
ABSTRACT
Introduction: The biological heterogeneity of acute myeloid leukemia (AML) complicates personalized medicine. Individual prognosis is typically based on the presence of chromosomal and genetic lesions. Nevertheless, these classifications often lack a priori information about response to therapy. Since the protein expression landscape reflects the functional activity state of cells, we hypothesize that analyzing this can be used for the identification of protein activity markers to provide better risk stratification as well as may provide targeted therapeutic guidance in AML.
Areas covered: Herein, we review recently new adopted drugs in the treatment for AML and discuss how quantitative proteomic techniques may contribute to better therapeutic selection in AML.
Expert commentary: The net functional state of the cell is defined by the activity of protein within all the pathways that are active in the cell. Recognition of the proteomic profile of the leukemic blast could, therefore, complement current classification systems by providing a better a priori description of what pathways are important within a cell as a guide to the selection of therapy for the patient.
1. Acute myeloid leukemia
1.1. Introduction
Acute myeloid leukemia (AML) refers to the malignant clonal evolution of hematopoietic stem cells in the bone marrow affecting normal blood formation. Although AML is relatively rare with ~20,000 new cases in the United States per year [Citation1], without treatment it is a rapidly fatal disease with median survival between 10 and 15 months [Citation2]. Over the past 50 years, due to significant improvements in chemotherapeutic strategies, hematopoietic stem cell transplantations (HSCT) and supportive care, AML outcome has slowly improved and currently 70% of pediatric patients, 35–40% of adults younger than 60, and 5–15% of the elderly (>60 years) can be cured [Citation2–Citation4]. These numbers are obviously still unsatisfying and underline the need to investigate treatment opportunities beyond the existing.
In this review, we will first discuss currently used therapeutic approaches in AML, including targeted treatment strategies that have recently entered the clinic. Then, we will introduce the general concept of quantitative proteomics and debate why we think this technology may hold promise in identifying new diagnostic and prognostic biomarkers and in providing new therapeutic leads, especially in a heterogeneous disease as AML.
1.2. Current treatment strategies in AML
Current treatment approaches in AML focus on resetting the bone marrow with the essential goal of eradicating the malignant leukemic clone and restoring normal blood production. Standard therapy involves one or two cycles of conventional induction chemotherapy based on cytarabine and an anthracycline in adults, i.e. 3 days anthracycline followed by 7 days cytarabine intravenously (‘3 + 7’), and with the addition of etoposide as the third drug in the pediatric population. These regimes result in complete remission (CR) in 85–90% of treated children, 60–80% of adults <60 years and in 40–60% of adults >60 years [Citation2,Citation3,Citation5]. More recently the recognition of some commonly occurring mutations and the development of newer agents that target the mutated proteins arising from these affected genes has led to the incorporation of those agents into induction regimens which will be further discussed in paragraph 1.3. Induction therapy is followed by several, typically 3–6, courses of consolidation to minimize the risk of relapse. The choice of post-remission consolidation depends on the response to induction therapy and the presence of molecular genetic and cytogenetic abnormalities (, modified from the European LeukemiaNet (ELN) risk stratification by genetics for adults [Citation5] and genetically defined prognostic groups in pediatric AML [Citation6]). For example, favorable risk group patients are often treated with a high dose of cytosine arabinoside post-remission and patients with a more unfavorable cytogenetic profile are more typically referred for HSCT in the first remission.
Table 1. AML risk classification by genetics.
1.3. Targeted therapeutic approaches in AML
1.3.1. Targeted drugs for subsets mutated AML
Cytogenetic status is often considered as the most significant prognostic factor in AML. However, some leukemias (about 25% of pediatric and almost 50% of adult AML cases) have normal cytogenetics (CN-AML) and outcomes within this group are highly variable. Risk stratification and treatment choice in this group often depend on the presence of one or more driver mutations [Citation2,Citation3]. On average, an adult AML patient has 13 mutations including 2–4 driver mutations, some of which already listed in [Citation7,Citation8]. The majority of these mutated genes are not targetable themselves and only serve as a prognostic factor, but there are a few noteworthy exceptions.
Mutations in the FMS-like tyrosine kinase 3 (FLT3) are found in ~30% of AML patients and yield poor prognostic significance at all ages since these patients often experience relapse after obtained CR [Citation9,Citation10]. Recently midostaurin and gilteritinib, which target the activating FLT3 mutated receptor regardless of mutation status, have been developed and have received FDA approval [Citation11]. The kinase inhibitor midostaurin, first synthesized in 1986, was initially developed to target protein kinase C (PKC) and vascular endothelial growth factor receptor (VEGFR), but limited efficiency was found based on clinical trials in various diseases. It was recognized later that midostaurin inhibits FLT3 and KIT tyrosine kinases and was considered as a treatment for FLT3 mutated AML [Citation12].
Another frequently occurring and targetable mutation in AML is the IDH mutation. An IDH1 or IDH2 mutation happens in 15–20% of newly diagnosed AML patients, most commonly in the normal karyotype-classified cases. Mutant forms are associated with abnormal enzymatic activity that results in the production of an atypical metabolite (2-hydroxyglutarate, "2-HG") which poisons TET2 leading to epigenetic dysregulation and a hematopoietic differentiation block at an early stage in the AML development [Citation13,Citation14]. Small molecule inhibitors have shown to reverse these effects and in 2017 the first IDH2 inhibitor (enasidenib) received FDA approval for the treatment of relapsed or refractory adult AML patients with the IDH2 mutation [Citation15]. This was followed by approval of the IDH1 inhibitor ivosidenib in 2018 [Citation16].
