoms of schizophrenia are a part of Marfan syndrome, and later papers have suggested that the cause of schizophrenia should be linked to the FBN1 gene. However, a linkage study failed to demonstrate a common genetic factor [Citation328]. In conclusion, for most of the papers, diagnostic information is sparse and the diagnosis of Marfan syndrome appears questionable.
16.3. Depression
Eight papers described the coexistence of Marfan syndrome and depression [Citation329,Citation335–Citation341]. However, again, most are case reports with questionable clinical data regarding the diagnosis of Marfan syndrome. Of the five papers presenting groups of patients, one is based on self-reported diagnosis of Marfan syndrome [Citation337]; the other four studies based on the diagnosis of Marfan syndrome on Ghent-1 or Ghent-2. DNA investigations are not presented. The studies use different methods to assess depression. The reported prevalence of depression is diverging from ‘no difference between Marfan syndrome and control [Citation339], “within normal range” [Citation340] to “depression ever in 24,8%” [Citation341] and “depressive symptoms” in 44%’ [Citation337].
In conclusion, the prevalence of depression among people with Marfan syndrome is probably similar as in the general population. To assess the true prevalence of psychiatric problems including schizophrenia and depression in Marfan syndrome, larger studies in patients with confirmed Marfan syndrome should be performed.
16.4. Fatigue
Patients with Marfan syndrome have reported fatigue as a severe problem. Six reports focused on fatigue [Citation168,Citation268,Citation323,Citation340,Citation342–Citation345]. In all reports, the diagnosis of Marfan syndrome was based on Ghent-1, except for one paper in which the revised Ghent nosology was used [Citation345]. None of the papers report DNA investigations. All reports document a higher level of fatigue in Marfan syndrome than in the general population. Two papers even report ‘severe fatigue’ in around 40-50% of the patients [Citation342,Citation345].
In conclusion, fatigue should be considered as an important feature among patients with Marfan syndrome which needs to be acknowledged in the life-long follow-up.
16.5. Pain
In our personal experience, some patients with Marfan syndrome experience pain as a severe and chronic problem. Of these, some individuals had undergone cardiac surgery, some were permanent opiate users and others were addictive users also of other pain medications. In the literature, the prevalence of pain in Marfan syndrome ranges from 47% to 91%, and mean values of pain are reported higher than in the general population.
Eighteen papers, published before 15 May 2015, presenting data about pain in Marfan syndrome, are included in a systematic review by Velvin et al. [Citation346]. Of these, only the study by Nelson et al. had its primary focus on pain [Citation347]. Across the various studies, the methods of patient sampling, Marfan syndrome diagnostics, and tools for measuring pain were diverging, and the prevalence, extent, and type of pain varied accordingly. However, chronic pain is reported to be more prevalent in Marfan syndrome than in the general population. Back pain, neck pain, shoulder pain, chest pain, knee pain, and headache/migraine (see above) are the most commonly observed types.
After May 2015, another relevant paper was published: Speed et al. reported on a questionnaire-based study in 245 people with self-reported Marfan syndrome [Citation348]. Pain, often throughout the body, is reported to result in profound disability and psychological burden.
Pain treatment in Marfan syndrome is not easy – Nelson and Speed report low satisfaction with pain treatment [Citation347,Citation348].
In conclusion, pain assessment must be included in all consultations for patients with Marfan syndrome. As the pain is chronic, focus must be on treatment modalities without risk of addiction. Cognitive therapy, mental and physical activities and transcutaneous nerve stimulation (TNS) must be considered.
16.6. Expert opinion on patients’ reported symptoms ()
Autism, Asperger syndrome, Attention Deficit Syndrome (ADS) and Developmental delay are mentioned in the table ‘patients‘ symptoms according to the German Marfan Organization’ (). These conditions are relatively common in the general population. The prevalence of each of them among persons with Marfan syndrome is not known. Larger studies in persons with clinically and genetically confirmed Marfan syndrome must be done. Joint pain is also mentioned in . ‘Pain’ and ‘chronic pain’, often in more than one body area, is reported more often by persons with Marfan syndrome than by persons in the general population. It is difficult to distinguish joint pain from musculoskeletal pain caused by other structures (ligaments, tendons muscles, etc.). The prevalence of joint pain in Marfan syndrome is unknown.
17. Hemostatic features by Katharina Kornhuber and Harald Kaemmerer
17.1. Activated fibrinolysis, thrombin and platelet function
So far, there are only few studies reporting on hemostasis in patients with Marfan syndrome. Early single case reports presented Marfan patients with pathological postoperative bleeding disorders [Citation349], as well as thromboembolic events [Citation350,Citation351] and assumed an association with the inherited disease. Since then, case-control studies on hemostasis have been performed in Marfan patients including up to 50 patients. Touat et al. revealed increased thrombin and platelet activation in Marfan patients with an aortic diameter above 45 mm, but not below 45 mm [Citation352]. Kornhuber et al. found increased levels of D-dimers in Marfan patients compared to healthy controls and a correlation between D-dimers and the diameter of the aortic root [Citation353]. This is consistent with other studies on thoracic aortic aneurysms of other etiologies [Citation354,Citation355] and with studies on abdominal aortic aneurysms [Citation356,Citation357].
Bridges et al. detected increased factor VIII von Willebrand factor antigen (fVIIIvVVFAg) levels and thrombomodulin levels in Marfan patients [Citation358]. They postulated that fVIIIvVVFAg deviations were secondary to endothelial injury caused by impaired tensile strength of blood vessels and microvascular damage stimulating the release of fVIIIvVVFAg [Citation358,Citation359]. They postulated that endothelial injury also results in elevated thrombomodulin levels [Citation358,Citation359].
17.2. Acquired von Willebrand disease type 2a
Recently, Kornhuber et al. suspected the occurrence of an acquired von Willebrand syndrome type 2A in a subgroup of investigated Marfan patients [Citation353]. They assumed that aortic dilatation and other frequent cardiovascular manifestations of Marfan syndrome, such as heart valve abnormalities or dysfunction, could lead to increased shear stress and altered rheology [Citation360–Citation363], and subsequently to hemostatic changes [Citation352,Citation364]. Other studies show that an increased shear stress affects the von Willebrand factor, which becomes more susceptible to proteases, resulting in a dysfunctional interaction with platelets [Citation365–Citation367]. Such effects have been described in patients with acquired von Willebrand syndrome type 2a [Citation366,Citation368] and could also be found in patients with aortic valve stenosis [Citation369–Citation371] or dysfunctional heart valve prostheses [Citation372].
17.3. Platelet dysfunction
In addition, it is interesting to note that the Ehlers-Danlos syndrome, another connective tissue disease which may be accompanied by aortic dilatation, can be associated with platelet dysfunction [Citation373–Citation375]. Platelet dysfunction could be found in Marfan patients as well, analyzed by the Multiplate and PFA 100 tests [Citation353].
17.4. Expert opinion on patients’ reported symptoms ()
Being asked to list unusual symptoms and complaints noticed in themselves Marfan patients reported either on recurrent thrombosis or on coagulation disorders. Both entities encompass a broad spectrum of potential causes but also occur frequently in the normal population. Whether there is actually a causal relationship with Marfan syndrome is certainly speculative, and certainly requires further clarification in the individual case. However, our comments indicate that the literature refers to a causal relationship between Marfan syndrome and recurrent thrombosis and coagulation disorders. This should be taken into account in the long-term care of affected patients, who often have to undergo surgery.
17.5. Conclusion
Increased fibrinolysis, thrombin, and platelet activation has been described in Marfan patients, and Marfan syndrome may be associated with acquired von Willebrand disease type 2a. The relevance for diagnostic and therapeutic consequences needs further definition.
18. Eye features by Stephan Linke
The role of the precise ocular status in diagnosing and defining the progression of Marfan syndrome has gained prominence with the revised Ghent criteria, as the presence of ectopia lentis with aortic dilation (Z-score ≥2) is currently sufficient to establish the final diagnosis [Citation16]. In vivo biomechanical dynamic Scheimpflug-based analyses revealed pathologic corneal deformation in patients with Marfan syndrome having an advanced systemic score [Citation376]. The potential for ocular abnormalities to aid in the diagnosis of Marfan syndrome may be much greater than currently acknowledged due to the aforementioned modern screening techniques. Investigation of biomechanical and dynamic changes in tissues that contain abnormal microfibrils is a reasonable next step in refining the approach to diagnosing Marfan syndrome and monitor its progression.
18.1. Expert opinion on patients’ reported symptoms ()
Patients mention two distinct ophthalmic features that are not listed as features of Marfan syndrome in the Ghent nosologies, which we will briefly discuss here:
Early onset cataract as the major symptom from the patients perspective is interesting and to my opinion due to the combination of ectopia lentis and cataract. Both are lens related and result in visual disturbance (increase of refractive error and optical aberrations by ectopia lentis) and reduced visual acuity (loss of transparency by early onset cataract). The patient (and doctor) cannot unambiguously differentiate these entities since both result in reduced visual acuity. Lens replacement (cataract) surgery is the common treatment option.
Prolonged conjunctival wound healing from the patients’ perspective is surprising for me and probably a consequence of retinal detachment surgery. Due to altered scleral tissue buckling surgery can be more complex. It would be interesting to correlate the patient-reported prolonged conjunctival wound healing to the complexity of the surgery.
19. Discussion
The purpose of this article was to bring the far side of the Marfan syndrome closer to the awareness of scientists, physicians, and persons who are affected by the disease. We present the patients’ perspective together with experts’ systematic mapping of the landscape of the less well-known features of the Marfan syndrome. Indeed, we came up with an impressive list of 14 major organ features of Marfan syndrome that require future scientific and clinical work-up.
After having mapped a landscape, the next task usually is to check how to make use of the grounds being mapped. To our view, this condenses down to three questions as listed below. Rather than profound answers, we only can provide some evaluative estimates, which we summarize in , and which we comment along briefly in the following:
19.1. Which features are manifestations of Marfan syndrome?
Which of the 14 major organ features manifest because of Marfan syndrome, and which occur in Marfan syndrome, mostly as a consequence of aging, medical therapy, lifestyle, or because the individual with Marfan syndrome has an additional, independent disease that origins from other etiologies than Marfan syndrome [Citation377]?
Defining features of Marfan syndrome that affect the various organ systems listed are fraught with several issues. First, it is difficult to separate true manifestations from incidental occurrences. In many cases, reports of one or a few co-occurrences exist, but no studies quantify the prevalence to determine if these are random events. Second, some features emerge with age, and since people with Marfan syndrome have only recently been living to near-normal life expectancies, accurate estimates of, say, atrial fibrillation or diverticulosis will only emerge in years to come. On the other hand, there is a tendency to report occurrences of common conditions in young patients with Marfan syndrome. Finally, reports of the clinical association from more than a decade ago are complicated by related conditions being lumped with true Marfan syndrome.
provides our estimation of the degree to which organ features in Marfan syndrome are caused by the disease itself, and therefore tend to qualify as manifestations of the syndrome ().
shows our evaluative literature analysis and personal estimate of the existence or strength of association of clinical features of Marfan syndrome in overlapping clinical syndromes such as Loeys-Dietz syndrome and vascular Ehlers-Danlos syndrome ().
