
ABSTRACT
Introduction: In 2018, an estimated 377,000 people developed multidrug-resistant tuberculosis (MDR-TB), urging for new effective treatments. In the last years, it has been accepted that efflux pumps play an important role in the evolution of drug resistance. Strategies are required to mitigate the consequences of the activity of efflux pumps.
Areas covered: Based upon the literature available in PubMed, up to February 2020, on the diversity of efflux pumps in Mycobacterium tuberculosis and their association with drug resistance, studies that identified efflux inhibitors and their effect on restoring the activity of antimicrobials subjected to efflux are reviewed. These support a new strategy for the development of anti-TB drugs, including efflux inhibitors, using in silico drug repurposing.
Expert opinion: The current literature highlights the contribution of efflux pumps in drug resistance in M. tuberculosis and that efflux inhibitors may help to ensure the effectiveness of anti-TB drugs. However, despite the usefulness of efflux inhibitors in in vitro studies, in most cases their application in vivo is restricted due to toxicity. In a time when new drugs are needed to fight MDR-TB and extensively drug-resistant TB, cost-effective strategies to identify safer efflux inhibitors should be implemented in drug discovery programs.
1. Introduction
Tuberculosis (TB) remains a serious Public Health problem worldwide, causing millions of deaths per year and it is estimated that one-third of the world population is latently infected [Citation1]. Recently, the World Health Organization (WHO) has launched the ‘End TB’ strategy, which aims to reduce TB deaths by 95% and to cut new cases by 90% between 2015 and 2035 [Citation2]. Although there has been a decrease in global TB incidence rates and mortality, the present rate of such decline is insufficient to meet the 2035 goals mainly due to several challenges such as the rise of multidrug-resistant TB (MDR-TB) and extensively drug-resistant TB (XDR-TB) [Citation2]. Treatment of M/XDR-TB is extremely difficult since it requires more expensive drugs with a higher toxicity profile, during a longer period (up to 24 months), resulting in patient noncompliance and poorer outcomes. Therefore, there is a need to identify new drugs and new combined therapies that are better tolerated with the possibility to treat both drug-susceptible and drug-resistant TB alike, faster and efficiently.
Over the years, it has become widely accepted that the overall resistance of mycobacteria to any antimicrobial agent is due to a synergy between intrinsic and genetic resistance [Citation3,Citation4]. Since no horizontal transfer of resistance genes has been reported in Mycobacterium tuberculosis, genetic drug resistance is mainly acquired by spontaneous mutations in chromosomal genes that cause modification or overproduction of the drug target, provide the ability to inactivate drugs, or decrease the activation of the drug into its active form [Citation5,Citation6]. In M. tuberculosis, intrinsic resistance is a result of the interplay between the impermeable cell wall, which limits drug uptake, and the activity of membrane proteins, also known as efflux pumps, that transport a variety of substrates from the interior to the exterior of the cell [Citation3,Citation4,Citation7,Citation8]. Several studies have proposed that efflux activity emerges before the acquisition of mutations in drug target-coding genes. Overexpression of efflux pumps in M. tuberculosis previously exposed to subinhibitory concentrations of isoniazid and ethambutol resulted in a low-level resistance phenotype, which allowed the bacteria to survive in the presence of the antibiotic until the acquisition of mutations in drug target genes that confer high-level resistance [Citation9–12]. This has also been hypothesized for the emergence of resistance to bedaquiline, a recent anti-TB drug recommended for use in the treatment of M/XDR-TB. It was demonstrated that mutations in rv0678, a regulator of the MmpS5-MmpL5 efflux pump, might be the first step in low-level resistance, followed by high-level resistance due to atpE mutations [Citation13].
This may reflect what occurs during a long-term therapy, as in the case of TB, where an inappropriate therapy may result from incorrect prescription, poor compliance by the patient and inadequate drug supply and quality. An unsuitable treatment can cause a sustained pressure of subinhibitory concentrations of an anti-TB drug that may cause increased efflux activity and survival of a low/moderate-level resistant population from which results the selection and expansion of mutation based drug-resistant M. tuberculosis variants – the evolution of resistance arrow-of-time in mycobacteria [Citation3,Citation4].
Therefore, it is crucial to understand the mechanism(s) that cause efflux-mediated resistance in order to find appropriate ways to prevent it. The use of an efflux inhibitor as adjuvant to therapy would help lower the frequency of mutant selection and, thus, prevent the development of resistance. In addition, given the urgency for new therapeutic regimens for the control of M/XDR-TB, new drug discovery strategies are needed in order to find new drugs or repurpose existing drugs, including efflux inhibitors, in a cost-effective way. After a literature search in PubMed, covering the studies published on efflux in M. tuberculosis up to February 2020, this review will give a general overview of what is currently known about the contribution of efflux pumps in drug resistance in M. tuberculosis and how efflux inhibitors may help to ensure the effectiveness of anti-TB drugs and prevent the emergence of M/XDR-TB. Finally, we will present our experience and recent findings in using a new drug discovery strategy to identify new anti-TB drugs that may be used as efflux inhibitors.
2. Efflux pumps and drug resistance in M. tuberculosis
Efflux pump systems are membrane proteins that are widespread in many organisms, including Gram-positive and -negative bacteria and eukaryotes. These transporters are categorized in different families according to their structural characteristics and energy requirements: ATP-binding cassette (ABC) superfamily; the major facilitator superfamily (MFS); the multidrug and toxic compound extrusion (MATE) family; the small multidrug resistance (SMR) family; the proteobacterial antimicrobial compound efflux (PACE) superfamily; and the resistance nodulation division (RND) superfamily. The ABC superfamily are classified as primary transporters since they use the hydrolysis of ATP as energy source, whereas the other families of efflux pumps are defined as secondary transporters, because they use the proton (or sodium in the case of MATE family) gradient [Citation14–16]. The efflux pumps that have been associated with drug resistance in M. tuberculosis are summarized in .
Table 1. List of efflux pumps associated with drug resistance in M. tuberculosis.
The natural role of efflux pumps is to intervene in several physiologic processes. For example, Rv1410c (P55) is involved in the maintenance of normal growth characteristics and in the oxidative stress response and Rv1258c is important for normal growth kinetics and cell morphology and it appears to play a relevant role in the stationary phase of growth [Citation17–19]. Other transporters like the mycobacterial membrane proteins large (MmpL) are involved in the transport of lipids and promote cell wall biosynthesis [Citation20,Citation21]. In particular, MmpL3 is involved in the export of mycolates, one of the key components of the cell wall and MmpL7 exports phthiocerol dimycocerosate, a lipid component of the outer membrane [Citation22–26]. In addition to their natural role in the cell, most efflux pumps have the ability to transport a diversity of unrelated compounds. For example, Mmr plays a role in the resistance to tetraphenylphosphonium, ethidium bromide, erythromycin, acriflavine, safranin O, pyronin Y and cetyltrimethylammonium bromide and Rv1217c-Rv1218c has been associated with the efflux of several compounds and resistance to biarylpiperazines, bisanilinopyrimidines, novobiocins, pyrazolones, pyridines and pyrroles [Citation27–29].
However, in the last years, efflux pumps have become more relevant due to their association with resistance to antimicrobials, namely first-line anti-TB drugs. For example, Rv1410c, Rv2936 and Rv0783 may be responsible for low-level resistance to rifampicin and Rv1258c has been reported to confer rifampicin tolerance during macrophage infection [Citation18,Citation19,Citation30–32]. Recently, it was demonstrated that point mutations in Rv1258c in clinical isolates can confer clinically relevant drug resistance to pyrazinamide, streptomycin and isoniazid [Citation33]. Exposure of M. tuberculosis to isoniazid caused overexpression of mmpL7 and mmr, the latter also in the presence of ethambutol [Citation22,Citation34–36]. The doxorubicin-resistance operon, drrABC, has also been implicated in resistance to ethambutol [Citation37]. In 2017, Zhang and collaborators identified four putative efflux proteins in M. tuberculosis: Rv0191, Rv3756c, Rv3008, and Rv1667c. They demonstrated that overexpression of the genes coding for these proteins caused resistance to pyrazinamide that was reduced by efflux inhibitors [Citation38].
Efflux pumps have been associated with resistance to all classes of antibiotics, including drugs used in the treatment of drug-resistant TB. The ABC transporter coded by the Rv2686c-Rv2687c-Rv2688c operon is responsible for fluoroquinolone efflux, while DrrABC is involved in resistance to several antibiotics, including tetracycline, erythromycin, norfloxacin, streptomycin and chloramphenicol [Citation37,Citation39]. Recently, it was demonstrated that MDR-TB isolates showed increased overexpression of drrA and drrB [Citation40]. Rv0194 has been associated with resistance to β-lactams, streptomycin, tetracycline, chloramphenicol and vancomycin, while Rv1258c mediates low-level resistance to tetracycline and aminoglycosides [Citation19,Citation30,Citation41,Citation42]. Other transporters implicated in drug resistance are Rv2477 (ofloxacin and streptomycin) and Rv1473 (macrolides) [Citation41,Citation43]. The Rv2994 is a MFS efflux pump that is associated with resistance to streptomycin and fluoroquinolones [Citation34]. The Rv2333c has been shown to confer low-level resistance to tetracycline and spectinomycin, whereas Rv1410c has been associated with resistance to clofazimine [Citation18,Citation44]. Recently, MmpL5 was demonstrated to be involved in the resistance to bedaquiline, clofazimine and other antibiotics [Citation45–49].
Overall, the described studies have highlighted the contribution of efflux pumps to the development of drug resistance and reinforced the urgency to develop strategies that prevent efflux-mediated resistance.
3. Efflux inhibitors as a strategy to prevent efflux-mediated resistance in M. tuberculosis
In the last years, there has been an increased interest in the search and development of compounds that act as efflux inhibitors. The use of these compounds in combination with currently used anti-TB drugs has the potential to increase the intracellular concentration of these drugs, thus improving or restoring the activity of standard antimycobacterials in resistant strains and reduce the duration of the treatment. Moreover, efflux inhibitors could help overcome M. tuberculosis intrinsic resistance to antibiotics and prevent the emergence of mutations conferring drug resistance.
Although some efflux inhibitors have been identified in the last years (), most of them have only been studied in vitro and ex vivo, few moved to in vivo animal studies, with even fewer reports of their use for the clinical management of M/XDR-TB patients under compassionate therapy regimens [Citation50,Citation51]. The reason for the rare use of these compounds is mainly due to concerns with toxicity, since these agents can target both prokaryote and eukaryote transporters. In fact, the clinical development of several efflux inhibitors had to be put on hold due to toxicity problems [Citation52]. In order to be used for therapeutic purposes, an efflux inhibitor should present limited toxicity to the host’s cells at the dosage required to achieve an effect against the infectious agent. A way to overcome this problem would be to use selective inhibitors that specifically target efflux pumps that are not present in human cells. A drug discovery strategy that could help overcome this limitation is drug repurposing [Citation53]. In many cases, the pharmacokinetics and toxicity data are already available for a repurposed drug, allowing it to follow directly to pre-clinical testing and clinical trials, thus reducing time, costs and risks. In fact, some compounds known to be efflux inhibitors are repurposed drugs that were already approved for the treatment of other diseases. These compounds and others that demonstrated inhibitory activity against M. tuberculosis efflux pumps are described in the following sections.