1.3.2. AML-based immunotherapy
In hematological diseases, significant improvement in outcomes has been shown with immunotherapy-based regimes. The first FDA approved antibody-based drug in cancer was rituximab, which targets the antigen CD20 on the surface of specific B-cell non-Hodgkin lymphoma (NHL) cells and is part of the gold standard ever since [Citation16,Citation17]. Moreover, in B-cell acute lymphoid leukemia (B-ALL), success has been booked with chimeric antigen receptor (CAR)-T-cell therapy. CAR-T-cells are genetically engineered patient cells that express CARs to target specific antigens on cancer cells. Tisagenlecleucel targeting the CD19 antigen was the first CAR-T cell drug that received FDA approval. In 2018, Maude et al. published their phase II study of Tisagenlecleucel in pediatric B-ALL that showed a complete remission rate of 81% [Citation18]. The relapse rate, however, remained high, especially with antigen-negative leukemia [Citation19]. The key to success in immunotherapy lies within the identification of a target antigen that is predominantly expressed on the surface of malignant cells, such as CD20 on B-cell NHL and CD19 on B-ALL cells. Expression of such target should preferably be restricted to malignant cells, and expression should be present on mature malignant cells as well as on malignant stem cells. For AML the identification of leukemia-specific antigens has been less successful and current therapeutic antibody and CAR-T approaches are directed against CD33 and CD123, both of which are also present on normal hematopoietic stem cells; thus caution in their usage is warranted [Citation20].
Gemtuzumab ozogamicin (GO) is a drug that targets the CD33 transmembrane protein and has been approved for relapsed AML after a promising phase II trial in 2000. Ten years later GO was voluntarily withdrawn when results from a randomized trial did not show improved efficacy between treatment arms. However, in this study, patients received lower dose chemotherapy when GO was added to their treatment compared to patients who only received chemotherapy. Subsequent studies have shown the efficacy of adding GO to chemotherapy leading to its reapproval in 2017 [Citation21,Citation22].
1.3.3. Targeting signaling pathways in AML
The inhibition of a single protein in clinical practice with regards to AML has been very limited, until last year. In November 2018, the BCL-2 inhibitor venetoclax (ABT-199) was approved in combination with azacytidine, decitabine, or cytarabine for the treatment of newly diagnosed AML patients 1) older than 75 years or 2) with comorbidities that excluded them from induction therapy. BCL-2 is an apoptotic enzyme that prevents cell death by regulating the intrinsic apoptotic, so-called mitochondrial pathway. Via this pathway, apoptosis depends on the activation of caspases via mitochondrial outer membrane permeabilization (MOMP). BCL-2 prevents the MOMP and apoptosis by binding and sequestering effector pro-apoptotic proteins such as BIM, PUMA, BAD, BID, and BIK [Citation23]. Since BCL-2 was recognized as protein to prevent cell death, it became an interesting target in cancer. BCL-2 expression was first found in lymphoid cells and therefore studies began to investigate the potential of targeting BCL-2 in chronic lymphoid leukemia (CLL) and lymphomas, resulting in the first FDA approval for venetoclax in 2016 for CLL [Citation23]. Venetoclax as monotherapy showed biological activity in AML patients as well, but responses remained limited. Resistance and disease progression can be acquired via the upregulation of alternative anti-apoptotic proteins, such as MCL-1, that also could bind the effectors. And since, it is not the expression quantity that sensitizes cells after BCL-2 inhibition but rather the amount of protein that is bound to the pro-apoptotic effectors that drive MOMP [Citation24]. A breakthrough was achieved when the BCL-2 inhibitor was combined with the hypomethylating agent 5-azacitidine. Complete remission rates for patients treated with the combination were 67% compared to earlier described 28% when treated with a hypomethylation agent only. Moreover, the combination improved overall survival to a median of 17.5 months compared to 10.4 months in 5-azacitidine monotherapy [Citation25]. Similar clinical results were achieved when venetoclax was combined with low-dose cytarabine [Citation26,Citation27].
Despite the success of venetoclax, targeting single proteins is often not sufficient to kill the leukemic cell as they can bypass the inhibition by alternative escape routes [Citation28]. Targeting these routes is often performed by combining available drugs to target multiple proteins of the intracellular signaling pathway or by targeting the upstream trigger that activates the pathway. For example, patients with chronic myeloid leukemia (CML) express the fusion protein BCR-ABL1 due to the chromosomal Philadelphia translocation. The discovery of a drug (imatinib) that targets the kinase activity of BCR-ABL1, thereby preventing the downstream activation of the proteins that actuate the leukemic physiology, improved 10 years overall survival of CML to 83% [Citation29]. AML holds a more complex etiology. In AML it is not one but multiple signal transduction pathways that are recognized to promote leukemogenesis and affect proliferation and cell survival [Citation30]. The idea that simultaneous activation of various signaling pathways influence outcome and response to therapy in AML has been studied extensively and has led to the development of numerous agents that target components of activated signaling pathways. The mitogen-activated protein kinase (MAPK)/extracellular-regulated kinase (ERK) is an example of a frequently dysregulated pathway in AML. Inhibitors against compounds of this pathway have been developed and trials with MEK inhibitors are underway. Although the use of MEK inhibitors has been reported as safe and tolerable, limited efficiency was recently observed in a phase 2 clinical trial [Citation31]. It is likely that only therapeutics that target multiple activated signaling pathways will be beneficial for patients with AML.
Another pathway that is aberrantly upregulated in myeloid leukemia is the hedgehog signaling pathway. Normally, the hedgehog pathway is activated during embryogenesis and silenced after birth [Citation32]. The intracellular hedgehog pathway is activated upon stimulation of an integral membrane protein Smoothened (SMO). If activated, SMO triggers translocation of the glioma-associated proteins (GLI) to the nucleus which will result in target gene transcription. Both overexpression of SMO and GLI are associated with resistance to therapy and hedgehog pathway inhibition indeed increases drug sensitivity in AML [Citation33]. These findings prompted doctors to combine pharmacological inhibition of the hedgehog pathway with chemotherapy in the clinical setting. The SMO inhibitor glasdegib (PF‐04449913) prevents downstream activation of the hedgehog pathway and was well tolerated and safe when combined with chemotherapy, including low-dose cytarabine and decitabine, in AML and MDS patients in a phase I clinical trial [Citation34]. Promising results from a phase II study were recently published where glasdegib combined with low-dose cytarabine significantly improved overall survival and complete remission rates in adult AML and MDS patients who were ineligible for high-dose chemotherapy compared to who only received low-dose cytarabine [Citation35]. Notably, improvement of outcomes with the combination therapy was independent of cytogenetic group or mutational status in this phase II study. The use of glasdegib in combination with high-dose chemotherapy or azacytidine is currently under investigation in a multicenter phase III clinical trial (ClinicalTrials.gov, NCT03416179) for previously untreated AML patients.