19.2. Which features require change of clinical management?
Whereas question (i) reflects the etiology of organ features in Marfan syndrome, question (ii) more pragmatically deals with the clinical consequences for Marfan care. Which features require assessment in a baseline evaluation performed in a Marfan center, which features need regular monitoring once the diagnosis of Marfan syndrome is established, which features should be checked for, only in case of specific symptoms of complications in Marfan syndrome, and which features may not play a role in clinical care at all? provides some evaluative estimates of the clinical consequences of each organ feature ().
19.3. How can we keep in touch with changes of Marfan syndrome?
Patients change with increases of age, changing lifestyles, new professional requirements, and new social and environmental conditions [Citation378]. Medicine also changes with refined diagnostic modalities, new or modified therapies, changing medical guidelines, directives, reimbursement rules, governments, and public fashions. Therefore, the face of Marfan syndrome will keep on changing, and scientists and physicians should not lock in on a specific picture of Marfan syndrome simply because they fall in love with what appears familiar.
Among many ways of keeping in touch with change, we wish to highlight the importance of listening to our patients. As is the case in many disorders, there is a discrepancy between what patients perceive as problems related to their condition and what is available in the medical literature or commonly known and looked for by professionals. Although major advances have already been made in the past decade, more efforts to empower patients and involve them in both clinical management and research are needed. To this end, international networks- and consortia such as the European Reference Network on Rare multisystemic vascular diseases (VASCERN) and the GenTAC alliance may offer structured platforms. Indeed, with regard to ERNs, two key drivers have been identified:, on the one hand, medical experts and scientists (because of the existing interdependence of research and care in this highly specialized field) and on the other hand patients and their families, including the patient organizations [Citation379]. In the set-up of clinical trials, patient reported outcomes (PRO) are also increasingly gaining importance [Citation380].
Finally, the broad spectrum of manifestations mentioned in this work clearly bodes for a multidisciplinary and holistic approach of the Marfan patient. Ideally, a central physician/caregiver, aware of the many possible features should coordinate the care and relay to appropriate specialists whenever needed. The important role of multidisciplinary teams in the management and treatment of Marfan syndrome has been described previously [Citation381].
In summary, the data presented in this manuscript provide an elaborate overview of several clinical features of Marfan syndrome that are commonly not in the spotlight. Both from a patient perspective and from a clinical point of view though, a number of these characteristics certainly deserve more attention and we hope we have raised awareness for this. Future research, nosologies, and guidelines may consider some of these less well-known features of Marfan syndrome.
20. Expert opinion
I. Reviews and guidelines on Marfan syndrome tend to lock in on the prototypical manifestations of the disease. In this review, we draw attention to less well-known clinical features that may be of importance for Marfan patients nevertheless. Based on our review, we classify these clinical features as follows:
(1) Clinical features that are unlikely to be manifestations of the Marfan syndrome:
bicuspid aortic valve
aneurysm of the cerebral arteries
cognitive dysfunction
schizophrenia
(2) Clinical features that are most likely to be considered as manifestations of Marfan syndrome:
tricuspid valve prolapse
cardiomyopathy
sleep apnea
vascular disease of the aortic branch vessels
liver- and kidney cysts
(3) Clinical features that are potential complications of natural or medical course of Marfan syndrome:
heart failure
supraventricular and ventricular arrhythmia
restrictive lung disease
spontaneous cerebrospinal fluid leakage
fatigue
chronic pain
II. In addition to these more singular features of Marfan syndrome, the image and perception of Marfan syndrome as a whole appears to transform as well:
From a disease of the young and slender to a disease of the old and obese
From a monogenetic disease of isolated organ system involvement (‘multi-systemic’) to a multifactorial generalized atherosclerotic and degenerative (‘omni-systemic’) disease
From isolated aortic disease to a more generalized arterial disease
III. Taking these insights into account, we propose some recommendations for medical guidelines and clinical care, such as
Baseline and follow-up echocardiographic evaluation of tricuspid aortic valve prolapse and regurgitation
Baseline and follow-up echocardiographic and N-terminal pro-brain natriuretic peptide-based monitoring of left- and right ventricular function
Baseline and follow-up 24-h ambulatory electrocardiography, at least in individuals with symptoms of arrhythmia
Baseline and follow-up assessment of pulmonary symptoms with the initiation of functional and imaging tests, following general guidelines of respiratory medicine
Ambulatory, unattended respiratory polysomnography screening of sleep apnea may be considered in adults with Marfan syndrome
Baseline imaging of abdominal organs for organ cysts
Baseline and follow-up clinical evaluation for urinary incontinence in adult women
Baseline clinical evaluation of muscle strength and risk factors and symptoms of reduced bone mineral density
Baseline dental and orthodontic examination and dental follow-up
Baseline and follow-up assessment and documentation of pain location, severity, and character, careful evaluation of potential causes of pain and symptomatic treatment, where appropriate
IV. However, there are gaps of knowledge, where we observe the following most urgent needs for prospective, multicenter clinical studies of the natural history, risk factors and treatment options of
Myocardial dysfunction and arrhythmia
Sleep-disordered breathing
Vascular manifestations
V. On the road to improved care for clinical features of Marfan patients there are some structural weaknesses to be overcome:
Lack of awareness of clinical features of rare disorders such as Marfan syndrome leading to delayed diagnosis of the disease
Economic disparities between countries and regions obviate access to equal medical care
Bureaucracy, severe legal regulations including data protection laws and exaggerated escalation of ethical requirements prevent multicenter research collaboration of experts for rare diseases who do not have enough resources to ensure compliance with the growing burden of legal demands
Basic researchers tend to cage themselves into an isolated and disconnected world of their labs to stay remote from patients’ real needs and physicians’ pressing questions.
Adequate management of patients and families with Marfan syndrome necessitates a multidisciplinary and holistic approach
Multicenter – patient empowered international collaborations between clinicians and researchers should be encouraged to explore modalities for improved management and new targets for tailored treatment
In summary, the data presented in this review translate easily and quickly into clinical practice. This translation is likely to improve Marfan care. The review offers numerous clues for future research, both clinical and basic, that uses as the point of departure the serious consideration of features of those individuals who are affected by the Marfan syndrome.
Article highlights
We performed a review of clinical features of the Marfan syndrome that are not listed in the Ghent nosology. This review yielded the following major results:
Patients report symptoms and complaints that experts may not have in their focus of study and research.
Bicuspid aortic valve, aneurysm of the cerebral arteries, cognitive dysfunction, and schizophrenia may be removed from the list of potential manifestations of the Marfan syndrome.
Tricuspid valve prolapse, cardiomyopathy, sleep apnea, vascular disease of the aortic branching vessels, and liver and kidney cysts emerge as manifestations of the Marfan syndrome.
Heart failure, supraventricular and ventricular arrhythmia, restrictive lung disease, headache, fatigue, and chronic pain are features that complicate the natural or medical course of Marfan syndrome.
The image of Marfan syndrome as a whole is in transition from a monogenetic disease of isolated organ system involvement (‘multi-systemic’) of the young and slender to a multifactorial generalized atherosclerotic and degenerative (‘generalized’) disease of the older and obese.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Supplemental Material
Download MS Word (577.2 KB)Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Marfan AB-J. Un cas de déformation congènital des quatre membres plus pronouncée aux extrémitiés charactérisée par l’allongement des os avec un certain degré d’amonassesment. Bull Mem Soc Med Hop (Paris) Ser. 1896;3(13):220–226.
- Marfan AB-J. La dolichoste´nome´lie (dolichome´lie arachnodactylie). Ann Med. 1938;44:5–29.
- Piper RK, Irvine-Jones E. Arachnodactylia and its association with congenital heart disease: report of a case and review of the literature. Am J Dis Children. 1926;31(6):832–839.
- Börger F. Über zwei Fälle von Arachnodaktylie. Z Kinder-Heilk. 1914;12(2–3):161–184.
- von Pfaundler M. Arachnodaktylie. Münchener Medizinische Wochenschrift. 1914;61(5):280.
- Weve H. Über Arachnodakylie (Dystrophia mesodermalis congenita, Typus Marfan). Arch Augenheilkd. 1931;104:1–46.
- Jéquier PM. Observations sur le syndrome de Marfan (dolichosténomelié ou arachnodactylie). Helv Med Acta. 1943;10:233–236.
- Etter LE, Glover LP. Arachnodactyly complicated by dislocated lens and death from rupture of dissecting aneurysm of the aorta. JAMA. 1943;123:88–89.
- von Kodolitsch Y, De Backer J, Schüler H et al. Perspectives on the revised ghent criteria for the diagnosis of Marfan syndrome. The Application of Clinical Genetics;2015. [cited 2015 Jun 8], 137–155.
- Mckusick VA. The cardiovascular aspects of Marfan’s syndrome: a heritable disorder of connective tissue. Circulation. 1955;11(3):321–342.
- Sakai LY, Keene DR, Engvall E. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol. 1986;103:2499–2509.
- Dietz HC, Cutting CR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337–339.
- Likert R. A technique for the measurement of attitudes. Arch Psychol. 1932;22:5–55.
- Das Marfan-Syndrom. ed. (Deutschland), MH). Berlin, Heidelberg: Springer Berlin Heidelberg; 2017.
- Marfan Hilfe (Deutschland) e.V. Das Marfan-Syndrom. Berlin, Heidelberg: Springer. 2017; Available from: https://link.springer.com/book/10.1007/978-3-662-53259-1
- Loeys BL, Dietz HC, Braverman AC, et al. The revised ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476–485.
- Beroukhim RS, Roosevelt G, Yetman AT. Comparison of the pattern of aortic dilation in children with the Marfan’s syndrome versus children with a bicuspid aortic valve. Am J Cardiol. 2006;98(8):1094–1095.
- Detaint D, Michelena HI, Nkomo VT, et al. Aortic dilatation patterns and rates in adults with bicuspid aortic valves: a comparative study with Marfan syndrome and degenerative aortopathy. Heart. 2014;100(2):126–134.
- Mueller GC, Stark V, Steiner K, et al. Impact of age and gender on cardiac pathology in children and adolescents with Marfan syndrome. Pediatr Cardiol. 2013;34(4):991–998.
- Wozniak-Mielczarek L, Sabiniewicz R, Drezek-Nojowicz M, et al. Differences in cardiovascular manifestation of Marfan syndrome between children and adults. Pediatr Cardiol. 2019;40(2):393–403.
- Itagaki S, Chikwe JP, Chiang YP, et al. Long-term risk for aortic complications after aortic valve replacement in patients with bicuspid aortic valve versus Marfan syndrome. J Am Coll Cardiol. 2015;65(22):2363–2369.
- Nistri S, Porciani MC, Attanasio M, et al. Association of Marfan syndrome and bicuspid aortic valve: frequency and outcome. Int J Cardiol. 2012;155(2):324–325.
- Milleron O, Ropers J, Arnoult F, et al. Clinical significance of aortic root modification associated with bicuspid aortic valve in Marfan Syndrome. Circ Cardiovasc Imaging. 2019;12(3):e008129.
- Ellis PR, Cooley DA, De Bakey ME. Clinical considerations and surgical treatment of annulo-aortic ectasia. report of successful operation. J Thorac Cardiovasc Surg. 1961;42:363–370.
