
ABSTRACT
Introduction: Cancer changes the proteome in complex ways that reach well beyond simple changes in protein abundance. Genomic and transcriptional variations and post-translational protein modification create functional variants of a protein, known as proteoforms. Childhood cancers have fewer genomic alterations but show equally dramatic phenotypic changes as malignant cells in adults. Therefore, unraveling the complexities of the proteome is even more important in pediatric malignancies.
Areas covered: In this review, the biological origins of proteoforms and technological advancements in the study of proteoforms are discussed. Particular emphasis is given to their implication in childhood malignancies and the critical role of cancer-specific proteoforms for the next generation of cancer therapies and diagnostics.
Expert opinion: Recent advancements in technology have led to a better understanding of the underlying mechanisms of tumorigenesis. This has been critical for the development of more effective and less harmful treatments that are based on direct targeting of altered proteins and deregulated pathways. As proteome coverage and the ability to detect complex proteoforms increase, the most need for change is in data compilation and database availability to mediate high-level data analysis and allow for better functional annotation of proteoforms.
1. Introduction
Cancer is the leading cause of death in children in developing countries [Citation1,Citation2]. Most childhood cancers have treatment options with survival rates over 80% [Citation3]. Chemotherapy and radiation, the most common treatment methods, are aggressive and may lead to long term side effects ranging from mild to severe complications that negatively impact the overall quality of life of patients [Citation2]. In addition, these cancers generally have a high incidence of relapse that is often refractory to the traditional chemotherapeutics [Citation4]. Furthermore, cure rates for pediatric solid tumors have not improved much in the past decade with patients having recurrent disease performing particularly poorly [Citation5]. This has led to an increased interest in developing new therapies that are more targeted and less toxic. Most new therapies, including immunotherapies and targeted therapies, rely on the increased abundance or altered activity of proteins or specific proteoforms [Citation6]. For these new therapies to be successful, a comprehensive understanding of the proteoform landscape and the underlying mechanisms is essential.
Whole genome sequencing is now feasible for large-scale studies, and a number of initiatives aim to identify possible mutations in cancer patients that can be the targets for treatment [Citation7,Citation8]. This genomic information has contributed tremendously to our understanding of the dysregulated pathways underlying tumorigenesis, identified genetic alterations associated with specific disease phenotypes, and showed that each person’s individual cancer is unique. Thanks to large whole-genome sequencing studies the specifics of childhood cancers are becoming increasingly apparent [Citation2,Citation9]. Ma et al. published 142 driver genes that had recurring mutations in childhood cancers of which only 45% overlapped with adult cancers. Their study further determined that pediatric cancers had a lower mutational burden (0.17 mutations per million bases compared to 1–10 mutations per million bases found in adult cancers) [Citation9]. Grobner et al. published similar findings and showed that the overall mutational burden was 14 times lower in pediatric cancers compared to adult cancers [Citation2]. Through these studies, we now know that childhood and adult cancers are dissimilar at the molecular level. Genomic and transcriptomic studies identify variations that may lead to cancer initiation and progression, however, there is still very little known about how these tumor genomes shape the functional and targetable proteome [Citation10].
DNA and mRNA sequences dictate the amino acid sequences of proteins. However, they do not accurately predict the expression of the protein or whether the protein is stable and functional. Several studies have shown there is only a partial concordance between transcripts and their associated proteins [Citation11–Citation14]. Studies performed by Ning et al. and Akbani et al. found concordance of protein to mRNA to be in the range of 0.5–0.55 [Citation15,Citation16].
In addition to only moderate correlation of gene, transcript and protein level, variations on the whole genome and proteome level further contribute to this discordance. There are approximately 20,000 known protein-coding genes but over a hundred thousand protein proteoforms have been identified. Some speculate upwards of a million exist that have not yet been identified, eliminating the once popular idea that one gene encodes one protein [Citation17,Citation18]. There are several terms to describe these variants, such as ‘protein species’ or ‘isoforms’ [Citation19,Citation20]. However, the term ‘protein species’ does not differentiate between proteins that originated from the same gene or proteins from an entirely different gene and the term ‘isoforms’ specifically refers to different proteins arising from alternative splicing of a single gene. The term ‘proteoform’ applies to all the different proteins derived from a single gene, including all forms of genetic variation, alternative splicing, and post-translational modifications [Citation21]. Groups of related proteoforms that are derived from a single gene and share a similar combination of modifications and variants are termed ‘proteoform families’ [Citation17]. The existence of proteoforms creates molecularly distinct proteins that modulate a wide variety of biological processes including cell signaling, gene regulation, and activation [Citation21]. Proteoforms can have distinct functions and can differ in their subcellular localization, binding partners, structure, and kinetics, among others. In-depth characterization has only been done for few of the known proteoforms and remains an important research topic [Citation22].
Proteins are essential effectors of cellular function and can act as biomarkers or potential drug targets [Citation23]. Identification of genomic consequence on the proteome and protein pathways responsible for malignant transformation may lead to the development of more effective treatments. In addition, specific proteoforms can be detected and targeted for treatment. The process of identifying aberrantly expressed proteins and disease-associated proteoforms has significantly improved due to advancements in technology, specifically mass spectrometry [Citation24]. Mass spectrometry is a versatile tool that allows for the analysis of several aspects of proteins including structure, sequence, quantity, and importantly, modifications [Citation25].
In this review, we first introduce different types of proteoforms and then discuss their relevance in the development, progression and diagnosis or treatment of childhood malignancies. We conclude with an overview of proteoform detection methods and discuss past successes, challenges, and advantages of bringing proteoform detection and targeting to the clinic.
2. Origin of proteoforms
This section details the range of mechanisms that generate proteoforms which have been associated with pediatric malignancies. For an in-depth overview of the general underlying genomic and enzymatic processes, we refer the reader to recent reviews [Citation6,Citation17,Citation20,Citation21,Citation26]. Where established we point out the clinical relevance or translational potential of the described proteoforms.
2.1. Genomic variations leading to distinct proteoforms
2.1.1. SNPs, SNVs, and indels
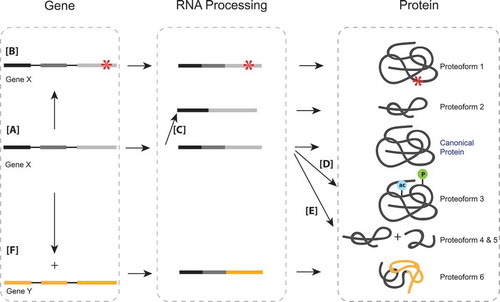
There are different types of mutations that may occur in the genetic sequence. Single nucleotide polymorphisms (SNPs) are alterations in the coding region of DNA. Non-synonymous SNPs change the codon to one that codes for a different amino acid, resulting in changes to the protein sequence ()). SNPs can be classified as either passenger or driver mutations [Citation27]. Driver mutations often lead to a significant change in protein expression and activation or suppression of functional activity [Citation6]. Single nucleotide variants (SNVs) are similar to SNPs but refer to variants found at the individual level, whereas SNPs refer to variants on a population level (i.e. the variant is found in >1% of the population) [Citation27]. Insertions or deletions (indels) of short nucleotide sequences often have a similar effect as SNPs and may lead to alterations in amino acid sequences. Proteoforms resulting from non-synonymous SNPs, SNVs, or indels are critical in the study of cancer, especially when tumor suppressors or oncogenes are directly involved.
Figure 1. (a) The canonical pathway of the gene, to transcript, to final protein. (b) Single nucleotide polymorphisms on the genomic level can translate to an altered amino acid sequence and therefore an altered protein or proteoform. (c) Alternative splicing at the mRNA level leads to exclusion of exons, creating different proteoforms that originated from the same original genomic sequence. (d) Post-translational modifications at the protein level create proteoforms that may have altered functions from the canonical protein. (e) Proteolysis, a protein level modification that cleaves proteins, creates proteoforms that are distinct from the original protein they are cleaved from. (f) The fusion of two different genes may lead to the formation of an entirely new protein.
Several studies have characterized common genomic variations in cancer, but only more recently some of these studies focused specifically on pediatric cancers [Citation28–Citation30]. The most notable among these are by Grobner (2018) and Ma (2018) identifying 149 commonly mutated genes in 961 pediatric tumors and 142 commonly mutated genes in 1,699 pediatric tumors, respectively [Citation2,Citation9]. Another study on recurrent pediatric solid tumors investigated 19 tumor pairs using a targeted sequencing panel for 381 genes and detected 173 SNVs/InDels [Citation31]. The most commonly altered genes found were TP53, PKHD1, BRCA2, INSR, CDK12, LRP1B, NOTCH1, EPHA5, RBI, NOTCH3, and ARID1A. Poor clinical outcome in patients was further explained by the presence of concordant mutations in original and recurrent tumours. Although childhood cancers generally have a lower mutational burden, these studies have indicated several variants that could be participating in oncogenesis.
SNP proteoforms often affect tumor suppressor pathways leading to malignant transformation [Citation32]. For example, phosphatase and tensin homolog (PTEN) is a tumor suppressor that plays a key role in tumorigenesis in a variety of cancers and is predominantly mutated in childhood T-cell acute lymphoblastic leukemia (T-ALL) [Citation4]. Mutations in the catalytic domain of this protein can cause severe phenotypes that are even worse than the loss of function (LOF) mutations [Citation32,Citation33]. In particular, the PTEN A126G mutation, also occurring in the catalytic domain, creates a proteoform with a completely different function, converting a tumor suppressor into a tumor driver [Citation34]. Characterization of how these proteoforms can alter their normal activity of tumor suppression is critical to understanding disease progression.
Similarly, variants in oncogenes often lead to pathogenesis. A commonly mutated pathway in pediatric cancers is the receptor tyrosine kinase/ras (RTK/RAS) pathway, and mutations in this pathway are considered possible targets for treatment [Citation35]. This is exemplified by an in-depth study on clonal evolution in high hyperdiploid acute lymphoblastic leukemia, a subtype of the most common childhood cancer [Citation36]. The most commonly recurring mutations were detected in the RTK/RAS pathway, specifically in KRAS. The majority of mutations occurred in the signal transduction domains, therefore drastically affecting the ability of proteins to carry out their normal cell signaling functions [Citation37] ()). Similar findings were noted in a study of 23 primary neuroblastomas in which several activating mutations in the RAS-mitogen-activated protein kinase (RAS-MEK) pathway were identified. Furthermore, 11 mutations were detected in primary tumors that were also detected in corresponding relapse tumors, and six mutations were unique to the relapse samples. All SNVs detected in ALK were associated with constitutive activation of this receptor tyrosine kinase that is known to activate RAS-mitogen-activated protein kinase (MAPK) signaling [Citation38]. These studies not only indicate the role of RAS in primary tumor initiation but its potential role in relapse.