An overview of the above discussed therapeutic targets that recently have been approved for subgroups of AML is shown in . The integration of these compounds as optional treatments is a major breakthrough for AML, especially for those with very poor prognosis (relapsed and refractory AML) and the elderly. Many more clinical trials are currently ongoing testing different combinations of therapeutic possibilities for different subtypes of patients at different stages of disease.
Table 2. Summary of new approved therapeutic drugs for AML.
2. Utilizing proteomics in AML
2.1. Why should we use proteomics in AML?
The aforementioned, current risk classification in AML is based on the presence of chromosomal abnormalities and recurrent driver mutations in the leukemic cell. Both classifications yield great prognostic relevance and have contributed to disease classification and risk stratification [Citation7]. However, chromosomal and molecular abnormalities are difficult to treat themselves. Of note is that the majority of drugs against genes that are frequently mutated in AML target all forms of the target proteins and are not specific for inhibiting the mutated form (e.g. midostaurin and gilteritinib in FLT3), with drugs targeting the IDH mutations as notable exception. Many developed drug target proteins that are not mutated in AML, but which are functionally ‘downstream’ of mutated genes. Moreover, leukemic blasts are affected by external and environmental factors including cytokine exposure, hypoxia, and mesenchymal stromal cell (MSC) interaction and it is believed that the combination of the (epi)genetic changes and microenvironmental influences determines leukemic cell fate. The net effect of these combined influences is predominantly displaced on the protein level and their affected signaling pathways [Citation42]. We, therefore, argue that characterization of differentially expressed proteins and recognition of proteomic signatures within (subgroups of) AML may facilitate and improve treatment stratification.
Multiple signaling transduction pathways are involved in the development and progression of AML. While these pathways are typically studied independent of each other, they actually interact with each other to direct the happening of the leukemic cell. Strategies that simultaneously look at all of these pathways, and that are capable of integrating the net effect of all pathways on the entire spectrum of downstream protein effectors and actuators would provide a more complete ‘systems biology' view of the function of the leukemic cell. Crucial to the improving success of cancer therapy has been the realization that combination therapy is typically required to improve outcome. An approach that simultaneously assesses the activity of multiple pathways and multiple effectors could suggest logical points for combination targeted therapy. To achieve this, assessment of a broad spectrum of the proteome is required. The two main techniques that are capable of analyzing the proteome in AML are mass-spectrometry (MS)- and antibody-based techniques.
2.2. High-throughput proteomic techniques used in AML
2.2.1. Mass spectrometry
based proteomics is a well-established technique and commonly used for identification and quantitative assessment of proteins within complex samples. MS is based on the measurement of charged ions from a protein analyte. To increase the sensitivity of the measurement, different compounds in very complex samples can be separated by gel electrophoresis, liquid chromatography (LC-MS), gas chromatography (GC-MS), or another mass spectrometer (MS-MS) prior to analysis. Historically, there are two ways to detect proteins by MS including using a ‘top-down’ or ‘bottom-up’ approach [Citation43,Citation44]. After protein extraction, the top-down approach separates and quantitates purified intact proteins (ions) using 2D gel electrophoresis or MS-MS and enables the characterization of unique proteoforms including degradation products, protein isoforms, post-translational modifications (PTMs), as well as low-mass proteins. The usage of the top-down approach is however often limited to low-throughput individual protein studies. In contrast, the bottom-up approach is more broadly used for analyzing more complex protein mixtures. Bottom-up proteomics using LC-MS is also known as ‘shotgun proteomics’ [Citation45]. With this technique, the proteins are digested into peptides by enzymes (e.g. trypsin) before analysis by MS. Since only one fraction of all produced peptides (ions) is restored into a protein, PTMs and alternative isoform information will not be recovered in bottom-up proteomics. Both approaches are thus associated with advantages and disadvantages and choice of technique depends on the research aim of the study.
In both approaches, before MS analysis, ionization of the sample is performed to break molecules into charged ions. These ions then pass through an electric or magnetic field which sorts them based on mass-to-charge (m/z) ratio. Relative abundance of the separated ions is often presented in a mass spectrum to decipherer the identity of the proteins. The most frequently used techniques for ionization of analytes are ‘matrix-assisted laser desorption/ionization’ (MALDI) or electrospray ionization (ESI). Using MALDI, an energy-absorbing-matrix is added to the analyte and when the sample is irradiated with laser energy, the matrix will ionize the analyte [Citation46]. MALDI usually produces single-charged ions (z = 1) with a maximum mass of 350 kDa. The ion sorting process according to the mass-to-charge ratio is then often performed by a ‘time of flight’ (TOF) analyzer. This technique simply measures the travel time of the ion before it reaches the detector [Citation47]. The lighter the ion, the faster and vice versa. A variation of MALDI is ‘surface-enhanced laser desorption/ionization’ (SELDI) [Citation48]. With this technique, proteins are not bound to species in the matrix but to the surface. Another technique to ionize an analyte is with ESI. The protein analyte is dissolved in a conductive solvent and high voltage is then applied to the sample to create aerosols. While MALDI produces single-charged ions, ESI can produce multicharged ions (up to ~75) and can be applied to in particular proteins with high molecular masses [Citation49].
