
ABSTRACT
Introduction: Advances in mass spectrometry (MS)-based proteomic strategies have resulted in robust protein biomarker discovery studies often performed on high resolution accurate mass (HRAM) platforms. For successful translation of promising protein biomarkers into useful clinical tests, trans-sector networks and collaboration among stakeholders involved in the biomarker pipeline are urgently needed.
Areas covered: In this perspective, literature- and empirical evidence is combined with author’s opinions to discuss the progress of ultrahigh resolution MS and provide insight in its potential for validation and development of clinical tests.
Expert commentary: Thus far two ‘low resolution’ MS strategies have been implemented in the clinic: quantification of proteins using triple quadrupole instruments and identification of unknown microorganisms using comparative analysis with spectral libraries on MALDI-TOF instruments. The rise of HRAM technology further boosts the potential of MS-based tests for detection and quantitation of disease-specific biomarkers which meet the analytical performance specifications needed for clinical assays.
1. Introduction
Soon after the advent of mass spectrometry (MS)-based proteomics, it became clear that many applications would benefit from higher resolution instruments since these provide improved mass precision and accuracy and consequently yield more confident identifications [Citation1]. In the early days of biomolecular MS, and in fact long before the term ‘high-resolution accurate mass (HRAM)’ was coined, mass spectrometers with ultrahigh resolving powers (i.e. >100,000) were used by a small number of dedicated groups to characterize for example proteins and carbohydrates [Citation2–Citation4]. Meanwhile hybrid-type quadrupole time-of-flight (qTOF) mass spectrometers have evolved to currently available resolving powers up to 50,000 [Citation5,Citation6]. Both these high- and ultrahigh resolution mass spectrometers are referred to as HRAM platforms that have matured into a widely applied high-quality and large-scale procedure for structural analysis of almost any type of biomolecule. To a large extent, the increased popularity of HRAM is the result of the introduction of the Orbitrap™ mass spectrometer followed by the emergence of a ‘family’ of related instruments [Citation7,Citation8]. Moreover, important developments in Fourier transform ion cyclotron resonance (FTICR) MS have contributed to the wide availability of mass analyzers with ultrahigh resolving powers [Citation3,Citation9,Citation10]. It is outside the scope of this perspective to reiterate performance metrics in great detail [Citation3,Citation7]. Here, we limit the discussion on opportunities of HRAM in clinical laboratories in areas of unmet clinical needs to ultrahigh resolution platforms (i.e. FTICR and Orbitrap™ MS), although qTOF systems also play an important role in HRAM applications.
The technological advances in ultrahigh resolution MS have lead over the last two decades to numerous ‘-omics’ studies, including proteomics and metabolomics. Multiple reports have been published on the identification of biomarkers using such omics-based approaches, however, sadly, none of these markers has been translated for clinical application [Citation11] and as such, have no clinical value as of yet. This can be partially ascribed to the satisfactory use of current immunoassays both from the laboratory and clinical perspective. In addition, both the fields of MS-based proteomics as well as clinical chemistry have experienced that there is a disconnect between the strategies and technologies used for – omics studies (focused mostly on identification and relative quantitation) and those used in the clinical diagnostics setting (focused on absolute quantitation and throughput) [Citation12]. In order to bridge this gap, these efforts of biomarker translation have converged into a novel field of clinical MS. Recently, a fit-for-purpose approach was proposed for the transfer from MS-based biomarker discovery (or exploratory) studies to the use of MS-based technologies in clinical tests [Citation13]. It describes strategies in a tier system, in which biomarker discovery can be developed as tier 3 type measurements and biomarker validation can be performed using tier 2 type assays. These are then improved gradually to tier 1 tests, which may have potential as medical tests. In tier 1 tests, absolute quantitation has to be in line with the ISO 15189:2012 requirements on metrological traceability, meaning that test results should be traceable to standards of higher order, when available. Furthermore, the analytical sensitivity of the test as well as its selectivity for individual molecular compounds has to be specified. In this perspective, we will discuss the advances of HRAM technology and its potential for biomarker validation and further development into a medical test.
2. Protein applications in clinical MS
In general, genomics is considered to be the method of choice for the detection of an individual’s state of health or disease. It is a powerful approach to detect genetic differences, however, does not provide information at the phenotypic level. Proteins are regarded as the ‘workhorses’ and as such provide an excellent measure of an individual’s (patho)physiological state. Since the emergence of MS-based proteomics, numerous improvements in sample preparation strategies in combination with a wide range of instrumental developments have resulted in robust and optimized MS-based proteomics pipelines [Citation14]. These results have attracted increasing interest toward the use of mass spectrometric techniques for large-scale clinical applications. Although MS strategies have been widely applied for measurement of small molecules (e.g. toxins, drugs, metabolites) since the 1980s [Citation15–Citation18], it is relatively recent that proteins were added as a clinical application [Citation19–Citation21]. Several reviews have highlighted the use of MS for the quantitation of proteins in the clinical setting, including strategies for preanalysis, protein digestion, and the use of internal standards (e.g. [Citation12,Citation22–Citation24]); however, these are typically addressing standard MS-based proteomics approaches using triple quadrupole LC-MS/MS instruments. In this perspective, we focus on the advantages of HRAM MS and its potential for translating promising protein biomarkers into diagnostic protein tests.