Table 2. List of most frequently used efflux inhibitors in M. tuberculosis.
3.1. Phenothiazines
The phenothiazines are antipsychotic drugs that have long been known to have in vitro and in vivo antimycobacterial activity. Chlorpromazine, the first antipsychotic drug developed, was found to have anti-TB activity in vitro. However, the interest in chlorpromazine as a possible anti-TB agent decreased due to the side-effects observed in patients [Citation54]. Other phenothiazines were found to have anti-TB activity, but thioridazine (TZ) was the most promising, presenting a dual effect on M. tuberculosis: i) inhibition of the activity of efflux pumps; and ii) enhancement of macrophage killing activity through the inhibition of potassium and calcium channels, causing pH decrease in the phagolysosome, activation of the hydrolases and consequent killing of the mycobacteria [Citation4,Citation54–57]. This fact is particularly important since pulmonary TB is considered mainly an intracellular infection of the alveolar macrophage. Despite these promising results, there are only two reports of clinical management of patients with TZ to date: in Argentina, XDR-TB patients were successfully treated with the combination of TZ and other antibiotics and, in India, where this drug has been used for compassionate purposes in the treatment of XDR-TB patients failing other treatment regimens [Citation50,Citation51]. The reason is mainly due to concerns with toxicity. Although very small concentrations of TZ are required to kill the bacilli inside the macrophages, there is still the concern of adverse side-effects in the central nervous system and cardiotoxicity, which may occur over a prolonged administration period such as the one required for TB treatment. Efforts have been made to reduce toxicity, such as the synthesis of TZ analogs or the encapsulation of the drug in nanoparticles, but further pre-clinical studies and clinical trials are needed [Citation58,Citation59]. It has been suggested that TZ indirectly inhibits efflux pumps or ion channels by blocking their energy supply, ATP or proton motive force (PMF), required for substrate transport. This is a result of the inhibition of the type-II NADH menaquinone oxidoreductase (NDH-2), a key protein of the oxidative phosphorylation pathway [Citation60]. The importance of this pathway as a novel target pathway for drug discovery was recently supported by the development of bedaquiline, which targets the mycobacterial ATP synthase, another oxidative phosphorylation component [Citation61]. Unfortunately, bedaquiline, which revealed to be highly potent against M/XDR-TB strains in vivo and an excellent drug against all forms of rifampicin resistant TB, also suffers from the same adverse side-effects in the central nervous system and cardiotoxicity reported for phenothiazines [Citation62]. Therefore, given the demonstrated potential of TZ and the scarcity of new drug targets for TB, it is of extreme importance to further explore the drug’s mode of action in order to identify potential targets for the development of new and less toxic analogues.
3.2. Phenylalkylamines
Phenylalkylamines are calcium channel blockers and are often used to treat various disorders, such as angina, hypertension and cardiac arrhythmia. The classical example of this group is verapamil (VP), which is also a substrate and inhibitor of the mammalian drug efflux pump P-glycoprotein and of uptake transporters such as the organic cation transporter protein [Citation63–65]. Over the years, several studies have demonstrated that VP can strongly inhibit efflux of ethidium bromide in mycobacteria [Citation66,Citation67]. In addition, VP was found to partially restore the in vitro activity of rifampicin, isoniazid, ethambutol and fluoroquinolones in drug-resistant M. tuberculosis isolates [Citation9,Citation11,Citation35,Citation36,Citation68–71]. Moreover, this compound also prevented the acquisition of drug tolerance inside macrophages and accelerated the sterilization of infected lungs in mice treated with a first-line drug regimen [Citation32,Citation72]. In addition, co-administration of VP with subinhibitory doses of bedaquiline had the same bactericidal effect in mice as the bedaquiline dose used in humans and also protected against the development of resistant mutants in vivo [Citation72]. Other studies demonstrated that VP decreased the MIC of bedaquiline and clofazimine in M. tuberculosis both in drug-susceptible and drug-resistant clinical isolates [Citation73,Citation74]. Moreover, VP also reduced macrophage-induced drug tolerance to bedaquiline and moxifloxacin [Citation75]. However, another study showed that VP reduced the MIC of bedaquiline and clofazimine in vitro but failed to increase the bactericidal effect of bedaquiline in vivo [Citation47]. Nevertheless, other studies described that VP increased the sterilizing activity of a regimen composed of bedaquiline, clofazimine, and pyrazinamide in vivo, but had no significant effect on bedaquiline pharmacokinetics [Citation76,Citation77]. Preclinical studies also indicate that VP potentiates the activity of bedaquiline against M. tuberculosis, reducing the bedaquiline dose required for cure, which could be a promising strategy for improving MDR-TB treatment [Citation78–81]. In addition, VP may be cardioprotective, reducing the risk of QT prolongation and the dose related toxicity [Citation78]. However, the effect of VP in vivo is probably due to a higher bioavailability of the co-administered drugs through an effect on the mammalian drug transporters, rather than by the inhibition of mycobacterial efflux pumps [Citation76,Citation82–84]. Recently, it has been shown that VP does not affect intracellular drug uptake in M. tuberculosis through direct inhibition of efflux pumps, but it indirectly alters efflux pump function by interfering with membrane energetics [Citation85]. Nevertheless, VP concentrations required to achieve bactericidal activity against M. tuberculosis are very high and likely to be toxic to human cells. Therefore, further studies are needed to design more effective and selective VP analogs.
3.3. Protonophores
These compounds, also known as proton translocators or uncouplers, dissipate the PMF and electrochemical gradient, preventing further synthesis of ATP and, consequently, the normal function of cell processes that depend on this energy gradient such as efflux pumps [Citation4,Citation56,Citation86]. Carbonyl cyanide m-chlorophenyl hydrazone (CCCP), 2,4-dinitrofenol (DNP) and valinomycin are examples of such compounds that have been widely used to study the role of efflux pumps in mycobacteria. For example, CCCP decreased the MIC of tetracycline in a M. smegmatis strain expressing Rv1258c and decreased the activity of this efflux pump in a M. bovis BCG overexpressing strain [Citation19,Citation30]. Moreover, CCCP was one of the efflux inhibitors that abolished the decreased susceptibility to several compounds, such as ethidium bromide, acriflavine and safranin O in M. tuberculosis strains overexpressing jefA and mmr (Rv3065) (Rv2459) [Citation28,Citation87]. CCCP and valinomycin reduced novobiocin and rifampicin resistance mediated by P55 [Citation18]. It was also demonstrated in several studies that CCCP and DNP reduce efflux pump activity associated with fluoroquinolone resistance [Citation39,Citation68,Citation87]. However, due to their mechanism of action, these compounds are highly cytotoxic and, thus, further studies are needed to determine the concentrations that are simultaneously the safest and most effective.
3.4. Plant-derived compounds
In the last years, several efflux inhibitors of plant origin have been described. Reserpine, piperine, farnesol, luteolin and biochanin A are among the most used in M. tuberculosis. The alkaloid reserpine was used for many years as an antihypertensive, but its toxicity limited its clinical use [Citation56,Citation86]. Nevertheless, this compound has been widely used in the in vitro evaluation of the activity of antimicrobial agents, namely in mycobacteria. For example, reserpine reduced ciprofloxacin resistance mediated by Rv2686 and decreased the MIC of isoniazid in M. smegmatis expressing mmpL7 [Citation22,Citation39]. In addition, reserpine increased susceptibility to isoniazid in M. bovis BCG and in M. tuberculosis susceptible and resistant strains [Citation88,Citation89]. This compound has also been shown to inhibit the pyrazinoic acid efflux pump, thus, increasing susceptibility to pyrazinamide in M. tuberculosis [Citation90].
Piperine, an organic alkaloid compound found in Piper nigrum and Piper longum, was reported to inhibit the Rv1258c efflux pump in M. tuberculosis [Citation91]. Recently, it was demonstrated that piperine had a synergistic effect with rifampicin and streptomycin, but low effect in enhancing the killing in several strains of M. tuberculosis with different resistance profiles [Citation92]. Therefore, further studies are needed to clarify if piperine may be used as an adjunctive drug in TB treatment. Other natural products such as farnesol, biochanin A and luteolin have been shown to inhibit efflux of ethidium bromide in M. smegmatis [Citation93,Citation94]. Recently, modified biochanin A derivatives inhibited efficiently M. avium efflux pumps in a synergistic activity with macrolides and fluoroquinolones [Citation95].
3.5. Other efflux inhibitors
Other compounds with efflux inhibitor activity are Phe-Arg-β-naphtylamide (PAβN) and timcodar. PaβN is known to be a broad-spectrum efflux inhibitor of several Gram-negative bacteria and was reported to be active against M. tuberculosis [Citation96–98]. However, there have been evidences that PaβN may not be the most adequate tool to study efflux mechanisms since it is also an efflux pump substrate and presents a dual mode of action: efflux inhibition and membrane-permeabilizing activity [Citation97–99].
Timcodar is a mammalian efflux pump inhibitor that showed synergism when used in combination with rifampicin, bedaquiline and clofazimine [Citation100]. When assessing the activity of this compound against intracellular M. tuberculosis in host macrophages, it was demonstrated that timcodar caused approximately a 10-fold increase in the growth inhibition of M. tuberculosis and presented synergy with rifampicin, moxifloxacin and bedaquiline [Citation100]. Moreover, in a mouse model of infection, timcodar potentiated the efficacy of rifampicin and isoniazid. However, no additive effect of timcodar was observed when used in combination with moxifloxacin and linezolid [Citation100,Citation101]. Therefore, further studies are required to evaluate the effect of timcodar in the potentiation of efficacy of anti-TB drugs.
In order to potentiate the activity at lower concentrations and decrease the associated toxicity of efflux inhibitors targeting energy in M. tuberculosis, lipophilic compounds that present concomitant anti-TB and efflux inhibitory activity were designed. This is the case of the very promising 2-aminothiazole UPAR-174, a glycolytic pathway enolase inhibitor that also acts as efflux inhibitor, active against replicating, nonreplicating and intracellular M. tuberculosis, dissipating membrane potential and causing ATP depletion [Citation102–104].
4. An in silico repurposing strategy to identify potential efflux inhibitors in M. tuberculosis
Overall, the majority of the known mycobacterial efflux inhibitors are derived from existing drugs developed for other therapeutic usages that revealed high efflux inhibitory activity in vitro. These compounds are good examples of a rational approach that accelerates drug discovery and makes it cost effective – drug repositioning or repurposing [Citation53]. It has been observed that the most effective ones are those that have a direct effect on the mycobacterial energy metabolism [Citation86,Citation103,Citation105]. Energy metabolism and limiting ATP production, as well as the PMF balance, are the most efficient ways to inhibit mycobacterial efflux and are critical for antibiotic susceptibility of M. tuberculosis [Citation105].