The clinical applications of MS in acute leukemia include the identification of diagnostic biomarkers. Xu et al. for example developed a proteomic-classification system by analyzing 151 de novo acute leukemia patients using SELDI-TOF MS. The proteomic-subtypes correlated with the type of leukemia (acute promyelocytic leukemia (APL), granulocytic AML, monocytic AML, and acute lymphocytic leukemia (ALL)). The authors suggest that this proteomic classification holds promise to identify potential protein biomarkers for each specific subgroup of acute leukemia [Citation50]. Protein biomarkers are also identified to distinguish between malignant AML and the premalignant myelodysplastic syndrome (MDS). Clinically, measurement of the percentage of bone marrow blasts remains the method of monitoring disease progression. A patient is diagnosed with MDS if the bone marrow contains <20% blasts, but the diagnosis changes into AML if 21% or more blasts are found. It is however clear that this cutoff between premalignant to malignant disease does not precisely reflect the degree of malignancy. It would be of clinical interest if a reliable biomarker could predict which MDS patients have a higher chance to progress into AML. Using MS, serum protein CXC chemokine ligands 4 and 7 (CXCL4, CXCL7) have been identified as proteins with decreased expression in advanced MDS [Citation51]. Another study using MALDI-TOF MS identified that MOES, EZRI, and AIFM1 could be considered as AML-specific expressing proteins and therefore may serve as diagnostic biomarkers to distinguish MDS and AML [Citation52].
MS has also been used to identify prognostic biomarkers in AML. SELDI-TOF MS analysis of 54 samples from newly diagnosed AML patients revealed two prognostically different protein profiles. The authors identified a marker with high discriminative potential that predicted poor survival independent from cytogenetics in ~70%. MS peptide sequencing could identify the marker as the mature granulocyte marker S100A8. Additional Western blot analysis could confirm that S100A8 at the time of diagnoses was highest expressed in patients with a poor prognosis [Citation53]. Another member of the S100 family with prognostic value in AML was recently discovered by Alanazi et al. Using LC-MS/MS, 110 aberrant expressed nuclear proteins were identified in AML patients compared to normal CD34+ cells. S100A4 was the highest overexpressed protein and additional research showed that its expression is required for blast cell survival but not for normal cell development suggesting this could be a new therapeutic target [Citation54].
The prognostic significance of driver mutations in AML often depends on co-occurring circumstances. For example, the impact of an IDH mutation itself remains controversial [Citation55]. It became however clear that the level of the produced oncometabolite 2-HG actually has prognostic value. Measurement of the 2-HG levels with reverse-phase LC-MS in 223 de novo AML samples showed that patients with 2-HG levels above 700 ng/ml were highly susceptible to have an IDH mutation (~87%). Pretreatment levels were not predictive for outcomes, but 2-HG levels >200 ng/ml after achieving CR predicted worse overall survival in this population [Citation56]. In line with these findings, Janin et al. published that low 2-HG serum levels after induction therapy indeed correlated with a better outcome in terms of overall and disease-free survival [Citation57].
Although many targets have been identified, AML is known as a phenotypically and functionally distinct disease and intratumor heterogeneity is one reason that may delay the translation of these targets to the clinic. A better understanding of the intratumor heterogeneity of leukemic blasts might improve this process. Combining MS and flow cytometry (mass cytometry or cytometry by time-of-flight, ‘CyTOF’) enables measurement of protein levels in single cells including PTMs and proteolysis products [Citation58]. Levine et al. have used this technique in 16 pediatric AML patients to analyze surface and intracellular signaling protein levels in millions of leukemic and nonmalignant cells. It is remarkable that the authors report substantial decoupled profiles between surface markers and intracellular proteins suggesting that not all surface markers necessarily reflect the intracellular protein expression state [Citation59].
2.2.2. Antibody-based protein microarrays
One other approach that enables us to detect and to quantify proteins is by the use of antibodies. The advantage of antibody-based proteomic techniques is that antibodies against whole proteins as well as against their isoforms (e.g. alternative splicing variants, PTMs) can be used. Among antibody-based techniques, the protein microarray is frequently the technique of choice to study proteomics in leukemia. Functional protein microarrays are generally used in basic research laboratories to investigate the biochemical activities of proteins such as their interactions with other proteins, DNA, RNA, small molecules, and lipids. Discussing the applications of these arrays in leukemia is however beyond the scope of this manuscript.
For clinical purposes, analytical protein microarrays have been used for large-scale protein expression profiling as well as for biomarker discovery. The main two types of analytical protein microarrays are the forward-phase protein array (FPPA) and the reverse-phase protein array (RPPA). In FPPA, multiple solid-phased antibodies are printed on an array with known positions. A patient protein lysate can then be printed on the array and will bind to the particular antibody if the protein is expressed. After exposure to a secondary antibody, protein expressions are measured. The advantage of this technique is that expressions of multiple proteins are measured simultaneously per sample [Citation60]. However, finding two highly specific, non-overlapping antibodies complicates the generalized usage of this technique. Another disadvantage of FPPA is that a relatively high amount of protein lysate is needed for analysis, which is certainly not always available in the clinical setting.
Both limitations can be overcome by RPPA. With RPPA, a significantly lower amount of protein lysate is needed and instead of antibodies, protein lysates of multiple samples are printed onto a slide. The expression of a single protein is then measured simultaneously in all samples by antibody detection. RPPA is unique as it can assess protein expression levels including PTMs in >1000 (patient) samples at the same time. The limitation of this technique compared to MS-based techniques is that only strictly validated antibodies can be used, as it is crucial to minimize nonspecific binding on the array to prevent false-positive protein expressions. Antibody-based arrays, therefore, limit the coverage of the proteome and lack the ability to identify de novo proteins that are expressed. However, on the other hand, if validated, measured expression of the protein of interest and PTMs are highly accurate and sensitive. We and others have shown the high inter- and intra-slide reproducibility, precision, throughput, and reliability of RPPA [Citation61,Citation62]. In hematological malignancies, these arrays have been generated for adult and pediatric AML [Citation42,Citation63], APL [Citation64], adult and pediatric ALL [Citation65], and AML derived MSC [Citation66] and a CLL array is underway.