In medical laboratories, MS is not commonly used as a technology for protein measurements (yet). Thus far, there are two typical MS strategies that are used for protein tests in so-called specialty labs. The first one involves quantification of proteotypic peptides on a triple quadrupole mass spectrometer in combination with liquid chromatography (LC) and electrospray ionization (ESI). Here, proteins are digested according to a standard bottom-up proteomics workflow followed by quantification of proteotypic peptides applying an isotopically labeled peptide analog in a so-called multiple reaction monitoring (MRM) experiment [Citation25,Citation26]. In the context of clinical MS, the application of such a protein quantification approach was recently named quantitative Clinical Chemistry Proteomics (qCCP) [Citation22]. Our aim is to develop and evaluate specific qCCP tests in domains with unmet clinical needs since qCCP tests should have the potential to outperform immunoassays that can be disturbed by interfering antibodies [Citation22]. With the development and introduction of qCCP tests, the analytical specificity is enhanced and moreover, any clinically relevant isoform (proteoform) [Citation27] can be detected and quantified as will be further discussed and exemplified in the section ‘Protein applications in clinical MS…’. Essential elements for proper qCCP standardization, in line with the metrological traceability concept, are: defining the measurands; selecting suitable proteotypic peptides; manufacturing labeled internal standards; optimizing the trypsin digestion so that equimolarity between protein and peptide is guaranteed, and calibration using well-defined internal standards and/or external calibrators. According to the international metrology vocabulary (VIM), metrological traceability is defined as: ‘property of a measurement result whereby the result can be related to a reference through a documented unbroken chain of calibrations, each contributing to the measurement uncertainty’ [Citation28]. Highly accurate results are obtained from the direct analysis of proteotypic peptides derived from well-defined measurands at the proteoform level (further discussed in the section on metrological traceability). The improved accuracy of LC-MS/MS tests for thyroglobulin as compared to immunoassays was recently reported and provides evidence thereof [Citation29]. The second MS strategy that is routinely applied in the clinic involves the measurement of protein signatures in combination with a comparative analysis with reference spectra (libraries). To this end, a time-of-flight (TOF) mass analyzer is used in combination with matrix-assisted laser desorption/ionization (MALDI). This MALDI-TOF-MS approach is applied worldwide as a routine identification tool for typing and identifying microbial species in clinical microbiology laboratories and is known as Biotyper™ [Citation30]. For this purpose, alternative ambient ionization methods are also applied, such as rapid evaporative ionization MS (REIMS) [Citation31]. In both approaches, the accurate identification of unknown microorganisms is achieved by comparing protein profiles obtained from cultured colonies to spectra in a library without any a priori knowledge [Citation32–Citation34].
MS applications allow multiplexed protein quantification and protein isoform (proteoform) differentiation, for example for the purpose of phenotyping. Multiplex protein analysis has been recognized as a key feature for the next generation of protein technology, similarly to the multiplexed analyses already common in genomics [Citation35]. Such tests will allow multiplexing of clinically relevant, disease- or organ-specific proteins in one assay, thus facilitating personalized medicine. Therefore, it is believed that qCCP is an important technology that has potential for precision medicine [Citation36]. In recent years, several multiplexed protein quantification tests have been developed as exemplified below. Although these examples are not based on MS (yet), the benefits of applying HRAM-based proteomics strategies for multiplexed protein analysis in a clinical test will be discussed throughout this review. The first example is the FDA-approved OVA1® ovarian cancer triage test, which comprises the analysis of 5 serum proteins as biomarkers to assist physicians in determining whether an ovarian mass has a high likelihood of malignancy [Citation37]. The second example is the so-called Nephrocheck® that has recently been approved for the risk assessment of acute kidney injury (AKI) in intensive care patients that have undergone cardiac surgery or had respiratory compromise. In this test, two small tubular cell-derived proteins are measured: TIMP2 and IGFBP-7, both markers of cell cycle arrest [Citation38]. When combined, these two proteins currently provide the highest sensitivity for the detection of AKI at an early stage [Citation38,Citation39].
3. Dynamic range of protein concentrations in clinical samples
For this audience, it is clear that the biggest challenge of MS-based proteomics assays comprises the up-front separations that are needed to tackle the enormous dynamic range of protein concentrations in any clinical sample, be it a body fluid, a tissue section, or a cell lysate. For this reason, the multiplexed and high-throughput MS-based measurement and quantification of specific protein(s) or protein panel(s) is severely hampered by this dynamic concentration range, for example in serum or plasma the protein concentrations differ more than 12 orders of magnitude [Citation40]. Generally, this range is subdivided into a ‘high,’ ‘medium,’ and ‘low’ protein concentration range, respectively, representing micromole per liter, nanomole per liter, and picomole per liter endogenous protein concentrations. While accurate quantification of a set of proteins in body fluids is possible within each of these three concentration ranges, provided that suitable sample preparation strategies have been applied, a simultaneous procedure for the full concentration range is not possible. Coincidently, all high-end MS systems have a dynamic measurement range of 3–4 orders of magnitude, which matches with the rough separation of three concentration ranges of body fluids in ‘high,’ ‘medium,’ and ‘low’ [Citation6]. The first three to four orders of magnitude (high concentration) can be covered without extensive up-front purification. The second three to four orders of magnitude (medium concentration) are usually covered after application of off-line chromatographic fractionation steps, or after depletion of high concentration proteins. The third and lowest part of the concentration range can be covered exclusively in combination with immunocapture procedures.