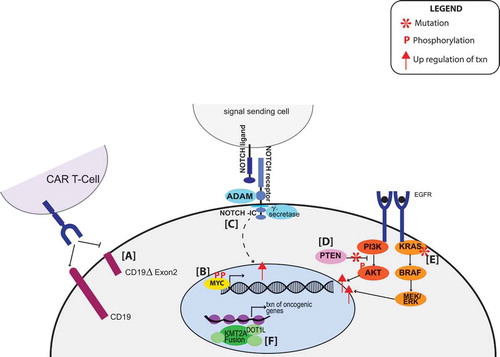
Figure 2. (a) Alternative splicing of CD19 leads to a proteoform that is no longer recognizable by CAR-T cells. (b) The phosphoproteoform of MYC leads to prolonged half-life of MYC and increased transcription of MYC target genes. (c) Mutations of NOTCH increase NOTCH-IC (created by proteolytic cleavage of the NOTCH receptor) and increase transcription of cell survival genes. (d) PTEN is a phosphatase that inhibits PI3K/AKT pathway. When it is deleted, there is aberrant phosphorylation of downstream targets leading to activation of transcription. (e) SNPs found in KRAS lead to constitutive activation of the pathway and increased transcription. (f) KMT2A fusions recruit transcription cofactors such as DOT1L and increase transcription.
Genomic variations may alter proper cell signaling in many ways. Mutations in the janus kinase/signal transducer and activator of transcription proteins (JAK/STAT) pathway have also been implicated in the oncogenesis of many cancers. The R683 JAK mutant has been uniquely identified in pediatric B-ALL and has been shown to lead to constitutive activation of the JAK/STAT pathway leading to overactive cell proliferation in BaF3 cells [Citation39]. Similarly, gain-of-function somatic mutations in tyrosine-protein phosphatase non-receptor type 11 (PTPN11) are associated with many pediatric malignancies such as B-ALL and AML but are very rarely seen in adults. These mutations affect the protein tyrosine phosphatase domains, disrupting their normal inhibitory function leading to hyperactivation of the catalytic activity [Citation40]. Investigating mutations in these cancer-associated pathways are essential to identifying proteoforms that could be biomarkers or targets from treatment.
It is also important to probe other less-studied pathways when trying to find novel proteoforms. Many notable studies have identified lesser-known variants that could be playing a role in tumour progression. St Jude’s children hospital analyzed 127 pediatric high-grade gliomas (HGGs), including diffuse intrinsic pontine gliomas (DIPGs) and non-brainstem HGGs (NBS-HGGs) [Citation41]. Using multiple sequencing platforms, they detected 39,590 SNVs and indel sequence mutations, most notable was the identification of recurrent somatic mutations in activin A receptor, type I (ACVR1) exclusively in DIPGs. All SNVs or indels detected in ACVR1 occurred near the inhibitory glycine-serine-rich (GS) domain or ATP binding pocket of the kinase domain, indicating that these mutations may shift the kinase into a constitutively active state. These findings suggest a potential gain of function activity that could be targeted for treatment.
These studies indicate the implications genomic variants have on their respective proteins and how this can alter proper cellular functions. This is critical to identifying new targets for treatment or improving already existing treatments.
2.1.2. Gene fusions
Structural rearrangements in the chromosome can lead to the exchange of DNA coding information between genes, causing a fused gene. Gene fusions are a unique type of proteoform as they do not originate from the same gene but occur when two distinct genes combine to form a new gene ()). This leads to altered functions of their respective proteins, for example, constitutive activation of signaling pathways, which may drive oncogenic activity [Citation42]. The clinical detection of gene fusions may offer clarification during diagnosis, aid in risk stratification, and may provide targets for molecularly guided treatments [Citation43].
Often, a single gene fuses with one of the multiple partners within a type of cancer, making that single gene a good potential target for treatment [Citation44]. For example, in Philadelphia-like acute lymphoblastic leukemia (Ph-like ALL), JAK2 is rearranged to at least 14 different fusion partners. Extensive preclinical studies show activation of signaling pathways downstream of JAK2 in these cases due to overactive kinase activity. Thus, several studies have investigated the efficacy of tyrosine kinase inhibitors in Ph-like ALL [Citation45]. Lysine Methyltransferase 2A (KMT2A), an activator of promoter regions in the genome has been reported to have 80 different direct fusion partners in patients with acute leukemia [Citation42,Citation46]. Some form of the fusion has been reported in 18% of the pediatric AML patients and 6–8% pediatric ALL patients, respectively, [Citation47,Citation48]. In a study on 13 patients with KMT2A fusions, the KMT2A/MLLT3 gene fusion was present in 38% of the patients [Citation49]. Small-molecule inhibitors, such as DOT1L, against KMT2A fusion-interacting proteins, are producing promising results in mixed lineage leukemia (MLL) [Citation50] ()). Additionally, because KMT2A is a methyltransferase, there is potential to block KMT2A activity by the peptidomimetic MM401 [Citation51]. Fusions can be great targets for treatment because they often involve genes that already have inhibitors developed and may have different treatment options depending on the partner genes that are involved.
Furthermore, Palvick et al. (2017) demonstrated the importance of targeting commonly occurring neurotrophic tyrosine kinase receptor (NTRK) fusions [Citation52]. In nine pediatric to young adult patients with mesenchymal cancers harboring NTRK fusions, five different breakpoints were found in two NTRK family members, NTRK1 and NTRK3. The inhibitors LOXO-1 and entrectinib have been successful in adult cancer patients harboring these fusions. A pan-pediatric solid tumor trial is open for entrectinib which may provide a treatment option for pediatric patients with these fusions [Citation53–Citation55].
Fusions can also act as prognostic factors. For example, the ETV6-RUNX1 fusion which occurs in approximately 25% of the childhood B cell precursor-acute lymphoblastic leukemia (pB-ALL), is generally associated with good prognosis [Citation56]. The TCF3-PBX1 associated ALL previously had an intermediate or unfavorable prognosis; however, advancements in therapeutic options have improved the outcomes of these cases [Citation57]. Although gene fusions are not considered proteoforms in the traditional sense, they can have a significant impact on their transcripts and provide either additional information about a specific cancer or be a potential target for treatment.
2.2. Alternative splicing
Isoforms constitute the most established subgroup of proteoforms. Isoforms are generated when particular exons are either included or excluded from final processed mRNA due to alternative splicing. This results in proteins with slightly different amino acid sequences even though they are coded by the same gene ()). Alternative mRNA splicing is the main source of tissue-specific proteoform diversity [Citation58]. In cancer, splicing events are frequently dysregulated, resulting in aberrant proteoforms that have been implicated in disease initiation and progression [Citation59]. Splicing changes are emerging as potential targets for therapy, and the splicing mechanisms of a tumor in addition to the mutational profile of splicing factors can be informative for selection of effective therapies [Citation60].
Numerous splice variants can exist for a protein. P53 is one of the most studied tumor suppressor proteins that exist in different isoforms and several of its functional variants have been characterized [Citation61]. One variant resulting in a truncated N-terminal arises from an internal transcriptional start site in the first exon. Several other proteoforms with an N-terminal deletion have also been discovered, usually due to an internal promoter before exon 5 [Citation62]. In addition, truncating the transactivating domain, N-terminal variants likely become dominant-negative regulators [Citation63]. Bourdon et al. specifically interrogated the p53γ isoform (truncated c-terminal) and found that patients with loss of p53γ showed poor survival, whereas those who co-expressed p53γ had lower rates of recurrence, with survival similar to patients with functional wild-type p53 [Citation64], thus indicating the variable roles of distinct P53 isoforms.
It is important to understand how the different isoforms vary in function, or even if they do have different functions. There are several splice variants of the epidermal growth factor receptor (EGFRI) that contribute to tumorigenesis. The most common splice variant in glioblastoma is EGFRvIII, which skips exons 2–7 excluding the extracellular ligand-binding domain, leading to constitutive tyrosine phosphorylation and consequent activation of several downstream signaling pathways [Citation65]. Furthermore, as demonstrated by Inda et al., tumor heterogeneity involving both wild type and EGFRvIII isoform can have a variety of implications for tumorigenesis, including activation of proliferation and cell survival pathways [Citation66]. Zhang et al. demonstrated that alternative splicing of myocyte enhancer factor 2C (MEF2C) promotes tumorigenicity in rhabdomyosarcoma (RMS) cells by showing that proteoform MEF2Cα1 was ubiquitously expressed with no myogenic activity, whereas the MEF2Cα2 proteoform was required for efficient differentiation. Additionally, overexpression of MEF2Cα2 in RMS cells increased oncogenic activity and promoted differentiation in RMS cells [Citation67]. These studies fortify the importance of understanding the uniqueness of each proteoform and their biological functions.
Isoforms can also affect drug metabolism. For example, alternative splicing of BRAF affects the kinase domain, making it resistant to vermurafenib [Citation63] a popular BRAF inhibitor included in many treatment programs for childhood cancers, including the NCI-COG Pediatric match trial [Citation68]. This can be accounted for by treating with spliceostatin A, or its analog meayamycin B (MAMB), which slows down the growth of vemurafenib-resistant tumors by decreasing the amount of the resistant BRAF3-9 splice variant [Citation69]. In another example, alternative splicing of the CD19 antigen, specifically a proteoform lacking exon 2, is associated with resistance to CAR-T cell therapy in B-ALL [Citation70] ()). Because distinct isoforms can alter the response to treatment, it is essential to understand variants and proteoforms on a global level.
Though isoforms are the most commonly studied type of proteoform, there is still much to uncover. They do not only play a role in disease initiation and progression but also affect drug efficacy. It is important to characterize these proteoforms on a large scale in different disease types to effectively target them.