Since RPPA measures protein expression in multiple samples at the same time, this technique can be used to identify proteins that are differentially expressed in leukemia compared to protein expression in nonmalignant bone marrow and blood cells. Abnormal high or low expression of a protein of interest could potentially be considered as diagnostic biomarker which may be useful in early disease detection. Recognition of specific protein expression patterns could furthermore improve subcategorization within the heterogeneous landscape of AML. Kornblau et al. have shown distinct proteomic profiles across FAB defined morphological categories within 256 adult AML patients using RPPA. There were 24 proteins out of 51 tested that were distinctly expressed between myeloid (M0-M2) or monocytic (M4-M5) AML. Patients in M0-M5, in addition, had a different proteomic profile than M6 and M7 categorized patients. In addition, two individual proteins, TP53 and BAX, showed different expressions across different cytogenetic abnormalities. Protein expression also correlated with mutational status. FLT3 mutated patients had a higher expression of 13 proteins, including phosphorylated STAT5 and PKCα. Interestingly, these findings were in line with previously higher described higher mRNA expression of these proteins in FLT3-AML [Citation62].
RPPA-based proteomic profiles to distinguish between different leukemic subtypes are also defined. Hoff et al. identified different protein expression signatures between AML and ALL [Citation63,Citation65], as well as between AML and APL [Citation64]. Interestingly, comparable protein profiles between pediatric AML and ALL patients were also defined. For example, one protein constellation with both AML as ALL patients showed upregulation of SMAD proteins as well as proteins acting in the mTOR signaling pathway. Therapeutic targets against these proteins could potentially be effective in both leukemias with this protein profile. The patients did not share clinical features or demographics suggesting that protein patterns are not driven by genetic events only [Citation65].
Besides the identification of diagnostic markers within and between specific types of leukemia, RPPA provides a platform to identify prognostic biomarkers that predict outcome in (subgroups of) AML.
For instance, Quintas-Cardama et al. investigated TRIM62 levels in adult AML patients and were the first to describe a tumor suppressor role for TRIM62 in AML [Citation67]. Low TRIM62 protein expression was identified as an adverse prognostic factor for survival and complete remission in this population which was supported by higher white blood count and higher levels of bone marrow blasts and monocytes. The loss of TRIM62 was prominent in normal karyotype AML and the prognostic significance was notably independent of NPM1 or FLT3 mutation. Since current post-remission treatment stratification in normal karyotype depends on NPM1 and FLT3 status, this report highlights the relevance of analyzing the proteome to further refine treatment stratification.
While studying the same population, high phosphorylation of serine 318–321 on FOXO3 was identified as an independent adverse prognostic factor [Citation68] and low expression of ASH2L was related to poorer outcomes as well [Citation69]. Other individual prognostic protein biomarkers that have been discovered in AML include LGALS3 [Citation70], TGM2 [Citation71], and FLI1 [Citation72].
2.3. Translation to the clinic
2.3.1. Prediction of therapy sensitivity
In this manuscript, some identified protein activity biomarkers in diagnostics and prognosis in AML have been described. Although the impact of these markers on outcome can be clear, the translation of protein activity markers to the clinic remains relatively limited. One reason being that they do not impact the choice of treatment. In a malignant disease such as AML characterized by a chaos of combination of genetic events, doctors rather overtreat a patient instead of using a prognostic marker with not completely perfect prediction [Citation73]. However, since a finite number of recurrent protein expression signatures exist, we argue that these patterns surely suggest targets to aim for targeted therapy. A vast and growing list of inhibitors against proteins is available and identification of clinical scenarios in which these inhibitors may be more sensitive may help bring highly predictive and validated protein markers to the clinic. Proteomic profiling can assist in identifying situations in which specific protein inhibitors may be more sensitive.
For example, a variation of BET inhibitors is currently under investigation in clinical trials in AML. In leukemia, inhibition of the BET-member bromodomain containing 4 (BRD4) prevents transcription of oncogenic genes (e.g. MYC) through inhibiting BRD4 protein binding to acetylated lysine residues of histone tails [Citation74,Citation75]. Interestingly, this discovery was based on an RNA interference screening, and although leukemic cells are more sensitive to BRD4 inhibition than normal hematopoietic cells, the impact of BRD4 mutations or aberrant protein expression remained unknown for many years. In 2017, our group analyzed and reported BRD4 protein expression in AML using RPPA from 511 newly diagnosed adult AML patients [Citation76]. Patients who expressed higher BRD4 protein levels compared to normal, nonmalignant, bone marrow-derived CD34+ cells had inferior remission durations and overall survival. These analyses retained statistical significance in patients with unfavorable cytogenetics. The adverse prognostic impact of high BRD4 expression was again observed when the analysis was restricted to only the freshly prepared samples (upon arrival in the laboratory, n = 205/511) with a median survival of 103 vs. 53 weeks (P = 0.04). Median event-free survival was also significantly shorter for high BRD4 expressors (58 vs. 87 weeks, P = 0.02). In line with these findings, high classified BRD4 expressors had a higher percentage of bone marrow blasts (67% vs. 51%, P < 0.001) and peripheral blood blasts (51% vs. 33%, P < 0.001). Morphologically, high BRD4 expressors had more often undifferentiated (M0) or minimal maturation (M1) AML as defined by the FAB classification compared to low-classified patients (26% of high vs. 7% of low BRD4 expressors). If BRD4 is higher expressed in undifferentiated and minimal maturation AML, then these patients, in particular, may potentially benefit from the addition of BET inhibitors to their therapeutic regime. If we could develop a test that measures BRD4 expression in a fast, sensitive, and clinical-applicable approach, we can easily select patients that may be the ultimate candidates for BET inhibitors.
