
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The initial steps of Cu2O sulphidation to Cu2S have been studied using plane-wave density functional theory at the PBE-D3+U level of sophistication. Surface adsorption and dissociation of H2S and H2O, as well as the replacement reaction of lattice oxygen with sulphur, have been investigated for the most stable (111) and (100) surface facets under oxygen-lean conditions. We find that the (100) surface is more susceptible to sulphidation than the (111) surface, promoting both H2S adsorption, dissociation and the continued oxygen–sulphur replacement. The results presented in this proceeding bridge previous results from high-vacuum experiments on ideal surface to more realistic corrosion conditions and set the grounds for future mechanistic studies. Potential implications on the long-term final disposal of spent nuclear fuel are discussed.
This paper is part of a supplement on the 6th International Workshop on Long-Term Prediction of Corrosion Damage in Nuclear Waste Systems.
GRAPHICAL ABSTRACT
Introduction
Regardless of the choice of concept, the safe disposal of spent nuclear fuel resides on a number of critical premises. In the KBS-3 model for final disposal under Swedish bedrock conditions, one such premise is the slow and uniform, sulphide-controlled corrosion of the copper waste canister [Citation1,Citation2]. This type of behaviour is believed to dominate during the main part of the repository life-time. The corrosion is assumed to be uniform since the sulphide film (mainly chalcocite, Cu2S) is active under the predicted steady-state conditions [Citation3–6]. However, in the initial phase of the disposal, the canister’s surface will be covered by passivating (hydr)oxide films, predominantly in the form of cuprite (Cu2O) and other corrosion products, owing to surface oxidation during the prehandling process and residual O2 trapped in the bore holes.
While the above could potentially pose a problem in the nuclear waste disposal, numerous studies have shown that the sulphidation process(1)
(1) occurs readily in aqueous environments and that the initial oxide film will be converted to the sulphide chalcocite (Cu2S) [Citation7–9]. The mechanism for this conversion, which is an important part in the understanding of sulphur-assisted degradation of passive films [Citation10], is, nevertheless, not as well understood as the corrosion process. A correct description of the mechanism is necessary in order to obtain a comprehensive picture of the performance of the copper canister and to rule out the possibility of the formation of a passive film that may result in localised corrosion. It should be noted here that the present study assumes a somewhat simplified picture of the canister film composition: for instance are chloride-containing Cu(II) hydroxides, such as para-atacamite, also expected to form under early repository conditions.
Herein, we present results on the initial stage of the cuprite sulphidation process from density functional theory (DFT) calculations on the Cu2O(111) and (100) low-index crystal surfaces. Computational chemistry has previously been successful in modelling similar problems [Citation11,Citation12] and allows for the study at the necessary atomic resolution. The current study should be seen as a first attempt to model the complex mechanism of Cu2O sulphidation, setting the grounds for continued studies. We, therefore, include a perspective over the route ahead by describing the required future steps for understanding the sulphidation mechanism under different conditions. We also highlight the complementary roles of experiments and theoretical computations, where computational methods reinforce experiments by their ability to provide an atomic scale rationale to experimental results and by offering the possibility to make elaborate estimations into areas yet to be explored experimentally. Although our results are discussed in the perspective of the nuclear waste disposal, we also anticipate them to be of broader relevance, e.g. in heterogeneous catalysis, corrosion in the petroleum and chemical industry as well as for the corrosion of electronic materials [Citation13].
Results and discussion
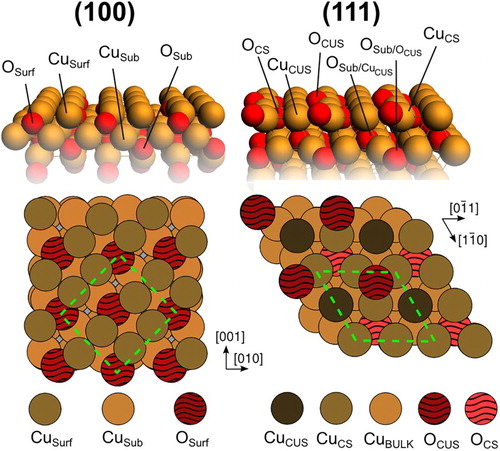
DFT calculations at the PBE-D3+U level of theory [Citation14–17] were conducted on periodic slab models for (111) and (100) surfaces of Cu2O as described in the computational methods section of the supplemental material. Both surface facets were represented by their most stable terminations under oxygen-lean conditions: the non-polar, stoichiometric O-terminated (111) surface and the Cu-terminated (100) surface, as shown in [Citation18–21]. In the present study, we investigate the early stages of sulphidation of cuprous oxide under vacuum conditions. This is a necessary first step to understand the atomistic mechanism of sulphidation and ultimately provides us with phase diagrams for the surface sulphidation on the (111) and (100) low-index surface facets. By the use of thermodynamic relations and complementary experimental and computational data, these diagrams can be extended to cover also wet conditions, the effect of pH, sulphide concentration and impurities, to mention a few examples. Figures of all the optimised structures considered herein are included in the supplemental material.