- Golden RL, Lakin H. The forme fruste in Marfan’s syndrome. N Engl J Med. 1959;260(16):797–801.
- Pepe G, Nistri S, Giusti B, et al. Identification of fibrillin 1 gene mutations in patients with bicuspid aortic valve (BAV) without Marfan syndrome. BMC Med Genet. 2014;15:23.
- Girdauskas E, Geist L, Disha K, et al. Genetic abnormalities in bicuspid aortic valve root phenotype: preliminary results. Eur J Cardiothorac Surg. 2017;52(1):156–162.
- Beighton P, De Paepe A, Danks D, et al. International nosology of heritable disorders of connective tissue, Berlin, 1986. Am J Med Genet A. 1988;29(3):581–594.
- De Paepe A, Devereux RB, Dietz HC, et al. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet. 1996;62(4):417–426.
- Phornphutkul C, Rosenthal A, Nadas AS. Cardiac manifestations of Marfan syndrome in infancy and childhood. Circulation. 1973;47(3):587–596.
- Pyeritz RE, Wappel MA. Mitral valve dysfunction in the Marfan syndrome. clinical and echocardiographic study of prevalence and natural history. Am J Med. 1983;74(5):797–807.
- Sisk HE, Zahka KG, Pyeritz RE. The Marfan syndrome in early childhood: analysis of 15 patients diagnosed at less than 4 years of age. Am J Cardiol. 1983;52(3):353–358.
- Chen S, Fagan LF, Nouri S, et al. Ventricular dysrhythmias in children with Marfan’s syndrome. Am J Dis Child. 1985;139(3):273–276.
- Geva T, Sanders SP, Diogenes MS, et al. Two-dimensional and doppler echocardiographic and pathologic characteristics of the infantile Marfan syndrome. Am J Cardiol. 1990;65(18):1230–1237.
- Morse RP, Rockenmacher S, Pyeritz RE, et al. Diagnosis and management of infantile marfan syndrome. Pediatrics. 1990;86(6):888–895.
- Brown OR, DeMots H, Kloster FE, et al. Aortic root dilatation and mitral valve prolapse in Marfan’s syndrome: an echocardiographic study. Circulation. 1975;52(4):651–657.
- Roberts WC, Honig HS. The spectrum of cardiovascular disease in the Marfan syndrome: a clinico-morphologic study of 18 necropsy patients and comparison to 151 previously reported necropsy patients. Am Heart J. 1982;104(1):115–135.
- Pini R, Roman MJ, Kramer-Fox R, et al. Mitral valve dimensions and motion in Marfan patients with and without mitral valve prolapse. comparison to primary mitral valve prolapse and normal subjects. Circulation. 1989;80(4):915–924.
- Rybczynski M, Mir TS, Sheikhzadeh S, et al. Frequency and age-related course of mitral valve dysfunction in the Marfan syndrome. Am J Cardiol. 2010;106(7):1048–1053.
- Rybczynski M, Treede H, Sheikhzadeh S, et al. Predictors of outcome of mitral valve prolapse in patients with the marfan syndrome. Am J Cardiol. 2011;107(2):268–274.
- Kühne K, Keyser B, Groene EF, et al. FBN1 gene mutation characteristics and clinical features for the prediction of mitral valve disease progression. Int J Cardiol. 2013;168(2):953–959.
- Aydin A, Adsay BA, Sheikhzadeh S, et al. Observational cohort study of ventricular arrhythmia in adults with Marfan syndrome caused by FBN1 mutations. PLoS One. 2013;8(12):e81281.
- Rybczynski M, Bernhardt AM, Rehder U, et al. The spectrum of syndromes and manifestations in individuals screened for suspected Marfan syndrome. Am J Med Genet A. 2008;146A(24):3157–3166.
- Sheikhzadeh S, De Backer J, Gorgan N, et al. The main pulmonary artery in adults: a controlled multicenter study with assessment of echocardiographic reference values, and the frequency of dilatation and aneurysm in Marfan syndrome. Orphanet J Rare Dis. 2014;9(1):203.
- Stark VC, Huemmer M, Olfe J, et al. The pulmonary artery in pediatric patients with Marfan syndrome: an underestimated aspect of the disease. Pediatr Cardiol. 2018;39(6):1194–1199.
- Nollen GJ, van Schijndel KE, Timmermans J, et al. Magnetic resonance imaging of the main pulmonary artery: reliable assessment of dimensions in Marfan patients on a simple axial spin echo image. Int J Cardiovasc Imaging. 2003;19(2):141–147. discussion 149-150.
- Espinola-Zavaleta N, Iqbal FM, Nanda NC, et al. Echocardiographic study of a Mestizo-Mexican population with Marfan syndrome. Echocardiography. 2010;27(8):923–930.
- Joyce F, Tingleff J, Pettersson G. Expanding indications for the Ross operation. J Heart Valve Dis. 1995;4(4):352–363.
- Sievers HH, Hanke T, Stierle U, et al. A critical reappraisal of the ross operation: renaissance of the subcoronary implantation technique? Circulation. 2006;114(1 Suppl):I504–511.
- Gu X, He Y, Li Z, et al. Echocardiographic versus histologic findings in Marfan syndrome. Tex Heart Inst J. 2015;42(1):30–34.
- Alpendurada F, Wong J, Kiotsekoglou A, et al. Evidence for Marfan cardiomyopathy. Eur J Heart Fail. 2010;12(10):1085–1091.
- Gott VL, Greene PS, Alejo DE, et al. Replacement of the aortic root in patients with Marfan’s syndrome. N Engl J Med. 1999;340:1307–1313.
- Diller GP, Kempny A, Alonso-Gonzalez R, et al. Survival prospects and circumstances of death in contemporary adult congenital heart disease patients under follow-up at a large tertiary centre. Circulation. 2015;132(22):2118–2125.
- Hetzer R, Siegel G, Delmo Walter EM. Cardiomyopathy in Marfan syndrome. Eur J Cardiothorac Surg. 2016;49(2):561–567.
- Yetman AT, Bornemeier RA, McCrindle BW. Long-term outcome in patients with Marfan syndrome: is aortic dissection the only cause of sudden death? J Am Coll Cardiol. 2003;41:329–332.
- Alpendurada F, Wong J, Kiotsekoglou A, et al. Evidence for Marfan cardiomyopathy. Eur J Heart Fail. 2010;12:1085–1091.
- Savolainen A, Nisula L, Keto P, et al. Left ventricular function in children with the marfan syndrome. Eur Heart J. 1994;15(5):625–630.
- Das BB, Taylor AL, Yetman AT. Left ventricular diastolic dysfunction in children and young adults with Marfan syndrome. Pediatr Cardiol. 2006;27:256–258.
- De Backer JF, Devos D, Segers P, et al. Primary impairment of left ventricular function in Marfan syndrome. Int J Cardiol. 2006;112(3):353–358.
- Rybczynski M, Koschyk DH, Aydin MA, et al. Tissue Doppler imaging identifies myocardial dysfunction in adults with Marfan syndrome. Clin Cardiol. 2007;30:19–24.
- Kiotsekoglou A, Sutherland GR, Moggridge JC, et al. Impaired right ventricular systolic function demonstrated by reduced atrioventricular plane displacement in adults with Marfan syndrome. Eur J Echocardiography. 2009;2:295–302.
- Meijboom LJ, Timmermans J, Van Tintelen JP, et al. Evaluation of left ventricular dimensions and function in Marfan’s syndrome without significant valvular regurgitation. Am J Cardiol. 2005;95(6):795–797.
- Rouf R, MacFarlane EG, Takimoto E, et al. Nonmyocyte ERK1/2 signaling contributes to load-induced cardiomyopathy in Marfan mice. JCI Insight. 2017;2(15):e91588.
- Cook JR, Carta L, Bénard L, et al. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J Clin Investig. 2014;124(3):1329–1339.
- Steijns F, van Hengel J, Sips P, et al. A heart for fibrillin: spatial arrangement in adult wild-type murine myocardial tissue. Histochem Cell Biol. 2018;150(3):271–280.
- Humphrey JD, Schwartz MA, Tellides G, et al. Role of mechanotransduction in vascular biology: focus on thoracic aortic aneurysms and dissections. Circ Res. 2015;116(8):1448–1461.
- Aalberts JJJ, van Tintelen JP, Meijboom LJ, et al. Relation between genotype and left-ventricular dilatation in patients with Marfan syndrome. Gene. 2014;534:40–43.
- Chen C, Fagan LF, Nouri S, et al. Ventricular dysrhythmias in children with Marfan’s Syndrome. Am J Dis Children. 1985;139(3):273–276.
- Aydin A, Adsay BA, Sheikhzadeh S, et al. Observational cohort study of ventricular arrhythmia in adults with Marfan syndrome caused by FBN1 mutations. PLoS ONE. 2013;8:e81281.
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the anticoagulation and risk factors in atrial fibrillation (ATRIA) study. J Am Med Assoc. 2001;285(18):2370–2375.
- Rowland D, Mace J. Coincidence of Wolff-Parkinson-White and Marfan syndromes. J Pediatr. 1975;86(3):482.
- Johnson CD. The Wolff-Parkinson-White syndrome associated with Marfan’s syndrome. Bol Asoc Med P R. 1989;81:361-364.
- Savolainen A, Kupari M, Toivonen L, et al. Abnormal ambulatory electrocardiographic findings in patients with the Marfan syndrome. J Intern Med. 1997;241:225–230.
- Hoffmann BA, Rybczynski M, Rostock T, et al. Prospective risk stratification of sudden cardiac death in Marfan’s syndrome. Int J Cardiol. 2013;167:2539–2545.
- Cameron DE, Alejo DE, Patel ND, et al. Aortic root replacement in 372 Marfan patients: evolution of operative repair over 30 years. Ann Thorac Surg. 2009;87:1344–1350.
- Bear ES, Tung MY, Bordiuk J. Marfan’s syndrome with complete heart block and junctional rhythm. JAMA. 1971;217:335.
- Chopra KL, Krishnan S, Joseph PP. Intermittent bundle branch block and complete heart block in Marfan’s syndrome. Indian Heart J. 1970;22:53–56.
- Arunamata AA, Nguyen CT, Ceresnak SR, et al. Utility of serial 12-lead electrocardiograms in children with Marfan syndrome. Cardiol Young. 2018;28(8):1009–1013.
- Becker AE, van Mantgem JP. The coronary arteries in Marfan’s syndrome. A morphologic study. Am J Cardiol. 1975;36:315–321.
- Mortensen KH, Thuesen L, Kristensen IB, et al. Spontaneous coronary artery dissection: A western Denmark heart registry study. Catheterization Cardiovasc Interventions. 2009;74(5):710–717.
- Vanzetto G, Berger-Coz E, Barone-Rochette G, et al. Prevalence, therapeutic management and medium-term prognosis of spontaneous coronary artery dissection: results from a database of 11,605 patients. Eur J Cardiothorac Surg. 2009;35:250–254.
- Henkin S, Negrotto SM, Tweet MS, et al. Spontaneous coronary artery dissection and its association with heritable connective tissue disorders. Heart. 2016;102:876–881.