4. Metrological traceability
Clinicians largely base their medical decisions on medical test results produced in medical laboratories (for example, Rohr et al. reported a value of 66% [Citation41]). For clinicians, it is essential that test results are accurate and accompanied by suitable reference values and/or decision limits, to enable correct interpretation. To that end, clinical chemistry tests should be standardized. Universal standardization can be reached when the entire reference system and metrological traceability chain are in place. After all, metrological traceability is defined as the property of a measurement result whereby the result can be related to a reference through a documented unbroken chain of calibrations, each contributing to the measurement uncertainty [Citation42]. When the full metrological traceability chain is in place for an analyte, with limited measurement uncertainty, equivalence of test results is ensured between clinical laboratories independent of location or time.
So far, standardization is only achieved for a relatively small number of protein analytes, because of heterogeneity of the measurands, lack of commutable, value-assigned reference materials, and lack of internationally recognized reference measurement procedures [Citation43] leading to inconsistent results between laboratories around the globe. Worldwide, proteins in body fluids are generally measured indirectly using antibody-based immunoassays. Immunoassays recognize heterogeneous mixtures of protein isoforms (proteoforms), either biologically active, inactive, or mixed. As the measurand is often undefined, standardization of immunoassays is difficult to achieve. Moreover, immunoassays can also be flawed by heterophilic antibodies or autoantibodies, giving inaccurate results [Citation20,Citation44]. Thyroglobulin tests based on immunoassay formats are a striking example, where interference of autoantibodies results in flawed test results with a deleterious impact on patient outcome [Citation45]. The direct effect of these nonideal immunoassays is the potentially high rate of diagnostic mistakes.
In our laboratory, we have evaluated the potential of MS applications within a clinical setting with a proof-of-principle study and assessed the feasibility of developing and standardizing qCCP tests [Citation46,Citation47]. More specifically, serum apolipoproteins A-I, B, C-I, C-II, C-III and E and apo E polymorphism were targeted, with the aim to improve cardiovascular risk assessment as compared to standard serum lipids. Analytical performance specifications for serum apolipoproteins were predefined based on biological variation data [Citation42,Citation48]. We demonstrated that it was feasible to fulfill the analytical performance specifications required to make a qCCP test fit-for-clinical purpose.
As a strategy, qCCP enables the unraveling of underlying pathology of specific diseases by detection and quantitation of clinically relevant proteoforms at the molecular level. This enables detection of disease-specific subgroups and selection of targeted categories of patients which will truly benefit from expensive interventions or treatments, boosting precision medicine. Both manufacturers of in vitro diagnostics (IVD) and academic laboratories have a huge interest in this technology as it enables introducing promising protein biomarkers in clinical practice in a direct, multiplexed, and antibody-independent way.
5. Recent advances in HRAM technologies and applications
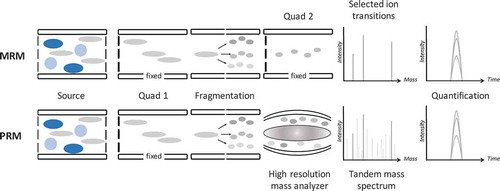
Advances and alternative acquisition strategies for the Orbitrap™ in relation to clinical MS have been reviewed recently [Citation49]. The targeted analysis of compounds (peptides) using a hybrid-type Orbitrap™ instrument was coined parallel reaction monitoring (PRM) [Citation50,Citation51]. Of particular interest within the field of clinical chemistry is the ability to capture ‘high accuracy signals’ of all the fragment ions of a precursor in a measurement using Orbitrap™ technology (). In contrast to an MRM method, the PRM strategy allows for method development without a priori knowledge on fragmentation patterns of the peptide of interest. In addition, in data analysis, peptide identification (confirmation) can easily be accomplished, and fragment selection can be performed based on performance. Moreover, application of HRAM for quantification purposes compared to MRM typically performed on a triple quadrupole LC-MS/MS instrument provides improved selectivity due to the narrow mass window that can be selected during post-acquisition data analysis of tandem MS-data [Citation52]. In other words, although the isolation window of a peptide in MS-1 is the same for q-ultrahigh resolution and for a triple quadrupole, it is the second part of the HRAM platform that makes the differences and that comes with clear advantages for selection of transitions (post-acquisition) [Citation49]. While not for all proteins and all conditions, it was shown that for peptide NVNDVIAPAFVK from urine the limit-of-quantification was improved for 4 out of the 5 transitions when using PRM compared to MRM. This shows that the increased selectivity obtained by HRAM can be beneficial, particularly in the analysis of lower abundance proteins in a complex matrix, where interferences are likely to occur [Citation53,Citation54].
Figure 1. Analytical pathways of MRM (top) versus PRM (bottom). The crucial difference is that in MRM only selected fragment ions are monitored, while in PRM all fragment ions are monitored, out of which during data analysis the best performing fragment ions are used for quantification.