Figure 1. Atomic models showing the top and side views of the ideal p(1×1) (111) and the copper terminated and c(2×2) ridge-dimer reconstructed (100) surface facet of Cu2O. CS(CUS) = coordinatively (un)saturated atom.
Surface adsorption and reactions
shows the very initial stage of H2S sulphidation of cuprite: the adsorption and dissociation of H2S onto and at the surface. The equivalent processes are also shown for H2O. These processes are experimentally relatively well studied by e.g. photoelectron spectroscopy and surface tunnelling microscopy on the Cu2O(111) low-index surface under ultra-high vacuum conditions for both H2O [Citation22,Citation23] and H2S [Citation24]. Although no quantitative adsorption data are available, Lin et al. [Citation24] have shown that, at low coverage, H2S completely dissociates on the Cu2O(111) surface to form S2− ad and 2Had, where the latter are adsorbed on the surface O atoms. Water adsorbs molecularly on the ideal (111) surface [Citation23,Citation25–27], but as the mixture of H2Oad and OHad + Had on the oxygen vacant surface [Citation22,Citation26,Citation27]. As can be seen in , experimental and computational data are in qualitative agreement: DFT shows that H2S adsorbs stronger than H2O and its dissociation is more thermodynamically favourable than that of H2O on the cuprite (111) surface (assuming identical H2S and H2O partial pressures). Where available, the computed DFT adsorption energies are in line with previous DFT calculations, see the supplemental material [Citation23,Citation25–30]. Note also that the relations of can be converted to arbitrary pressures via ΔG = −RTlnK.
Figure 2. Gibbs free energy surfaces for H2X (X = O,S) adsorption and dissociation to Had + XHad and 2Had + Xad on the (111) and (100) surfaces of Cu2O at 298.15 K, pH2O = pH2S = 1 bar and pH2 = 531 nbar (i.e. tropospheric annual mean [Citation34]).
![Figure 2. Gibbs free energy surfaces for H2X (X = O,S) adsorption and dissociation to Had + XHad and 2Had + Xad on the (111) and (100) surfaces of Cu2O at 298.15 K, pH2O = pH2S = 1 bar and pH2 = 531 nbar (i.e. tropospheric annual mean [Citation34]).](/cms/asset/308034ec-b5c2-41af-ab1f-fea74e345aa1/ycst_a_1284393_f0002_c.jpg)
The (111) surface is the most stable [Citation19,Citation20] low-index surface of cuprite under various conditions and is accordingly the most common surface facet. However, many surface-related properties will be controlled by other, more reactive, surface facets. Therefore, we have also studied the less stable [Citation19,Citation20] low-index Cu2O(100) surface. H2O adsorption and dissociation have been studied both experimentally and computationally on the (100) surface [Citation30–32]. In contrast, no data are available for H2S interactions on the same surface.
also includes computed data for H2O and H2S on the (100) surface. We find similar trends as for the (111) surface, namely that H2S adsorbs stronger and dissociates further than H2O. In comparison to (111), it is evident that the (100) surface generally disfavours molecular adsorption but significantly promotes adsorbate dissociation. For the sulphidation process, the formation of S2− ad is likely a key step and the fact that it is more exothermic on the (100) surface than on the (111) surface is a strong indicator with regard to at which surface the oxide-to-sulphide conversion process is expected to dominate.