- Vandamme M, De Backer J, De Backer T, et al. The spectrum of spontaneous coronary artery dissection: illustrated review of the literature. Acta Cardiol. 2017;72:599–609. (Ed.^(Eds).
- Krittanawong C, Kumar A, Johnson KW, et al. Conditions and factors associated with spontaneous coronary artery dissection (from a national population-based cohort study). Am J Cardiol. 2019;123:249–253.
- Palank EA, Dawson JT, Cowen GD, et al. Primary dissecting aneurysm of the right coronary artery. Chest. 1977;72:774–776.
- Corrado D, Thiene G, Cocco P, et al. Non-atherosclerotic coronary artery disease and sudden death in the young. Heart. 1992;68(6):601–607.
- Bateman AC, Gallagher PJ, Vincenti AC. Sudden death from coronary artery dissection. J Clin Pathol. 1995;48:781–784.
- Bonacchi M, Prifti E, Giunti G, et al. Emergency management of spontaneous coronary artery dissection. J Cardiovasc Surg. 2002;43(2):189–193.
- Angiolillo DJ, Moreno R, Macaya C. Isolated distal coronary dissection in Marfan syndrome. Ital Heart J Suppl. 2004;5:305–306.
- Wain-Hobson J, Roule V, Dahdouh Z, et al. Spontaneous coronary artery dissection: one entity with several therapeutic options. Cardiovasc Revascularization Med. 2012;13(3):203.e201–204.
- Tokura M, Taguchi I, Kageyama M, et al. Clinical features of spontaneous coronary artery dissection. J Cardiol. 2014;63(2):119–122.
- Sao C, Wakabayashi K, Suzuki H. Natural course of isolated spontaneous coronary artery dissection in Marfan syndrome. Int J Cardiol. 2014;177:20–22.
- Kaadan MI, MacDonald C, Ponzini F, et al. Prospective cardiovascular genetics evaluation in spontaneous coronary artery dissection. Circ Genom Precis Med. 2018;11(4):e001933.
- Lovitt WVJ, Corzine WJJ. Dissecting intramural hemorrhage of anterior descending branch of left coronary artery. AMA Arch Pathol. 1952;54:458–462.
- Boschetti AE, Levine A. Cystic medionecrosis with dissecting aneurysm of coronary arteries. AMA Arch Intern Med. 1958;141(5):428.
- Duarte SBCP, Beraldo DO, Cesar LAM, et al. anomalous coronary artery origin in a young patient with Marfan syndrome. Case Rep Cardiol. 2017;3861923:5.
- Zanotti G, Gupta R, Reece TB, et al. Aortic valve sparing root replacement with unroofing and reconstruction of an anomalous right coronary artery. Ann Thorac Surg. 2016;101(5):1980–1982.
- Brothers JA, Harris MA, Paridon SM. Anomalous aortic origin of a coronary artery in siblings with Marfan syndrome. Cardiol Young. 2011;21:238–240.
- Papadopoulos DP, Moyssakis I, Votteas VE. Coexistence of anomalous origin of the coronary arteries and severe aortic regurgitation in Marfan syndrome. Clin Rheumatol. 2006;25:737–738.
- Imperadore F, Girardini D, Vassanelli C, et al. Dissection of the ascending aorta complicated by acute myocardial infarction in a patient with Marfan’s syndrome and anomalous origin of the left circumflex artery. description of a case. G Ital Cardiol. 1994;24(4):435–440.
- Menon VK, Alurkar VM, Durairaj M. Anomalous origin of the coronary artery in Marfan’s syndrome. Indian Heart J. 1983;35:176–177.
- Winter MM, Koolbergen DR, Groenink M, et al. Computed tomography coronary angiography should be performed in all patients with Marfan Syndrome prior to aortic root replacement. Int J Cardiol. 2014;177(2):e70–71.
- Jegatheeswaran A, Devlin PJ, McCrindle BW, et al. Features associated with myocardial ischemia in anomalous aortic origin of a coronary artery: a congenital heart surgeons’ society study. J Thorac Cardiovasc Surg. 2019;158(3):822–834.e823.
- Yetman AT, McCrindle BW. The prevalence and clinical impact of obesity in adults with Marfan syndrome. Can J Cardiol. 2010;26:e137-e139.
- Szeberin Z, Fehérvári M, Krepuska M, et al. Fetuin-A serum levels in patients with aortic aneurysms of Marfan syndrome and atherosclerosis. Eur J Clin Invest. 2011;41(2):176–182.
- Van Herck JL, De Meyer GRY, Martinet W, et al. Impaired fibrillin-1 function promotes features of plaque instability in apolipoprotein E-deficient mice. Circulation. 2009;120(24):2478–2487.
- Van der Donckt C, Van Herck JL, Schrijvers DM, et al. Elastin fragmentation in atherosclerotic mice leads to intraplaque neovascularization, plaque rupture, myocardial infarction, stroke, and sudden death. Eur Heart J. 2015;36(17):1049–1058.
- Shah H, Jan MU, Altaf A, et al. Correlation of hyper-homocysteinemia with coronary artery disease in absence of conventional risk factors among young adults. J Saudi Heart Assoc. 2018;30(4):305–310.
- Benke K, Ágg B, Mátyás G, et al. Gene polymorphisms as risk factors for predicting the cardiovascular manifestations in Marfan syndrome: role of folic acid metabolism enzyme gene polymorphisms in Marfan syndrome. Thromb Haemost. 2015;114(4):748–756.
- Giustia B, Porciania MC, Brunelli T, et al. Phenotypic variability of cardiovascular manifestations in Marfan Syndrome. possible role of hyperhomocysteinemia and C677T MTHFR gene polymorphism. Eur Heart J. 2003;24(22):2038–2045.
- Wilson DG, Bellamy MF, Ramsey MW, et al. Endothelial function in Marfan syndrome: selective impairment of flow- mediated vasodilation. Circulation. 1999;99:909–915.
- Kang SS, Rosenson RS. Analytic approaches for the treatment of hyperhomocysteinemia and Its Impact on vascular disease. Cardiovasc Drugs Ther. 2018;32:233–240. (Ed.^(Eds).
- Yang HHC, van Breemen C, Chung AWY. Vasomotor dysfunction in the thoracic aorta of Marfan syndrome is associated with accumulation of oxidative stress. Vascul Pharmacol. 2010;52(1–2):37–45.
- Angeloni E, Vitaterna A, Pirelli M, et al. Effects of statin therapy on ascending aorta aneurysms growth: a propensity-matched analysis. Int J Cardiol. 2015;191:52–55.
- McLoughlin D, McGuinness J, Byrne J, et al. Pravastatin reduces marfan aortic dilation. Circulation. 2011;124(11Suppl):S168–173.
- Boucker P, Li WP, Matz RL, et al. LRP1 functions as an atheroprotective integrator of TGFβ and PDGF signals in the vascular wall: implications for Marfan syndrome. PLoS ONE. 2007;2(5):e448.
- Morris SA, Orbach DB, Geva T, et al. Increased vertebral artery tortuosity index is associated with adverse outcomes in children and young adults with connective tissue disorders. Circulation. 2011;124:388–396.
- Croisile B, Deruty R, Pialat J, et al. Aneurysm of the internal carotid artery and cervical mega-dolicho-arteries in Marfan syndrome. Neurochirurgie. 1988;34:342-347.
- Kasirajan K, Matteson B, Marek JM, et al. Covered stents for true subclavian aneurysms in patients with degenerative connective tissue disorders. J Endovascular Ther. 2004;10:647-652.
- Kono AK, Higashi M, Morisaki H, et al. High prevalence of vertebral artery tortuosity of Loeys-Dietz syndrome in comparison with Marfan syndrome. Jpn J Radiol. 2010;28:273–277.
- Mariucci EM, Lovato L, Rosati M, et al. Dilation of peripheral vessels in Marfan syndrome: importance of thoracoabdominal MR angiography. Int J Cardiol. 2013;167:2928–2931.
- Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281.
- Franken R, El Morabit A, De Waard V, et al. Increased aortic tortuosity indicates a more severe aortic phenotype in adults with Marfan syndrome. Int J Cardiol. 2015;194:7–12.
- Celestin B, Jondeau G, Milleron O. Increased vertebral arterial tortuosity index in adult marfan FBN1 associated with adverses outcomes. Arch Cardiovasc Dis Suppl. 2018;10(2):254.
- Schoenhoff FS, Yildiz M, Langhammer B, et al. The fate of nonaortic arterial segments in Marfan patients. J Thorac Cardiovasc Surg. 2019;157:2150–2156.
- Yetman AT, Roosevelt GE, Veit N, et al. Distal aortic and peripheral arterial aneurysms in patients with Marfan syndrome. J Am Coll Cardiol. 2011;58(24):2544–2545.
- Groner LK, Lau C, Devereux RB, et al. Imaging of the postsurgical aorta in Marfan syndrome. Curr Treat Options Cardiovasc Med. 2018;20(10):80.
- Latter DA, Ricci MA, Forbes RDC, et al. Internal carotid artery aneurysm and Marfan’s syndrome. Canadian Journal of Surgery. 1989;6:463-466.
- Aschwanden M, Wegmann W. Dissecting aneurysm of the common iliac artery in Marfan syndrome: rupture following sports activities. Schweizerische Medizinische Wochenschrift. 1990;120(22):836–839.
- Aghoutane N, Bakkali T, Zoulati M, et al. Un anévrisme de l’artère axillo-sous-clavière en pré-rupture révélant un syndrome de Marfan. J Med Vasc. 2018;44(1):71–75.
- Flanagan PV, Geoghegan J, Egan TJ. Iliac artery aneurysm in Marfan’s Syndrome. Eur J Vasc Surg. 1990;4(3):323–324.
- Mounier-Vehier C, Millaire A, Vrtovsnik F, et al. Dissecting aneurysm of the left iliac artery disclosing Marfan’s disease. Annales De Cardiologie Et D’angeiologie. 1991;40:537–540.
- Hatrick AG, Malcolm PN, Burnand KG, et al. A superficial femoral artery aneurysm in a patient with Marfan’s syndrome. Eur J Vasc Endovascular Surg. 1998;15:459–460.
- Safioleas MC, Kakisis D, Evangelidakis EL, et al. Thromboendarterectomy of the right common iliac artery in a patient with Marfan’s syndrome and restoration with a new technique. Int Angiology. 2001;20:241-243.
- Ito M, Kuroda S, Nakayama N, et al. Extracranial internal carotid artery aneurysm associated with marfan syndrome: case report. Neurol Surg. 2007;35;793-797.
- González JMD, García BÁ, Lebrun JM, et al. Combined surgery for the treatment of bilateral subclavian artery aneurysm in Marfan syndrome. J Vasc Surg. 2007;45(1):180–182.
- Awais M, Williams DM, Deeb GM, et al. Aneurysms of medium-sized arteries in Marfan syndrome. Ann Vasc Surg. 2013;27:1188.e5–1188.e7.
- Patra S, Singh AP, Srinivas BC. Marfan syndrome with spontaneous rupture of aneurysm of common iliac artery. Indian Pediatr. 2013;50:507-508.
- Gaertner S, Alembik Y, Cordeanu EM, et al. Should we systematically screen for peripheral arterial aneurysms in all patients with Marfan syndrome? Int J Cardiol. 2014;172:e94-e95.
