
1. Introduction
How proteins are selected and secreted from the crowded intracellular space is a key question in biology [Citation1]. Besides the classical ER-Golgi exocytic route, which still dominates the detailed characterization of secretomes (all proteins secreted by a cell), three additional unconventional secretory pathways have been described: (i) Golgi-bypassing of membrane proteins, (ii) pore-mediated secretion (lipid or proteinaceous pore), and (iii) vesicle-mediated secretion (secretory lysosomes, endosomes and multivesicular bodies) [Citation1,Citation2]. The main reasons for the dominance of classical secretion for the full characterization of secretomes are, besides its partially higher protein abundance, (i) the concept of classical secretion relies on a well-established signal hypothesis is [Citation3], (ii) classically secreted proteins can be predicted due to their N-terminal signal peptide and thereby differentiated from intracellular ‘contaminants’ [Citation4], and (iii) analytical tools (e.g. specific secretion inhibitors brefeldin A and monensin) are available that allow validation of the classical secretory pathway. However, a growing body of experimental data shows that a large proportion of secreted proteins are released by unconventional secretory pathways and are not intracellular contaminants, as considered previously. Particularly, data from proteomic studies contribute to this observation [Citation5]. In contrast to classical protein secretion, for proteins released by unconventional secretory pathways, a signal motif has not yet been described, and analytical tools that allow identification of the underlying secretory pathways remain scarce. Due to these factors, full characterization of the ‘unconventional secretome’ has not yet been realized. Thus, alternative approaches have been developed and applied to unravel these mechanisms of protein secretion. In this context, comparative secretomics has proven to be a versatile tool to allow full characterization of the secretome [Citation6].
2. Comparative secretomics: the method
Here, comparative secretomics is defined as the parallel analysis of extracellular and intracellular proteomes by quantitative mass spectrometry (MS) () to fully characterize the secretome without prediction tools. Several approaches have been applied with different frequencies, and these studies mainly differ in the quantification methods (e.g. isotope labeling, label-free, or peptide spectrum matches) and data analysis approaches (e.g. fold change, significance analysis of microarrays (SAM), or localized statistics of protein abundance distribution (LSPAD)). However, all take advantage of the unbiased identification of proteins by MS [Citation7–9]. Although mostly applied in cell culture models using serum-free medium, comparative secretomics is not restricted to specific cell culture conditions and should be compatible with approaches based on click-chemistry [Citation10] or comparisons of isotope-labeled amino acid incorporation rates (CILAIR) [Citation11].
Figure 1. Overview of the comparative secretomics workflow. Any kind of cell culture model can be analyzed. Therefore, the supernatant (secretome) and cells (intracellular proteome) are collected and prepared according to the media conditions. After proteolytic digestion, the peptides are analyzed by LC-MS/MS. The quantitative analysis will give access to bona fide secreted proteins and the qualitative peptide matching allows to draw conclusion about ectodomain shedding
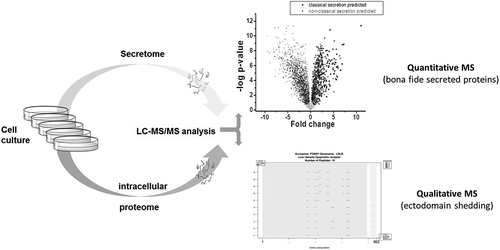
3. Comparative secretomics gives access to high-confident secretome
To date, the major challenge of secretome characterization is the differentiation of actively secreted proteins from contaminants originating from cell culture medium or dead cells. Common approaches are simply based on the identification of ‘secreted’ proteins by shotgun MS, mRNA sequencing, or immunological detection (e.g. antibody arrays). Depending on whether the methods are robust against the identification of false-positives (contaminants), these approaches strongly depend on the prediction of protein secretion, which has been shown only to be accurate for classically secreted proteins [Citation12,Citation13]. Therefore, comparative secretomics was developed to allow the direct experimental identification of secreted proteins by omitting bioinformatic annotations as a decisive step. Comparative secretomics have shown to be robust and give access to high-confident data with a high proportion (>80%) of bona fide secreted proteins and a five-fold higher identification rate if consideration was not limited to proteins exclusively identified in the secretome [Citation6]. Nevertheless, further research is necessary to better understand the composition of the secretome, because it depends on different criteria, such as cell type and culture condition. Further characterization of the high-confident secretome has revealed that proteins released by unconventional vesicle-mediated secretion processes are difficult to detect and that this protein group is not accessible without additional sample preparation protocols (unpublished data).
4. Comparative secretomics gives access to unconventionally secreted proteins
Comparative secretomics provides unbiased access to the otherwise neglected unconventionally secreted proteins, without prior in-depth functional validation. With the help of comparative secretomics, it was possible to increase the number of candidate proteins that are secreted that lack a signal peptide and to develop a novel prediction tool for protein secretion [Citation13]. In contrast, existing computational tools for predicting unconventional protein secretion, such as SecretomeP [Citation14] and SPRED [Citation15], make use of classical secretory proteins by removing their signal peptides, based on the hypothesis that all secretory proteins share common features, regardless of specific pathways [Citation13]. In the first version, it was shown that OutCyte exhibits a higher accuracy for the prediction of unconventionally secreted proteins (AUC = 0.801). In line with this, eight features were extracted as important for the prediction of unconventional protein secretion, including a high frequency of arginine residues and positively charged amino acids. These examples verified that comparative secretomics provides a better understanding of unconventional non-vesicle-mediated secretory pathways.