As previously mentioned, the oxidative phosphorylation pathway has emerged as a novel target pathway for TB drug discovery, due to the development of bedaquiline, which targets the mycobacterial ATP synthase [Citation61]. The inhibition of the different components of this pathway prevents respiratory electron transport, abolishes the PMF and stops production of ATP, thus inactivating several physiological processes including the activity of efflux pumps and ion channels active transport [Citation106]. An example of this is TZ, the antipsychotic drug that indirectly inhibits efflux pumps or ion channels as a result of the inhibition of NDH-2 that intervenes in the oxidative phosphorylation pathway [Citation57,Citation60]. Considering the above premises, compounds that block the oxidative phosphorylation pathway may have a similar activity or even surpass the potential of TZ. Therefore, it is of great importance to explore oxidative phosphorylation as a target for the development of new drugs against TB. In the following subsections, we describe and review the basis of a new strategy for the identification of drugs with potential inhibitory activity against efflux pumps in M. tuberculosis – a target-based approach directed to proteins involved in energy metabolism and membrane transport in M. tuberculosis, based upon in silico drug repurposing.
4.1. Generation of an in silico library of approved drugs with potential activity against M. tuberculosis
The rational workflow of the strategy is summarized in and is inspired in the methodology previously described by Bispo et al. and Neves et al. and can be easily adapted to other metabolic pathways and other microorganisms [Citation107,Citation108]. A list of M. tuberculosis proteins involved in energy metabolism and membrane transport was compiled using the TDR Targets Database and Mycobrowser [Citation109,Citation110]. In the case of TDR Targets Database the strategy was as follows: i) Enter TDR targets database, http://www.tdrtargets.org/, select targets [Citation111,Citation112]; ii) Select M. tuberculosis; iii) Targets: Select ‘energy metabolism’, ‘oxidative phosphorylation’ and ‘membrane transport’ under the parameter ‘KEGG high-level pathway’. The search results obtained with TDR targets were crosschecked and complemented with a search in Mycobrowser: i) Enter Mycobrowser website https://mycobrowser.epfl.ch/ [112]; select Advanced Search; Enter protein function and free text search: ‘energy’, ‘oxidative phosphorylation’, ‘membrane’, ‘transporter’. This search revealed 668 genes that encode proteins involved in energy metabolism and membrane transport (Table S1). The corresponding protein sequences were used to interrogate two publicly available web-based databases that provide detailed information on drugs and their targets: DrugBank and STITCH 5.0 [Citation113,Citation114]. The search strategy used by DrugBank relies on the principle of homology, where each query (query = protein of M. tuberculosis) results in a comparison by similarity with all drug targets known to the databases. Only proteins with a statistical similarity value of E-value ≤ 10−20 were considered potential targets [Citation108]. STITCH defines a group of interactions of variable statistical confidence between drugs and targets. In this case, only targets with a score ≥ 0.8 were considered for further studies [Citation108]. From the targets selected from both databases only the ones predicted to interact with approved drugs were selected. The sequence similarity screening in these databases predicted 69 targets associated with 246 approved drugs. Examples of the protein targets and their respective functions are presented in (the complete data can be found in Table S2). DrugBank and STITCH 5.0 exclusively predicted 216 (87.8%) and 12 (4.9%) of the approved drugs, respectively, whilst 18 (7.3%) drugs were predicted by both databases. The functional regions of the approved drug targets and M. tuberculosis targets were compared using The ConSurf Server, a bioinformatics tool for estimating the evolutionary conservation of amino acid positions in a protein based on the phylogenetic relationships between homologous sequences [Citation115]. This procedure was used to estimate the conservation of active sites between the proteins and the preservation of affinity for the predicted M. tuberculosis drugs. The parameters for the analysis were selected as previously described [Citation108]. The results obtained were classified as functional residues with high (≥ 80%) or moderate conservation (60-79%). When the conservation between functional residues is less than 59%, the putative targets were excluded from further analyzes. This strategy resulted in a list of 19 potential druggable M. tuberculosis targets (2.8% of the interrogated targets) that could interact with 24 approved drugs. presents these targets and their potential drugs (detailed data provided by Table S2). These drugs are approved for a variety of indications, such as treatment of epilepsy, chronic immune thrombocytopenia, arrhythmia and cancer (). The PubMed database was used to verify if the identified drugs had already been tested against M. tuberculosis [Citation116]. Drugs were classified as ‘tested’ when any in vitro and/or in vivo tests were already performed in M. tuberculosis and ‘not tested’ when no publication could be found. According to this search, the following drugs were never tested against M. tuberculosis: acetyldigitoxin, bretylium, deslanoside, dexlansoprazole, enasidenib, esomeprazole, ethacrynic acid, fomepizole, halothane, isoflurane, ivosidenib, levoleucovorin, rabeprazole and tipiracil.
Figure 1. Flowchart summarizing the in silico drug repurposing strategy and corresponding results
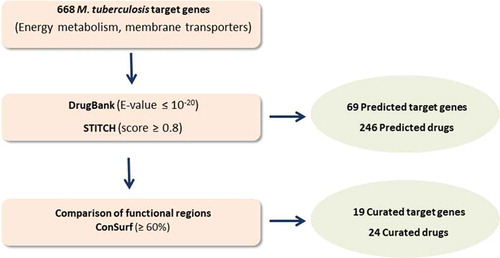
Figure 2. Distribution of the predicted drugs according to their original indication
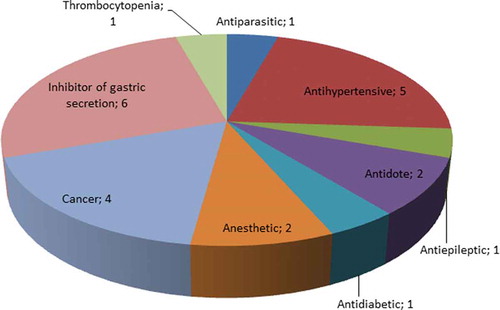
Table 3. Potential anti-TB drugs and their predicted targets identified in this study
4.2. Drugs already tested against M. tuberculosis
From the 24 drugs identified using this strategy, 10 were already tested for activity against M. tuberculosis. As proof-of-concept, this strategy successfully identified bedaquiline, recommended by WHO as part of combination therapy in adults with pulmonary MDR-TB [Citation117]. Since bedaquiline targets the mycobacterial ATP synthase, an enzyme that intervenes in the oxidative phosphorylation and essential for the generation of energy in M. tuberculosis, this provides an adequate validation of the strategy.
Doxorubicin was also one of the drugs identified by this method. This drug is an antineoplastic in the anthracycline class, used to produce regression in solid tumors and disseminated neoplastic conditions and as a component of adjuvant chemotherapy in breast cancer. However, doxorubicin can cause toxicity in multiple organs, including cardiotoxicity, which has to do with its mechanism of action [Citation118,Citation119]. Doxorubicin binds to nucleic acids, presumably by specific intercalation of the planar anthracycline nucleus with the DNA double helix. It may also inhibit polymerase activity, affecting regulation of gene expression, and producing free radical damage to DNA [Citation120]. In mycobacteria, previous studies have found evidences that doxorubicin is a potent inhibitor of the DNA primase (DnaG), with an MIC of 5 μM in M. tuberculosis [Citation121]. Our analysis revealed the NADH dehydrogenase I chain B (Rv3146) and chain D (Rv3148) as potential targets of doxorubicin in M. tuberculosis. However, it has been previously demonstrated that doxorubicin is a substrate of efflux pumps, particularly the DrrAB transporter, which undermines its potential as anti-TB drug [Citation37]. Therefore, further studies are necessary in order to clarify the mode of action of doxorubicin in M. tuberculosis and to develop safer derivatives that are not subject to efflux.
Fostamatinib is indicated for the treatment of rheumatoid arthritis, chronic thrombocytopenia and autoimmune diseases and acts as an inhibitor of the tyrosine kinase. Fostamatinib is a pro-drug, requiring hydrolysis by the intestine’s alkaline phosphatase that generates R406, which is the active metabolite [Citation122]. Neither fostamatinib nor R406 were found to be carcinogenic or mutagenic. Serious adverse effects include hypertension, neutropenia, diarrhea, and hepatotoxicity in the recommended human dose [Citation123–125]. In 2018, Kanehiro et al. described R406 as an inhibitor of the mycobacterial serine/threonine-protein kinase PknG. The same study demonstrated that R406 showed bactericidal activity against M. bovis BCG in infected human macrophages without cytotoxicity [Citation126]. Fostamatinib was revealed by our in silico method to have several potential targets in M. tuberculosis: transmembrane serine/threonine-protein kinase B (Rv0014c); transmembrane serine/threonine-protein kinase A PknA (Rv0015c); probable transmembrane serine/threonine-protein kinase E PknE (Rv1743) and anchored-membrane serine/threonine-protein kinase PknF (Rv1746). Interestingly, Rv0014c and Rv0015c are essential genes.
Thiabendazole is a fungicide and parasiticide mostly used for the treatment of strongyloidiasis, cutaneous larva migrans, visceral larva migrans, and trichinosis. Thiabendazole is a known inhibitor of tubulin polymerization. It selectively binds to nematode ß-tubulin, inhibiting polymerization, thus preventing the formation of microtubules and preventing cell division [Citation127,Citation128]. Thiabendazole has also been shown to inhibit the helminth-specific enzyme, fumarate reductase [Citation129]. Previous studies examined the effect of thiabendazole in M. tuberculosis and demonstrated that this drug prevented FtsZ polymerization, causing septum formation inhibition and, thus, abolishing cell division [Citation130,Citation131]. Our strategy identified a probable succinate dehydrogenase (Rv0248c) and a probable fumarate reductase (Rv1552) as potential targets of thiabendazole in M. tuberculosis. These enzymes are involved in interconversion of fumarate and succinate in aerobic respiration. Experimental studies are now needed in order to determine if these proteins are indeed targets of thiabendazole.
Valproic acid, or valproate, is a fatty acid derivative and anticonvulsant used in the treatment of epilepsy and bipolar disorders [Citation132]. This drug has been also investigated for neuroprotective, anti-manic, and anti-migraine effects. Currently, it is a compound of interest in oncology drug research for its anti-proliferative effects and is the subject of many clinical trials. The mechanism of action of valproate is relatively unknown, but it is suggested that the drug’s action is a result of several pathways: i) inhibition of succinic semialdehyde dehydrogenase and increasing GABAergic neurotransmission; impact on fatty acid metabolism, which alters membrane fluidity; and noncompetitive inhibition of brain microsomal long-chain fatty acyl-CoA synthetase, which decreases available arichidonyl-CoA, a substrate in the production of inflammatory prostaglandins [Citation133–135]. Valproate has been previously tested in M. tuberculosis. In 2015, Schieber et al. demonstrated that this drug stimulates the autophagosome formation in vitro and, in 2018, Rao and collaborators showed that it improves the action of isoniazid and rifampicin in the macrophage [Citation136,Citation137]. However, the specific target in M. tuberculosis has not been identified to date. Our study predicted a possible acyl-CoA dehydrogenase FadE19 (Rv2500c) as a potential M. tuberculosis target.