- Speciali JG, Lison MP, Junqueira GL. Aneurisma intracraniano na síndrome de Marfan: a case report. Arq Neuropsiquiatr. 2013;29(4):453–457.
- Jourdan C, Artru F, Convert J, et al. Intracranial aneurysm and dysplasia of elastic tissue: pre- and postoperative problems. Agressologie. 1990;31(6):405–408.
- Stehbens WE, Delahunt B, Hilless AD. Early berry aneurysm formation in marfan’s syndrome. Surg Neurol. 1989;31(3):200–202.
- Schievink WI, Parisi JE, Piepgras DG, et al. Intracranial aneurysms in Marfan’s syndrome: an autopsy study. Neurosurgery. 1997;41(4):866–871.
- Kubo Y, Ogasawara K, Kurose A, et al. Ruptured cerebral fusiform aneurysm with mucopolysaccharide deposits in the tunica media in a patient with Marfan syndrome. J Neurosurg. 2008;110(3):518–520.
- Finney HL, Roberts TS, Anderson RE. Giant intracranial aneurysm associated with Marfan’s syndrome. J Neurosurg. 2009;45(3):342–347.
- Matsuda M, Matsuda I, Handa H, et al. Intracavernous giant aneurysm associated with Marfan’s syndrome. Surg Neurol. 1979;12:119-121.
- Ohtsuki H, Sugiura M, Iwaki K, et al. A case of Marfan’s syndrome with a ruptured distal middle cerebral aneurysm. Neurol Surg. 1984;12:983-985.
- Resende LA, Asseis EA, Costa LS, et al. Síndrome de marfan e aneurismas intracranianos gigantes: relato de um caso. Arq Neuropsiquiatr. 1984;42:294–297.
- Narycheva IA, Romkin MA, Sokolina NA, et al. [Neurologic manifestations of Ehlers-Danlos syndrome] Nevrologicheskie proiavleniia sindroma Elersa-Danlosa. ZhNevropatolPsikhiatrIm SSKorsakova. 1989;89:48-53. (Ed.^(Eds).
- Higashida RT, Halbach VV, Hieshima GB, et al. Cavernous carotid artery aneurysm associated with Marfan’s syndrome: treatment by balloon embolization therapy. Neurosurgery. 1988;22(2):297–300.
- Hainsworth PJ, Mendelow AD. Giant intracranial aneurysm associated with Marfan’s syndrome: a case report. J Neurol Neurosurg Psychiatry. 1991;54:471–472. (Ed.^(Eds).
- Rose BS, Pretorius DL. Dissecting basilar artery aneurysm in Marfan syndrome: case report. Am J Neuroradiol. 1991;12:503-504.
- Van Den Berg JSP, Limburg M, Hennekam RCM. Is Marfan syndrome associated with symptomatic intracranial aneurysms? Stroke. 1996;27(1):27:10–12.
- Conway JE, Hutchins GM, Tamargo RJ. Marfan syndrome is not associated with intracranial aneurysms. Stroke. 1999;30(8):1632–1636.
- Wityk RJ, Zanferrari C, Oppenheimer S. Neurovascular complications of Marfan Syndrome. Stroke. 2002;33:680–684.
- Kim ST, Brinjikji W, Kallmes DF. Prevalence of intracranial aneurysms in patients with connective tissue diseases: A retrospective study. Am J Neuroradiol. 2016;37(8):1422–1426.
- Debette S, Goeggel Simonetti B, Schilling S, et al. Familial occurrence and heritable connective tissue disorders in cervical artery dissection. Neurology. 2014;83(22):2023–2031.
- Kim ST, Brinjikji W, Lehman VT, et al. Association between carotid artery tortuosity and carotid dissection: a case-control study. J Neurosurg Sci. 2018;62(4):413–417.
- Brandt T, Orberk E, Weber R, et al. Pathogenesis of cervical artery dissections: association with connective tissue abnormalities. Neurology. 2001;57(1):24–30.
- Alurkar A, Karanam LS, Oak S, et al. Carotid dissection in Marfan’s syndrome. Neurol India. 2013;61(2):206–207.
- Zielinski T, Wolkanin-Bartnik J, Janaszek-Sitkowska H, et al. Persistent dissection of carotid artery in patients operated on for type A acute aortic dissection–carotid ultrasound follow-up. Int J Cardiol. 1999;70(2):133–139.
- Giossi A, Ritelli M, Costa P, et al. Connective tissue anomalies in patients with spontaneous cervical artery dissection. Neurology. 2014;83(22):2032–2037.
- Dittrich R, Heidbreder A, Rohsbach D, et al. Connective tissue and vascular phenotype in patients with cervical artery dissection. Neurology. 2007;68(24):2120–2124.
- Keane MG, Pyeritz RE. Medical management of Marfan syndrome. Circulation. 2008;117(21):2802–2813.
- Grotemeyer D, Duran M, Park EJ, et al. Visceral artery aneurysms - follow-up of 23 patients with 31 aneurysms after surgical or interventional therapy. Langenbecks Arch Surg. 2009;394(6):1093–1100.
- Powell RO, Babu SB, Bommayya G. Left gastric artery aneurysm in Marfan syndrome: A unique case. BMJ Case Rep. 2015;29:1-3.
- Hagerty T, Geraghty P, Braverman AC. Abdominal aortic aneurysm in Marfan Syndrome. Ann Vasc Surg. 2017;40:294.e1–294.e6.
- Van Dijk N, Boer MC, Mulder BJM, et al. Is fatigue in Marfan syndrome related to orthostatic intolerance? Clin Auton Res. 2008;18(4):187–193.
- Tanoue LT. Pulmonary involvement in collagen vascular disease: A review of the pulmonary manifestations of the Marfan syndrome, ankylosing spondylitis, sjögren’s syndrome, and relapsing polychondritis. J Thorac Imaging. 1992;7:62.
- Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476–485.
- Corsico AG, Grosso A, Tripon B, et al. Pulmonary involvement in patients with Marfan syndrome. Panminerva Med. 2014;56:177-182.
- De Maio F, Fichera A, De Luna V, et al. Orthopaedic aspects of marfan syndrome: the experience of a referral center for diagnosis of rare diseases. Adv Orthop. 2016;2016:1–6. 8275391.
- Mayer O, Campbell R, Cahill P, et al. Thoracic Insufficiency Syndrome. Curr Probl Pediatr Adolesc Health Care. 2016;46(3):72–97.
- Pyeritz RE, Francke U. The second international symposium on the Marfan syndrome. Am J Med Genet A. 1993;47(1):1–160.
- Qiao J, Xu L, Liu Z, et al. Surgical treatment of scoliosis in Marfan syndrome: outcomes and complications. Eur Spine J. 2016;25(10):3288–3293.
- Fraser S, Child A, Hunt I. Pectus updates and special considerations in Marfan syndrome. Pediatr Rep. 2017;9(4):7277.
- Kelly Jr RE Jr, Obermeyer RJ, Nuss D. Diminished pulmonary function in pectus excavatum: from denying the problem to finding the mechanism. Ann Cardiothorac Surg. 2016;5(5):466–475.
- Kanagaratnam A, Phan S, Tchantchaleishvilli V, et al. Ravitch versus nuss procedure for pectus excavatum: systematic review and meta-analysis. Ann Cardiothorac Surg. 2016;5(5):409–421.
- Mao YZ, Tang ST, Li S. Comparison of the nuss versus ravitch procedure for pectus excavatum repair: an updated meta-analysis. J Pediatr Surg. 2017;52(10):1545–1552.
- Yoon WW, Sedra F, Shah S, et al. Improvement of pulmonary function in children with early-onset scoliosis using magnetic growth rods. Spine (Phila Pa 1976). 2014;39:1196–1202.
- Nossov SB, Curatolo E, Campbell RM, et al. VEPTR: are we reducing respiratory assistance requirements? J Pediatr Orthop. 2019;39(1):28–32.
- Gadepalli SK, Hirschl RB, Tsai WC, et al. Vertical expandable prosthetic titanium rib device insertion: does it improve pulmonary function? J Pediatr Surg. 2011;46(1):77–80.
- Gawkrodger DJ. Marfan’s syndrome presenting as bilateral spontaneous pneumothorax. Postgrad Med J. 1981;57:240–241.
- Hall JR, Pyeritz RE, Dudgeon DL, et al. Pneumothorax in the Marfan Syndrome: prevalence and therapy. Ann Thorac Surg. 1984;37:500–504.
- Hao W, Fang Y, Lai H, et al. Marfan syndrome with pneumothorax: case report and review of literatures. J Thorac Dis. 2017;9(12):E1100–E1103.
- Fortea-Sanchis C, Ángel Yepes V, Priego Jiménez P, et al. Marfan Syndrome and pneumothorax. Cirugía Española (English Edition). 2015;93(8):e87–e88.
- Viveiro C, Rocha P, Carvalho C, et al. Spontaneous pneumothorax as manifestation of Marfan syndrome. BMJ Case Rep. 2013;2013:bcr2013201697-bcr2013201697.
- Dugan KC, Laxmanan B, Murgu S, et al. Management of persistent air leaks. Chest. 2017;152(2):417–423.
- Pelizzo G, Arbustini E, Pasqua N, et al. Thoracoscopic treatment of pneumothorax in marfan syndrome: hemostatic patch to support lung resection recovery. Case Rep Surg. 2018;7597215:5.
- Booms P, Cisler J, Mathews KR, et al. Novel exon skipping mutation in the fibrillin-1 gene: two ‘hot spots’ for the neonatal Marfan syndrome. Clin Genet. 1999;55(2):110–117.
- Milewicz DM, Duvic M. Severe neonatal Marfan syndrome resulting from a de novo 3-bp insertion into the fibrillin gene on chromosome 15. Am J Hum Genet. 1994;54(3):447–453.
- Robbesom AA, Koenders MMJF, Smits NC, et al. Aberrant fibrillin-1 expression in early emphysematous human lung: A proposed predisposition for emphysema. Mod Pathol. 2008;21(3):297–307.
- Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121.
- König P, Boxer R, Morrison J, et al. Bronchial hyperreactivity in children with Marfan syndrome. Pediatr Pulmonol. 1991;11:29‐36.
- Frischmeyer-Guerrerio PA, Guerrerio AL, Oswald G, et al. Allergy: TGFβ receptor mutations impose a strong predisposition for human allergic disease. Sci Transl Med. 2013;5(195):195ra194.
- Foster ME, Foster DR. Bronchiectasis and Marfan’s syndrome. Postgrad Med J. 1980;56:718–719.
- Teoh PC. Bronchiectasis and spontaneous pneumothorax in Marfan’s syndrome. Chest. 1977;72:672–673.
- Chisholm JC, Cherniack NS, Carton RW. Results of pulmonary function testing in 5 persons with the Marfan syndrome. J Lab Clin Med. 1968;71:25–28.
- Cistulli PA, Sullivan CE. Sleep disorders in Marfan’s syndrome. Lancet. 1991;337(8753):1359–1360.
- Cistulli PA, Sullivan CE. Sleep-disordered breathing in Marian’s syndrome. Amer Rev Respir Dis. 2013;147(3):645–648.