Sulphide-oxide replacement
The full sulphidation process is anticipated to pass through numerous elementary steps, with the possibility of different competing mechanistic routes [Citation7]. The comprehensive investigation of all of these steps and mechanisms is beyond the scope of the present study. This will, however, be the topic of future studies. Nevertheless, the most fundamental knowledge is, arguably, understanding of the overall process leading from nH2S to nH2O with n lattice O2− replaced by n S2−. This is studied in the following and will provide us the thermodynamic driving force for the surface sulphidation reaction.
summarises our results for the H2Sad + Cu2Osurf → H2Oad + Cu2Ssurf reaction at low degrees of sulphidation. Sulphidation leading to S2− at different lattice positions, including the first subsurface layers, was considered. We find from this that sulphidation of the top layer is beneficial over sulphidation of the subsurface layer for both the (111) and (100) surface facets. The result is reasonable since Cu2O and Cu2S have dissimilar crystal structures and thus placing the larger S2− ion in a subsurface layer will add residual strain to the structure. The result is also reasonable since the sulphidation energy is expected to converge towards that of the bulk reaction for subsurface replacement. From our study, we can, furthermore, conclude that the (100) surface is more easily sulphidised than the (111) surface, which may most conveniently be explained by the fact that the (100) surface atoms have a lower degree of saturation (i.e. lower coordination number) than the (111) surface atoms. Additionally, the results show that the adsorbed H2O molecule formed during the replacement process has a stabilising effect on adjacent S2−, and more so on the surface exchange than for subsurface exchange. For the (100) surface, the water stabilisation has the effect of retaining the sulphur atom inside the lattice, whereas without the (H2O)ad the sulphur atom instead prefers to migrate to a site on top of the surface.
Table 1. Relative electronic energies, ΔEsulph, in kJ mol−1 for the low degree (25%) surface and subsurface Cu2O sulphidation process defined by: H2Sad + Cu2Osurf/sub → H2Oad + Cu2Ssurf/sub.
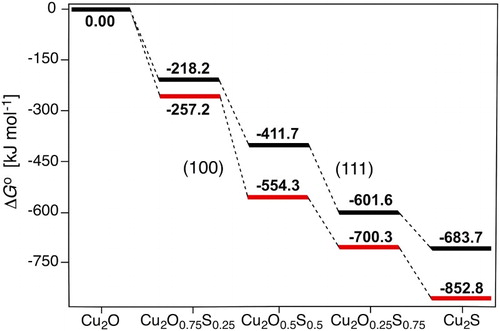
Larger degrees of sulphidation were also studied via the reaction nH2S(g) + nCu2Osurf → nH2O(g) + nCu2Ssurf. Based on the conclusions from the low degree of sulphidation, we assumed that the process would first proceed to a full top layer of Cu2S before the subsurface layer is sulphidised. Thus only top layer surface replacement is considered in the following. We also conclude that, although the H2O adsorbent does have a stabilising effect on the replacement, the effects are small compared to other aspects and the overall trends can be recovered by considering H2O (and H2S) in gas phase. All unique combinations of replacement to surface layer site(s) were considered, where the lowest energy structures for each degree of sulphidation were used in the analysis below. The full results are reported in the supplemental material.
shows the free energy profile for the successive sulphidation of the (111) and (100) surfaces at 298.15 K, assuming standard pressures of 1 bar for all species. At these conditions, full sulphidation of the top layer is beneficial for both the surface facets. By converting the free energies to arbitrary pressures of H2O and H2S at 298.15 K, we obtain the phase diagrams (Figure S5) in the supplemental information. These pinpoint that the stability region for the sulphidised surface is large on both surfaces: a fully Cu2S terminated Cu2O surface is beneficial under essentially all realistic, oxygen-lean, conditions for both the (111) and (100) surfaces, in agreement with experimental findings [Citation24,Citation33]. We again find that the (100) surface is more readily sulphidised than the (111) surface. Note that at high pressures, cooperative adsorbate effects may alter the relative stabilities of the sulphidised surfaces.
Figure 3. Standard Gibbs free energy profile for step-wise (0, 25, 50, 75 and 100%) surface sulphidation of the (111) and (100) Cu2O surface facets at 298.15 K.
Conclusion
The results presented herein on the initial sulphidation process of the Cu2O(111) and Cu2O(100) surfaces aim to bridge our knowledge from experimental ultra-high vacuum conditions to realistic conditions applicable to the predicted conditions in nuclear waste repository vaults. H2O and H2S adsorption and dissociation have been studied by DFT, indicating that H2S adsorption and dissociation are more beneficial than the corresponding processes for H2O on the (111) surface, in line with experimental results. We find the same trends for the (100) surface, where H2S interactions have not yet been studied experimentally. We have also established that the (100) surface adsorbs both H2O and H2S stronger than the (111) surface and that adsorption is molecular for H2O onto (111) and dissociative to OH− ad on (100). H2Sad dissociates to S2− ad on both surface with the dissociation being more exothermic on (100).