Ouabain is a cardiac glycoside similar to digitoxin that is used to treat congestive heart failure, supraventricular arrhythmias and to control ventricular rate in the treatment of chronic atrial fibrillation [Citation138,Citation139]. This compound inhibits the Na+/K+-ATPase membrane transporter, causing an increase in intracellular sodium and calcium concentrations, which may promote activation of contractile proteins such as actin and myosin [Citation140]. Ouabain has been demonstrated to potentiate the intracellular killing of M. tuberculosis by macrophages [Citation141,Citation142]. Recently, Kaur and collaborators performed a virtual screening to search for novel drugs against secretory lipid-catabolizing enzymes in M. tuberculosis. Ouabain was identified as a suitable drug for the enzyme LipU [Citation143]. We have predicted a probable metal cation transporter P-type ATPase A (Rv1997) as a possible target for ouabain. These evidences suggest that this compound affects several cell processes and, thus, more studies are needed to clarify its mode of action.
Metformin is an antihyperglycemic agent used for the management of type II diabetes and is prescribed to at least 120 million people worldwide [Citation144,Citation145]. It is suggested that metformin inhibits the mitochondrial complex I, preventing the production of mitochondrial ATP leading to increased cytoplasmic ADP:ATP and AMP:ATP ratios. These changes activate the AMP-activated protein kinase, an enzyme that plays an important role in the regulation of glucose metabolism [Citation146,Citation147]. Our analysis identified Rv0235c, annotated as a probable conserved transmembrane protein involved in cell wall and cell processes, as a potential target for metformin in M. tuberculosis. However, this drug has been the subject of several studies evaluating the use of metformin in TB treatment. It was demonstrated that the use of metformin in diabetes mellitus patients reduced the risk of subsequent active TB and that it has potentially beneficial effects in innate host responses to M. tuberculosis [Citation148,Citation149]. A study using the mouse model of TB infection found that mice treated with metformin and isoniazid showed a considerable reduction in the bacillary load in the lungs and reduced areas of lung tissue damage compared with isoniazid monotherapy [Citation150,Citation151]. A clinical trial is being conducted to assess the efficacy and safety of metformin-containing anti-TB treatment in comparison to the standard 6-month regimen in patients with newly diagnosed sputum smear-positive drug-sensitive pulmonary TB [Citation152].
Our in silico strategy also identified several compounds used to treat gastric acid-related disorders. They are known to be inhibitors of the parietal gastric cell H+/K+-ATPase pump, hence their use in gastroesophageal reflux disease, peptic ulcer disease, and other diseases characterized by the oversecretion of gastric acid. Our analysis predicted a probable metal cation transporter P-type ATPase A CtpF (Rv1997) as a potential target in M. tuberculosis. In the case of omeprazole and pantoprazole, a previous study has revealed that these drugs have no effect in M. tuberculosis [Citation153]. On the other hand, the same study showed that lansoprazole has intracellular activity against M. tuberculosis. Target identification studies revealed that lansoprazole kills M. tuberculosis by targeting its cytochrome bc1 complex leading to disruption of the mycobacterial respiratory chain and ATP depletion [Citation153].
The described body of work already performed, provide useful data about the identified drugs that may be used as a starting point for future studies by the scientific and pharmaceutical community. In this case, information concerning toxicity and pharmacokinetics properties will have to be considered before selecting the most promising compounds.
4.3. Drugs not yet tested against M. tuberculosis
We predicted 14 drugs that, to our knowledge, have not yet been experimentally tested against M. tuberculosis. Levoleucovorin is indicated for use as rescue therapy following high-dose methotrexate in the treatment of osteosarcoma or for diminishing the toxicity associated with inadvertent overdosage of folic acid antagonists. Levoleucovorin has an additional indication for use in combination chemotherapy with 5-fluorouracil in the palliative treatment of patients with advanced metastatic colorectal cancer [Citation154,Citation155]. The predicted M. tuberculosis target is the serine hydroxymethyltransferase GlyA2 (Rv0070c).
Our study identified two drugs that are used for the treatment of adult patients with relapsed or refractory acute myeloid leukemia, ivosidenib and enasidenib. These agents are inhibitors of isocitrate dehydrogenases 1 and 2, mitochondria-localized enzymes involved in diverse cellular processes [Citation156–158]. The potential M. tuberculosis target identified in this study for both drugs is a probable isocitrate dehydrogenase (Rv3339c).
Tipiracil is a thymidine phosphorylase inhibitor that is used in combination with trifluridine for the treatment of refractory metastatic colorectal cancer [Citation159]. The predicted target in M. tuberculosis is the thymidine phosphorylase DeoA (Rv3314c).
Dexlansoprazole, esomeprazole and rabeprazole are inhibitors of the human H+/K+-ATPase, that to our knowledge have not been tested in M. tuberculosis. The predicted target identified by our method is a probable metal cation transporter P-type ATPase A CtpF (Rv1997). Further studies are needed to evaluate the effect of these compounds in M. tuberculosis. However, the adverse effect of these drugs needs to be further clarified due to recent evidence that prescription of acid-suppressive agents seems to be associated with TB infection/activation [Citation160,Citation161].
This strategy also identified several antiarrhythmic, antihypertensive drugs that potentially target a probable metal cation transporter P-type ATPase A CtpF (Rv1997) in M. tuberculosis. Deslanoside is a cardiac glycoside used to treat congestive heart failure, supraventricular arrhythmias and to control ventricular rate in the treatment of chronic atrial fibrillation. This compound inhibits the Na+/K+-ATPase pump, resulting in an increase in intracellular sodium and calcium concentrations that may promote activation of contractile proteins, such as actin and myosin [Citation162]. Acetyldigitoxin is a cardioactive derivative of lanatoside A or of digitoxin used for fast digitalization in congestive heart failure. The proposed mode of action is similar to deslanoside: inhibition of the Na+/K+ pump that leads to increased amounts of calcium [Citation163]. Ethacrynic acid is used in the treatment of high blood pressure and edema caused by diseases like congestive heart failure, liver failure, and kidney failure. It inhibits the symport of sodium, potassium, and chloride primarily in the ascending limb of Henle. This pharmacological action results in excretion of these ions, increased urinary output, and reduction in extracellular fluid [Citation164]. Finally, the last drug in this group is bretylium, used in emergency medicine, cardiology, and other specialties for the acute management of ventricular tachycardia and ventricular fibrillation. The main mode of action for bretylium is thought to be inhibition of voltage-gated potassium channels and the Na+/K+-ATPase [Citation165].
Fomepizole is used as an antidote in confirmed or suspected methanol or ethylene glycol poisoning. It is a competitive inhibitor of alcohol dehydrogenase, the enzyme that catalyzes the initial steps in the metabolism of ethylene glycol and methanol to their toxic metabolites [Citation166]. The predicted target in M. tuberculosis is the zinc-type alcohol dehydrogenase AdhD (Rv3086).
Halothane and isoflurane are general inhalation anesthetics used for induction and maintenance of general anesthesia [Citation167]. Halothane causes general anesthesia due to its actions on multiple ion channels, which ultimately lowers nerve conduction, breathing and cardiac contractility. Its immobilizing effects have been attributed to its binding to potassium channels in cholinergic neurons [Citation168]. Isoflurane’s anesthetic effect is thought to be the result of: i) the reduction in junctional conductance by decreasing gap junction channel opening times and increasing gap junction channel closing times; ii) the activation of calcium dependent ATPase in the sarcoplasmic reticulum by increasing the fluidity of the lipid membrane; iii) the binding to the D subunit of ATP synthase and NADH dehydrogenase; iv) the binding to the GABA receptor, the large conductance calcium-activated potassium channel, the glutamate receptor and the glycine receptor [Citation169]. The potential M. tuberculosis target identified in this study is a probable NADH dehydrogenase I (chain H) (Rv3152). We did not find any published study of these agents as antimycobacterials and little is known about the antibacterial mechanisms of anesthetics, with previous studies reporting contradictory results [Citation170–172]. The technical difficulties associated with the study of these agents may be responsible for the variety of results obtained.
Experimental evidence is needed to clarify if any of these identified drugs presents antimycobacterial activity and if their mechanism of action involves the suggested targets. In addition, it is also imperative to determine if their absorption, distribution, metabolism, excretion and toxicity (ADMET) properties could affect their activity in vivo. If their activity is revealed to be promising, these compounds could be important leads in TB drug discovery.
4.4. Experimental strategies for validation of activity of identified drugs
To confirm the efficacy of these drugs, in vitro and in vivo studies will have to be performed. This can be applied even to the drugs that have already been tested against M. tuberculosis, since these compounds may be interesting lead molecules and become the starting point for the design of new improved molecules. The identified drugs will need to be sequentially evaluated using different methodologies in order to be classified as potential antimycobacterials or as potential efflux inhibitors [Citation35,Citation36,Citation55,Citation173]. However, this classification may be confusing as the same compound can be a good efflux inhibitor and restore the activity of established antibiotics, while presenting antimicrobial activity by itself at concentrations near its MIC. Determination of MICs in vitro in extracellular (bacteria in liquid culture) and intracellular conditions (infected macrophages) will give information concerning the activity of these compounds against M. tuberculosis and their ability to enhance the killing of intracellular mycobacteria. In addition, the evaluation of synergistic interactions between these drugs and currently used anti-TB drugs (e.g. checkerboard method) will allow the identification of potential adjuvants. In this case, further studies would have to be performed in order to clarify if a given compound acts as adjuvant by inhibition of efflux or by other mechanism(s). Efflux activity can be studied using a fluorescent marker dye, such as ethidium bromide, as an efflux pump substrate in several bacteria [Citation35,Citation36,Citation66,Citation67]. In addition to the assays described above, the predicted target on M. tuberculosis must be confirmed, for example, through the selection of resistant mutants in the presence of the compound or by the construction of knockdown and overexpression mutants and the following comparison of the MIC of these strains with the wild-type strain. However, even if a given drug proves itself to be promising after these studies, the issue of toxicity must be addressed. Therefore, evaluation of drug cytotoxicity in specific cell-lines (e.g., human macrophages and others) must be carried out in parallel with the determination of antimycobacterial activity. Only the compounds that can be used at safe concentrations should be selected. Other studies could also involve the determination of in vitro and ex vivo drug activity against drug-susceptible and M/XDR-TB M. tuberculosis strains. Furthermore, efficient penetration through the lipidic mycobacterial cell wall of new anti-TB drugs must be guaranteed and lipophilicity is a very important feature to consider in designing new or repurposing old drugs as effective agents against M. tuberculosis [Citation103]. Finally, the compounds that prove to be the most interesting, with the desired antimycobacterial and synergistic properties, as well as demonstrable inhibitory activity at low nontoxic concentrations, will be selected for further research, namely in the mouse model and pharmacokinetic and pharmacodynamic studies in preparation for potential clinical trials [Citation36,Citation58,Citation173].