- Kohler M, Blair E, Risby P, et al. The prevalence of obstructive sleep apnoea and its association with aortic dilatation in Marfan’s syndrome. Thorax. 2009;64:162–166.
- Rybczynski M, Koschyk D, Karmeier A, et al. Frequency of sleep apnea in adults with the marfan syndrome. Am J Cardiol. 2010;105(12):1836–1841.
- Cistulli PA, Sullivan CE. Sleep apnea in Marfan’s syndrome: increased upper airway collapsibility during sleep. Chest. 1995;108:631-635.
- Cistulli PA, Gotsopoulos H, Sullivan CE. Relationship between craniofacial abnormalities and sleep-disordered breathing in Marfan’s syndrome. Chest. 2001;120:1455–1460.
- Muiño Mosquera L, Bauters F, Dhondt K. Sleep apnea and the impact on cardiovascular risk in patients with Marfan syndrome. Mol Genet Genomics. 2019;7:e805.
- Peppard PE, Young T, Palta M, et al. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med. 2002;342(19):1378–1384.
- O’Connor GT, Caffo B, Newman AB, et al. Prospective study of sleep-disordered breathing and hypertension: the sleep heart health study. Am J Respir Crit Care Med. 2009;179:1159-1164.
- Lee W, Nagubadi S, Kryger MH, et al. Epidemiology of obstructive sleep apnea: A population-based perspective. Expert Rev Respir Med. 2008;2(3):349–364.
- Cano-Pumarega I, Barbé F, Esteban A, et al. Sleep apnea and hypertension: are there sex differences? The vitoria sleep cohort. Chest. 2017;152(4):742–750.
- Bauters F, Rietzschel ER, Hertegonne KBC, et al. The link between obstructive sleep apnea and cardiovascular disease. Curr Atheroscler Rep. 2016;18(1):8.
- Serizawa N, Yumino D, Takagi A, et al. Obstructive sleep apnea is associated with greater thoracic aortic size. J Am Coll Cardiol. 2008;52:885–886.
- Meuleman C, Boccara F, Nguyen XL, et al. Is the aortic root dilated in obstructive sleep apnoea syndrome? Arch Cardiovasc Dis. 2008;101:391–397.
- Cicek D, Lakadamyali H, Yaǧbasan BD, et al. Obstructive sleep apnoea and its association with left ventricular function and aortic root parameters in newly diagnosed, untreated patients: a prospective study. J Int Med Res. 2011;39(6):2228–2238.
- Baguet J-P, Minville C, Tamisier R, et al. Increased aortic root size is associated with nocturnal hypoxia and diastolic blood pressure in obstructive sleep apnea. Sleep. 2011;34(11):1605–1607.
- Kohler M, Pitcher A, Blair E, et al. The impact of obstructive sleep apnea on aortic disease in marfan’s syndrome. Respiration. 2013;86:39–44.
- Chow K, Pyeritz RE, Litt HI. Abdominal visceral findings in patients with Marfan syndrome. Genet Med. 2007;9:208–212.
- Merza AP, Raiser MW. Biliary tract manifestations of the Marfan syndrome. Am J Gastroenterol. 1987;82:779-782.
- Stheneur C, Faivre L, Collod-Béroud G, et al. Prognosis factors in probands with an FBN1 mutation diagnosed before the age of 1 year. Pediatr Res. 2011;69(3):265–270.
- Ayse Esin K, Bahar C, Fatih A, et al. Delayed presentation of morgagni hernia in a patient with Marfan syndrome. Pediatr Int. 2010;52:e17-e19.
- Beck TF, Campeau PM, Jhangiani SN, et al. FBN1 contributing to familial congenital diaphragmatic hernia. Am J Med Genet A. 2015;167:831–836.
- Broad JB, Wu Z, Clark TG, et al. Diverticulosis and nine connective tissue disorders: epidemiological support for an association. Connect Tissue Res. 2019;60(4):389–398.
- Romere C, Duerrschmid C, Bournat J, et al. Asprosin, a fasting-induced glucogenic protein hormone. Cell. 2016;165(3):566–579.
- Duerrschmid C, He Y, Wang C, et al. Asprosin is a centrally acting orexigenic hormone. Nat Med. 2017;23(12):1444–1453.
- Ghaben AL, Scherer PE. Adipogenesis and metabolic health. Nat Rev Mol Cell Biol. 2019;20(4):242–258.
- Brownstein AJ, Bin Mahmood SU, Saeyeldin A, et al. Simple renal cysts and bovine aortic arch: markers for aortic disease. Open Heart. 2019;6(1):e000862.
- Hibender S, Wanga S, van der Made I, et al. Renal cystic disease in the Fbn1 C1039G/+ Marfan mouse is associated with enhanced aortic aneurysm formation. Cardiovasc Pathol. 2019 [38(January–February)];38:1–6.
- Bouleti C, Flamant M, Escoubet B, et al. Risk of ascending aortic aneurysm in patients with autosomal dominant polycystic kidney disease. Am J Cardiol. 2019;123(3):482–488.
- Biermann CW, Gasser TC, Breuer C, et al. [Marfan syndrome and cystic kidneys of the adult type]. Helv Chirurgica Acta. 1992;59(3):513–515.
- Hateboer N, Buchalter M, Davies SJ, et al. Co-occurrence of autosomal dominant polycystic kidney disease and Marfan syndrome in a kindred. Am J Kidney Dis. 2000;35(4):753–760.
- Somlo S, Rutecki G, Giuffra LA, et al. A kindred exhibiting cosegregation of an overlap connective tissue disorder and the chromosome 16 linked form of autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1993;4(6):1371–1378.
- Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009;60:321–337.
- Al-Haggar M, Bakr A, Wahba Y, et al. A novel fibrillin-1 mutation in an egyptian marfan family: A proband showing nephrotic syndrome due to focal segmental glomerulosclerosis. Saudi J Kidney Dis Transpl. 2017;28(1):141–148.
- Gupta A, Gaikwad J, Khaira A, et al. Marfan syndrome and focal segmental glomerulosclerosis: a novel association. Saudi J Kidney Dis Transpl. 2010;21:754–755..
- Sbar GD, Venkataseshan VS, Huang Z, et al. Renal disease in marfan syndrome. Am J Nephrol. 1996;16:320–326.
- Hartner A, Schaefer L, Porst M, et al. Role of fibrillin-1 in hypertensive and diabetic glomerular disease. Am J Physiol Renal Physiol. 2005;290(6):F1329–1336.
- Baum MA, Harris HW, Burrows PE, et al. Renovascular hypertension in Marfan syndrome. Pediatr Nephrol. 1997;11(4):499–501.
- Loughridge LW. Renal abnormalities in the marfan syndrome. QJM. 1959;28:531-544.
- Schoeneman MJ, Plewinska M, Mucha M, et al. Marfan syndrome and medullary sponge kidney: case report and speculation on pathogenesis. Int J Pediatr Nephrol. 1984;5:103-104.
- Civilibal M, Caliskan S, Numan F, et al. Renovascular hypertension in a child with Marfan syndrome. Anatol J Cardiol. 2010;10:E11-E11.
- Curry RA, Gelson E, Swan L, et al. Marfan syndrome and pregnancy: maternal and neonatal outcomes. BJOG. 2014;121(5):610–617.
- Liang ST. Marfan syndrome, recurrent preterm labour and grandmultiparity. Aust N Z J Obstet Gynaecol. 1985;25(4):288–289.
- Anum EA, Hill LD, Pandya A, et al. Connective tissue and related disorders and preterm birth: clues to genes contributing to prematurity. Placenta. 2009;30(3):207–215.
- Lind J, Wallenburg HC. The Marfan syndrome and pregnancy: a retrospective study in a Dutch population. Eur J Obstet Gynecol Reprod Biol. 2001;98(1):28–35.
- Rossiter JP, Repke JT, Morales AJ, et al. A prospective longitudinal evaluation of pregnancy in the Marfan syndrome. Am J Obstet Gynecol. 1995;173(5):1599–1606.
- Lipscomb KJ, Smith JC, Clarke B, et al. Outcome of pregnancy in women with Marfan’s syndrome. Br J Obstet Gynaecol. 1997;104(2):201–206.
- Meijboom LJ, Drenthen W, Pieper PG, et al. Obstetric complications in Marfan syndrome. Int J Cardiol. 2006;110(1):53–59.
- Chan SSC, Chan DKH, Pang SMW, et al. Urinary incontinence should be added to the manifestation in women with Marfan syndrome. Int Urogynecol J. 2010;21(5):583–587.
- Child AH. Non-cardiac manifestations of Marfan syndrome. Ann Cardiothorac Surg. 2017;6:599–609.
- Carley ME, Schaffer J. Urinary incontinence and pelvic organ prolapse in women with Marfan or Ehlers-Danlos syndrome. Am J Obstet Gynecol. 2000;182(5):1021–1023.
- Minassian VA, Yan X, Lichtenfeld MJ, et al. The iceberg of health care utilization in women with urinary incontinence. Int Urogynecol J. 2012;23(8):1087–1093.
- Harris SS, Link CL, Tennstedt SL, et al. Care seeking and treatment for urinary incontinence in a diverse population. J Urol. 2007;177(2):680–684.
- Hannestad YS, Rortveit G, Hunskaar S. Help-seeking and associated factors in female urinary incontinence. the norwegian epincont study. epidemiology of incontinence in the county of Nord-Trøndelag. Scand J Prim Health Care. 2002;20:102–107.
- Wu JM, Vaughan CP, Goode PS, et al. Prevalence and trends of symptomatic pelvic floor disorders in U.S. women. Obstet Gynecol. 2014;123:141–148.
- O’Halloran T, Bell RJ, Robinson PJ, et al. Urinary incontinence in young nulligravid women: A cross-sectional analysis. Ann Intern Med. 2012;157(2):87–93.
- Jabs C, Child AH. Genitourinary tract in women with Marfan syndrome. In: Child AH, editor. Diagnosis and management of Marfan syndrome. London: Springer. 2016. p. 219–225.
- Hentzen C, Turmel N, Chesnel C, et al. urinary disorders and Marfan syndrome: a series of 4 cases. Urol Int. 2018;101(3):369–371.
- Epenetos AA, Collis CH. Seminoma in Marfan’s syndrome. Postgrad Med J. 1979;55(648):762–764.
- Berkowitz GS, Lapinski RH, Dolgin SE, et al. Prevalence and natural history of cryptorchidism. Pediatrics. 1993;92(1):44–49.
- Schneider MB, Dittmar S, Boxer RA. Anterior sacral meningocele presenting as a pelvic/abdominal mass in a patient with Marfan syndrome. J Adolesc Health. 1993;14(4):325–328.
- Sahin N, Genc M, Kasap E, et al. Anterior sacral meningocele masquerading as an ovarian cyst: a rare clinical presentation associated with Marfan Syndrome. Clin Pract. 2015;5(2):752.
- Burks TN, Cohn RD. Role of TGF-β signaling in inherited and acquired myopathies. Skelet Muscle. 2011;1(1):19.
- Cohn RD, Van Erp C, Habashi JP, et al. Angiotensin II type 1 receptor blockade attenuates TGF-β-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13(2):204–210.
- Jones KB, Sponseller PD, Erkula G, et al. Symposium on the musculoskeletal aspects of marfan syndrome: meeting report and state of the science. J Orthop Res. 2007;25(3):413–422.