2.3. Post-translational modifications
2.3.1. Overview
While proteoforms defined by SNVs, structural re-arrangement, or splicing originate from processes at the DNA or RNA level, co- and post-translational protein modification result in new proteoforms at the protein level [Citation71] (-e)). Proteoforms originating from post-translational modifications (PTMs) are invisible to genomic and transcriptomic analysis. The UniProt database includes over 450 types of PTMs [Citation72]. Post-translational modifications are capable of generating a functional change in the protein that can affect localization, abundance, catalytic activity, protein–protein interactions, and overall aberrant cell signaling [Citation73]. Aberrant post-translational modification of some oncogenic proteins results in tumor initiation and are therefore increasingly recognized as potential targets for anti-cancer drugs [Citation74]. In normal cells, the formation of PTMs is typically tightly controlled and often act as signals to trigger or terminate downstream regulatory processes. However, in diseases such as cancer, these reactions can become constitutively activated or deactivated, contributing to dysregulation of the normal processes [Citation74,Citation75]. Modified proteoforms, or modified members of proteoform families, have far-reaching effects including altered gene expression, activation or de-activation of enzymes, perturbation of signaling pathways and cell morphology compounding to phenotypic changes including proliferation, apoptosis, and migration [Citation6]. In the following, we will focus on four well studied PTMs, namely phosphorylation, acetylation, methylation, and proteolysis, all of which have well-established relevance in pediatric malignancies. For a more general introduction into the molecular properties, detection, and clinical relevance of specific PTMs, we refer to recent reviews [Citation17,Citation76–Citation78].
2.3.2. Phosphorylation
One of the most commonly occurring and well-studied post-translational modifications is phosphorylation [Citation79]. Many proteins involved in cell signaling pathways, especially those that regulate cell cycle, are activated or deactivated by kinases and phosphatases, respectively, [Citation80]. Signaling pathways regulated by protein kinases contribute to the initiation and progression of several types of cancer, thus leading to a significant effort in developing kinase inhibitors to intercept these pathways [Citation81,Citation82].
2.3.2.1. Aberrant phosphoproteoforms
Post-translationally modified proteoforms may have the same global effect as genome alterations. For example, MYC activation characterizes a subgroup of medulloblastomas [Citation83]. While MYC-amplification is considered a ‘hallmark’ of MYC activation, Archer et al.’s study of medulloblastomas showed that the majority of cases in subgroup 3 did not show amplification of MYC. Instead, they observed a characteristic increase in post-translational modification of the MYC protein at multiple sites and predominantly but not exclusively by phosphorylation. A few of the phosphorylated sites are known to regulate the transcriptional activity and half-life of MYC [Citation84] ()). This illustrates the importance to incorporate protein level assessment of oncogenic drivers such as MYC as genome level analysis is blind to modifications occurring at the protein level.
Similar to MYC, the phosphorylation state of AKT also has implication in disease. Amanda Li et al. investigated childhood ependymoma, to identify potential new biomarkers, including phosphorylated AKT (pAKT Ser-473). In 180 patients, they detected pAKT in 46% of the intracranial ependymoma tumors and 76% of the supratentorial ependymomas, and 35% of the posterior fossa ependymomas. While pAKT did not correlate significantly to overall survival, it was associated with a poorer 5-year progression-free survival in posterior fossa ependymomas, indicating not only its potential as a biomarker but also a vital role to risk stratification [Citation85]. Additionally, it has been shown that high levels of phosphorylated protein kinase PKB (AKT) and low levels of EGFR expression in glioblastoma patients decreased the response to erlotinib, an inhibitor of epidermal growth factor receptors (EGFR) [Citation86]. These studies demonstrate the diversity that can be caused by one type of modification in one single protein.
Understanding phospho-proteoforms has the potential to elucidate unknown regulatory mechanisms involved in cancer initiation [Citation87]. For example, while it is known that the phosphatase CD45 (also known as protein tyrosine phosphatase, receptor type, C, PTPRC) regulates the activation of lymphocytes [Citation88,Citation89], the specific molecules that mediate this activity are still unknown. The lymphocyte phosphatase-associated phosphoprotein (LPAP) is a good candidate for being one of these molecules because it is tightly regulated with CD45 and it has been noted that LPAP phosphorylation levels change upon activation of lymphocytes [Citation90,Citation91]. Filatov showed that LPAP has at least four sites of phosphorylation and that resting (non-activated) cells express at least six different LPAP proteoforms depending on phospho-state. Additionally, T and B cells differed in the relative abundance of the various phospho-proteoforms, indicating possible differences in their roles [Citation92].
2.3.2.2. Aberrant enzymatic activity
As previously mentioned, the regulators of signaling pathways can also have mutations that alter either kinase or phosphatase activity causing aberrant signaling. PTEN regulates the phosphoinositide3-kinase/protein kinase B/mechanistic target of rapamycin (PI3K/Akt/mTOR) cascade, by dephosphorylating phosphatidylinositol (3,4,5)-trisphosphate (PIP3) and blocking PI3K activity [Citation93] ()). However, the deletion of this gene, which is frequently seen in T-ALL, causes overactivation of this pathway [Citation94]. Furthermore, wildtype PTEN is phosphorylated at a collection of residues near the C terminus, resulting in the downregulation of PTEN phosphatase activity [Citation95]. Casein kinase 2 (CK2), a kinase responsible for PTEN phosphorylation and typically overexpressed in T-ALL, presents as a potential target for treatment [Citation96].
Several cancers are characterized by aberrant expression of various receptor tyrosine kinases (RTK), leading to extensive research of how these activated RTKs could be targets for new drugs [Citation97]. Activating mutations of the anaplastic lymphoma kinase (ALK) gene which encodes an RTK, are a major oncogenic driver of neuroblastomas [Citation98]. These mutations lead to constitutive phosphorylation of ALK and downstream signaling molecules that are required for cell proliferation and survival [Citation99]. These studies demonstrate how kinase and phosphatase inhibitors can be effective drugs.
2.3.3. Acetylation
Acetylation has been studied to a much lesser extent than phosphorylation. Although it is most often associated with histone modification, it plays a vital role in the regulation of many different types of proteins as first demonstrated on a large scale by Mann et al. [Citation100]. The number of acetylated proteins identified to be involved in various cellular processes establishes acetylation as a globally important PTM [Citation101].
Dysregulation of a normal tumor suppressing activity is a common hallmark of cancer. Sonic hedgehog medullablastoma (SHH-MB) is characterized by aberrant sonic hedgehog/GLI signaling but mechanisms of this de-regulation were unknown until Miele et al.’s investigation of this pathway. They found that miR-326 associated Arrb1 negatively regulates two components of the hedgehog pathway through mediating p300 acetylation, leading to repressed transcriptional activity of GLI1 [Citation102]. Because acetylation is a key regulatory modification, the study of acetylation and the enzymes that promote acetylation are under crucial investigation.
For example, acetyltransferases play a vital role in cancer initiation and progression and are potentially targetable. A classical role of acetylation is exemplified by the protein SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 (SMARCB1). Homozygous deletion or inactivation of SMARCB1 is commonly associated with malignant rhabdoid tumors, an aggressive tumor of young children [Citation103]. SMARCB1 also plays a role in histone acetylation making it a target for histone deacetylase inhibitors. Several studies have shown HDACi mimic acetylation function of SMARCB1 in SMARCB1-null cells, leading to clinical trials of HDACi [Citation104]. Similarly, the N-terminal acetyltransferase ARD1, a regulator of caspase activity, has been suggested as a proapoptotic enzyme required for activation of caspases [Citation105]. Knockdown of ARD1 in response to DNA damage suppressed caspase-2, caspase-3/-7, and caspase-9, presenting a potentially targetable pathway [Citation106].
2.3.4. Methylation
Similar to acetylation, methylation has in the past been generally associated with histone modifications [Citation107]; however, its role in the regulation of other proteins is becoming more widely appreciated. These non‐histone targets represent transcription factors, cell signaling molecules, and tumor suppressor proteins [Citation108]. Additionally, the clinical implications of dysregulation of methyltransferases and demethylases in the development of many diseases, including cancer, have instigated an effort to design inhibitors targeting these enzymes [Citation109].
The success of methyltransferase inhibitors was demonstrated by Mohammad et al. (2017) in his study of cyclin-dependent kinase Inhibitor 2A (CDKN2A) H3K27me3 in diffuse intrinsic pontine glioma (DIPG). As much as 80% of the DIPGs harbor a mutation in histone 3 leading to expression of H3K27M, causing global reduction of H3K27me3 levels [Citation110]. Mohammad et al. developed and studied a mouse model of DIPG in which H3K27M initiates tumorigenesis. They discovered that all H3K27M expressing cells retained H3K72me3 on the CDKN2A locus, which is an important checkpoint to prevent cells from oncogenic transformation [Citation111]. Therefore, silencing of this locus by H3K72me3 could be an important event in tumorigenesis of DIPGs. They also used this model to show that EZH2 inhibitors, an already common drug therapy in adult cancers [Citation112,Citation113], can abolish tumor cell growth by acting on the CDKN2A pathway.
Regulation of important cellular processes by methylated proteoforms is also exemplified by the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB). The transcription factor NF-kB has been found in many cancers to be constitutively active therefore contributing to oncogenesis [Citation114]. Several methylation sites on this protein have been identified, but Tao Lu et al. found that the nuclear receptor binding SET domain protein 1 (NSD1) and the demethylase F-box leucine-rich protein 11 (FBXL11) regulate NF-kB through the reversible methylation of K218 and K221 of the p65 subunit [Citation115] Methylation of these two residues leads to activated NF-kB. Overexpression of FBXL11 decreased NF-kB activity; however, when NSD1 was overexpressed, it reversed this inhibitory effect. Thus, demonstrating how PTMs on one proteoform often cooperate together to induce a phenotype and understanding of this relationship could be important for determining how to target these pathways.