5. Conclusion
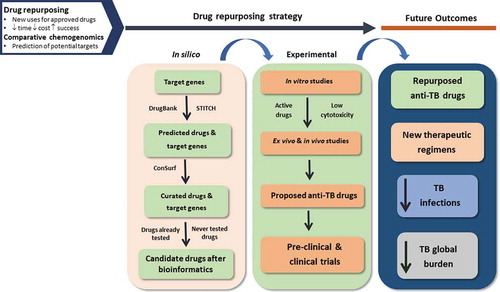
It is now widely accepted and demonstrated that mycobacterial efflux pumps contribute to drug resistance by allowing the bacteria to survive in the presence of subinhibitory concentrations of antibiotics until the emergence of mutations, as reviewed in this work. Several evidences support the use of efflux inhibitors as a strategy to overcome efflux of anti-TB drugs in mycobacteria, shortening the treatment regimens and preventing the emergence of resistance, as aimed by the WHO End TB Strategy [Citation2,Citation174]. In addition, several of these efflux inhibitors also act as enhancers of the macrophage activity, which makes them especially useful in the case of TB, since the bacteria reside inside the pulmonary macrophage [Citation4]. However, despite their promising results in vitro, none of them have been used in clinical practice for TB management, with the exception of TZ used in compassionate therapy for particular XDR-TB cases [Citation50,Citation51]. Their application in vivo is restricted mostly due to toxicity concerns, since most of these compounds are able to target both prokaryote and eukaryote transporters. In a time when new drugs are needed to fight M/XDR-TB, cost-effective strategies to identify safer efflux inhibitors should be implemented in drug discovery programs. This review has highlighted a new strategy for the development of anti-TB drugs, including efflux inhibitors, using in silico drug repurposing (). This approach has been successfully used for the identification of new hits for malaria, schistosomiasis, human african trypanosomiasis and leishmaniasis [Citation107,Citation108,Citation175]. Concerning the identified potential anti-TB drugs, if promising activities are revealed, they could constitute important starting points for lead identification and optimization and, ultimately, for the development of new antimycobacterials, including efflux inhibitors.
Figure 3. Graphical summary of the proposed strategy for the development of anti-TB drugs, using in silico drug repurposing. TB, tuberculosis
In conclusion, it is scientifically evident that efflux inhibitors should be exploited for synergistic use with current therapeutic regimens to improve the efficacy of TB treatment. If efflux inhibitors can be used as adjuvants of currently used anti-TB drugs, they could not only restore the activity of such drugs, but also modulate the killing activity of the infected macrophage. However, there is still the need for clinical trials that indisputably demonstrate the importance of efflux inhibitors as adjuvant compounds for the current recommended therapies for TB and M/XDR-TB. Several efforts have been made to address specific issues such as toxicity, lipophilicity and selectivity of these compounds. The synthesis of analogues of known efflux inhibitors (e.g. VP and TZ analogues), the study of certain natural derived products (e.g. berberine, piperine and other berberflavonoids) and promising drug repurposing strategies are important directions for the future development of the ideal efflux inhibitor [Citation92,Citation103,Citation104,Citation176].
6. Expert opinion
TB is one of the top ten causes of death and the leading cause from a single infectious agent (above HIV/AIDS). M. tuberculosis has the ability to acquire resistance to all anti-TB drugs known, leaving very limited therapeutic options. In order to prevent the selection of resistant variants of M. tuberculosis, TB treatment consists of a combination of three or more drugs, given simultaneously for a long period of time to promote their synergistic activity and increase the probability of a successful outcome. M. tuberculosis is equipped with other drug resistance mechanisms that act in synergy with drug resistance mutations, namely efflux pump systems that allow the bacteria to survive in the presence of subinhibitory concentrations of antibiotics until the selection of resistant mutants. This fact is particularly relevant in the case of TB, due to its long-term therapeutic regimens that frequently cause poor patient adherence facilitating acquisition of resistance. The inhibition of efflux pumps through the action of efflux inhibitors could be a way to restore the activity of antibiotics subject to efflux by decreasing the mutation frequency and, thus, prevent the evolution toward resistance. Therefore, efflux pumps are attractive drug targets and efflux inhibitors could be used in combination with current anti-TB drugs, in new therapeutic regimens, to help increase treatment efficacy. The major limitation to this approach has to do with toxicity, since most of these compounds have an effect on both prokaryotic and eukaryotic cells. In particular, their side effects could be an issue in combined therapy, emphasizing the need for more advanced studies concerning the dose of efflux inhibitor used in combination with existing first-line anti-TB drugs and the subsequent side effects. It is crucial to tailor the pharmacokinetics of the efflux inhibitor to the pharmacokinetics of the antibiotic in order to optimize synergy. This applies even to drugs that are already approved for clinical use for other diseases (e.g. TZ and VP and analogues), which have shown promising in vitro and in vivo activity against TB. Although the use of efflux inhibitors as adjuvant therapeutic agents still faces many challenges, the evidences show that they provide many advantages. Future studies should continue to focus on the design and development of new efflux inhibitors and new anti-TB drugs to be used in combination of multiple drugs in new therapeutic regimens. In this case, drug repurposing provides several advantages, namely reducing costs, risk and time associated with drug discovery. This approach combined with in silico computer-aided drug design strategies can greatly improve the drug discovery paradigm and help identify promising drug leads for TB. Also, new drugs and drug development pipelines should always take into account the effluxability of the drug as an important variable in its pharmacodynamics and possible mechanism of resistance, as recently happened with bedaquiline, and design strategies to minimize these undesirable events that reduce tremendously its efficacy and lifetime [Citation13,Citation47].
Ending the TB epidemic by 2030 is one of the health targets of the UN Sustainable Development Goals (UN-SDGs). Therefore, it is expected that many efforts will continue to be made in the next five years in order to achieve this goal. This includes the design of novel antimycobacterial agents and the development of accelerated drug discovery strategies. Efflux inhibitors provide the attractive possibility of restoring the activity of already available antibiotics, saving time, effort and financial investment that is usually required for drug development. There is still a long way to go before efflux inhibitors can be routinely used in clinical practice. Future studies will continue to address the issues of selectivity and safe usage of these compounds. Hopefully, advancements in in silico and computer-aided drug design strategies will help to achieve the rapid discovery of novel, effective and safe efflux inhibitors drugs and ultimately decrease the global burden of TB and contribute to achieve the 2030 UN-SDGs.
Article highlights
Tuberculosis (TB) control faces many challenges, including the increase of drug-resistant forms of Mycobacterium tuberculosis that require new effective therapeutic regimens.
Efflux pumps play a significant role in the development of drug resistance in M. tuberculosis during treatment; efflux inhibitors could prevent this outcome.
Scientific evidence supports the use of efflux inhibitors as therapeutic adjuvants to increase the effectiveness of anti-TB drugs.
Drugs that target energy metabolism components may inhibit several bacterial physiological processes, namely the activity of efflux pumps.
Drug repurposing can accelerate drug discovery and make it cost effective, by finding new uses for existing approved drugs. This can be achieved by using an in silico approach.
New cost-effective strategies to identify safer efflux inhibitors should be implemented in drug discovery programs.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Supplemental Material
Download Zip (245.7 KB)Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- World Health Organization. Global tuberculosis report 2019. Geneva: World Health Organization; 2019.
- World Health Organization. Global strategy and targets for tuberculosis prevention, care and control after 2015. Geneva: World Health Organization; 2015.
- Louw GE, Warren RM, Gey van Pittius NC, et al. Balancing act: efflux/influx in mycobacterial drug resistance. Antimicrob Agents Chemother. 2009;53(8):3181–3189.
- Viveiros M, Martins M, Rodrigues L, et al. Inhibitors of mycobacterial efflux pumps as potential boosters for anti-tubercular drugs. Expert Rev Anti Infect Ther. 2012;10(9):983–998.
- Almeida Da Silva PE, Palomino JC. Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis: classical and new drugs. J Antimicrob Chemother. 2011;66(7):1417–1430.
- Al-Saeedi M, Al-Hajoj S. Diversity and evolution of drug resistance mechanisms in Mycobacterium tuberculosis. Infect Drug Resist. 2017;10:333–342.
- De Rossi E, Aínsa JA, Riccardi G. Role of mycobacterial efflux transporters in drug resistance: an unresolved question FEMS. Microbiol Rev. 2006;30(1):36–52.
- Rodrigues L, Parish T, Balganesh M, et al. Antituberculosis drugs: reducing efflux=increasing activity. Drug Discov Today. 2017;22(3):592–599.
- Srivastava S, Musuka S, Sherman C, et al. Efflux-pump-derived multiple drug resistance to ethambutol monotherapy in Mycobacterium tuberculosis and the pharmacokinetics and pharmacodynamics of ethambutol. J Infect Dis. 2010;201(8):1225–1231.
- Pasipanodya JG, Gumbo T. A new evolutionary and pharmacokinetic–pharmacodynamic scenario for rapid emergence of resistance to single and multiple anti-tuberculosis drugs. Curr Opin Pharmacol. 2011;11(5):457–463. .
- Machado D, Couto I, Perdigão J, et al. Contribution of efflux to the emergence of isoniazid and multidrug resistance in Mycobacterium tuberculosis. PLoS One. 2012;7(4):e34538.
- Schmalstieg AM, Srivastava S, Belkaya S, et al. The antibiotic resistance arrow of time: efflux pump induction is a general first step in the evolution of mycobacterial drug resistance. Antimicrob Agents Chemother. 2012;56(9):4806–4815.
- Ismail N, Ismail NA, Omar SV, et al. In vitro study of stepwise acquisition of rv0678 and atpE mutations conferring bedaquiline resistance. Antimicrob Agents Chemother. 2019;63(8):pii: e00292-19.
- Piddock LJ. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin Microbiol Rev. 2006;19(2):382–402.
- Du D, Wang-Kan X, Neuberger A, et al. Multidrug efflux pumps: structure, function and regulation. Nat Rev Microbiol. 2018;16(9):523–539.
- Hassan KA, Liu Q, Henderson PJ, et al. Homologs of the Acinetobacter baumannii AceI transporter represent a new family of bacterial multidrug efflux systems. mBio. 2015 Feb 10;6(1):pii: e01982-14.
- Silva PE, Bigi F, Santangelo MP, et al. Characterization of P55, a multidrug efflux pump in Mycobacterium bovis and Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2001;45(3):800–804.
- Ramón-García S, Martín C, Thompson CJ, et al. Role of the Mycobacterium tuberculosis P55 efflux pump in intrinsic drug resistance, oxidative stress responses, and growth. Antimicrob Agents Chemother. 2009;53(9):3675–3682.
- Ramón-García S, Mick V, Dainese E, et al. Functional and genetic characterization of the tap efflux pump in Mycobacterium bovis. BCG Antimicrob Agents Chemother. 2012;56(4):2074–2083.