- Haine E, Salles JP, Khau Van Kien P, et al. Muscle and bone impairment in children with Marfan syndrome: correlation with age and FBN1 genotype. J Bone Miner Res. 2015;30:1369–1376.
- Delaney K, Kasprzycka P, Ciemerych MA, et al. The role of TGF-β1 during skeletal muscle regeneration. Cell Biol Int. 2017;41(7):706–715.
- Kim J, Lee J. Role of transforming growth factor-β in muscle damage and regeneration: focused on eccentric muscle contraction. J Exerc Rehabil. 2017;13(6):621–626.
- Giske L, Stanghelle JK, Rand-Hendrikssen S, et al. Pulmonary function, working capacity and strength in young adults with Marfan syndrome. J Rehabil Med. 2003;35:221–228.
- Percheron G, Fayet G, Ningler T, et al. Muscle strength and body composition in adult women with Marfan syndrome. Rheumatology. 2007;46(6):957–962.
- Behan WMH, Longman C, Petty RKH, et al. Muscle fibrillin deficiency in Marfan’s syndrome myopathy. J Neurol Neurosurg Psychiatry. 2003;74(5):633–638.
- Krueger C, Hoffmann FM. Identification of retinoic acid in a high content screen for agents that overcome the anti-myogenic effect of TGF-beta-1. PLoS ONE. 2010;5:e15511.
- Kohlmeier L, Gasner C, Marcus R. Bone mineral status of women with Marfan syndrome. Am J Med. 1993;95:568–572.
- Kohlmeier L, Gasner C, Bachrach LK, et al. The bone mineral status of patients with marfan syndrome. J Bone Miner Res. 1995;10:1550-1555.
- Le Parc JM, Plantin P, Jondeau G, et al. Bone mineral density in sixty adult patients with Marfan syndrome. Osteoporos Int. 1999;10(6):475–479.
- Carter N, Duncan E, Wordsworth P. Bone mineral density in adults with Marfan syndrome. Rheumatology (Oxford). 2000;39:307–309.
- Moura B, Tubach F, Sulpice M, et al. Bone mineral density in Marfan syndrome. A large case-control study. Joint Bone Spine. 2006;73:733–735.
- Grover M, Brunetti-Pierri N, Belmont J, et al. Assessment of bone mineral status in children with Marfan syndrome. Am J Med Genet A. 2012;158A:2221–2224.
- Trifirò G, Marelli S, Viecca M, et al. Areal bone mineral density in children and adolescents with Marfan syndrome: evidence of an evolving problem. Bone. 2015;73:176–180.
- Gray JR, Bridges AB, Mole PA, et al. Osteoporosis and the Marfan syndrome. Postgrad Med J. 1993;69:373–375.
- Tobias JH, Dalzell N, Child AH. Assessment of bone mineral density in women with marfan syndrome. Rheumatology. 1995;34(6):516–519.
- Giampietro PF, Peterson M, Schneider R, et al. Assessment of bone mineral density in adults and children with Marfan syndrome. Osteoporos Int. 2003;14:559-563.
- Smaldone S, Clayton NP, Del Solar M, et al. Fibrillin-1 regulates skeletal stem cell differentiation by modulating TGFβ activity within the marrow niche. J Bone Miner Res. 2016;31(1):86–97.
- Nistala H, Lee-Arteaga S, Smaldone S, et al. Fibrillin-1 and −2 differentially modulate endogenous TGF-beta and BMP bioavailability during bone formation. J Cell Biol. 2010;190:1107–1121.
- Rifkin DB, Todorovic V. Bone matrix to growth factors: location, location, location. J Cell Biol. 2010;190(6):949–951.
- Chen S, Grover M, Sibai T, et al. Losartan increases bone mass and accelerates chondrocyte hypertrophy in developing skeleton. Mol Genet Metab. 2015;115(1):53–60.
- Nistala H, Lee-Arteaga S, Carta L, et al. Differential effects of alendronate and losartan therapy on osteopenia and aortic aneurysm in mice with severe Marfan syndrome. Hum Mol Genet. 2010;19(24):4790–4798.
- Dolci C, Pucciarelli V, Gibelli DM, et al. The face in marfan syndrome: a 3D quantitative approach for a better definition of dysmorphic features. Clin Anat. 2018;31(3):380–386.
- Ting BL, Mathur D, Loeys BL, et al. The diagnostic value of the facial features of Marfan syndrome. J Children’s Orthopaedics. 2010;4:545–551.
- Westling L, Mohlin B, Bresin A. Craniofacial manifestations in the Marfan syndrome: palatal dimensions and a comparative cephalometric analysis. J Craniofac Genet Dev Biol. 1998;18:211–218.
- Cistulli PA, Sullivan CE. Influence of maxillary morphology on nasal airway resistance in Marfan’s syndrome. Acta Otolaryngol. 2000;120:410-413.
- Rybczynski M, Bernhardt AMJ, Rehder U, et al. The spectrum of syndromes and manifestations in individuals screened for suspected Marfan syndrome. Am J Med Genet A. 2008;146A(24):3157–3166.
- De Coster P, Pauw GD, Martens L, et al. Craniofacial structure in Marfan syndrome: a cephalometric study. Am J Med Genet A. 2004;131(3):240–248.
- Child AH. Non-cardiac manifestations of Marfan syndrome. Ann Cardiothorac Surg. 2017;6(6):599–609.
- Garreau de Loubresse C, Mullins MM, Moura B, et al. Spinal and pelvic parameters in Marfan’s syndrome and their relevance to surgical planning. J Bone Joint Surg Br. 2006;88(4):515–519.
- Grahame R, Pyeritz RE. The Marfan syndrome: joint and skin manifestations are prevalent and correlated. Br J Rheumatol. 1995;34:126–131.
- Ledoux M, Beauchet A, Fermanian C, et al. A case-control study of cutaneous signs in adult patients with Marfan disease: diagnostic value of striae. J Am Acad Dermatol. 2011;64:290–295.
- Ziv R, Schewach-Millet M, Trau H, et al. Alopecia universalis in a patient with Marfan’s syndrome. Israel J Med Sci. 1990;26:705–707.
- Herman KL, Salman K, Rose LI. White forelock in Marfan’s syndrome: an unusual association, with review of the literature. Cutis. 1991;48:82-84.
- Harper JI, Sidwell RU. Vermiculate atrophoderma in a boy with Marfan syndrome. Br J Dermatol. 1999;141(4):750–752.
- Nallegowda M, Yadav SL, Singh U, et al. An unusual nail presentation in Marfan’s syndrome. J Dermatol. 2002;29(3):164–167.
- Graul-Neumann LM, Kienitz T, Robinson PN, et al. Marfan syndrome with neonatal progeroid syndrome-like lipodystrophy associated with a novel frameshift mutation at the 3ʹ terminus of the FBN1-gene. Am J Med Genet A. 2010;152A(11):2749–2755.
- De Coster PJA, Martens LCM, De Paepe A. Oral manifestations of patients with Marfan syndrome: A case-control study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;93(5):564–572.
- Rahman A, Danne N, Geurtsen W, et al. Kariesprävalenz bei kindern mit Marfan-Syndrom. Oralprophylaxe Und Kinderzahn Hkd. 2014;36:3.
- Staufenbiel I, Hauschild C, Kahl-Nieke B, et al. Periodontal conditions in patients with Marfan syndrome - a multicenter case control study. BMC Oral Health. 2013;13:1-5.
- Bauss O, Neter D, Rahman A. Prevalence of pulp calcifications in patients with Marfan syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol. 2008;106(6):e56–e61.
- Bauss O, Sadat-Khonsari R, Fenske C, et al. Temporomandibular joint dysfunction in Marfan syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97:592–598.
- Martin VT, Neilson D. Joint hypermobility and headache: the glue that binds the two together - part 2. Headache. 2014;54(8):1403–1411.
- Koppen H, Vis JC, Gooiker DJ, et al. Aortic root pathology in Marfan syndrome increases the risk of migraine with aura. Cephalalgia. 2012;32(6):467–472.
- Tronvik E, Stovner LJ, Helde G, et al. Prophylactic treatment of migraine with an angiotensin II receptor blocker: a randomized controlled trial. J Am Med Assoc. 2003;289(1):65–69.
- Davenport RJ, Chataway SJS, Warlow CP. Spontaneous intracranial hypotension from a CSF leak in a patient with Marfan’s syndrome. J Neurol Neurosurg Psychiatry. 1995;59(5):516–519.
- Schievink WI, Gordon OK, Tourje J, et al. Connective tissue disorders with spontaneous spinal cerebrospinal fluid leaks and intracranial hypotension: a prospective study. Neurosurgery. 2004;54(1):65–71.
- Milledge JT, Ades LC, Cooper MG, et al. Severe spontaneous intracranial hypotension and Marfan syndrome in an adolescent. J Paediatr Child Health. 2005;41:68–71.
- Puget S, Kondageski C, Wray A, et al. Chiari-like tonsillar herniation associated with intracranial hypotension in Marfan syndrome. J Neurosurg Pediatr. 2008;106(1 Suppl):48–52.
- Albayram S, Baş A, Ozer H, et al. Spontaneous intracranial hypotension syndrome in a patient with Marfan syndrome and autosomal dominant polycystic kidney disease. Headache. 2008;48(4):632–636.
- Cheuret E, Edouard T, Mejdoubi M, et al. Intracranial hypotension in a girl with Marfan syndrome: case report and review of the literature. Child’s Nerv Syst. 2008;24:509–513.
- Bassani L, Graffeo C, Behrooz N, et al. Noninvasive diagnosis and management of spontaneous intracranial hypotension in patients with marfan syndrome: case report and review of the literature. Surg Neurol Int. 2014;5(1):8.
- Uysal S, Albayram S, Ercan ET. Spontaneous intracranial hypotension: A case report. J Child Neurol. 2008;23:1312–1315.
- Schievink WI, Maya MM, Louy C, et al. Diagnostic criteria for spontaneous spinal CFS leaks and intracranial hypotension. Am J Neuroradiol. 2008;29(5):853–856.
- Low PA, Tomalia VA. Orthostatic hypotension: mechanisms, causes, management. J Clin Neurol. 2015;11(3):220–226.
- De Paepe A, Devereux RB, Dietz HC, et al. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet A. 1996;62(4):417–426.
- Hofman KJ, Bernhardt BA, Pyeritz RE. Marfan syndrome: neuropsychological aspects. Am J Med Genet A. 1988;31:331–338.
- Handisides JC, Hollenbeck-Pringle D, Uzark K, et al. Health-related quality of life in children and young adults with Marfan Syndrome. J Paediatr. 2019;204:250–255.e251.
- Lannoo E, Paepe AD, Leroy B, et al. Neuropsychological aspects of Marfan syndrome. Clin Genet. 2008;65–9(2):65–69.
- Rand-Hendriksen S, Sørensen I, Holmström H, et al. Fatigue, cognitive functioning and psychological distress in Marfan syndrome, a pilot study. Psychol Health Med. 2007;12:305–313.
- Leone JC, Swigar ME. Marfan’s Syndrome and neuropsychiatric symptoms: case report and literature review. Compr Psychiatry. 1986;27(3):247–250.