2.3.5. Proteolysis
Most post-translational modification reactions are reversible. Proteolysis, in contrast, is the irreversible cleavage of the protein backbone in 2 fragments ()). Proteolysis is frequently a tightly regulated, sequence and substrate-specific process that results in one or both of the cleaved fragments remaining as stable proteoforms with often altered properties [Citation116]. Cleavage results in the formation of new protein N and C termini on the proteoforms that, in turn, can be modified leading to additional functional diversity [Citation78]. Aberrant proteolysis and the generation of alternative stable, truncated proteoforms has been linked to all hallmarks of cancer [Citation117]. Proteases are, therefore, an attractive drug target, yet realization of this potential has proven challenging as proteases and their inhibitors are highly interconnected in the protease web [Citation11]. While it remains to be seen if the proteolytic activity will be targetable in the future proteolytic proteoforms and proteolytic neo termini are already being used as process specific biomarkers [Citation118]
Proteolytic processes have several implications in cancer. The Notch signaling pathway is one of the most commonly overactivated signaling pathways in cancer, and mutations in NOTCH family proteins are detected in approximately 70% of the T-cell acute lymphoblastic leukemias [Citation119]. The pathway is regulated by two proteolytic processes by ADAM family proteins and gamma-secretases which releases the NOTCH intracellular domain leading to subsequent activation of the transcriptional complex [Citation120] ()). Because proteolytic processing of Notch by γ-secretase is essential for activation, γ-secretase inhibitors can be used to block activation. Several clinical trials for gamma-secretase inhibitors are currently open [Citation121]. Additionally, the protease dipeptidyl peptidase 4 (DDP4) has been shown to inhibit the chemokine C-X-C motif chemokine 10 (CXCL10), preventing recruitment of lymphocytes to the tumor. Inhibition of this proteolytic activity and increasing immune response to the tumor has potential in the clinic [Citation122], thus exemplifying how proteases can effectively be targeted for treatment.
As noted in the hallmarks of cancer [Citation117], cancer cells have signaling proteins that disrupt normal immune cell function, leading to improper immune responses to tumor cells [Citation123]. One of the molecules associated with this aberrant process is programmed death-ligand-1 (PD-L1) which is expressed on T-cells, B-cells, dendritic cells and natural killer T-cells to suppress anti-cancer immunity [Citation124]. The soluble form of PD-L1 (sPD-L1), created by cleavage of the membrane-bound protein, has been found in several human cancer cell lines including a glioblastoma cell line [Citation123]. Additionally, increased sPD-L1 levels in the blood are associated with metastasis and poor prognosis in diffuse large B-cell lymphoma [Citation125,Citation126]. A better understanding of the different forms of this important immune response regulator can provide insights into more successful treatments.
Another example of how proteoforms generated from proteolytic cleavage can sometimes have distinct functions was demonstrated by Casar et al. (2014) and his study of CUB domain-containing protein-1 (CDCP1). The cleavage of CDCP1 facilitates early stages of spontaneous metastasis [Citation127] and they identified the membrane-retained 70-kDa CDCP1 newly formed protein as a preferential binding partner of β1 integrin. This complex induces intracellular phosphorylation signaling involving focal adhesion kinase-1 (FAK) and PI3K-dependent AKT activation [Citation127]. Therefore, CDCP1 cleavage may represent a potential target for CDCP1-positive cancers.
As exemplified by the mentioned studies, post-translational modifications further increase the complexity of the protoeome in several contexts. As our understanding of how these modifications arise and how they alter protein function expands, more specific treatments targeting these modifications can be developed.
3. Proteogenomics
Proteome complexity drives the need to unify genomic and proteomic information. Genetics has pioneered the effort towards precision medicine, but advanced proteomic technology can further elucidate tumor biology and improve protein level detection of molecular responses. As previously indicated, genomic and transcriptomic analysis can identify potential proteoforms [Citation128], however actual abundance and additional features of these proteoforms can only be detected on the protein level. Integrating genomic and proteomic information will provide a more accurate description of molecular phenotypes. Furthermore, most MS identification methods rely on reference databases, such as Ensembl, UniProt, or RefSeq, for peptide matching which are valuable resources [Citation129]. However, these databases do not account for heterogeneity of tumor proteomes, therefore, making variant specific peptide identification difficult.
Proteogenomic workflows allow for more detailed information than attainable from either genomics or proteomics alone. This is exemplified by Yadav et al.’s work to better classify peptides bound to and presented by major histocompatibility complex (MHC) of tumor cells to be recognized by cytotoxic T cells. They developed an approach combining transcriptome sequencing and whole-exome analysis with mass spectrometry to identify neo-epitopes in two widely used murine tumor models. Of the >1,300 amino acid changes identified, ~13% were predicted to bind MHCI, a small fraction of which were confirmed by mass spectrometry [Citation130]. As immunotherapies progress, this level of classification is becoming more important.
Several proteogenomics experiments have enabled further clarification of the relationship between genomic, transcriptomic and proteomic information. In performing global proteomics on 95 colon and rectal tumors that had matched RNA and transcriptomic data, Zhang et al. first found that mRNA transcript abundance did not correlate to protein abundance differences between tumors. Additionally, they noted that copy number alterations showed strong cis- and trans- effects on mRNA but this did not translate to the protein level. Furthermore, they utilized their proteomics data to prioritize candidate driver genes, specifically within the chromosome 20q amplicon which was associated with the largest global changes at both the mRNA and protein levels [Citation131]. Another group implemented a proteogenomic workflow in PDX tumor cell lines by creating patient-specific databases using the tool Quantitative Integrated Library of Translated SNPs/Splicing (QUILTS), developed specifically for cancer proteome analysis. They detected 772 unique variant peptides across all 48 sample process replicates. Interestingly approximately 20% lacked mRNA evidence [Citation132]. Combining the genomic, transcriptomic, and proteomic information provides a more global view of identified variants.
Proteogenomics studies can also provide in-depth resolution of mechanisms within a single type of cancer. A proteogenomic study on medulloblastoma, a major pediatric brain cancer, was performed using an adapted form of a labeled MS-based technique called stable isotope labeling by/with amino acids in cell culture (SILAC) [Citation133]. A special atlas containing labeled proteins from eight medulloblastoma cell lines was developed for this study and used to comparatively analyze protein networks between the four genomic subgroups of medulloblastoma. They identified notable regulators common to all subgroups such as EGFR or proteins specific to one group such as the tumor suppressor mir-122. Therefore, exemplifying protein level information can further elucidate tumor heterogeneity.
Moving towards the incorporation of genomic and proteomic data will reveal new pathways of disease progression and possible treatment. The proteogenomics approach will also elucidate previously characterized pathways to improve variant identification and new drug targets.
4. Detection and characterization of proteoforms
So far, we have highlighted the important roles proteoforms play in cancers, focusing on childhood malignancies, and how some studies are attempting to target specific proteoforms. It is becoming apparent that the unique physiology of a cancer cell and its microenvironment can lead to specific proteoforms and/or proteoform families only found in a cancer context. However, we have yet to assemble a complete picture of all relevant proteoforms in pediatric cancer, or in fact any disease. In order for proteoforms to reach their full clinical potential, it is necessary to be able to accurately detect and quantify all the various proteoforms. In the following, we will discuss the two most widespread approaches to study proteoforms, namely antibody- and mass spectrometry-based methods [Citation6].
4.1. Antibody-based methods
The most traditional methods for direct measurement of proteins involve the use of antibodies. They are routinely used for Western blot (WB), immunoprecipitation (IP), enzyme-linked immunosorbent assays (ELISA), quantitative immunofluorescence (QIF), and immunohistochemistry (IHC) [Citation134]. These methods can prove to be useful in the clinic for in-depth characterization of various proteoforms due to their quick and reliable workflow. Western blot allows for the simultaneous assessment of the presence and quantity of individual proteoform features, such as phosphorylation, and the size of the possibly truncated proteoform [Citation135].
Antibodies can be raised to specifically recognize sequence variations or PTMs on selected proteoforms [Citation136]. For example, one of the most common mutations in oncogenic BRAF is V600F and small molecule inhibitors targeting this specific mutation are currently involved in clinical studies [Citation137]. These mutations are most commonly detected by DNA-based technologies, such as sequencing [Citation138,Citation139]. However, Capper et al. (2011) developed a monoclonal antibody for BRAF V600E to differentiate between this oncogenic proteoform and the wild-type form. However, these assays are limited to the specific variants they are designed for and require highly selective antibodies to avoid off-target capture [Citation140].
IHC assays can also be a valuable tool in the clinic due to their high sensitivity and high throughput ability. IHC assays are used to detect protein markers in biopsies to support cancer diagnosis, prognosis, and monitoring [Citation141]. For example, Mino-kinudson et al. (2010) utilized IHC assays to assess specific ALK rearrangements in adenocarcinomas [Citation142]. These EML4-ALK fusions are found in approximately 5% of the lung adenocarcinomas and could be a potential target for therapy. Using novel highly sensitive antibody ALK they were able to detect ALK rearrangements with increased sensitivity and reliability that has potential to be routinely applied in the clinic [Citation142]. Although they have many advantages, some limitations of IHC assays include the efficiency of antibodies, limited specificity, and cross-reactivity of the antibodies [Citation140].
Protein microarrays, a staple of proteomics studies, have been a successful tool for studying various protein-related events, protein expression, interaction, function, and post-translational modifications [Citation143]. Arrays can be designed to cover thousands of proteins and have several purposes including glycopeptide microarrays [Citation144], protease activity [Citation145], and activity of small molecule inhibitors [Citation146]. Microarrays can be an important tool for looking at specific known modifications or as a screen for potential drugs, but they are constricted by library size and potential changes from the fixation process to the chip are a cause for concern [Citation143].
Although not an antibody-based assay, fluorescence in situ hybridization (FISH) is commonly used in the clinic to detect structural abnormalities such as chromosomal amplifications or fusions [Citation147]. FISH probes can be designed for specific variants or gene fusions that are already known, which makes for an efficient assay and helpful diagnostic. For example, it can detect multiple partners of the mixed lineage leukemia gene [Citation44]. Similar to antibody-based assays, FISH cannot detect novel variants and is dependent on the quality and targets of existing probes [Citation42].
Fuzery et al. do an excellent job of outlining clinically validated assays for protein biomarker identification, the majority of which are immunoassays. These assays are applicable to a wide variety of clinical samples including plasma and FFPE tissues [Citation148]. While antibody-based methods are still a gold standard to distinguish proteoforms, the limitations of antibody sensitivity in addition to the lack of resolution to accurately detect proteoforms have led to increased efforts in utilizing mass spectrometry (MS) for proteoform detection.
4.2. Mass spectrometry-based methods
Mass spectrometry (MS) is gaining increasing interest in its ability to accurately detect and distinguish between proteoforms based on molecular mass and other physicochemical parameters [Citation149]. Traditionally, bottom-up proteomics has been used for global and targeted proteomics assays because it is highly sensitive and most MS technology is developed for this workflow [Citation150] ()). However, in many instances of proteoform detection, a more comprehensive analysis is needed, which has led to increased use of top-down and middle-down proteomic methods [Citation151,Citation152] ()). First, we will discuss how more traditional bottom-up MS techniques are being implemented by looking at a few notable cases, then focus on how proteomics techniques are improving with the use of top-down and middle-down proteomics.