- Domenech P, Reed MB, Barry CE 3rd. Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance. Infect Immun. 2005;73(6):3492–3501.
- Ma S, Huang Y, Xie F, et al. Transport mechanism of Mycobacterium tuberculosis MmpL/S family proteins and implications in pharmaceutical targeting. Biol Chem. 2020;401(3):331–348.
- Pasca MR, Guglierame P, De Rossi E, et al. mmpL7 gene of Mycobacterium tuberculosis is responsible for isoniazid efflux in Mycobacterium smegmatis. Antimicrob Agents Chemother. 2005;49(11):4775–4777.
- Tullius MV, Harmston CA, Owens CP, et al. Discovery and characterization of a unique mycobacterial heme acquisition system. Proc Natl Acad Sci USA. 2011;108(12):5051–5056.
- Grzegorzewicz AE, Pham H, Gundi VA, et al. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat Chem Biol. 2012;8(4):334–341.
- Varela C, Rittmann D, Singh A, et al. MmpL genes are associated with mycolic acid metabolism in mycobacteria and corynebacteria. Chem Biol. 2012;19(4):498–506.
- Owens CP, Chim N, Graves AB, et al. The Mycobacterium tuberculosis secreted protein Rv0203 transfers heme to membrane proteins MmpL3 and MmpL11. J Biol Chem. 2013;288(30):21714–21728.
- Balganesh M, Kuruppath S, Marcel N, et al. Rv1218c, an ABC transporter of Mycobacterium tuberculosis with implications in drug discovery. Antimicrob Agents Chemother. 2010;54(12):5167–5172.
- Rodrigues L, Villellas C, Bailo R, et al. Role of the Mmr efflux pump in drug resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2013;57(2):751–757.
- De Rossi E, Branzoni M, Cantoni R, et al. mmr, a Mycobacterium tuberculosis gene conferring resistance to small cationic dyes and inhibitors. J Bacteriol. 1998;180(22):6068–6071.
- Ramón-García S, Martín C, Aínsa JA, et al. Characterization of tetracycline resistance mediated by the efflux pump Tap from Mycobacterium fortuitum. J Antimicrob Chemother. 2006;57(2):252–259.
- Pang Y, Lu J, Wang Y, et al. Study of the rifampin monoresistance mechanism in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2013;57(2):893–900.
- Adams KN, Takaki K, Connolly LE, et al. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell. 2011;145(1):39–53.
- Liu J, Shi W, Zhang S, et al. Mutations in efflux pump Rv1258c (Tap) cause resistance to pyrazinamide, isoniazid, and streptomycin in M tuberculosis. Front Microbiol. 2019;10:216.
- Gupta AK, Katoch VM, Chauhan DS, et al. Microarray analysis of efflux pump genes in multidrug-resistant Mycobacterium tuberculosis during stress induced by common anti-tuberculous drugs. Microb Drug Resist. 2010;16(1):21–28.
- Rodrigues L, Machado D, Couto I, et al. Contribution of efflux activity to isoniazid resistance in the Mycobacterium tuberculosis complex. Infect Genet Evol. 2012;12(4):695–700.
- Machado D, Pires D, Perdigão J, et al. Ion channel blockers as antimicrobial agents, efflux inhibitors, and enhancers of macrophage killing activity against drug resistant Mycobacterium tuberculosis. PLoS One. 2016;11(2):e0149326.
- Choudhuri BS, Bhakta S, Barik R, et al. Overexpression and functional characterization of an ABC (ATP-binding cassette) transporter encoded by the genes drrA and drrB of Mycobacterium tuberculosis. Biochem. 2002;J367(Pt 1):279–285.
- Zhang Y, Zhang J, Cui P, et al. Identification of novel efflux proteins Rv0191, Rv3756c, Rv3008, and Rv1667c involved in pyrazinamide resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2017;61(8):pii: e00940-17.
- Pasca MR, Guglierame P, Arcesi F, et al. Rv2686c-Rv2687c-Rv2688c, an ABC fluoroquinolone efflux pump in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2004;48(8):3175–3178.
- Khosravi AD, Sirous M, Absalan Z, et al. Comparison of drrA and drrB efflux pump genes expression in drug-susceptible and-resistant Mycobacterium tuberculosis strains isolated from tuberculosis patients in Iran. Infect Drug Resist. 2019;12:3437–3444.
- Danilchanka O, Mailaender C, Niederweis M. Identification of a novel multidrug efflux pump of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2008;52(7):2503–2511.
- Aínsa JA, Blokpoel MC, Otal I, et al. Molecular cloning and characterization of Tap, a putative multidrug efflux pump present in Mycobacterium fortuitum and Mycobacterium tuberculosis. J Bacteriol. 1998;180(22):5836–5843.
- Duan W, Li X, Ge Y, et al. Mycobacterium tuberculosis Rv1473 is a novel macrolides ABC Efflux Pump regulated by WhiB7. Future Microbiol. 2019;14:47–59.
- Ramón-García S, Martín C, De Rossi E, et al. Contribution of the Rv2333c efflux pump (the Stp protein) from Mycobacterium tuberculosis to intrinsic antibiotic resistance in Mycobacterium bovis. BCG J Antimicrob Chemother. 2007;59(3):544–547.
- Briffotaux J, Huang W, Wang X, et al. MmpS5/MmpL5 as an efflux pump in Mycobacterium species. Tuberculosis (Edinb). 2017;107:13–19.
- Li B, Ye M, Guo Q, et al. Determination of MIC distribution and mechanisms of decreased susceptibility to bedaquiline among clinical isolates of Mycobacterium abscessus. Antimicrob Agents Chemother. 2018;62(7):pii: e00175–18.
- Andries K, Villellas C, Coeck N, et al. Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PLoS One. 2014;9(7):e102135.
- Hartkoorn RC, Uplekar S, Cole ST. Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2014;58(5):2979–2981.
- Narang A, Garima K, Porwal S, et al. Potential impact of efflux pump genes in mediating rifampicin resistance in clinical isolates of Mycobacterium tuberculosis from India. PLoS One. 2019;14(9):e0223163.
- Abbate E, Vescovo M, Natiello M, et al. Successful alternative treatment of extensively drug-resistant tuberculosis in Argentina with a combination of linezolid, moxifloxacin and thioridazine. J Antimicrob Chemother. 2012;67(2):473–477.
- Udwadia ZF, Sen T, Pinto LM. Safety and efficacy of thioridazine as salvage therapy in Indian patients with XDR-TB. Recent Pat Antiinfect Drug Discov. 2011;6(2):88–91.
- Opperman TJ, Nguyen ST. Recent advances toward a molecular mechanism of efflux pump inhibition. Front Microbiol. 2015;6:421.
- Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3(8):673–683.
- Kristiansen JE, Dastidar SG, Palchoudhuri S, et al. Phenothiazines as a solution for multidrug resistant tuberculosis: from the origin to present. Int Microbiol. 2015;18(1):112.
- Rodrigues L, Aínsa JA, Amaral L, et al. Inhibition of drug efflux in mycobacteria with phenothiazines and other putative efflux inhibitors. Recent Pat Antiinfect Drug Discov. 2011;6(2):118–127.
- Viveiros M, Martins M, Couto I, et al. Molecular tools for rapid identification and novel effective therapy against MDRTB/XDRTB infections. Expert Rev Anti Infect Ther. 2010;8(4):465–480.
- Amaral L, Viveiros M. Thioridazine: A non-antibiotic drug highly effective, in combination with first line anti-tuberculosis drugs, against any form of antibiotic resistance of Mycobacterium tuberculosis due to its multi-mechanisms of action. Antibiotics (Basel). 2017;6(1):pii: E3.
- Pieroni M, Machado D, Azzali E, et al. Rational design and synthesis of thioridazine analogues as enhancers of the antituberculosis therapy. J Med Chem. 2015;58(15):5842–5853.
- Vibe CB, Fenaroli F, Pires D, et al. Thioridazine in PLGA nanoparticles reduces toxicity and improves rifampicin therapy against mycobacterial infection in zebrafish. Nanotoxicology. 2016;10(6):680–688.
- Weinstein EA, Yano T, Li LS, et al. Inhibitors of type II NADH: menaquinoneoxidoreductase represent a class of antitubercular drugs. Proc Natl Acad Sci USA. 2005;102(12):4548–4553.
- Andries K, Verhasselt P, Guillemont J, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307(5707):223–227.
- Cohen K, Maartens G. A safety evaluation of bedaquiline for the treatment of multi-drug resistant tuberculosis. Expert Opin Drug Saf. 2019;18(10):875–882.
- Speelmans G, Staffhorst RW, De Wolf FA, et al. Verapamil competes with doxorubicin for binding to anionic phospholipids resulting in increased internal concentrations and rates of passive transport of doxorubicin. Biochim Biophys Acta. 1995;1238(2):137–146.
- Pereira E, Teodori E, Dei S, et al. Reversal of multidrug resistance by verapamil analogues. Biochem Pharmacol. 1995;50(4):451–457.
- Boxberger KH, Hagenbuch B, Lampe JN. Common drugs inhibit human organic cation transporter 1 (OCT1)-mediated neurotransmitter uptake. Drug Metab Dispos. 2014;42(6):990–995.
- Rodrigues L, Wagner D, Viveiros M, et al. Thioridazine and chlorpromazine inhibition of ethidium bromide efflux in Mycobacterium avium and Mycobacterium smegmatis. J Antimicrob Chemother. 2008;61(5):1076–1082.
- Rodrigues L, Sampaio D, Couto I, et al. The role of efflux pumps in macrolide resistance in Mycobacterium avium complex. Int J Antimicrob Agents. 2009;34(6):529–533.
- Singh M, Jadaun GP, Ramdas, et al. Effect of efflux pump inhibitors on drug susceptibility of ofloxacin resistant Mycobacterium tuberculosis isolates. Indian J Med Res. 2011;133:535–540.
- Louw GE, Warren RM, Gey van Pittius NC, et al. Rifampicin reduces susceptibility to ofloxacin in rifampicin-resistant Mycobacterium tuberculosis through efflux. Am J Respir Crit Care Med. 2011;184(2):269–276.
- Li G, Zhang J, Guo Q, et al. Study of efflux pump gene expression in rifampicin-monoresistant Mycobacterium tuberculosis clinical isolates. J Antibiot (Tokyo). 2015;68(7):431–435.
- Sun Z, Xu Y, Sun Y, et al. Ofloxacin resistance in Mycobacterium tuberculosis is associated with efflux pump activity independent of resistance pattern and genotype. Microb Drug Resist. 2014;20(6):525–532.
- Gupta S, Tyagi S, Almeida DV, et al. Acceleration of tuberculosis treatment by adjunctive therapy with verapamil as an efflux inhibitor. Am J Respir Crit Care Med. 2013;188(5):600–607.
- Gupta S, Cohen KA, Winglee K, et al. Efflux inhibition with verapamil potentiates bedaquiline in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2014;58(1):574–576.