A second hybrid-type mass spectrometer that provides ultrahigh resolution is an FTICR-MS system [Citation3]. Although FTICR-MS is the superior technique when analyzing samples of high complexity in a single spectrum with simultaneous identifications based on accurate masses, with petroleomics and analysis of complex natural products being key examples [Citation55,Citation56], it has found little application in bottom-up proteomics workflows. This is due to the absence of fast MS/MS peptide identification tools in commercially available FTICR platforms, whereas most of the Orbitrap™ systems are equipped with ion traps and sophisticated acquisition software to switch from one mass analyzer to the other to perform simultaneous mass measurements. For middle-down and top-down proteomics approaches, FTICR however remains a key player [Citation2,Citation57,Citation58]. The basic design of FTICR is radial trapping of ions in a strong magnetic field combined with a weak axial electric field, followed by image current detection after coherently exciting the trapped ions. The resulting time-domain signal (transient) is digitized and converted (Fourier transformed) into the frequency-domain spectrum to obtain a mass spectrum [Citation59]. Over the years, FTICR-MS has undergone many technological improvements of its various components, both with regard to hardware and software. Where initially FTICR-MS has been the domain of the academic research environment, the current commercial instruments are available to a larger and less specialized group of users. Modern FTICR instruments have become robust platforms that provide high-quality mass spectral data sets. In our laboratory, we perform various analyses on an FTICR platform in combination with MALDI. One involves high-throughput MALDI-FTICR profiling of biomolecules in clinical cohorts aiming for case–control classifications. Species of interest are serum peptides and – proteins up to 15 kDa that are obtained after single-step sample cleanup procedures [Citation60,Citation61], released N-glycans that are derived and purified from serum proteins [Citation62], or complex glycopeptide mixtures that are obtained from various immunoglobulins [Citation63]. In addition, we have applied an MALDI-FTICR profiling method that builds on the standard MALDI-TOF approach (Biotyper™) for typing and identifying bacteria and yeasts as described in the previous section. With this platform, we could show that application of HRAM on the protein profiles improved strain classification and allowed for a more detailed analysis of the observed compounds [Citation64].
6. Protein applications in clinical MS that potentially benefit from HRAM
While levels of individual proteins or panels of proteins may serve as biomarkers, proteomics also studies specific proteoforms as markers of disease states. Although proteoform mapping may be accomplished using bottom-up procedures, the preferred strategy is top-down intact protein MS [Citation65–Citation68]. Proteoform-based disease studies could include genetic polymorphisms, such as the ApoE phenotypes we previously reported [Citation47], but also include posttranslational modifications such as phosphorylation, acetylation, ubiquitination and, notably, also glycosylation. An example of the clinical relevance of various proteoforms involves the biomarker prostate-specific antigen (PSA). It is well known that PSA exists in multiple proteoforms [Citation69], including the free pro-PSA [Citation70] and BPSA [Citation71] forms, which are associated with increased and decreased malignant potential, respectively. In-depth glycosylation analysis (mapping various ‘glycoforms’) of PSA has revealed that further stratification of patients and individuals at risk can be achieved using different glycoforms [Citation72]. In addition, the analysis of intact PSA glycoproteins was reported using a reversed phase LC separation method combined with high resolution TOF mass detection [Citation73]. Similarly, there is a large body of studies describing the differential glycosylation patterns of serum proteins in patients with different types of malignancies. These glycosylation patterns are often derived from high abundance proteins, and as such would be well targetable using clinical MS assays. Recently, MRM technology was developed and applied to assess the differential N-glycosylation patterns of serum immunoglobulins in ovarian cancer patients compared to controls [Citation74–Citation76]. In addition, a MALDI-FTICR system has been used to map a complex mixture of glycopeptides obtained from immunoglobulin A, exemplified in and showing the benefits of HRAM with regard to resolving overlapping species [Citation63]. While the role of protein glycosylation measurements in clinical chemistry is currently limited [Citation77], it is anticipated that improved technology, in combination with the availability of glycan, glycopeptide, or glycoprotein reference materials will enable clinical chemists to harvest on the potential that has been ascribed to protein glycosylation. This statement could also be expanded to other posttranslational modifications or families of proteoforms.
Figure 2. Example of the analysis of protein glycosylation patterns from large immunoglobulin A glycopeptides using high resolution MALDI-FTICR MS. An in-house example of a MALDI-FTICR mass spectrum that was used for the simultaneous analysis of N- and O-glycopeptides from serum IgA [Citation63]. Relative intensities are plotted in the y-direction (arbitrary units). Panel a shows the major part of the profile. Panels b and c exemplify the benefits of ultrahigh resolution. Here, symbols are used to indicate isotopic peaks that belong to the same species (i.e., measurand). In panel B four different species are clearly resolved (filled squares, open circles, stars and small triangles), whereas in panel c two different species are perfectly resolved (filled squares and stars). The isotopic distribution of species with near nominal mass are resolved as a result of the ultrahigh resolving power, and moreover, glycopeptides could be confidently assigned using the accurate masses.
![Figure 2. Example of the analysis of protein glycosylation patterns from large immunoglobulin A glycopeptides using high resolution MALDI-FTICR MS. An in-house example of a MALDI-FTICR mass spectrum that was used for the simultaneous analysis of N- and O-glycopeptides from serum IgA [Citation63]. Relative intensities are plotted in the y-direction (arbitrary units). Panel a shows the major part of the profile. Panels b and c exemplify the benefits of ultrahigh resolution. Here, symbols are used to indicate isotopic peaks that belong to the same species (i.e., measurand). In panel B four different species are clearly resolved (filled squares, open circles, stars and small triangles), whereas in panel c two different species are perfectly resolved (filled squares and stars). The isotopic distribution of species with near nominal mass are resolved as a result of the ultrahigh resolving power, and moreover, glycopeptides could be confidently assigned using the accurate masses.](/cms/asset/041d3947-f820-492f-b494-6df75572b75d/ieru_a_1253477_f0002_b.gif)
Although a discussion of the role of tissue MS-imaging approaches in clinical MS is outside the scope of this perspective, it is stressed that this type of spatially resolved protein measurements improves by applying HRAM analysis [Citation78,Citation79]. This was shown on MALDI-FTICR systems and will find wider usage provided data analysis pipelines are further developed [Citation80,Citation81].