5. Comparative secretomics gives access to candidate proteins released by ectodomain shedding
Although ectodomain shedding (enzymatic shaving) of membrane proteins is a well-known process that releases functionally active proteins into the extracellular space and significantly contributes to the secretome, it is not considered a secretory process. Nevertheless, the identification of membrane proteins released by ectodomain shedding is relevant for the full characterization of the secretome and exclusion of contaminants. The bottom-up approach (analysis of proteolytic peptides) applied to MS analysis, within comparative secretomics, offers the opportunity to match the identified peptides with the protein sequence and the respective intra- and extracellular domains (). Due to the availability of sequence data from the secretome and intracellular proteome, comparative secretomics allows meaningful conclusions to be made regarding membrane proteins released by ectodomain shedding or released due to cell death. For ectodomain shedding proteins, only peptides of the extracellular domain are identified in the secretome samples and peptides of both parts are identified in intracellular proteome samples. For proteins released due to cell death, peptides of both parts are identified in secretome samples. Thus, comparative studies provide information on ectodomain shedding without additional sample preparation.
6. Concluding remarks
Comparative secretomics offers, for the first time, an in-depth characterization of the human secretome through the identification of unconventionally secreted proteins (except vesicle-mediated protein secretion) and removal of intercellular contaminants by a direct experimental approach. Based on Outycyte’s prediction, the number of unconventionally secreted proteins has been estimated to be 3,475 proteins, exceeding the number of current annotations [Citation16] by factor 10, and demonstrates that the human secretome is far more complex than expected. Thus, a database that contains high-confident secretome data from comparative secretomics should be the next step to open new avenues to better understand the extracellular space, develop novel biomarkers, and identify drug targets. I am convinced that comparative secretomics will initiate further projects and contribute to unravel new mechanisms of protein secretion.
Declaration of interests statement
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Sitia R, Rubartelli A. Evolution, role in inflammation, and redox control of leaderless secretory proteins. J Biol Chem. 2020;295(22):7799–7811.
- Rabouille C, Malhotra V, Nickel W. Diversity in unconventional protein secretion. J Cell Sci. 2012;125(Pt 22):5251–5255.
- Matlin KS. Spatial expression of the genome: the signal hypothesis at forty. Nat Rev Mol Cell Biol. 2011;12(5):333–340.
- Almagro Armenteros JJ, Tsirigos KD, Sonderby CK, et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol. 2019;37(4):420–423.
- Villarreal L, Mendez O, Salvans C, et al. Unconventional secretion is a major contributor of cancer cell line secretomes. Mol Cell Proteomics. 2013;12(5):1046–1060.
- Poschmann G, Brenig K, Lenz T, et al. Comparative secretomics gives access to high confident secretome data: evaluation of different methods for the determination of bona fide secreted proteins. Proteomics. 2020;e2000178.
- Grube L, Dellen R, Kruse F, et al. Mining the secretome of C2C12 muscle cells: data dependent experimental approach to analyze protein secretion using label-free quantification and peptide based analysis. J Proteome Res. 2018;17(2):879–890.
- Loei H, Tan HT, Lim TK, et al. Mining the gastric cancer secretome: identification of GRN as a potential diagnostic marker for early gastric cancer. J Proteome Res. 2012;11(3):1759–1772.
- Luo X, Liu Y, Wang R, et al. A high-quality secretome of A549 cells aided the discovery of C4b-binding protein as a novel serum biomarker for non-small cell lung cancer. J Proteomics. 2011;74(4):528–538.
- Eichelbaum K, Winter M, Berriel Diaz M, et al. Selective enrichment of newly synthesized proteins for quantitative secretome analysis. Nat Biotechnol. 2012;30(10):984–990.
- Roelofsen H, Dijkstra M, Weening D, et al. Comparison of isotope-labeled amino acid incorporation rates (CILAIR) provides a quantitative method to study tissue secretomes. Mol Cell Proteomics. 2009;8(2):316–324.
- Nielsen H, Petsalaki EI, Zhao L, et al. Predicting eukaryotic protein secretion without signals. Biochim Biophys Acta Proteins Proteom. 2019;1867(12):140174.
- Zhao L, Poschmann G, Waldera-Lupa D, et al. OutCyte: a novel tool for predicting unconventional protein secretion. Sci Rep. 2019;9(1):19448.
- Bendtsen JD, Jensen LJ, Blom N, et al. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel. 2004;17(4):349–356.
- Kandaswamy KK, Pugalenthi G, Hartmann E, et al. SPRED: a machine learning approach for the identification of classical and non-classical secretory proteins in mammalian genomes. Biochem Biophys Res Commun. 2010;391(3):1306–1311.
- Uhlen M, Karlsson MJ, Hober A, et al. The human secretome. Sci Signal. 2019;12:609.