- Almeida D, Ioerger T, Tyagi S, et al. Mutations in pepQ confer low-level resistance to bedaquiline and clofazimine in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2016;60(8):4590–4599.
- Adams KN, Szumowski JD, Ramakrishnan RL. Verapamil, and its metabolite norverapamil, inhibit macrophage-induced, bacterial efflux pump-mediated tolerance to multiple anti-tubercular drugs. J Infect Dis. 2014;210(3):456–466.
- Xu J, Tasneen R, Peloquin CA, et al. Verapamil increases the bioavailability and efficacy of bedaquiline but not clofazimine in a murine model of tuberculosis. Antimicrob Agents Chemother. 2017;62(1):pii: e01692–17. .
- Kotwal P, Magotra A, Dogra A, et al. Assessment of preclinical drug interactions of bedaquiline by a highly sensitive LC-ESI-MS/MS based bioanalytical method. J Chromatogr B Analyt Technol Biomed Life Sci. 2019;1112:48–55.
- Srikshna G, Gupta S, Dooley KE, et al. Can the addition of verapamil to bedaquiline-containing regimens improve tuberculosis treatment outcomes? A novel approach to optimizing TB treatment. Future Microbiol. 2015;10(8):1257–1260.
- Ghajavan H, Kargarpour Kamakoli M, Khanipour S, et al. High prevalence of bedaquiline resistance in treatment-naive tuberculosis patients and verapamil effectiveness. Antimicrob Agents Chemother. 2019;63(3):pii: e02530–18.
- Scani JLR, Camargo AD, Seus VR, et al. Molecular modelling and competitive inhibition of a Mycobacterium tuberculosis multidrug-resistance efflux pump. J Mol Graph Model. 2019;87:98–108.
- Gupta S, Tyagi S, Bishai WR. Verapamil increases the bactericidal activity of bedaquiline against Mycobacterium tuberculosis in a mouse model. Antimicrob Agents Chemother. 2015;59(1):673–676.
- Seral C, Carryn S, Tulkens PM, et al. Influence of P-glycoprotein and MRP efflux pump inhibitors on the intracellular activity of azithromycin and ciprofloxacin in macrophages infected by Listeria monocytogenes or Staphylococcus aureus. J Antimicrob Chemother. 2003;51(5):1167–1173.
- Shugarts S, Benet LZ. The role of transporters in the pharmacokinetics of orally administered drugs. Pharm Res. 2009;26(9):2039–2054.
- Ughachukwu P, Unekwe P. Efflux pump-mediated resistance in chemotherapy. Ann Med Health Sci Res. 2012;2(2):191–198.
- Chen C, Gardete S, Jansen RS, et al. Verapamil targets membrane energetics in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2018;62(5):pii: e02107–17.
- Pule CM, Sampson SL, Warren RM, et al. Efflux pump inhibitors: targeting mycobacterial efflux systems to enhance TB therapy. J Antimicrob Chemother. 2016;71(1):17–26.
- Gupta AK, Reddy VP, Lavania M, et al. jefA (Rv2459), a drug efflux gene in Mycobacterium tuberculosis confers resistance to isoniazid & ethambutol. Indian J Med Res. 2010;132:176–188.
- Colangeli R, Helb D, Sridharan S, et al. The Mycobacterium tuberculosis iniA gene is essential for activity of an efflux pump that confers drug tolerance to both isoniazid and ethambutol. Mol Microbiol. 2005;55(6):1829–1840.
- Viveiros M, Portugal I, Bettencourt R, et al. Isoniazid-induced transient high-level resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2002;46(9):2804–2810.
- Zhang Y, Scorpio A, Nikaido H, et al. Role of acid pH and deficient efflux of pyrazinoic acid in unique susceptibility of Mycobacterium tuberculosis to pyrazinamide. J Bacteriol. 1999;181(7):2044–2049.
- Sharma S, Kumar M, Sharma S, et al. Piperine as an inhibitor of Rv1258c, a putative multidrug efflux pump of Mycobacterium tuberculosis. J Antimicrob Chemother. 2010;65:1694–1701.
- Hegeto LA, Caleffi-Ferracioli KR, Nakamura-Vasconcelos SS, et al. In vitro combinatory activity of piperine and anti-tuberculosis drugs in Mycobacterium tuberculosis. Tuberculosis (Edinb). 2018;111:35–40.
- Jin J, Zhang J-Y, Guo N, et al. Farnesol, a potential efflux pump inhibitor in Mycobacterium smegmatis. Mol Basel Switz. 2010;15:7750–7762.
- Lechner D, Gibbons S, Bucar F. Plant phenolic compounds as ethidium bromide efflux inhibitors in Mycobacterium smegmatis. J Antimicrob Chemother. 2008;62(2):345–348.
- Cannalire R, Machado D, Felicetti T, et al. Natural isoflavone biochanin A as a template for the design of new and potent 3-phenylquinolone efflux inhibitors against Mycobacterium avium. Eur J Med Chem. 2017;140:321–330.
- Balganesh M, Dinesh N, Sharma S, et al. Efflux pumps of Mycobacterium tuberculosis play a significant role in antituberculosis activity of potential drug candidates Antimicrob. Agents Chemother. 2012;56(5):2643–2651.
- Lomovskaya O, Bostian KA. Practical applications and feasibility of efflux pump inhibitors in the clinic – a vision for applied use. Biochem Pharmacol. 2006;71(7):910–918.
- Pagès JM, Amaral L. Mechanisms of drug efflux and strategies to combat them: challenging the efflux pump of Gram-negative bacteria. Biochim Biophys Acta. 2009;1794(5):826–833.
- Schuster S, Bohnert JA, Vavra M, et al. Proof of an outer membrane target of the efflux inhibitor phe-arg-ß-naphthylamide from random mutagenesis. Molecules. 2019;24(3):pii: E470.
- Grossman TH, Shoen CM, Jones SM, et al. The efflux pump inhibitor timcodar improves the potency of antimycobacterial agents. Antimicrob Agents Chemother. 2015;59(3):1534–1541.
- de Knegt GJ, van der Meijden A, de Vogel CP, et al. Activity of moxifloxacin and linezolid against Mycobacterium tuberculosis in combination with potentiator drugs verapamil, timcodar, colistin and SQ109. Int J Antimicrob Agents. 2017;49(3):302–307.
- Wescott HH, Zuniga ES, Bajpai A, et al. Identification of enolase as the target of 2-aminothiazoles in Mycobacterium tuberculosis. Front Microbiol. 2018;9:2542.
- Machado D, Girardini M, Viveiros M, et al. Challenging the drug-likeness dogma for new drug discovery in tuberculosis. Front Microbiol. 2018;9:1367.
- Machado D, Azzali E, Couto I, et al. Adjuvant therapies against tuberculosis: discovery of a 2-aminothiazole targeting Mycobacterium tuberculosis energetics. Future Microbiol. 2018;13:1383–1402.
- Black PA, Warren RM, Louw GE, et al. Energy metabolism and drug efflux in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2014;58(5):2491–2503.
- Bald D, Villellas C, Lu P, et al. Targeting energy metabolism in Mycobacterium tuberculosis, a new paradigm in antimycobacterial drug discovery. mBio. 2017;8(2):pii: e00272–17.
- Bispo NA, Culleton R, Silva LA, et al. A systematic in silico search for target similarity identifies several approved drugs with potential activity against the Plasmodium falciparum apicoplast. PLoS One. 2013;8(3):e59288.
- Neves BJ, Braga RC, Bezerra JC, et al. In silico repositioning-chemogenomics strategy identifies new drugs with potential activity against multiple life stages of Schistosoma mansoni. PLoS Negl Trop Dis. 2015;9(1):e3435.
- Urán Landaburu L, Berenstein AJ, Videla S, et al. TDR Targets 6: driving drug discovery for human pathogens through intensive chemogenomic data integration. Nucleic Acids Res. 2020;48(D1):D992–D1005.
- Kapopoulou A, Lew JM, Cole ST. The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis (Edinb). 2011;91(1):8–13.
- TDR Targets [Internet]. 2020 [cited 2020 Jan 17]. Available from: https://www.tdrtargets.org/
- Mycobrowser [Internet]. 2020 [cited 2020 Jan 17]. Available from: https://mycobrowser.epfl.ch/
- Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–82.
- Szklarczyk D, Santos A, von Mering C, et al. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016;44(D1):D380–4.
- Ashkenazy H, Abadi S, Martz E, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucl Acids Res. 2016;44(W1):W344–50.
- Pubmed [Internet]. 2020 [cited 2020 Jan 17]. Available from: https://www.ncbi.nlm.nih.gov/pubmed/
- World Health Organization. The use of bedaquiline in the treatment of multidrug-resistant tuberculosis - Interim policy guidance. Geneva: World Health Organization; 2013.
- Pugazhendhi A, Edison TNJI, Velmurugan BK, et al. Toxicity of doxorubicin (Dox) to different experimental organ systems. Life Sci. 2018;200:26–30.
- Mordente A, Meucci E, Silvestrini A, et al. New developments in anthracycline-induced cardiotoxicity. Curr Med Chem. 2009;16(13):1656–1672.
- Weiss RB. The anthracyclines: will we ever find a better doxorubicin. Semin Oncol. 1992;19(6):670–686.
- Gajadeera C, Willby MJ, Green KD, et al. Antimycobacterial activity of DNA intercalator inhibitors of Mycobacterium tuberculosis primase DnaG. J Antibiot (Tokyo). 2015;68(3):153–157.
- Braselmann S, Taylor V, Zhao H, et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J Pharmacol Exp Ther. 2006;319(3):998–1008.
- Sweeny DJ, Li W, Clough J, et al. Metabolism of fostamatinib, the oral methylene phosphate prodrug of the spleen tyrosine kinase inhibitor R406 in humans: contribution of hepatic and gut bacterial processes to the overall biotransformation. Drug Metab Dispos. 2010;38(7):1166–1176.
- Baluom M, Grossbard EB, Mant T, et al. Pharmacokinetics of fostamatinib, a spleen tyrosine kinase (SYK) inhibitor, in healthy human subjects following single and multiple oral dosing in three phase I studies. Br J Clin Pharmacol. 2013;76(1):78–88.
- Rolf MG, Curwen JO, Veldman-Jones M, et al. In vitro pharmacological profiling of R406 identifies molecular targets underlying the clinical effects of fostamatinib. Pharmacol Res Perspect. 2015;3(5):e00175.
- Kanehiro Y, Tomioka H, Pieters J, et al. Identification of novel mycobacterial inhibitors against mycobacterial protein kinase G. Front Microbiol. 2018;J9:1517.
- Desai A, Mitchison TJ. Tubulin and FtsZ structures: functional and therapeutic implications. Bioessays. 1998;20(7):523–527.
- Downing KH. Structural basis for the interaction of tubulin with proteins and drugs that affect microtubule dynamics. Annu Rev Cell Dev Biol. 2000;16:89–111.