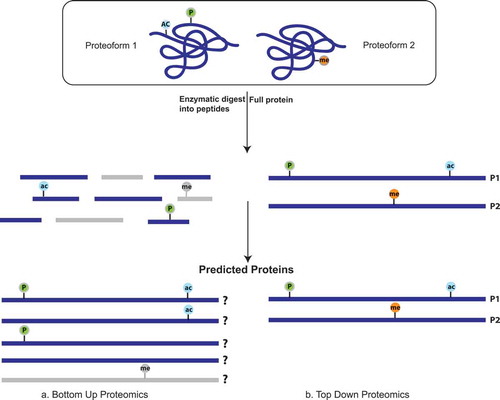
Figure 3. (a) In a bottom-up workflow, proteins are digested into peptides before sequencing by mass spectrometry and then analytically pieced back together when matched to a reference proteome. This decreases the resolution of proteoform detection because modifications may either be lost entirely (in gray) during digestion, or the peptides derived from the distinct proteoforms are indistinguishable from each other. (b) In top-down proteomics, there is no digestion before mass spectrometric analysis and therefore the full protein is sequenced allowing for increased coverage of each protein. In this way, it is possible to distinguish the various proteoforms within a sample.
4.2.1. Bottom-up proteomics
The bottom-up proteomics workflow involves enzymatic digestion of proteins into peptides before sequencing by mass spectrometry. The peptides are then computationally reassembled by matching to a reference proteome. Therefore, it is difficult to determine if modifications or variations found on separate peptides originate from the same proteoform or distinct proteoforms (-b)). Although detection of specific proteoforms is challenging, this method can be used to predict the presence of proteoform families, that may have distinct biological functions in a given condition. Unlike antibody-based methods, bottom-up proteomics is a more robust approach to study the features of proteoform families, including PTMs and expression level.
For targeted studies on proteoforms and proteoform families, multiple reaction monitoring (MRM) and parallel reaction monitoring (PRM) are the most commonly used bottom-up based techniques [Citation153]. They provide sensitive and specific quantification for a set of pre-defined proteins. Fu et al. used constrained MRM assays to quantitate distinct isoforms and PTMs implicated in various diseases [Citation154]. They outlined the development of their method including the selection of fragment ions, optimization of specific MS parameters, the establishment of calibration curves, among several other steps. Whiteaker developed a 62plex assay to quantify protein expression and PTMs such as phosphorylation and ubiquitination after induction of DNA damage demonstrating the utility of a quantitative multiplexed assay for studying cell signaling dynamics [Citation155,Citation156]. However, MRM and PRM methods fall short of identifying new proteoforms.
Shotgun or global proteomics is essential for full characterization of a proteome or deep interrogation of a specific pathway. Clinical application of these MS-based techniques was exemplified in the work by Doll et al.’s efforts to streamline a proteomic workflow [Citation157]. They took samples from a patient diagnosed with urachal carcinoma lung metastases. Utilizing a single vial sample preparation and label-free workflow on a quadrupole – Orbitrap mass spectrometer, followed by Maxquant for the quantification, they quantified 4857 proteins. Notably, LSD1, a druggable target, was discovered to be significantly upregulated. From biopsy to results of MS analysis took 6 days, and within 1 month they had approval for LSD1 inhibitor treatment, exemplifying how mass spectrometry can be translated to the clinic.
Another group took a computational approach to predict the occurrence of proteoforms [Citation158]. They focused on the limitation of existing protein quantification methods that have difficulty deciphering protein variation in MS shotgun data. They created a Bayesian model using statistically derived peptide signatures to segregate peptides that appear to be different than the canonical or most abundant form and assign these peptides to potential proteoform configurations. They further employed the over-expressed peptide signatures to estimate the relative abundance of identified proteoforms. To verify their approach, they performed a dilution study using shotgun proteomics on mouse plasma and found that their method performed with similar accuracy as current automated methods for proteoform detection.
4.2.2. Top-down proteomics
As mentioned, bottom-up proteomics has been successfully used for detection of proteoform families but is still limited by the loss of contextual information as a result of the required digestion into peptides. As technology advances top-down proteomics is emerging as a more popular tool for studying proteoforms. Top-down mass spectrometry allows for the determination of amino acid sequences and their PTMs without first digesting proteins into peptides. This provides more precise compositional information and adds molecular details which are typically lost when proteins are first digested into peptides as done in bottom-up proteomics [Citation22,Citation159] (). Mass spectrometry of complete proteins is the most optimal method to detect de novo proteoforms with complex PTM patterns [Citation6]. Several studies have already indicated clinical application for top-down proteomics, but most notable is its use for studying intact biomarkers that have been identified as candidate proteoform biomarkers in cerebrospinal fluid of pediatric brain tumor prognosis [Citation160].
Another exemplary clinical application is shown by Trenchevska’s method for proteoform detection using mass spectrometric immunoassays (MSIA) [Citation161]. This procedure utilizes a type of top-down proteomics, Matrix Assisted Laser Desorption/Ionization and mass spectrometry imaging (MALDI and MSI) combined with an immobilization capture that involves single or multiple antibodies towards the targeted protein(s). MSIA can overcome a few of the drawbacks associated with MS-based protein assays. The sample preparation is simple and high-throughput and analyzes intact proteins so there is high conservation of amino acid sequence. Furthermore, detection and identification are based solely on molecular mass which adds to specificity. To date, this assay has been shown to detect several diseases associated proteins, including biomarkers linked to type 2 diabetes and cardiovascular disease.
Additionally, the Kelleher lab has done exceptional work utilizing mass spectrometry to study proteoforms. Most notable is their in-depth analysis of KRAS, specifically the G13D proteoform (KRASb), which lead to the development of a targeted top-down proteomics assay to quantitate different KRAS proteoforms, thus demonstrating the potential clinical utility of top-down assays [Citation162].
There is currently one FDA approved top-down proteomics instrument, the Bruker MALDI Biotyper system. This is a MALDI Time-of-Flight (MALDI-TOF) system that is used by clinical laboratories to identify microorganisms by measuring their abundant proteins. This is a great step towards bringing mass spectrometry to the clinic, and future technological advances may allow for more clinically applicable instruments and assays.
Top-down proteomics has many advantages; however, it is still limited by suboptimal front-end separation of proteins and reduced sensitivity. To combine the sensitivity of bottom-up proteomics with the comprehensiveness of top-down proteomics, middle-down proteomics is becoming a prominent option for studying proteoforms [Citation163]. In this method, there is still a digestion step; however, the proteins are digested into much larger peptides than in the bottom-up workflows. The larger peptides allow for comprehensive sequence coverage and protein resolution in a more timely and efficient manner than top-down. Jiang et al. (2018) show the incredible utility and benefit of middle down proteomics in their characterization of Histone H4 [Citation164]. They quantified 233 proteoforms of H4 in two different breast cancer cell lines and were able to detect changes in phosphorylation, methylation, and acetylation throughout the cell cycle, demonstrating the high resolution of their workflow.
Antibody-based techniques for protein and proteoform detection have proven to be clinically useful over the decades and will continue to play a significant role in diagnostics and disease monitoring. However, the advancements in mass spectrometry techniques have paved way for unrivaled proteoform discrimination. As this technology continues to improve, the clinical applications of mass spectrometry will be infinite.
5. Challenges and prospects
Cancer-specific proteoforms that show altered properties compared to their canonical protein have the potential to be targeted by activating or deactivating agents. With the active development of technology and bioinformatics tools, existing approaches used to study aberrations at the level of proteins has greatly improved proteoform detection; however, there are still limits to what can be done.
For example, mass spectrometry has proven to be a valuable tool, especially for proteogenomic approaches. A few challenges remain that will likely improve in the coming years. For example, not all peptides in a sample can be detected due to the dynamic range in protein abundance and the lack of selection of all parent ions for fragmentation [Citation10]. Especially since many loss-of-function aberrations lead to improper folding, there is an increased chance these proteins will be degraded and therefore in low abundance [Citation10,Citation165]. New proteomic approaches to increase comprehensive coverage and sensitivity are evolving, including the previously mentioned top-down and middle-down proteomics in addition to data-dependent acquisition (DIA) [Citation26,Citation166].
Another challenge is differentiating between passenger and driver mutations and clinical relevance of potential proteoform biomarkers. For example, hundreds of biomarker candidates have been identified or proposed in various cancers; however, only a small number are clinically qualified. The majority of suggested biomarkers have either unknown or unclarified biological significance [Citation148]. Furthermore, for those that are theoretically suitable, they may not exhibit consistent or sufficient expression to warrant therapy development [Citation167]. Deep proteomic profiling combined with functional characterization studies on a large scale will aid in resolving these unknowns.
It becomes increasingly apparent that the onco-proteogenomic approach to combine genomics, transcriptomics, and proteomics on a patient-specific basis is the most comprehensive analysis to reveal potentially actionable proteoforms in childhood cancers. It is essential to understand the connection between genomic sequence and proteomic activity when trying to develop effective treatments [Citation129,Citation168,Citation169]. Furthermore, this sample specific approach utilizing custom search databases has shown lower false-positive and false-negative rates making mass spectrometry data increasingly reliable [Citation159].
Shifting towards mass spectrometry-based research methods will considerably simplify the process of a biomarker search and validation, especially for proteoforms that are difficult to detect otherwise [Citation170]. It will also facilitate the extension of a proteogenomic assessment by metabolite analysis based on the same technological platform. In the future, the development of mass spectrometry-based assays in the context of onco-proteogenomics has great potential to allow the evaluation of potential risks of disease, diagnose diseases, and monitor the efficiency of treatment. Overall, this will contribute to the more informative selection of proper therapies on an individual basis.
6. Conclusion
Proteoforms generated by either genome level, transcript level, or protein level alterations have various implications in childhood malignancies. As demonstrated by many pediatric focused studies, it is important to consider the unique molecular landscapes of pediatric cancers when determining biomarkers or potential targets for therapy. Current and expected technological improvements in next-generation sequencing and advanced mass spectrometry will advance our ability to elucidate new pathways for treatment and facilitate more widespread clinical applications of proteomics. This carries the promise to make precision medicine more accurate by adding information about the proteoforms that constitute the drug targets and downstream signaling pathways.