7. Expert commentary
In this perspective, two hybrid-types ultrahigh resolution MS systems were discussed, namely FTICR and Orbitrap™. Although both platforms can be used in both ‘ESI- and MALDI-mode,’ clinical MS applications that potentially benefit from HRAM are MALDI-FTICR and LC-ESI-Orbitrap™. The first one provides a powerful approach for mapping complex mixtures of (glyco)peptides and (small) proteins, the second one has great promise with regard to PRM strategies. Both platforms are suitable for top-down proteomics and proteoform mapping and it remains to be evaluated which one is preferred in which study. Gathering evidence on clinical performance, clinical efficacy, and cost-effectiveness become important when aiming at future clinical application of MS-basedtests [Citation82]. This is essential for demonstrating the clinical added value of this technology for precision medicine. At this moment, both HRAM platforms are expensive (>EUR 500,000), with higher operating costs for FTICR due to the need for liquid helium. However, lifetimes of more than 10 years are not uncommon for high field magnets, whereas long-term sustainability of the Orbitrap™ is not known yet since these earliest systems are from the late 2000s. It should also be kept in mind that (high) abundant proteins can be quantified in MS-based protein tests without the need of enrichment strategies. Thus, the application of specific monoclonal and/or polyclonal antibodies, which are essential in immunoassay-based tests, is not needed for such proteins, however, immunocapture remains a pivotal step when low abundant proteins are of interest.
8. Five-year view
Protein MS has found its way into clinical laboratories of academic and teaching hospitals. In this perspective, this was illustrated with two strategies, namely qCCP that applies bottom-up proteomics with LC-MRM-MS, and Biotyper™, which is based on protein profiling by MALDI-TOF-MS. The latter application is also pursued by other technologies, such as REIMS. Encouraged by the boosting interest in ‘The Association for Mass Spectrometry: Applications to the Clinical Laboratory’ (MSACL) and the increasing number of protein applications in this field we foresee that the implementation of both of these strategies will continue to grow [Citation83]. Furthermore, efforts to standardize qCCP tests are important, right from the development phase. In addition, the beneficial aspects of HRAM technology with regard to clinical MS enable enhanced selectivity of the quantities intended to be measured. It was shown that ultrahigh resolution instruments enable top-down proteoform mapping and provide high-quality protein-, glycan- and glycopeptide signatures that are suitable for case–control classifications or biomarker discovery studies. Moreover, upon careful evaluation of various assay characteristics, it becomes clear that HRAM technology can play an important role in improving existing, flawed immunoassays and in developing disease specific MS-based tests which give enhanced pathophysiological insight (overviewed in ). The advantages of MS-based assays in comparison to routine immunoassays are depicted and derived from Lehmann and coworkers [Citation12]. In some cases, our view on performance parameters of MS-based assays differs from the initial report. First of all, sensitivity levels and sample volume requirements of analytical MS-based workflows have improved and reduced the gap between immunoassays and MS. Second, we are more cautious with regard to robustness and cost-effectiveness of MS-based assays. Although optimistic, these aspects require further evaluation. In addition, in the comparison with Lehmann and coworkers, we have added three parameters that provide benefits with regard to MS analysis, namely standardization, selectivity, and (structural) characterization of the measurand. High-resolution methods inherently improve the analytical selectivity for the measurand(s) of interest and thus enables to define clearly the quantity intended to be measured. In addition, it has been shown that the use of HRAM results in improved analytical sensitivity and consequently provides a more robust platform that is less prone to matrix effects (i.e. co-eluting interferences, or overlapping species in single profiles). In line with Gallien and Domon, we anticipate that HRAM will be adopted in the nearby future and that low resolution instruments will only survive for specific applications that make use of relatively clean samples [Citation49].
Table 1. Comparison between immunoassays and MS-based assays for protein quantitation, together with potential improvements that may be achieved by implementation of HRAM a.
Key issues
HRAM strategies based on either FTICR or Orbitrap™ have matured into widely applied high-quality and large-scale procedures for structural analysis of almost any type of biomolecule.
The fields of MS-based proteomics as well as clinical chemistry have experienced that there is a disconnect between strategies and technologies used for protein identification and quantification, and those used in the clinical diagnostics setting. As a result, the efforts to translate biomarkers into a medical test have converged into a novel field of clinical MS.
MS applications allow multiplexed protein quantification and proteoform differentiation, for example for the purpose of phenotyping. Multiplexed protein analysis has been recognized as a key feature for the next generation of protein assays.
Living in a global world, medical tests should be well standardized, enabling equivalent results independent of location or time. We have evaluated the feasibility of standardizing qCCP tests with a proof-of-principle study on serum apolipoproteins including successful application of the concept of metrological traceability.
In contrast to an MRM method the Orbitrap™ PRM strategy allows for method development without a priori knowledge on fragmentation patterns of the peptide of interest. The capture of ‘high accuracy signals’ of all the fragment ions of a precursor has gained attention in the field of clinical chemistry.