- Köhler P, Bachmann R. The effects of the antiparasitic drugs levamisole, thiabendazole, praziquantel, and chloroquine on mitochondrial electron transport in muscle tissue from Ascaris suum. Mol Pharmacol. 1978;14(1):155–163.
- Slayden RA, Knudson DL, Belisle JT. Identification of cell cycle regulators in Mycobacterium tuberculosis by inhibition of septum formation and global transcriptional analysis. Microbiology. 1789–97;152(Pt6):2006.
- Knudson SE, Awasthi D, Kumar K, et al. A trisubstituted benzimidazole cell division inhibitor with efficacy against Mycobacterium tuberculosis. PLoS One. 2014;9(4):e93953.
- Lopez-Munoz F, Baumeister AA, Hawkins MF, et al. The role of serendipity in the discovery of the clinical effects of psychotropic drugs: beyond of the myth. Actas Esp Psiquiatr. 2012;40(1):34–42.
- El-Habr EA, Dubois LG, Burel-Vandenbos F, et al. A driver role for GABA metabolism in controlling stem and proliferative cell state through GHB production in glioma. Acta Neuropathol. 2017;133(4):645–660.
- Rosenberg G. The mechanisms of action of valproate in neuropsychiatric disorders: can we see the forest for the trees. Cell Mol Life Sci. 2007;64(16):2090–2103.
- Bazinet RP, Weis MT, Rapoport SI, et al. Valproic acid selectively inhibits conversion of arachidonic acid to arachidonoyl-CoA by brain microsomal long-chain fatty acyl-CoA synthetases: relevance to bipolar disorder. Psychopharmacology (Berl). 2006;184(1):122–129.
- Rao M, Valentini D, Zumla A, et al. Evaluation of the efficacy of valproic acid and suberoylanilide hydroxamic acid (vorinostat) in enhancing the effects of first-line tuberculosis drugs against intracellular Mycobacterium tuberculosis. Int J Infect Dis. 2018;69:78–84.
- Schiebler M, Brown K, Hegyi K, et al. Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol Med. 2015;7(2):127–139.
- Blaustein MP, Juhaszova M, Golovina VA. The cellular mechanism of action of cardiotonic steroids: A new hypothesis. Clin Exp Hypertens. 1998;20(5–6):691–703.
- Wasserstrom JA, Aistrup GL. Digitalis: new actions for an old drug. Am J Physiol Heart Circ Physiol. 2005;289(5):H1781–93.
- Nguyen AN, Wallace DP, Blanco G. Ouabain binds with high affinity to the Na,K-ATPase in human polycystic kidney cells and induces extracellular signal-regulated kinase activation and cell proliferation. J Am Soc Nephrol. 2007;18(1):46–57.
- Martins M, Viveiros M, Amaral L. Inhibitors of Ca2+ and K+ transport enhance intracellular killing of M. tuberculosis by non-killing macrophages. In Vivo. 2008;22(1):69–75.
- Amaral L, Martins M, Viveiros M. Enhanced killing of intracellular multidrug-resistant Mycobacterium tuberculosis by compounds that affect the activity of efflux pumps. J Antimicrob Chemother. 2007;59(6):1237–1246.
- Kaur G, Pandey B, Kumar A, et al. Drug targeted virtual screening and molecular dynamics of LipU protein of Mycobacterium tuberculosis and Mycobacterium leprae. J Biomol Struct Dyn. 2019;37(5):1254–1269.
- Viollet B, Guigas B, Sanz Garcia N, et al. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond). 2012;122(6):253–270.
- Lund SS, Tarnow L, Stehouwer CD, et al. Targeting hyperglycaemia with either metformin or repaglinide in non-obese patients with type 2 diabetes: results from a randomized crossover trial. Diabetes Obes Metab. 2007;9(3):394–407.
- Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60(9):1577–1585.
- Madiraju AK, Qiu Y, Perry RJ, et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat Med. 2018;24(9):1384–1394.
- Lee MC, Chiang CY, Lee CH, et al. Metformin use is associated with a low risk of tuberculosis among newly diagnosed diabetes mellitus patients with normal renal function: A nationwide cohort study with validated diagnostic criteria. PLoS One. 2018;13(10):e0205807.
- Lachmandas E, Eckold C, Böhme J, et al. Metformin alters human host responses to Mycobacterium tuberculosis in healthy subjects. J Infect Dis. 2019;220(1):139–150.
- Rodriguez-Carlos A, Valdez-Miramontes C, Marin-Luevano P, et al. Metformin promotes Mycobacterium tuberculosis killing and increases the production of human ß-defensins in lung epithelial cells and macrophages. Microbes Infect 2020;22(3):111–18.
- Singhal A, Jie L, Kumar P, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med. 2014;6(263):263ra159.
- Padmapriyadarsini C, Bhavani PK, Natrajan M, et al. Evaluation of metformin in combination with rifampicin containing antituberculosis therapy in patients with new, smear-positive pulmonary tuberculosis (METRIF): study protocol for a randomised clinical trial. BMJ Open. 2019;9(3):e024363.
- Rybniker J, Vocat A, Sala C, et al. Lansoprazole is an antituberculous prodrug targeting cytochrome bc1. Nat Commun. 2015;6:7659.
- Kovoor PA, Karim SM, Marshall JL. Is levoleucovorin an alternative to racemic leucovorin? A literature review. Clin Colorectal Cancer. 2009;8(4):200–206.
- Chuang VT, Suno M. Levoleucovorin as replacement for leucovorin in cancer treatment. Ann Pharmacother. 2012;46(10):1349–1357.
- Mondesir J, Willekens C, Touat M, et al. IDH1 and IDH2 mutations as novel therapeutic targets: current perspectives. J Blood Med. 2016;7:171–180.
- Medeiros BC, Fathi AT, DiNardo CD, et al. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia. 2017;31(2):272–281.
- Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant-IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–731.
- Puthiamadathil JM, Weinberg BA. Emerging combination therapies for metastatic colorectal cancer - impact of trifluridine/tipiracil. Cancer Manag Res. 2017;9:461–469.
- Hsu WH, Kuo CH, Wang SS, et al. Acid suppressive agents and risk of Mycobacterium tuberculosis: case-control study. BMC Gastroenterol. 2014;14:91.
- Cheng KC, Liao KF, Lin CL, et al. Correlation of proton pump inhibitors with pulmonary tuberculosis: a case-control study in Taiwan. Front Pharmacol. 2017;8:481.
- DrugBank: deslanoside [Internet].; 2020 Jan 3 [cited 2020 Jan 17]. Available from: https://www.drugbank.ca/drugs/DB01078
- DrugBank: acetyldigitoxin [Internet]. ; 2020 Jan 3 [cited 2020 Jan 17]. Available from: https://www.drugbank.ca/drugs/DB00511
- DrugBank: ethacrynic acid [Internet]. 2020 Jan 3[cited 2020 Jan 17]. Available from: https://www.drugbank.ca/drugs/DB00903
- DrugBank: bretylium [Internet]. 2020 Jan 3 [cited 2020 Jan 17]. Available from: https://www.drugbank.ca/drugs/DB01158
- DrugBank: fomepizole [Internet]. 2020 Jan 3 [cited 2020 Jan 17]. Available from: https://www.drugbank.ca/drugs/DB01213
- Bovill JG. Inhalation anaesthesia: from diethyl ether to xenon. Handb Exp Pharmacol. 2008;182:121–142.
- DrugBank: halothane [Internet]. 2020 Jan 3 [cited 2020 Jan 17]. Available from https://www.drugbank.ca/drugs/DB01159
- DrugBank: isoflurane [Internet]; 2020 Jan 3 [cited 2020 Jan 17]. Available from https://www.drugbank.ca/drugs/DB00753
- Slade JM. Bacterial growth in isoflurane vapour. Anaesthesia. 1993;48:1053–1054.
- Martínez-Serrano M, Gerónimo-Pardo M, Martínez-Monsalve A, et al. Antibacterial effect of sevoflurane and isoflurane. Rev Esp Quimioter. 2017;30(2):84–89.
- A-Rahman A, Pedler S, Bray R, et al. To the editor: effect of anesthetics gases on the growth of Pseudomonas aeruginosa, Haemophilus influenzae, and Staphylococcus aureus. Pediatr Pulmonol. 2002;34(3):226–227.
- Costa SS, Lopes E, Azzali E, et al. An experimental model for the rapid screening of compounds with potential use against mycobacteria. Assay Drug Dev Technol. 2016;14(9):524–534.
- Lienhardt C, Lönnroth K, Menzies D, et al. Translational research for tuberculosis elimination: priorities, challenges, and actions. PLoS Med. 2016;13(3):e1001965.
- Andrade CH, Neves BJ, Melo-Filho CC, et al. In silico chemogenomics drug repositioning strategies for neglected tropical diseases. Curr Med Chem. 2019;26(23):4355–4379.
- Solnier J, Martin L, Bhakta S, et al. Flavonoids as novel efflux pump inhibitors and antimicrobials against both environmental and pathogenic intracellular mycobacterial species. Molecules. 2020;25(3):E734.
- Li X, Li P, Ruan C, et al. Mycobacterium tuberculosis Rv0191 is an efflux pump of major facilitator superfamily transporter regulated by Rv1353c. Arch Biochem Biophys. 2019;667:59–66.
- Khanna A, Raj VS, Tarai B, et al. Emergence and molecular characterization of extensively drug-resistant Mycobacterium tuberculosis clinical isolates from the Delhi Region in India. Antimicrob Agents Chemother. 2010;54(11):4789–4793.
- De Rossi E, Arrigo P, Bellinzoni M, et al. The multidrug transporters belonging to major facilitator superfamily in Mycobacterium tuberculosis. Mol Med. 2002;8(11):714–724.
- Li XZ, Zhang L, Nikaido H. Efflux pump-mediated intrinsic drug resistance in Mycobacterium smegmatis. Antimicrob Agents Chemother. 2004;48(7):2415–2423.
- Doran JL, Pang Y, Mdluli KE, et al. Mycobacterium tuberculosis efpA encodes an efflux protein of the QacA transporter family. Clin Diagn Lab Immunol. 1997;4(1):23–32.
- Milano A, Pasca MR, Provvedi R, et al. Azole resistance in Mycobacterium tuberculosis is mediated by the MmpS5-MmpL5 efflux system. Tuberculosis (Edinb). 2009;89(1):84–90.
- Chung SY, Sung MK, Kim NH, et al. Inhibition of P-glycoprotein by natural products in human breast cancer cells. Arch Pharmacal Res. 2005;28(7):823–828.
- Rao PS, Satelli A, Moridani M, et al. Luteolin induces apoptosis in multidrug resistant cancer cells without affecting the drug transporter function: involvement of cell line-specific apoptotic mechanisms. Int J Cancer. 2012;130(11):2703–2714.
- Suzuki K, Tsuyuguchi K, Matsumoto H, et al. Effect of proton pump inhibitor alone or in combination with clarithromycin on mycobacterial growth in human alveolar macrophages. FEMS Microbiol Lett. 2000;182(1):69–72.