7. Expert opinion
Chemotherapy and radiation remain the most common methods of treatment for cancers. Recent advancements in technology have led to a better understanding of the underlying mechanisms of tumorigenesis which has allowed for increased efforts in developing more effective and less harmful treatments based on direct targeting of altered proteins and deregulated pathways. These advances would not have been possible without the tremendous improvements in next-generation sequencing over the past decade. There is now a variety of sequencing platforms that provide widespread access for a diverse set of needs at a reasonable cost. For example, while whole genome sequencing is traditionally the most informative method of sequencing, whole exome-sequencing, RNA-sequencing, and amplicon based targeted sequencing are able to provide important information at lower cost and shorter turnarounds times. Specifically, targeted sequencing is becoming more profoundly useful in the clinic due to its simple workflow, quick turnaround time, and high read depth of specific genes of interests [Citation171]. There are hundreds of panels commercially available that address a wide variety of clinical needs. These advances in sequencing now allow us to classify cancers based on genetic variation and not tissue of origin and reveal possible drug and cell therapy targets at an unprecedented rate.
As pointed out before, most new therapies target proteins and protein pathways, yet we currently predict their effectiveness based on variants detected in the genome or transcripts. It is no surprise that cancer affects the proteome in many different ways and the correlation between genome or transcriptome and proteome is often low. Resolving this complexity is thus critical to developing better treatments and a better understanding of treatment efficacy. Advancements in mass spectrometry have exponentially increased our knowledge of the proteome and the many proteoforms. Traditional bottom-up methods are still widely in use and will for the next years remain the method of choice for studies on defined proteoforms/proteoform families, such as fusions, CNVs or kinase activating phosphorylation in a clinical context. At the same time, newer methods such as top-down and middle down proteomics are making headway. These methods allow for in-depth coverage of the proteome and increase the ability to detect even complex combinatorial proteoforms [Citation22].
Where we see the most need for change is in data compilation and database availability. A few databases contain information on proteoforms for a given protein; however, the proteoforms often remain grouped under a single canonical protein rather than independent and unique proteoforms. It is essential to recognize that proteoforms often have distinct functions and should be treated as such [Citation17]. The inclusion of all proteoforms in the interpretation of proteomic analysis is paramount to obtain comprehensive and biologically accurate results. While the number of identified proteoforms increases exponentially, our ability to determine the specific function of each proteoform is not keeping up. This poses a significant challenge comparable to the time when new genes were identified at a rapid pace while the knowledge about the encoded proteins lagged behind. While some proteoforms differ fundamentally, most proteoforms will likely not differ in function. High-throughput functional characterization of all theoretically possible proteoforms, even if technically feasible in the future, would thus be futile if most theoretical proteoforms do not occur in vivo and do not differ in function. We and others are, therefore, working on computational methods to cluster proteoforms, deconvolute their functions, and provide databases that enable analysis and functional annotation of mass spectrometry data at proteoform resolution.
8. 5-year view
Along these lines, the proteogenomics approach, although relatively new has made significant strides in comprehensive coverage of the genome and proteome. This is largely due to the advancements in bioinformatic tools that allow for the integration of large and complex datasets [Citation74]. We foresee the field of biomarker identification and validation significantly improving over the next five years due to this approach. Studies interrogating biological and disease systems will see a boost from several bioinformatics developments that will not only improve analysis workflows but will provide extensive new information and advanced databases enabling functional interpretation of biological systems at an unprecedented level.
Implementing proteogenomics workflows in clinical settings has the ability to improve precision medicine and patient-specific selection of optimal treatment targets. Developing and validating assays that are applicable in a clinical setting will remain challenging. However, current developments in robust sample preparation methods, rapid liquid chromatography and dropping prices for current instrument technology beyond triple-quadrupoles will provide the basis for mass spectrometry to make a much larger impact in the clinic than we have seen in the past years.
Article highlights
Current treatments for childhood cancers are aggressive, and these cancers have a high incidence of relapse that is often refractory to traditional treatments, leading to increased efforts to develop new therapies.
Proteins are essential effectors of cellular function and are used as biomarkers and drug targets. Similarly, specific proteoforms can be detected and targeted for treatment.
The term ‘proteoform’ applies to all the different proteins derived from a single gene, including all forms of genetic variation, alternative splicing, and post-translational modifications.
We have yet to assemble a complete picture of all relevant proteoforms in pediatric cancer, or in fact any disease. In order for proteoforms to reach their full clinical potential, it is necessary to be able to accurately detect and quantify all the various proteoforms.
Mass spectrometry of complete proteins is the current best method for identifying distinct proteoforms with complex PTM patterns.
In the future, the development of mass spectrometry-based assays in the context of onco-proteogenomics has great potential to allow the evaluation of potential risks of disease, diagnose diseases, and monitor the efficiency of treatment.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
The authors would like to acknowledge Christopher Maxwell, PhD (BC Children’s Hospital Research Institute) as a co-supervisor of Amanda Lorentzian.
Additional information
Funding
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30.
- Gröbner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555(7696):321–327.
- Pui C-H, Gajjar AJ, Kane JR, et al. Challenging issues in pediatric oncology. Nat Rev Clin Oncol. 2011;8(9):540–549.
- Tran TH, Shah AT, Loh ML. Precision medicine in pediatric oncology: translating genomic discoveries into optimized therapies. Clin Cancer Res. 2017;23(18):5329–5338.
- Izquierdo E, Yuan L, George S, et al. Development of a targeted sequencing approach to identify prognostic, predictive and diagnostic markers in paediatric solid tumours. Oncotarget. 2017;8(67):112036–112050.
- Kiseleva OI, Lisitsa AV, Poverennaya EV. Proteoforms: methods of analysis and clinical prospects. Mol Biol (NY). 2018;52(3):335–349.
- Sireci AN, Aggarwal VS, Turk AT, et al. Clinical genomic profiling of a diverse array of oncology specimens at a large academic cancer center: identification of targetable variants and experience with reimbursement. J Mol Diagn. 2017;19(2):277–287.
- Ross DS, Zehir A, Cheng DT, et al. Next-generation assessment of human epidermal growth factor receptor 2 (ERBB2) amplification status: clinical validation in the context of a hybrid capture. J Mol. 2017;19(2)244-254.
- Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555(7696):371–376.
- Alfaro JA, Sinha A, Kislinger T, et al. Onco-proteogenomics: cancer proteomics joins forces with genomics. Nat Methods. 2014;11(11):1107–1113.
- Fortelny N, Overall CM, Pavlidis P, et al. Can we predict protein from mRNA levels? Nature. 2017;547(7664):E19–E20.
- Tuch BB, Laborde RR, Xu X, et al. Tumor transcriptome sequencing reveals allelic expression imbalances associated with copy number alterations. PLoS One. 2010;5(2):e9317.
- Kislinger T, Cox B, Kannan A, et al. Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell. 2006;125(1):173–186.
- Gygi SP, Rochon Y, Franza BR, et al. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19(3):1720–1730.
- Akbani R, Ng PKS, Werner HMJ, et al. A pan-cancer proteomic perspective on the cancer genome atlas. Nat Commun. 2014;5:3887.
- Ning K, Fermin D, Nesvizhskii AI. Comparative analysis of different label-free mass spectrometry based protein abundance estimates and their correlation with RNA-Seq gene expression data. J Proteome Res. 2012;11(4):2261–2271.
- Aebersold R, Agar JN, Amster IJ, et al. How many human proteoforms are there? Nat Chem Biol. 2018;14(3):206–214.
- Bamberger C, Martínez-Bartolomé S, Montgomery M, et al. Deducing the presence of proteins and proteoforms in quantitative proteomics. Nat Commun. 2018;9(1):2320.
- Jungblut P, Thiede B, Zimny-Arndt U, et al. Resolution power of two-dimensional electrophoresis and identification of proteins from gels. Electrophoresis. 1996;17(5):839–847.
- Ponomarenko EA, Poverennaya EV, Ilgisonis EV, et al. The size of the human proteome: the width and depth. Int J Anal Chem. 2016;7436849:2016.
- Smith LM, Kelleher NL. Consortium for top down proteomics. Proteoform: a single term describing protein complexity. Nat Methods. 2013;10(3):186–187.
- Toby TK, Fornelli L, Kelleher NL. Progress in top-down proteomics and the analysis of proteoforms. Annu Rev Anal Chem (Palo Alto, Calif). 2016;9(1):499–519.
- Rodriguez H, Pennington SR. Revolutionizing precision oncology through collaborative proteogenomics and data sharing. Cell. 2018;173(3):535–539.
- Drabovich AP, Martínez-Morillo E, Diamandis EP. Toward an integrated pipeline for protein biomarker development. Biochim Biophys Acta. 2015;1854(6):677–686.
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207.
- Uzozie AC, Aebersold R. Advancing translational research and precision medicine with targeted proteomics. J Proteomics. 2018;189:1–10.
- He Q, He Q, Liu X, et al. Genome-wide prediction of cancer driver genes based on SNP and cancer SNV data. Am J Cancer Res. 2014;4(4):394–410.
- Chmielecki J, Bailey M, He J, et al. Genomic profiling of a large set of diverse pediatric cancers identifies known and novel mutations across tumor spectra. Cancer Res. 2017;77(2):509–519.
- Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373(24):2336–2346.
- Lindqvist CM, Lundmark A, Nordlund J, et al. Deep targeted sequencing in pediatric acute lymphoblastic leukemia unveils distinct mutational patterns between genetic subtypes and novel relapse-associated genes. Oncotarget. 2016;7(39):64071–64088.
- Lee B, Lee JW, Shim JH, et al. Clinical relevance of genomic changes in recurrent pediatric solid tumors. Transl Oncol. 2018;11(6):1390–1397.
- Malaney P, Uversky VN, Davé V. PTEN proteoforms in biology and disease. Cell Mol Life Sci. 2017;74(15):2783–2794.
- Papa A, Wan L, Bonora M, et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell. 2014;157(3):595–610.
- Costa HA, Leitner MG, Sos ML, et al. Discovery and functional characterization of a neomorphic PTEN mutation. Proc Natl Acad Sci USA. 2015;112(45):13976–13981.
- Kiessling M, Rogler G. Targeting the RAS pathway by mitogen-activated protein kinase inhibitors. Swiss Med Wkly. 2015;145:w14207.
- C-H P, Mv R, Jr D. Acute lymphoblastic leukemia. N Engl J Med. 2004;350(15):1535–1548.
- Malinowska-Ozdowy K, Frech C, Schönegger A, et al. KRAS and CREBBP mutations: a relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia. 2015;29(8):1656–1667.