The use of HRAM strategies results in high accuracy fragment signals, which allow for the accurate identification of proteoforms and facilitate improved selectivity of targeted quantitative measurements.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
Notes on contributors
L. Renee Ruhaak
L.R. Ruhaak and Y. van der Burgt initiated the idea of writing this perspective together. C. M. Cobbaert has shared expertise on medical Test Evaluation and endorsed embedding of the concept of metrological traceability for standardization of MS-based qCCP tests. All three authors wrote various parts of the manuscript and have aligned this perspective to their common opinion.
References
- Mann M, Kelleher NL. Precision proteomics: the case for high resolution and high mass accuracy. Proc Natl Acad Sci U S A. 2008;105(47):18132–18138.
- Bogdanov B, Smith RD. Proteomics by FTICR mass spectrometry: top down and bottom up. Mass Spectrom Rev. 2005;24(2):168–200.
- Marshall AG. Milestones in fourier transform ion cyclotron resonance mass spectrometry technique development. Int J Mass Spectrom. 2000;200(1–3):331–356.
- Park Y, Lebrilla CB. Application of fourier transform ion cyclotron resonance mass spectrometry to oligosaccharides. Mass Spectrom Rev. 2005;24(2):232–264.
- Andrews GL, Simons BL, Young JB, et al. Performance characteristics of a new hybrid quadrupole time-of-flight tandem mass spectrometer (TripleTOF 5600). Anal Chem. 2011;83(13):5442–5446.
- Schilling B, MacLean B, Held JM, et al. Multiplexed, scheduled, high-resolution parallel reaction monitoring on a full scan QqTOF instrument with integrated data-dependent and targeted mass spectrometric workflows. Anal Chem. 2015;87(20):10222–10229.
- Makarov A. Electrostatic axially harmonic orbital trapping: a high-performance technique of mass analysis. Anal Chem. 2000;72(6):1156–1162.
- Michalski A, Damoc E, Hauschild JP, et al. Mass spectrometry-based proteomics using Q exactive, a high-performance benchtop quadrupole orbitrap mass spectrometer. Mol Cell Proteomics. 2011;10(9):M111 011015.
- Nyadong L, Inutan ED, Wang X, et al. Laserspray and matrix-assisted ionization inlet coupled to high-field FT-ICR mass spectrometry for peptide and protein analysis. J Am Soc Mass Spectrom. 2013;24(3):320–328.
- Hendrickson CL, Quinn JP, Kaiser NK, et al. 21 tesla fourier transform ion cyclotron resonance mass spectrometer: a national resource for ultrahigh resolution mass analysis. J Am Soc Mass Spectrom. 2015;26(9):1626–1632.
- Poste G. Bring on the biomarkers. Nature. 2011;469(7329):156–157.
- Lehmann S, Brede C, Lescuyer P, et al. Clinical mass spectrometry proteomics (cMSP) for medical laboratory: What does the future hold?. Clinica Chimica Acta. 2016;(16)30246–30247. doi: 10.1016/j.cca.2016.06.001.
- Carr SA, Abbatiello SE, Ackermann BL, et al. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol Cell Proteomics. 2014;13(3):907–917.
- Riley NM, Hebert AS, Coon JJ. Proteomics moves into the fast lane. Cell Systems. 2016;2(3):142–143.
- Thevis M, Schanzer W. Mass spectrometry in sports drug testing: structure characterization and analytical assays. Mass Spectrom Rev. 2007;26(1):79–107.
- Siuzdak G. The emergence of mass spectrometry in biochemical research. Proc Natl Acad Sci. 1994;91(24):11290–11297.
- Patti GJ, Yanes O, Siuzdak G. Innovation: metabolomics: the apogee of the omics trilogy. Nat Rev Mol Cell Biol. 2012;13(4):263–269.
- Kemna EH, Tjalsma H, Podust VN, et al. Mass spectrometry-based hepcidin measurements in serum and urine: analytical aspects and clinical implications. Clin Chem. 2007;53(4):620–628.
- Hoofnagle AN, Becker JO, Oda MN, et al. Multiple-reaction monitoring-mass spectrometric assays can accurately measure the relative protein abundance in complex mixtures. Clin Chem. 2012;58(4):777–781.
- Hoofnagle AN, Wener MH. The fundamental flaws of immunoassays and potential solutions using tandem mass spectrometry. J Immunol Methods. 2009;347(1–2):3–11.
- Scherl A. Clinical protein mass spectrometry. Methods. 2015;81:3–14.
- Lehmann S, Hoofnagle A, Hochstrasser D, et al. Quantitative clinical chemistry proteomics (qCCP) using mass spectrometry: general characteristics and application. Clin Chem Lab Med. 2013;51(5):919–935.
- Hoofnagle AN, Whiteaker JR, Carr SA, et al. Recommendations for the generation, quantification, storage, and handling of peptides used for mass spectrometry-based assays. Clin Chem. 2016;62(1):48–69.
- Percy AJ, Byrns S, Pennington SR, et al. Clinical translation of MS-based, quantitative plasma proteomics: status, challenges, requirements, and potential. Expert Rev Proteomics. 2016;13(7):673–684.
- Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods. 2012;9(6):555–566.
- Lange V, Picotti P, Domon B, et al. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst Biol. 2008;4:222.
- Smith LM, Kelleher NL. Consortium for top down p. proteoform: a single term describing protein complexity. Nat Methods. 2013;10(3):186–187.