- Romano J. Marfan’s Syndrome and Schizophrenia: a case report. Arch Gen Psychiatry. 2011;44(2):190–192.
- Gritti A, Pisano S, Catone G, et al. Psychiatric and neuropsychological issues in Marfan syndrome: A critical review of the literature. Int J Psychiatry Med. 2015;50:347–360. (Ed.^(Eds).
- Sirota P, Frydman M, Sirota L. Schizophrenia and Marfan syndrome. Br J Psychiatry. 1990;157:433–436.
- Kalsi G, Mankoo BS, Brynjolfsson J, et al. The Marfan syndrome gene locus as a favoured locus for susceptibility to schizophrenia. Psychiatr Genet. 1994;4(4):219–227.
- Mercuro G, Carpzniello B, Ruscazio M, et al. Association between psychiatric disorders and Marfan’s syndrome in a large sardinian family with a high prevalence of cardiac abnormalities. Clin Cardiol. 1997;20(3):243–245.
- Stramesi F, Politi P, Fusar-Poli P. Marfan syndrome and liability to psychosis. Med Hypotheses. 2007;68(5):1173–1174.
- Rao NP, Loganathan S, Prakash O, et al. Use of electroconvulsive therapy for schizophrenia with comorbid marfan syndrome. J Ect. 2009;25:276–277.
- Lemberg M, Thompson AW. Marfan syndrome and schizophrenia: a case report and literature review. Gen Hosp Psychiatry. 2010;32:228.e9–228.e10.
- Van Den Bossche MJA, Van Wallendael KLP, Strazisar M, et al. Co-occurrence of Marfan syndrome and schizophrenia: what can be learned? Eur J Med Genet. 2012;55(4):252–255.
- Sahadevan S, Nakulan A, John S, et al. Gender and psychiatric manifestations in Marfan syndrome-is there a link. Aust N Z J Psychiatry. 2014;48:588. (Ed.^(Eds).
- Van Tongerloo A, De Paepe A. Psychosocial adaptation in adolescents and young adults with Marfan syndrome: an exploratory study. J Med Genet. 1998;35:405–409.
- Wanson L, Godfroid IO. Psychiatric symptoms and Marfan: part of the syndrome or incidental to it? World J Biol Psychiatry. 2002;3:229–230.
- Peters KF, Kong F, Hanslo M, et al. Living with Marfan syndrome III. quality of life and reproductive planning. Clin Genet. 2002;62(2):110–120.
- Jha VN, Kumar M, Tarwani J. Co-occurrence of Marfan syndrome and bipolar disorder: A fifteen year follow up. Asian J Psychiatr. 2016;24:91–92.
- Benke K, Ágg B, Pólos M, et al. The effects of acute and elective cardiac surgery on the anxiety traits of patients with Marfan syndrome. BMC Psychiatry. 2017;17(1):253–260.
- Benninghoven D, Hamann D, Von Kodolitsch Y, et al. Inpatient rehabilitation for adult patients with Marfan syndrome: an observational pilot study. Orphanet J Rare Dis. 2017;12:127.
- Goldfinger JZ, Preiss LR, Hendershot TP, et al. Marfan Syndrome and quality of life in the genTAC registry. J Am Coll Cardiol. 2017;69(23):2821–2830.
- Bathen T, Velvin G, Rand-Hendriksen S, et al. Fatigue in adults with Marfan syndrome, occurrence and associations to pain and other factors. Am J Med Genet A. 2014;164:1931–1939.
- Velvin G, Bathen T, Rand-Hendriksen S, et al. Work participation in adults with Marfan syndrome: demographic characteristics, MFS related health symptoms, chronic pain, and fatigue. Am J Med Genet A. 2015;167A(12):3082–3090.
- Velvin G, Bathen T, Rand-Hendriksen S, et al. Satisfaction with life in adults with Marfan syndrome (MFS): associations with health-related consequences of MFS, pain, fatigue, and demographic factors. Qual Life Res. 2016;25(7):1779–1790.
- Rao SS, Venuti KD, Dietz H, et al. Quantifying health status and function in Marfan Syndrome. J Surg Orthop Adv. 2016;25:34–40.
- Velvin G, Bathen T, Rand-Hendriksen S, et al. Systematic review of chronic pain in persons with Marfan syndrome. Clin Genet. 2016;89(6):647–658.
- Nelson AM, Walega DR, McCarthy RJ. The incidence and severity of physical pain symptoms in marfan syndrome a survey of 993 patients. Clin J Pain. 2015;31:1080–1086. (Ed.^(Eds).
- Speed TJ, Mathur VA, Hand M, et al. Characterization of pain, disability, and psychological burden in Marfan syndrome. Am J Med Genet A. 2017;173(2):315–323.
- Mamiya S, Endo Y, Miura AB, et al. Disseminated intravascular coagulation accompanying thoracic and abdominal aortic aneurysm; Report of three cases. Jpn J Med. 2011;27(1):91–95.
- Alarcón-Segovia D, Javier Fierro F, de Jésus Villalobos J, et al. Bilateral renal vein thrombosis and nephrotic syndrome in a patient with the Marfan syndrome. Dis Chest. 2007;54(2):153–156.
- Humphries JE, Stouffer GA, Kelly TE, et al. Hypercoagulability in a patient with Marfan syndrome. J Med Genet. 1991;28(5):349–351.
- Touat Z, Lepage L, Ollivier V, et al. Dilation-dependent activation of platelets and prothrombin in human thoracic ascending aortic aneurysm. Arterioscler Thromb Vasc Biol. 2008;28:940–946.
- Kornhuber K, Seidel H, Pujol C, et al. Hemostatic abnormalities in adult patients with Marfan syndrome. Cardiovasc Diagn Ther. 2019;9(Suppl 2):S209–S220.
- Ihara A, Kawamoto T, Matsumoto K, et al. Relationship between hemostatic markers and circulating biochemical markers of collagen metabolism in patients with aortic aneurysm. Pathophysiol Haemost Thromb. 2003;33(4):221–224.
- Nomura F, Ihara A, Yoshitatsu M, et al. Relationship between coagulation cascade, cytokine, adhesion molecule and aortic aneurysm. Eur J Cardiothorac Surg. 2003;23(6):1034–1039.
- Sidloff DA, Stather PW, Choke E, et al. A systematic review and meta-analysis of the association between markers of hemostasis and abdominal aortic aneurysm presence and size. J Vasc Surg. 2014;59(2):528–535.e524.
- Yuan S-M, Shi Y-H, Wang -J-J, et al. Elevated plasma D-dimer and hypersensitive C-reactive protein levels may indicate aortic disorders. Braz J Cardiovasc Surg. 2011;26:573-581.
- Bridges AB, Gray JR, McLaren M, et al. Endothelial cell and platelet function in Marfan’s Syndrome. Endothelium. 1993;1(3):203–206.
- Hirata K, Triposkiadis F, Sparks E, et al. The marfan syndrome: abnormal aortic elastic properties. J Am Coll Cardiol. 1991;18(1):57–63.
- Bogren HG, Mohiaddin RH, Yang GZ, et al. Magnetic resonance velocity vector mapping of blood flow in thoracic aortic aneurysms and grafts. J Thorac Cardiovasc Surg. 1995;110(3):704–714.
- Geiger J, Hirtler D, Gottfried K, et al. Longitudinal evaluation of aortic hemodynamics in Marfan Syndrome: new Insights from a 4D flow cardiovascular magnetic resonance multi-year follow-up study. J Cardiovasc Magn Reson. 2017;19(1):33.
- Geiger J, Markl M, Herzer L, et al. Aortic flow patterns in patients with Marfan syndrome assessed by flow-sensitive four-dimensional MRI. J Magn Reson Imaging. 2012;35:594–600.
- Wang HH, Chiu HH, Tseng WYI, et al. Does altered aortic flow in marfan syndrome relate to aortic root dilatation? J Magn Reson Imaging. 2016;44(2):500–508.
- Hathcock JJ. Flow effects on coagulation and thrombosis. Arterioscler Thromb Vasc Biol. 2006;1729–37(8):1729–1737.
- Gogia S, Neelamegham S. Role of fluid shear stress in regulating VWF structure, function and related blood disorders. Biorheology. 2015;52(5–6):319–335.
- Horiuchi H. [A hemostatic disorder caused by high shear stress: acquired von Willebrand syndrome]. [Rinsho Ketsueki] Jpn J Clin Hematol. 2018;59:2233–2237.
- Petricevic M, Knezevic J, Samoukovic G, et al. Diagnosis and management of acquired von willebrand disease in heart disease: a review of the literature. Thorac Cardiovasc Surg. 2018. doi:https://doi.org/10.1055/s-0038-1673670.
- Fressinaud E. Von-Willebrand-syndrome type 2; 2009.
- Frank RD, Lanzmich R, Haager PK, et al. Severe aortic valve stenosis. Clin Appl Thromb Hemost. 2017;23:229–234.
- Iijima M, Itoh N, Murase R, et al. A surgical case of aortic stenosis with recurrent gastrointestinal bleeding: heyde syndrome. Int J Surg Case Rep. 2018;53:281–284.
- Vincentelli A, Susen S, Le Tourneau T, et al. Acquired von willebrand syndrome in aortic stenosis. N Engl J Med. 2003;349(4):343–349.
- Blackshear JL, McRee CW, Safford RE, et al. Von willebrand factor abnormalities and heyde syndrome in dysfunctional heart valve prostheses. JAMA Cardiol. 2016;1(2):198–204.
- Anstey A, Mayne K, Winter M, et al. Platelet and coagulation studies in Ehlers‐Danlos syndrome. Br J Dermatol. 1991;125(2):155–163.
- Artoni A, Bassotti A, Abbattista M, et al. Hemostatic abnormalities in patients with Ehlers–danlos syndrome. J Thromb Haemost. 2018;16(12):2425–2431.
- Busch A, Hoffjan S, Bergmann F, et al. Vascular type Ehlers-Danlos syndrome is associated with platelet dysfunction and low Vitamin D serum concentration. Orphanet J Rare Dis. 2016;11:111.
- Scheibenberger D, Frings A, Steinberg J, et al. Ocular manifestation in Marfan syndrome: corneal biomechanical properties relate to increased systemic score points. Graefe’s Arch Clin Exp Ophthalmol. 2018;256:1159–1163.
- Pyeritz RE. Marfan syndrome: improved clinical history results in expanded natural history. Genet Med. 2019;21(8):1683–1690.
- von Kodolitsch Y, Bernhardt AM, Kölbel T, et al. Maximizing therapeutic success: the key concepts of individualized medical strategy (IMS). Cogent Med. 2015;2(1):1109742.
- Héon-Klin V. European Reference networks for rare diseases: what is the conceptual framework? Orphanet J Rare Dis. 2017;12(1):137.
- Mercieca-Bebber R, King MT, Calvert MJ, et al. The importance of patient-reported outcomes in clinical trials and strategies for future optimization. Patient Relat Outcome Meas. 2018;9:53–367.
- von Kodolitsch Y, Rybczynski M, Vogler M, et al. The role of the multidisciplinary health care team in the management of patients with Marfan syndrome. J Multidiscip Healthc. 2016;9:587–614.