2.3.2. Clinical-applicable tools to predict protein inhibitor sensitivity
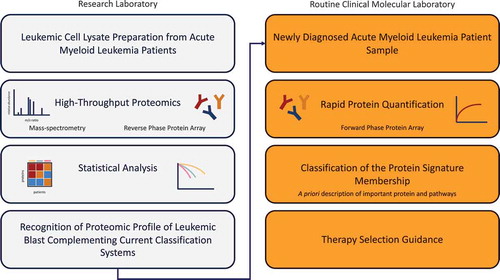
Techniques that rapidly provide information about relative protein expressions are enzyme-linked immunosorbent assay (ELISA) and immunohistochemistry (IHC). The advantage of ELISA above IHC is that quantification along all concentrations is possible while in IHC signal saturates and accurate quantification is limited [Citation77]. On the other hand, a disadvantage of ELISA is that a larger amount of protein lysate is needed which is not always available in the clinical setting. Another option to measure direct protein expression when a patient enters the clinic is the usage of FPPA. As discussed earlier, this technique allows the detection of multiple proteins in a single protein sample. We, therefore, believe that FPPA has the potential to become an important diagnostic tool in the clinical setting of AML. In the end, it is not the single protein expression, but the entire proteomic network that determines the net effect of the leukemic cell. Identification of sets of proteins instead of individual proteins that correlate with clinical characteristics in AML is still in its infancy but truly holds promise to bring personalized medicine and management to the next level. A schematic overview of how high-throughput proteomics could lead to specific pharmacological treatment for a subgroup of patients and how this could be used to a more personalized medicine approach is shown in .
Figure 1. Utilizing high-throughput proteomics in AML could lead to specific pharmacological treatment options for a subgroup of patients (light blue) which then could be used to a more personalized medicine approach (orange).
3. Future applications
3.1. RPPA analysis in multicenter clinical trials
The big advantage of RPPA over other proteomic techniques is that it is very sample sparing, requiring only 3 × 105 cells to analyze the expression of ~400 proteins. This makes RPPA a suitable approach for clinical applications. All protein samples are exposed to the same antibody at once allowing direct comparison of protein expression across the dataset. Therefore, we are currently exploring the usage of RPPA in multicenter clinical trials. Excitingly, patient samples can be obtained before and after treatment, and changes in protein expression over time can be analyzed by RPPA when samples are printed on the same slide. We hypothesize that changes in specific proteins may correlate with the resistance or sensitivity to certain treatment approaches retrospectively. Identification of these protein patterns may contribute to our understanding of the mechanism that drives chemoresistance. If we, for instance, could identify significant changes in protein expression upon 24 h after treatment that correlate with relapse at the long term, we may block that defensive adaptation by anticipate the changes and incorporating a specific inhibitor for that protein, or by early selecting of patients that may benefit from more intensive treatment regimens such as allogeneic HSCT.
3.2. Conclusion
For existing therapeutic strategies in AML, there is a lack of markers that are capable of reliably predicting prognosis or the therapeutic response prior to treatment. There is hope that elucidation of the AML-specific proteome will prompt the discovery of novel therapeutic targets and biomarkers. The big, unexpected success of BCL-2 protein inhibition in AML by venetoclax illustrates the potential impact of inhibition of key components of the proteome which encourages us to accelerate translation of validated proteins as diagnostic, prognostic, and/or therapeutic biomarker to the clinical setting. The development of an applicable clinical test that quickly provides information about relative protein expression of the patient may contribute to better stratification and eventually may be the next step in predictive and personalized medicine.
4. Expert commentary
AML is a complex disease and choosing the best treatment option for individual patients is an everyday challenge for hematologists. Patients are generally well characterized for the molecular events, translocations, and mutations that define their leukemia, but there is a disconnect between this information and the clinic as most of these cannot currently be targeted. Furthermore, these events occur in a nearly infinite number of combinations and the net effect of these changes on the physiology of the cell cannot easily be predicted just from the knowledge of which event is present. Concurrent with the explosion in genetic characterization, there is simultaneously an explosion in new therapeutic agents targeting specific critical components of various pathways affecting all components of the cell metabolism and physiology. These combined genetic events have a summary effect on the cell through the downstream effects they have. It may, therefore, be possible to use targeted agents aimed at a downstream consequence of a mutated gene, to interfere with that genes' ability to drive leukemia, even if the actual mutated protein cannot be targeted. As clinicians, we need a means to determine which targeted agent would have a beneficial effect in which situation. Since the net ‘summary’ functional state of the cell is defined by the activity of proteins within all the pathways that are active in the cell, we argue that recognition of which proteins and pathways is being utilized can provide this missing link between specific mutations, alone or in combination and the panoply of available targeted therapeutics. The recognition of the proteomic profile of the leukemic blast could, therefore, complement current classification systems by providing a better a priori description of what pathways are important within a cell as a guide to the selection of therapy for the patient. We and others have established that despite the chaos of all the combinations of genetic events that occur in leukemia, a finite number of recurrent protein expression signatures exist in leukemia and that these proteomic profiles correlate with response to treatment and that this can be independent of cytogenetic or molecular status. Additionally, these patterns suggest targets to aim for with targeted therapy.
The biggest remaining challenges to the routine clinical use of these are means of defining these patterns and verification of the predictive guidance they might provide toward therapy selection. Current platforms utilize methodologies appropriate to the research laboratory but not to the routine clinical molecular laboratory. Developing the means of rapidly performing tests that enable classification of the protein signature membership, which can be Clinical Laboratory Improvement Amendments (CLIA) certified, and therefore can be broadly utilized is half of the need. Performing validation studies that confirm that suggestions of which targeted agents would increase therapeutic efficacy when used in a particular signature is the other half. Knowledge of signature membership only has utility if a reliable knowledge based on which targeted agent should be used in each signature is available. Another weakness is that studies have looked at a finite portion of the proteome and there may be many other clinically relevant and targetable proteins that have not been recognized. Furthermore, as new agents that are not currently covered by the proteomic analyses performed to date are developed, there must be ways to incorporate that growing list of available targeted agents. The ability of these signatures to evolve as new drugs and targets are identified, studied and their relevance determined is required.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Article Highlights
AML is a very aggressive disease that occurs across all ages, and although survival is markedly better in the pediatric population, it remains a leading cause of childhood and adult cancer death.
For existing therapeutic strategies in AML, there is a lack of markers that are capable of reliably predicting prognosis or the therapeutic response prior to treatment.
Strategies that are capable of integrating the net effect of all pathways on the entire spectrum of downstream protein effectors and actuators would provide a more complete systems biology view of the function of the leukemic cell.