- JCGM200. International vocabulary of metrology – Basic and general concepts and associated terms. 2012. http://www.bipm.org/utils/common/documents/jcgm/JCGM_200_2012.pdf
- Netzel BC, Grant RP, Hoofnagle AN, et al. First steps toward harmonization of lc-ms/ms thyroglobulin assays. Clin Chem. 2016;62(1):297–299.
- Mellmann A, Cloud J, Maier T, et al. Evaluation of matrix-assisted laser desorption ionization-time-of-flight mass spectrometry in comparison to 16S rRNA gene sequencing for species identification of nonfermenting bacteria. J Clin Microbiol. 2008;46(6):1946–1954.
- Strittmatter N, Rebec M, Jones EA, et al. Characterization and identification of clinically relevant microorganisms using rapid evaporative ionization mass spectrometry. Anal Chem. 2014;86(13):6555–6562.
- Keys CJ, Dare DJ, Sutton H, et al. Compilation of a MALDI-TOF mass spectral database for the rapid screening and characterisation of bacteria implicated in human infectious diseases. Infect Genet Evol. 2004;4(3):221–242.
- Van Belkum A, Chatellier S, Girard V, et al. Progress in proteomics for clinical microbiology: MALDI-TOF MS for microbial species identification and more. Expert Rev Proteomics. 2015;12(6):595–605.
- Fournier PE, Drancourt M, Colson P, et al. Modern clinical microbiology: new challenges and . Nat Rev Microbiol. 2013;11(8):574–585.
- Kingsmore SF. Multiplexed protein measurement: technologies and applications of protein and antibody arrays nature reviews. Drug Discovery. 2006;5(4):310–320.
- Koomen JM, Haura EB, Bepler G, et al. Proteomic contributions to personalized cancer care. Mol Cell Proteomics . 2008;7(10):1780–1794.
- Fung ET. A recipe for proteomics diagnostic test development: the OVA1 test, from biomarker discovery to FDA clearance. Clin Chem. 2010;56(2):327–329.
- Kashani K, Al-Khafaji A, Ardiles T, et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit Care. 2013;17(1):R25.
- Gunnerson KJ, Shaw AD, Chawla LS, et al. TIMP2*IGFBP7 biomarker panel accurately predicts acute kidney injury in high-risk surgical patients. J Trauma Acute Care Surg. 2016;80(2):243–249.
- Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1(11):845–867.
- Rohr UP, Binder C, Dieterle T, et al. The value of in vitro diagnostic testing in medical practice: a status report. PLoS One. 2016;11(3):e0149856.
- Smit N, Van Den Broek I, Romijn FP, et al. Quality requirements for quantitative clinical chemistry proteomics. Adv Integr Med. 2014;2:1–13.
- www.jctlm.org/dbase.
- Netzel BC, Grebe SK, Algeciras-Schimnich A. Usefulness of a thyroglobulin liquid chromatography-tandem mass spectrometry assay for evaluation of suspected heterophile interference. Clin Chem. 2014;60(7):1016–1018.
- Hoofnagle AN, Roth MY. Clinical review: improving the measurement of serum thyroglobulin with mass spectrometry. J Clin Endocrinol Metab. 2013;98(4):1343–1352.
- Van Den Broek I, Nouta J, Razavi M, et al. Quantification of serum apolipoproteins A-I and B-100 in clinical samples using an automated SISCAPA-MALDI-TOF-MS workflow. Methods. 2015;81:74–85.
- Van Den Broek I, Romijn FP, Nouta J, et al. Automated multiplex LC-MS/MS assay for quantifying serum apolipoproteins A-I, B, C-I, C-II, C-III, and E with qualitative apolipoprotein E phenotyping. Clin Chem. 2016;62(1):188–197.
- Fraser C. Biological variation: from principles to practice . Washington, DC: AACC Press; 2001.
- Gallien S, Domon B. Advances in high-resolution quantitative proteomics: implications for clinical applications. Expert Rev Proteomics. 2015;12(5):489–498.
- Gallien S, Duriez E, Crone C, et al. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol Cell Proteomics. 2012;11(12):1709–1723.
- Peterson AC, Russell JD, Bailey DJ, et al. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics. 2012;11(11):1475–1488.
- Gallien S, Duriez E, Demeure K, et al. Selectivity of LC-MS/MS analysis: implication for proteomics experiments. J Proteomics. 2013;81:148–158.
- Abbatiello SE, Mani DR, Keshishian H, et al. Automated detection of inaccurate and imprecise transitions in peptide quantification by multiple reaction monitoring mass spectrometry. Clin Chem. 2010;56(2):291–305.
- Sherman J, McKay MJ, Ashman K, et al. How specific is my SRM?: the issue of precursor and product ion redundancy. Proteomics. 2009;9(5):1120–1123.
- Marshall AG, Rodgers RP. Petroleomics: chemistry of the underworld. Proc Natl Acad Sci U S A. 2008;105(47):18090–18095.
- Cho Y, Ahmed A, Islam A, et al. Developments in FT-ICR MS instrumentation, ionization techniques, and data interpretation methods for petroleomics. Mass Spectrom Rev. 2015;34(2):248–263.
- Johansson A, Enroth S, Palmblad M, et al. Identification of genetic variants influencing the human plasma proteome. Proc Natl Acad Sci U S A. 2013;110(12):4673–4678.