- Eleveld TF, Oldridge DA, Bernard V, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet. 2015;47(8):864–871.
- Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene. 2013;32(21):2601–2613.
- Liu X, Qu C-K. Protein tyrosine phosphatase SHP-2 (PTPN11) in hematopoiesis and leukemogenesis. J Signal Transduct. 2011;195239:2011.
- Wu G, Diaz AK, Paugh BS, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46(5):444–450.
- Mertens F, Tayebwa J. Evolving techniques for gene fusion detection in soft tissue tumours. Histopathology. 2014;64(1):151–162.
- Beadling C, Wald AI, Warrick A, et al. A multiplexed amplicon approach for detecting gene fusions by next-generation sequencing. J Mol Diagn. 2016;18(2):165–175.
- Mertens F, Johansson B, Fioretos T, et al. The emerging complexity of gene fusions in cancer. Nat Rev Cancer. 2015;15(6):371–381.
- Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol. 2017;35(9):975–983.
- Marschalek R. MLL-r Leukemia. J Leuk. 2016;4(3).
- Pui C-H, Carroll WL, Meshinchi S, et al. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol. 2011;29(5):551–565.
- Teachey DT, Hunger SP. Predicting relapse risk in childhood acute lymphoblastic leukaemia. Br J Haematol. 2013;162(5):606–620.
- Ney Garcia DR, de Souza MT, de Figueiredo AF, et al. Molecular characterization of KMT2A fusion partner genes in 13 cases of pediatric leukemia with complex or cryptic karyotypes. Hematol Oncol. 2016;35(4):760-768.
- Daigle SR, Olhava EJ, Therkelsen CA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122(6):1017–1025.
- Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015;15(6):334–346.
- Pavlick D, Schrock AB, Malicki D, et al. Identification of NTRK fusions in pediatric mesenchymal tumors. Pediatr Blood Cancer. 2017;64(8):e26433.
- Doebele RC, Davis LE, Vaishnavi A, et al. An oncogenic NTRK fusion in a soft tissue sarcoma patient with response to the tropomyosin-related kinase (TRK) inhibitor LOXO-101. Cancer Discov. 2015;5:1049–1057.
- Sartore-Bianchi A, Ardini E, Bosotti R, et al. Sensitivity to entrectinib associated with a novel LMNA-NTRK1 gene fusion in metastatic colorectal cancer. J Natl Cancer Inst. 2016;108(1).
- Farago AF, Le LP, Zheng Z, et al. Durable clinical response to entrectinib in NTRK1-rearranged non-small cell lung cancer. J Thorac Oncol. 2015;10(12):1670–1674.
- Sun C, Chang L, Zhu X. Pathogenesis of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia and mechanisms underlying its relapse. Oncotarget. 2017;8(21):35445–35459.
- Tasian SK, Hunger SP. Genomic characterization of paediatric acute lymphoblastic leukaemia: an opportunity for precision medicine therapeutics. Br J Haematol. 2017;176(6):867–882.
- Lee Y, Rio DC. Mechanisms and regulation of alternative Pre-mRNA splicing. Annu Rev Biochem. 2015;84:291–323.
- Sveen A, Kilpinen S, Ruusulehto A, et al. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene. 2016;35(19):2413–2427.
- Singh B, Eyras E. The role of alternative splicing in cancer. Transcription. 2017;8(2):91–98.
- Uversky VN. p53 proteoforms and intrinsic disorder: an illustration of the protein structure-function continuum concept. Int J Mol Sci. 2016;17(11):1874.
- Chen J, Weiss WA. Alternative splicing in cancer: implications for biology and therapy. Oncogene. 2015;34(1):1–14.
- Oltean S, Bates DO. Hallmarks of alternative splicing in cancer. Oncogene. 2014;33(46):5311–5318.
- Bourdon J-C, Fernandes K, Murray-Zmijewski F, et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005;19(18):2122–2137.
- Aldape K, Zadeh G, Mansouri S, et al. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):829–848.
- Inda -M-M, Bonavia R, Mukasa A, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010;24(16):1731–1745.
- Zhang M, Zhu B, Davie J. Alternative splicing of MEF2C pre-mRNA controls its activity in normal myogenesis and promotes tumorigenicity in rhabdomyosarcoma cells. J Biol Chem. 2015;290(1):310–324.
- MATCH Target and Agent Prioritization. Target and agent prioritization for the children’s oncology group—national cancer institute pediatric MATCH trial. National Cancer. 2017
- Abou-Fayçal C, Hatat A-S, Gazzeri S, et al. Splice variants of the RTK family: their role in tumour progression and response to targeted therapy. Int J Mol Sci. 2017;18(2):383.
- Sotillo E, Barrett DM, Black KL, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5(12):1282–1295.
- Duan G, Walther D. The roles of post-translational modifications in the context of protein interaction networks. PLoS Comput Biol. 2015;11(2):e1004049.
- The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45(D1):D158–D169.
- Filtz TM, Vogel WK, Leid M. Regulation of transcription factor activity by interconnected post-translational modifications. Trends Pharmacol Sci. 2014;35(2):76–85.
- Shukla HD. Comprehensive analysis of cancer-proteogenome to identify biomarkers for the early diagnosis and prognosis of cancer. Proteomes. 2017;5(4):28.
- Huang L-C, Ross KE, Baffi TR, et al. Integrative annotation and knowledge discovery of kinase post-translational modifications and cancer-associated mutations through federated protein ontologies and resources. Sci Rep. 2018;8(1):6518.
- Thygesen C, Boll I, Finsen B, et al. Characterizing disease-associated changes in post-translational modifications by mass spectrometry. Expert Rev Proteomics. 2018;15(3):245–258.
- Pagel O, Loroch S, Sickmann A, et al. Current strategies and findings in clinically relevant post-translational modification-specific proteomics. Expert Rev Proteomics. 2015;12(3):235–253.
- Lange PF, Overall CM, Protein TAILS. when termini tell tales of proteolysis and function. Curr Opin Chem Biol. 2013;17(1):73–82.
- Riley NM, Coon JJ. Phosphoproteomics in the age of rapid and deep proteome profiling. Anal Chem. 2016;88(1):74–94.
- Ardito F, Giuliani M, Perrone D. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy. Int J Mol Med. 2017;40(2):271-290.
- Hainaut P, Plymoth A. Targeting the hallmarks of cancer: towards a rational approach to next-generation cancer therapy. Curr Opin Oncol. 2013;25(1):50–51.
- Sawyers CL. Shifting paradigms: the seeds of oncogene addiction. Nat Med. 2009;15(10):1158–1161.
- Gibson P, Tong Y, Robinson G, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468(7327):1095–1099.
- Archer TC, Ehrenberger T, Mundt F, et al. Proteomics, post-translational modifications, and integrative analyses reveal molecular heterogeneity within medulloblastoma subgroups. Cancer Cell. 2018;34(3):396–410.e8.
- Li AM, Dunham C, Tabori U, et al. EZH2 expression is a prognostic factor in childhood intracranial ependymoma: a Canadian pediatric brain tumor consortium study. Cancer. 2015;121(9):1499–1507.
- Haas DA. Epidermal growth factor receptor, protein kinase B/ Akt,and glioma response to erlotinib. J Natl Cancer Inst. 2005;97(12):880–887.
- Singh V, Ram M, Kumar R, et al. Phosphorylation: implications in cancer. Protein J. 2017;36(1):1–6.
- Trowbridge IS, Thomas ML. CD45: an emerging role as a protein tyrosine phosphatase required for lymphocyte activation and development. Annu Rev Immunol. 1994;12:85–116.
- Ledbetter JA, Tonks NK, Fischer EH, et al. CD45 regulates signal transduction and lymphocyte activation by specific association with receptor molecules on T or B cells. Proc Natl Acad Sci USA. 1988;85(22):8628–8632.
- Takeda A, Wu JJ, Maizel AL. Evidence for monomeric and dimeric forms of CD45 associated with a 30-kDa phosphorylated protein. J Biol Chem. 1992;267(23):16651–16659.
- Mayya V, Lundgren DH, Hwang S-I, et al. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci Signal. 2009;2(84):ra46.
- Filatov A, Kruglova N, Meshkova T, et al. Lymphocyte phosphatase-associated phosphoprotein proteoforms analyzed using monoclonal antibodies. Clin Transl Immunology. 2015;4(10):e44.
- Haddadi N, Lin Y, Travis G, et al. PTEN/PTENP1: ’Regulating the regulator of RTK-dependent PI3K/Akt signalling’, new targets for cancer therapy. Mol Cancer. 2018;17(1):37.
- Evangelisti C, Chiarini F, McCubrey JA, et al. Therapeutic targeting of mTOR in T-cell acute lymphoblastic leukemia: an update. Int J Mol Sci. 2018;19(7):1878.
- Silva A, Yunes JA, Cardoso BA, et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest. 2008;118(11):3762–3774.
- Buontempo F, McCubrey JA, Orsini E, et al. Therapeutic targeting of CK2 in acute and chronic leukemias. Leukemia. 2018;32(1):1–10.
- Crose LES, Linardic CM. Receptor tyrosine kinases as therapeutic targets in rhabdomyosarcoma. Sarcoma. 2011;756982:2011.
- Matthay KK, Maris JM, Schleiermacher G, et al. Neuroblastoma. Nat Rev Dis Primers. 2016;2:16078.
- Matthay KK, George RE, Yu AL. Promising therapeutic targets in neuroblastoma. Clin Cancer Res. 2012;18(10):2740–2753.
- Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–840.
- Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. 2015;16(4):258–264.
- Miele E, Po A, Begalli F, et al. β-arrestin1-mediated acetylation of Gli1 regulates Hedgehog/Gli signaling and modulates self-renewal of SHH medulloblastoma cancer stem cells. BMC Cancer. 2017;17(1):488.
- Kim KH, Roberts CWM. Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet. 2014;207(9):365–372.
- Muscat A, Popovski D, Jayasekara WSN, et al. Low-dose histone deacetylase inhibitor treatment leads to tumor growth arrest and multi-lineage differentiation of malignant rhabdoid tumors. Clin Cancer Res. 2016;22(14):3560–3570.
- Yi CH, Sogah DK, Boyce M, et al. A genome-wide RNAi screen reveals multiple regulators of caspase activation. J Cell Biol. 2007;179(4):619–626.
- Zamaraev AV, Kopeina GS, Prokhorova EA, et al. Post-translational modification of caspases: the other side of apoptosis regulation. Trends Cell Biol. 2017;27(5):322–339.