Despite the chaos of all the combinations of genetic events that occur in AML, a finite number of recurrent protein expression signatures exist. These proteomic profiles correlate with response to treatment and prognosis, and can be independent of cytogenetic or molecular status.
Assessment of a broad spectrum of the proteome could suggest logical points for combination targeted therapy in AML.
Knowledge of a patient’s proteomic signature membership only has utility if a reliable knowledge based on which targeted agent should be used in each signature is available.
Additional information
Funding
References
- De Kouchkovsky I, Abdul-Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016;6(7):e441.
- Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–1152.
- Döhner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–474.
- de Rooij J, Zwaan C, van den Heuvel-eibrink M, et al. From biology to clinical management. J Clin Med Res . 2015;4(1):127–149.
- Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447.
- Creutzig U, van den Heuvel-eibrink MM, Gibson B, et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood. 2012;120(16):3187–3205.
- Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221.
- Di Nardo CD, Cortes JE. Mutations in AML: prognostic and therapeutic implications. Hematol. 2016;2016(1):348–355.
- Kottaridis PD, Gale RE, Langabeer SE, et al. Studies of FLT3 mutations in paired presentation and relapse samples from patients with acute myeloid leukemia: implications for the role of FLT3 mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood. 2002;100(7):2393–2398.
- Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99(12):4326–4335.
- Daver N, Schlenk RF, Russell NH, et al. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33(2):299–312.
- Stone RM, Manley PW, Larson RA, et al. Midostaurin: its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018;2(4):444–453.
- Losman J, Lee S, McMahon C, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;340(6127):1621–1625.
- Nassereddine S, Lap CJ, Haroun F, et al. The role of mutant IDH1 and IDH2 inhibitors in the treatment of acute myeloid leukemia. Ann Hematol. 2017;96(12):1983–1991.
- Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–731.
- Dotan E, Aggarwal C, Smith MR. Impact of Rituximab (Rituxan) on the treatment of B-cell non-Hodgkin’s lymphoma. P T. 2010;35(3):148–157.
- Jardin F. Improving R-CHOP in diffuse large B-cell lymphoma is still a challenge. Lancet Oncol. 2019;20(5):605–606.
- Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448.
- Brown CE, Mackall CL. CAR T cell therapy: inroads to response and resistance. Nat Rev Immunol . 2019;19(2):73–74.
- Hofmann S, Schubert ML, Wang L, et al. Chimeric antigen receptor (CAR) T cell therapy in acute myeloid leukemia (AML). J Clin Med Res. 2019;8:2.
- Rowe JM, Löwenberg B. Gemtuzumab ozogamicin in acute myeloid leukemia: a remarkable saga about an active drug. Blood. 2013;121(24):4838–4841.
- Appelbaum FR, Bernstein ID. Gemtuzumab ozogamicin for acute myeloid leukemia. Blood. 2017;130(22):2373–2376.
- Konopleva M, Letai A. BCL-2 inhibition in AML: an unexpected bonus? Blood. 2018;132(10):1007–1012.
- Pan R, Benito JM, Han L, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid Leukemia. Cancer Discov. 2014;4(3):362–675.
- DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17.
- Lin TL, Strickland SA, Fiedler W, et al. Phase Ib/2 study of venetoclax with low-dose cytarabine in treatment-naive patients age ≥ 65 with acute myelogenous leukemia. J Clin Oncol. 2016;34(15_suppl):7007.
- Wei AH, Tiong IS, Strickland SA, et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/II study. J Clin Oncol. 2019;37(15):1277–1284.
- Kampen KR, Ter Elst A, Mahmud H, et al. Insights in dynamic kinome reprogramming as a consequence of MEK inhibition in MLL-rearranged AML. Leukemia. 2014;28(3):589–599.
- Hochhaus A, Larson RA, Guilhot F, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376(10):917–927.
- Kornblau SM, Womble M, Qiu YH, et al. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood. 2006;108(7):2358–2365.
- Maiti A, Naqvi K, Kadia TM, et al. Phase II trial of MEK inhibitor binimetinib (MEK162) in RAS-mutant acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2019;19(3):142–148.
- Ok CY, Singh RR, Vega F. Aberrant activation of the hedgehog signaling pathway in malignant hematological neoplasms. Am J Pathol. 2012;180(1):2–11.
- Queiroz KCS, Ruela-de-Sousa RR, Fuhler GM, et al. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene. 2010;29(48):6314–6322.
- Savona MR, Pollyea DA, Stock W, et al. Phase Ib study of glasdegib, a Hedgehog pathway inhibitor, in combination with standard chemotherapy in patients with AML or high-risk MDS. Clin Cancer Res off J Am Assoc Cancer Res. 2018;24(10):2294–2303.
- Cortes JE, Heidel FH, Hellmann A, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia. 2019;33(2):379–389.
- Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454–464.
- Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3 -mutated AML. N Engl J Med. 2019;381(18):1728–1740.
- Roboz GJ, Rosenblat T, Arellano M, et al. International randomized phase III study of elacytarabine versus investigator choice in patients with relapsed/refractory acute myeloid leukemia. J Clin Oncol. 2014;32(18):1919–1926.
- Megías-Vericat JE, Ballesta-López O, Barragán E, et al. IDH1-mutated relapsed or refractory AML: current challenges and future prospects. Blood Lymphat Cancer. 2019;9:19–32.
- Lambert J, Pautas C, Terré C, et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica. 2019;104(1):113–119.
- Gamis AS, Alonzo TA, Meshinchi S, et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children’s Oncology Group trial AAML0531. J Clin Oncol. 2014;32(27):3021–3032.
- Hu CW, Qiu Y, Ligeralde A, et al. A quantitative analysis of heterogeneities and hallmarks in acute myelogenous leukaemia. Nat Biomed Eng. 2019;3(11):889–901.
- Chait BT. Mass spectrometry: bottom-up or top-down? Science. 2006;314(5796):65–66.
- Chait BT. Mass spectrometry in the postgenomic era. Annu Rev Biochem. 2011;80(1):239–246.