- Kalli A, Sweredoski MJ, Hess S. Data-dependent middle-down nano-liquid chromatography-electron capture dissociation-tandem mass spectrometry: an application for the analysis of unfractionated histones. Anal Chem. 2013;85(7):3501–3507.
- Comisarow MB, Marshall AG. Fourier transform ion cyclotron resonance spectroscopy. Chem Phys Lett. 1974;25(2):282–283.
- Nicolardi S, Velstra B, Mertens BJ, et al. Ultrahigh resolution profiles lead to more detailed serum peptidome signatures of pancreatic cancer. Translational Proteomics. 2014;2:39–51.
- Nicolardi S, Bogdanov B, Deelder AM, et al. Developments in FTICR-MS and its potential for body fluid signatures. Int J Mol Sci. 2015;16(11):27133–27144.
- Bladergroen MR, Reiding KR, Hipgrave Ederveen AL, et al. Automation of high-throughput mass spectrometry-based plasma n-glycome analysis with linkage-specific sialic acid esterification. J Proteome Res. 2015;14(9):4080–4086.
- Bondt A, Nicolardi S, Jansen BC, et al. Longitudinal monitoring of immunoglobulin A glycosylation during pregnancy by simultaneous MALDI-FTICR-MS analysis of N- and O-glycopeptides. Sci Rep. 2016;6:27955.
- Fleurbaaij F, Kraakman ME, Claas EC, et al. Typing pseudomonas aeruginosa isolates with ultrahigh resolution MALDI-FTICR mass spectrometry. Anal Chem. 2016;88(11):5996–6003.
- Zhang J, Guy MJ, Norman HS, et al. Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J Proteome Res. 2011;10(9):4054–4065.
- Whitelegge J. Intact protein mass spectrometry and top-down proteomics. Expert Rev Proteomics. 2013;10(2):127–129.
- Gault J, Malosse C, Machata S, et al. Complete posttranslational modification mapping of pathogenic neisseria meningitidis pilins requires top-down mass spectrometry. Proteomics. 2014;14(10):1141–1151.
- Lakshmanan R, Wolff JJ, Alvarado R, et al. Top-down protein identification of proteasome proteins with nanoLC-FT-ICR-MS employing data-independent fragmentation methods. Proteomics. 2014;14(10):1271–1282.
- De Angelis G, Rittenhouse HG, Mikolajczyk SD, et al. Twenty years of psa: from prostate antigen to tumor marker. Rev Urol. 2007;9(3):113–123.
- Pecoraro V, Roli L, Plebani M, et al. Clinical utility of the (−2)proPSA and evaluation of the evidence: a systematic review. Clin Chem Lab Med. 2016;54(7):1123–1132.
- Mikolajczyk SD, Millar LS, Wang TJ, et al. BPSA,” a specific molecular form of free prostate-specific antigen, is found predominantly in the transition zone of patients with nodular benign prostatic hyperplasia. Urology. 2000;55(1):41–45.
- Yoneyama T, Ohyama C, Hatakeyama S, et al. Measurement of aberrant glycosylation of prostate specific antigen can improve specificity in early detection of prostate cancer. Biochem Biophys Res Commun. 2014;448(4):390–396.
- Behnken HN, Ruthenbeck A, Schulz JM, et al. Glycan analysis of Prostate Specific Antigen (PSA) directly from the intact glycoprotein by HR-ESI/TOF-MS. J Proteome Res. 2014;13(2):997–1001.
- Ruhaak LR, Kim K, Stroble C, et al. Protein-specific differential glycosylation of immunoglobulins in serum of ovarian cancer patients. J Proteome Res. 2016;15(3):1002–1010.
- Hong Q, Lebrilla CB, Miyamoto S, et al. Absolute quantitation of immunoglobulin G and its glycoforms using multiple reaction monitoring. Anal Chem. 2013;85(18):8585–8593.
- Hong Q, Ruhaak LR, Stroble C, et al. A method for comprehensive glycosite-mapping and direct quantitation of serum glycoproteins. J Proteome Res. 2015;14:5179–5192.
- Ruhaak LR, Lebrilla CB. Applications of multiple reaction monitoring to clinical glycomics. Chromatographia. 2015;78(5–6):335–342.
- McDonnell LA, Heeren RM. Imaging mass spectrometry. Mass Spectrom Rev. 2007;26(4):606–643.
- Caprioli RM. Imaging mass spectrometry: molecular microscopy for the new age of biology and medicine. Proteomics. 2016;16(11–12):1607–1612.
- Spraggins JM, Rizzo DG, Moore JL, et al. MALDI FTICR IMS of intact proteins: using mass accuracy to link protein images with proteomics data. J Am Soc Mass Spectrom. 2015;26(6):974–985.
- Taban IM, Altelaar AF, Van Der Burgt YE, et al. Imaging of peptides in the rat brain using MALDI-FTICR mass spectrometry. J Am Soc Mass Spectrom. 2007;18(1):145–151.
- Horvath AR, Lord SJ, StJohn A, et al. From biomarkers to medical tests: the changing landscape of test evaluation. Clin Chim Acta. 2014;427:49–57.
- www.msacl.org.
- Palmblad M, Van Der Burgt YE, Mostovenko E, et al. A novel mass spectrometry cluster for high-throughput quantitative proteomics. J Am Soc Mass Spectrom. 2010;21(6):1002–1011.