By the study of O2− à S2− lattice replacement reaction, we have found that the (100) surface is more readily sulphidised than the (111) surface. Furthermore, it is more beneficial to sulphidise the surface top layer in comparison to the subsurface layer at low degrees of sulphidation for both surface facets. From an extended analysis at larger degrees of sulphidation, we conclude that complete sulphidation of the top layer is favourable under realistic Swedish nuclear waste repository conditions. In future studies, we intend to investigate the sulphidation mechanism, in further detail, on ideal as well as defective surfaces. We also intend to analyse a wider range of conditions, including the effects of aqueous and oxygen-rich environments.
Supplementary_material.docx
Download MS Word (2.4 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
J. H. Stenlid http://orcid.org/0000-0003-3832-2331
A. J. Johansson http://orcid.org/0000-0001-7686-7776
C. Leygraf http://orcid.org/0000-0002-9453-1333
T. Brinck http://orcid.org/0000-0003-2673-075X
Additional information
Funding
Related Research Data
References
- King F, Lilja C, Pedersen K, et al. An update of the state-of-the-art report on the corrosion of copper under expected conditions in a deep geologic repository. TR-10-67, SKB, 2010.
- King F, Lilja C, Vähänen M. Progress in the understanding of the long-term corrosion behaviour of copper canisters. J Nucl Mater. 2013;438:228–237. doi: 10.1016/j.jnucmat.2013.02.080
- Martino T, Partovi-Nia R, Chen J, et al. Mechanisms of film growth on copper in aqueous solutions containing sulphide and chloride under voltammetric conditions. Electrochim Acta. 2014;127:439–447. doi: 10.1016/j.electacta.2014.02.050
- Chen J, Qin Z, Shoesmith DW. Key parameters determining structure and properties of sulphide films formed on copper corroding in anoxic sulphide solutions. Corros Eng Sci Technol. 2014;49:415–419. doi: 10.1179/1743278214Y.0000000188
- King F, Lilja: C. Localised corrosion of copper canisters. Corros Eng Sci Technol. 2014;49:420–424. doi: 10.1179/1743278214Y.0000000182
- King F, Lilja: C. Localised corrosion of copper canisters in bentonite pore water. TR-13-27, SKB, 2013.
- Smith JM, Wren JC, Odziemkowski M, et al. The electrochemical response of preoxidized copper in aqueous sulfide solutions. J Electrochem Soc. 2007;154:C431–C438. doi: 10.1149/1.2745647
- Hollmark HM, Keech PG, Vegelius JR, et al. X-ray absorption spectroscopy of electrochemically oxidized Cu exposed to Na2S. Corros Sci. 2012;54:85–89. doi: 10.1016/j.corsci.2011.09.001
- Kristiansen PT, Massel F, Werme L, et al. Sulfidation of single-phase oxide on copper and as powder studied using soft X-ray spectroscopy. J Electrochem Soc. 2015;162:C785–C791. doi: 10.1149/2.0801514jes
- Marcus P. Corrosion mechanisms in theory and practice. 3rd ed. Boca Raton (FL): CRC Press; 2011. p. 395–417.
- Mayernick AD, Li R, Dooley KM, et al. Energetics and mechanism for H2S adsorption by ceria-lanthanide mixed oxides: implications for the desulfurization of biomass gasifier effluents. J Phys Chem C. 2011;115:24178–24188. doi: 10.1021/jp206827n
- Bouzoubaa A, Costa D, Diawara B, et al. Insight of DFT and atomistic thermodynamics on the adsorption and insertion of halides onto the hydroxylated NiO(111) surface. Corros Sci. 2010;52:2643–2652. doi: 10.1016/j.corsci.2010.04.014
- Leygraf C, Odnevall Wallinder I, Tidblad J, et al. Atmospheric corrosion. 2nd ed. Hoboken (NJ): John Wiley & Sons; 2016.
- Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865
- Grimme S, Antony J, Ehrlich S, et al. A consistent and accurate Ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys. 2010;132:154104. doi: 10.1063/1.3382344
- Grimme S, Ehrlich S, Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J Comput Chem. 2011;32:1456–1465. doi: 10.1002/jcc.21759
- Dudarev SL, Botton GA, Savrasov SY, et al. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U Study. Phys Rev B. 1998;57:1505–1509. doi: 10.1103/PhysRevB.57.1505
- Soldemo M, Stenlid JH, Besharat Z, et al. The surface structure of Cu2O(100). J Phys Chem C. 2016;120:4373–4381. doi: 10.1021/acs.jpcc.5b11350
- Soon A, Todorova M, Delley B, et al. Thermodynamic stability and structure of copper oxide surfaces: a first-principles investigation. Phys Rev B. 2007;75:125420. doi: 10.1103/PhysRevB.75.125420
- Bendavid LI, Carter: EA. First-principles predictions of the structure, stability, and photocatalytic potential of Cu2O surfaces. J Phys Chem B. 2013;117:15750–15760. doi: 10.1021/jp406454c
- Nilius N, Fedderwitz H, Gross B, et al. Incorrect DFT-GGA predictions of the stability of non-stoichiometric/polar dielectric surfaces: the case of Cu2O(111). Phys Chem Chem Phys. 2016;18:6729–6733. doi: 10.1039/C5CP06933E
- Önsten A, Weissenrieder J, Stoltz D, et al. Role of defects in surface chemistry on Cu2O(111). J Phys Chem C. 2013;117:19357–19364. doi: 10.1021/jp3112217
- Kronawitter CX, Riplinger C, He X, et al. Hydrogen-bonded cyclic water clusters nucleated on an oxide surface. J Am Chem Soc. 2014;136:13283–13288. doi: 10.1021/ja5056214
- Lin J, May JA, Didziulis SV, et al. Variable-energy photoelectron spectroscopic studies of hydrogen sulfide chemisorption on cuprous oxide and zinc oxide single-crystal surfaces: HS-bonding to copper(I) and zinc(II) sites related to catalytic poisoning. J Am Chem Soc. 1992;114:4718–4727. doi: 10.1021/ja00038a039
- Riplinger C, Carter EA. Cooperative effects in water binding to cuprous oxide surfaces. J Phys Chem C. 2015;119:9311–9323. doi: 10.1021/acs.jpcc.5b00383
- Yu X, Zhang X, Wang S, et al. A computational study on water adsorption on Cu2O(111) surfaces: the effects of coverage and oxygen defect. Appl Surf Sci. 2015;343:33–40. doi: 10.1016/j.apsusc.2015.03.065
- Zhang R, Li J, Wang B, et al. Fundamental studies about the interaction of water with perfect, oxygen-vacancy and pre-covered oxygen Cu2O(111) surfaces: thermochemistry, barrier, product. Appl Surf Sci. 2013;279:260–271. doi: 10.1016/j.apsusc.2013.04.078
- Casarin M, Maccato C, Vigato N, et al. A theoretical study of the H2O and H2S chemisorption on Cu2O(111). Appl Surf Sci. 1999;142:164–168. doi: 10.1016/S0169-4332(98)00668-0
- Zhang R, Liu H, Li J, et al. A mechanistic study of H2S adsorption and dissociation on Cu2O(111) surfaces: thermochemistry, reaction barrier. Appl Surf Sci. 2012;258:9932–9943. doi: 10.1016/j.apsusc.2012.06.053
- Li Y, Yan L, Wang G. Adsorption and dissociation of H2O on Cu2O(100): a computational study. J Nat Gas Chem. 2011;20:155–161. doi: 10.1016/S1003-9953(10)60162-4
- Stenlid JH, Soldemo M, Johansson AJ, et al. Reactivity at the Cu2O(100):Cu–H2O interface: a combined DFT and PES study. Phys Chem Chem Phys. 2016;18:30570–30584.
- Cox DF, Schulz KH. H2O adsorption on Cu2O(100). Surf Sci. 1991;256:67–76. doi: 10.1016/0039-6028(91)91200-H
- Galtayries A, Bonnelle: J-P. XPS and ISS studies on the interaction of H2S with polycrystalline Cu, Cu2O and CuO surfaces. Surf Interface Anal. 1995;23:171–179. doi: 10.1002/sia.740230308
- Novelli PC, Lang PM, Masarie KA, et al. Molecular hydrogen in the troposphere: global distribution and budget. J Geophys Res Atmospheres. 1999;104:30427–30444. doi: 10.1029/1999JD900788