- Wolters DA, Washburn MP, Yates JR 3rd. An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem. 2001;73(23):5683–5690.
- Hillenkamp F, Karas M, Beavis RC, et al. Matrix-assisted laser desorption/ionization mass spectrometry of biopolymers. Anal Chem. 1991;63(24):1193A–1203A.
- Tanaka K, Waki H, Ido Y, et al. Protein and polymer analyses up tom/z 100 000 by laser ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 1988;2(8):151–153.
- Issaq HJ, Veenstra TD, Conrads TP, et al. The SELDI-TOF MS approach to proteomics: protein profiling and biomarker identification. Biochem Biophys Res Commun. 2002;292(3):587–592.
- Fenn JB, Mann M, Meng CK, et al. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246(4926):64–71.
- Xu Y, Zhuo J, Duan Y, et al. Construction of protein profile classification model and screening of proteomic signature of acute leukemia. Int J Clin Exp Pathol. 2014;7(9):5569–5581.
- Aivado M, Spentzos D, Germing U, et al. Serum proteome profiling detects myelodysplastic syndromes and identifies CXC chemokine ligands 4 and 7 as markers for advanced disease. Proc Natl Acad Sci U S A. 2007;104(4):1307–1312.
- Braoudaki M, Tzortzatou-Stathopoulou F, Anagnostopoulos AK, et al. Proteomic analysis of childhood de novo acute myeloid leukemia and myelodysplastic syndrome/AML: correlation to molecular and cytogenetic analyses. Amino Acids. 2011;40(3):943–951.
- Nicolas E, Ramus C, Berthier S, et al. Expression of S100A8 in leukemic cells predicts poor survival in de novo AML patients. Leukemia. 2011;25(1):57–65.
- Alanazi B, Munje CR, Rastogi N, et al. Integrated nuclear proteomics and transcriptomics identifies S100A4 as a therapeutic target in acute myeloid leukemia. Leukemia. 2019.DOI:10.1038/s41375-019-0596-4.
- DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90(8):732–736.
- DiNardo CD, Propert KJ, Loren AW, et al. Serum 2-hydroxyglutarate levels predict isocitrate dehydrogenase mutations and clinical outcome in acute myeloid leukemia. Blood. 2013;121(24):4917–4924.
- Janin M, Mylonas E, Saada V, et al. Serum 2-hydroxyglutarate production in IDH1- and IDH2-mutated de novo acute myeloid leukemia: a study by the acute leukemia French association group. J Clin Oncol. 2014;32(4):297–305.
- Bandura DR, Baranov VI, Ornatsky OI, et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal Chem. 2009;81(16):6813–6822.
- Levine JH, Simonds EF, Bendall SC, et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. 2015;162(1):184–197.
- Haab BB. Methods and applications of antibody microarrays in cancer research. Proteomics. 2003;3(11):2116–2122.
- Tibes R, Qiu Y, Lu Y, et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5(10):2512–2521.
- Kornblau SM, Tibes R, Qiu YH, et al. Functional proteomic profiling of AML predicts response and survival. Blood. 2009;113(1):154–164.
- Hoff FW, Qiu Y, Kornblau SM, et al. Recognition of recurrent protein expression patterns in pediatric acute myeloid leukemia identified new therapeutic targets. Mol Cancer Res. 2018;16(8):1275–1286.
- Hoff FW, Hu CW, Qutub AA, et al. Proteomic profiling of acute promyelocytic leukemia identifies two protein signatures associated with relapse. ProteomicsClin Appl. 2019;13(4):e1800133.
- Hoff FW, Qiu Y, Kornblau SM, et al. Recurrent patterns of protein expression signatures in pediatric acute lymphoblastic leukemia: recognition and therapeutic guidance. Mol Cancer Res. 2018;16(8):1263–1274.
- Kornblau SM, Ruvolo PP, Wang RY, et al. Distinct protein signatures of acute myeloid leukemia bone marrow-derived stromal cells are prognostic for patient survival. Haematologica. 2018;103(5):810–821.
- Quintás-Cardama A, Zhang N, Qiu YH, et al. Loss of TRIM62 expression is an independent adverse prognostic factor in acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2015;15(2):115–127.
- Kornblau SM, Singh N, Qiu Y, et al. Highly phosphorylated FOXO3A is an adverse prognostic factor in acute myeloid leukemia. Clin Cancer Res. 2010;16(6):1865–1874.
- Butler JS, Qiu YH, Zhang N, et al. Low expression of ASH2L protein correlates with a favorable outcome in acute myeloid leukemia. Leuk Lymphoma. 2017;58(5):1207–1218.
- Ruvolo PP, Hu CW, Qiu Y, et al. LGALS3 is connected to CD74 in a previously unknown protein network that is associated with poor survival in patients with AML. E Bio Medicine. 2019;44:126–137.
- Pierce A, Whetton AD, Meyer S, et al. Transglutaminase 2 expression in acute myeloid leukemia: association with adhesion molecule expression and leukemic blast motility. Proteomics. 2013;13(14):2216–2224.
- Kornblau SM, Qiu YH, Zhang N, et al. Abnormal expression of FLI1 protein is an adverse prognostic factor in acute myeloid leukemia. Blood. 2011;118(20):5604–5612.
- Eleftherios Diamandis P. The failure of protein cancer biomarkers to reach the clinic: why, and what can be done to address the problem? BMC Med. 2012;10(1):87.
- Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–528.
- Pérez-Salvia M, Esteller M. Bromodomain inhibitors and cancer therapy: from structures to applications. Epigenetics. 2017;12(5):323–339.
- Bansal H, Kornblau S, Qiu Y, et al. Overexpression of BRD4 is an Adverse Prognostic Factor in Acute Myeloid Leukemia. Blood. 2017;130(Suppl_1):3794.
- Ferrier CM, de Witte HH, Straatman H, et al. Comparison of immunohistochemistry with immunoassay (ELISA) for the detection of components of the plasminogen activation system in human tumour tissue. Br J Cancer. 1999;79(9–10):1534–1541.