- Biggar KK, Li SS-C. Non-histone protein methylation as a regulator of cellular signalling and function. Nat Rev Mol Cell Biol. 2015;16(1):5–17.
- Carr SM, Poppy Roworth A, Chan C, et al. Post-translational control of transcription factors: methylation ranks highly. FEBS J. 2015;282(23):4450–4465.
- Hü K, Konze KD, Jin J. Selective inhibitors of protein methyltransferases. J Med Chem. 2015;58(4):1596–1629.
- Lewis PW, Müller MM, Koletsky MS, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340(6134):857–861.
- Mohammad F, Weissmann S, Leblanc B, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med. 2017;23(4):483–492.
- Knutson SK, Wigle TJ, Warholic NM, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8(11):890–896.
- McCabe MT, Ott HM, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–112.
- Karin M. NF-κB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1:a000141-a000141.
- Lu T, Jackson MW, Wang B, et al. Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc Natl Acad Sci USA. 2010;107(1):46–51.
- Klein T, Eckhard U, Dufour A, et al. Proteolytic cleavage-mechanisms, function, and “Omic” approaches for a near-ubiquitous posttranslational modification. Chem Rev. 2018;118(3):1137–1168.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674.
- Huesgen PF, Lange PF, Overall CM. Ensembles of protein termini and specific proteolytic signatures as candidate biomarkers of disease. Proteomics Clin Appl. 2014;8(5–6):338–350.
- Liu Y, Easton J, Shao Y, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49(8):1211–1218.
- Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling–are we there yet? Nat Rev Drug Discov. 2014;13(5):357–378.
- Yuan X, Wu H, Xu H, et al. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett. 2015;369(1):20–27.
- Barreira Da Silva R, Me L, Yatim N, et al. Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat Immunol. 2015;16(8):850–858.
- Chen J, Jiang CC, Jin L, et al. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol. 2016;27(3):409–416.
- Sharpe AH, Wherry EJ, Ahmed R, et al. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8(3):239–245.
- Rossille D, Gressier M, Damotte D, et al. High level of soluble programmed cell death ligand 1 in blood impacts overall survival in aggressive diffuse large B-Cell lymphoma: results from a French multicenter clinical trial. Leukemia. 2014;28(12):2367–2375.
- Junjun C, Hongbing S, Xiao Z, et al. Detection of soluble B7-H4 molecules in serum of patients with breast cancer and its clinical significance. J Int. 2013;1(4):215-218.
- Casar B, Rimann I, Kato H, et al. In vivo cleaved CDCP1 promotes early tumor dissemination via complexing with activated β1 integrin and induction of FAK/PI3K/Akt motility signaling. Oncogene. 2014;33(2):255–268.
- Dimitrakopoulos L, Prassas I, Diamandis EP, et al. Onco-proteogenomics: multi-omics level data integration for accurate phenotype prediction. Crit Rev Clin Lab Sci. 2017;54(6):414–432.
- Woo S, Cha SW, Na S, et al. Proteogenomic strategies for identification of aberrant cancer peptides using large-scale next-generation sequencing data. Proteomics. 2014;14(23–24):2719–2730.
- Yadav M, Jhunjhunwala S, Phung QT, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515(7528):572–576.
- Zhang B, Wang J, Wang X, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513(7518):382–387.
- Ruggles KV, Tang Z, Wang X, et al. An analysis of the sensitivity of proteogenomic mapping of somatic mutations and novel splicing events in cancer. Mol Cell Proteomics. 2016;15(3):1060-1071.
- Rivero-Hinojosa S, Lau LS, Stampar M, et al. Proteomic analysis of Medulloblastoma reveals functional biology with translational potential. Acta Neuropathol Commun. 2018;6(1):48.
- Solache R, Brockett T, Chuang DR, et al. Evolving strategies for application-specific validation of research use antibodies. Drug. 2013.
- Sawasdikosol S. Detecting tyrosine-phosphorylated proteins by Western blot analysis. Curr Protoc Immunol 2010. Chapter 11, Unit 11.3.1-11.
- Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol. 2003;21(3):255–261.
- Arkenau HT, Kefford R, Long GV. Targeting BRAF for patients with melanoma. Br J Cancer. 2011;104(3):392–398.
- Arcila M, Lau C, Nafa K, et al. Detection of KRAS and BRAF mutations in colorectal carcinoma roles for high-sensitivity locked nucleic acid-PCR sequencing and broad-spectrum mass spectrometry genotyping. J Mol Diagn. 2011;13(1):64–73.
- Capper D, Preusser M, Habel A, et al. Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation-specific monoclonal antibody. Acta Neuropathol. 2011;122(1):11–19.
- Fredolini C, Byström S, Pin E, et al. Immunocapture strategies in translational proteomics. Expert Rev Proteomics. 2016;13(1):83–98.
- Duraiyan J, Govindarajan R, Kaliyappan K, et al. Applications of immunohistochemistry. J Pharm Bioallied Sci. 2012;4(Suppl 2):S307–9.
- Mino-Kenudson M, Chirieac LR, Law K, et al. A novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistry. Clin Cancer Res. 2010;16(5):1561–1571.
- Sun H, Chen GYJ, Yao SQ. Recent advances in microarray technologies for proteomics. Chem Biol. 2013;20(5):685–699.
- Westerlind U, Schröder H, Hobel A, et al. Tumor-associated MUC1 tandem-repeat glycopeptide microarrays to evaluate serum- and monoclonal-antibody specificity. Angew Chem Int Ed Engl. 2009;48(44):8263–8267.
- Na Z, Li L, Uttamchandani M, et al. Microarray-guided discovery of two-photon (2P) small molecule probes for live-cell imaging of cysteinyl cathepsin activities. Chem Commun. 2012;48(58):7304–7306.
- Wu H, Ge J, Yao SQ. Microarray-assisted high-throughput identification of a cell-permeable small-molecule binder of 14-3-3 proteins. Angew Chem Int Ed Engl. 2010;49(37):6528–6532.
- Ratan ZA, Zaman SB, Mehta V, et al. Application of fluorescence in situ hybridization (FISH) technique for the detection of genetic aberration in medical science. Cureus. 2017;9(6):e1325.
- Füzéry AK, Levin J, Chan MM, et al. Translation of proteomic biomarkers into FDA approved cancer diagnostics: issues and challenges. Clin Proteomics. 2013;10(1):13.
- Li H, Nguyen HH, Ogorzalek Loo RR, et al. An integrated native mass spectrometry and top-down proteomics method that connects sequence to structure and function of macromolecular complexes. Nat Chem. 2018;10(2):139–148.
- Mann M, Kulak NA, Nagaraj N, et al. The coming age of complete, accurate, and ubiquitous proteomes. Mol Cell. 2013;49(4):583–590.
- Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537(7620):347–355.
- Tsiatsiani L, Heck AJR. Proteomics beyond trypsin. FEBS J. 2015;282(14):2612–2626.
- Vidova V, Spacil Z. A review on mass spectrometry-based quantitative proteomics: targeted and data independent acquisition. Anal Chim Acta. 2017;964:7–23.
- Fu Q, Chen Z, Zhang S, et al. Multiple and selective reaction monitoring using triple quadrupole mass spectrometer: preclinical large cohort analysis. Methods Mol Biol. 2016;1410:249–264.
- Whiteaker JR, Zhao L, Saul R, et al. A multiplexed mass spectrometry-based assay for robust quantification of phosphosignaling in response to DNA damage. Radiat Res. 2018;189(5):505–518.
- Whiteaker JR, Zhao L, Ivey RG, et al. Targeted mass spectrometry enables robust quantification of FANCD2 mono-ubiquitination in response to DNA damage. DNA Repair (Amst). 2018;65:47–53.
- Doll S, Kriegmair MC, Santos A, et al. Rapid proteomic analysis for solid tumors reveals LSD1 as a drug target in an end-stage cancer patient. Mol Oncol. 2018;12(8):1296–1307.
- B-Jm W-R, Matzke MM, Datta S, et al. Bayesian proteoform modeling improves protein quantification of global proteomic measurements. Mol Cell Proteomics. 2014.
- Alfaro JA, Ignatchenko A, Ignatchenko V, et al. Detecting protein variants by mass spectrometry: a comprehensive study in cancer cell-lines. Genome Med. 2017;9(1):62.
- Desiderio C, D’Angelo L, Rossetti DV, et al. Cerebrospinal fluid top-down proteomics evidenced the potential biomarker role of LVV- and VV-hemorphin-7 in posterior cranial fossa pediatric brain tumors. Proteomics. 2012;12(13):2158–2166.
- Trenchevska O, Nelson RW, Nedelkov D. Mass spectrometric immunoassays in characterization of clinically significant proteoforms. Proteomes. 2016;4(1):13.
- Ntai I, Fornelli L, DeHart CJ, et al. Precise characterization of KRAS4b proteoforms in human colorectal cells and tumors reveals mutation/modification cross-talk. Proc Natl Acad Sci USA. 2018;115(16):4140–4145.
- Cristobal A, Marino F, Post H, et al. Toward an optimized workflow for middle-down proteomics. Anal Chem. 2017;89(6):3318–3325.
- Jiang T, Hoover ME, Holt MV, et al. Middle-down characterization of the cell cycle dependence of histone H4 posttranslational modifications and proteoforms. Proteomics. 2018;18(11):e1700442.
- Wang X, Slebos RJC, Wang D, et al. Protein identification using customized protein sequence databases derived from RNA-Seq data. J Proteome Res. 2012;11(2):1009–1017.
- Sajic T, Liu Y, Aebersold R. Using data-independent, high-resolution mass spectrometry in protein biomarker research: perspectives and clinical applications. Proteomics Clin Appl. 2015;9(3–4):307–321.
- Tighe PJ, Ryder RR, Todd I, et al. ELISA in the multiplex era: potentials and pitfalls. Proteomics Clin Appl. 2015;9(3–4):406–422.
- Low TY, Heck AJ. Reconciling proteomics with next generation sequencing. Curr Opin Chem Biol. 2016;30:14–20.
- Kalia M. Biomarkers for personalized oncology: recent advances and future challenges. Metab Clin Exp. 2015;64(3 Suppl 1):S16–21.
- Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;24(8):971–983.
- So AP, Vilborg A, Bouhlal Y, et al. A robust targeted sequencing approach for low input and variable quality DNA from clinical samples. NPJ Genom Med. 2018;3:2.