
ABSTRACT
Introduction
Proteomic analysis has contributed significantly to the study of the neurodegenerative disease amyotrophic lateral sclerosis (ALS). It has helped to define the pathological change common to nearly all cases, namely intracellular aggregates of phosphorylated TDP-43, shifting the focus of pathogenesis in ALS toward RNA biology. Proteomics has also uniquely underpinned the delineation of disease mechanisms in model systems and has been central to recent advances in human ALS biomarker development.
Areas covered
The contribution of proteomics to understanding the cellular pathological changes, disease mechanisms, and biomarker development in ALS are covered.
Expert opinion
Proteomics has delivered unique insights into the pathogenesis of ALS and advanced the goal of objective measurements of disease activity to improve therapeutic trials. Further developments in sensitivity and quantification are expected, with application to the presymptomatic phase of human disease offering the hope of prevention strategies.
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that causes progressive weakness due to death of ventral horn motor neurons within the spinal cord and pyramidal cells of the motor cortical areas [Citation1]. ALS is aggressive, leading to death within 3 years of symptom onset in most cases, though its progression is highly variable [Citation1]. Although most cases of ALS are sporadic, around 10% of cases report a family history; most of these, and a small proportion of sporadic cases, are attributable to variants in one of a handful of genes, though variants in over 40 genes have been implicated in ALS [Citation2]. Beyond monogenic causes, ALS shows significant heritability in twin studies, and recent research indicates an oligogenic contribution to ALS susceptibility [Citation3,Citation4]. ALS has clinical and pathological overlap with frontotemporal dementia (FTD) and shares genetic risk, primarily through pleiotropic effects of hexanucleotide repeat expansion in an intronic region of the C9orf72 gene, which constitutes the most common monogenic cause of ALS [Citation1,Citation5].
The plethora of perturbations in intracellular pathways that has been implicated through different monogenic causes of ALS suggests that motor neuron degeneration occurs as the result of the final common pathway of many upstream molecular alterations such as defects in RNA processing, protein homeostatic processes, oxidative stress, and cytoskeletal perturbations [Citation6,Citation7]; epidemiological evidence suggests that there may be a summation of insults that lead to catastrophic neurodegeneration [Citation8].
In common with other neurodegenerative diseases, neuronal loss accompanied by insoluble protein inclusions are core pathological features of ALS, occurring primarily in the motor cortical regions, brainstem motor nuclei, and ventral horn of the spinal cord [Citation9]. Our knowledge of major aggregate components owes much to proteomics: the identification of TDP-43 as the major component of inclusions in over 95% of ALS cases (excepting those with genetic ALS due to SOD1 or FUS mutation) and 50% of FTD cases was achieved using liquid chromatography-tandem mass spectrometry (LC-MS/MS) of urea-soluble brain fractions [Citation9].
The consequences of this landmark finding shifted etiological hypotheses of ALS toward mechanisms in which TDP-43 plays a central role, particularly relating to its functions in transcription, translation, and splicing, the stress response, mitochondrial function, and the inherent aggregation properties of TDP-43 that might contribute to non-cell autonomous mechanisms of neurodegeneration [Citation6]. It also led to the identification of mutations in TARDBP, encoding TDP-43, as a cause of a small proportion of ALS cases, spawned novel ALS disease models, and led to refocusing of biomarker study toward measuring and characterizing full-length, truncated, and phosphorylated TDP-43 forms.
Whether ALS occurs through alterations in protein–protein interactions leading to aberrant stress response, excitotoxicity, decreases in protein degradation mechanisms leading to aggregation, changes in axonal transport mechanisms or gene splicing, or due to non-cell autonomous mechanisms – as the major biological effector molecules, proteins are implicated in every proposed pathological mechanisms in ALS [Citation6]. Proteomics is therefore ideally placed to disentangle these mechanisms by providing a platform to identify individual protein or coordinated protein network changes [Citation10] that can be applied to tissue, disease models, and directly in ALS patients.
This review highlights the contribution of proteomics to the study of ALS, in pathological tissue specimens, in disease models, and in the biomarker field (highlighted in ). It also discusses the potential future contributions of proteomic techniques at the leading edge.
Figure 1. Insights into ALS pathophysiology from proteomics
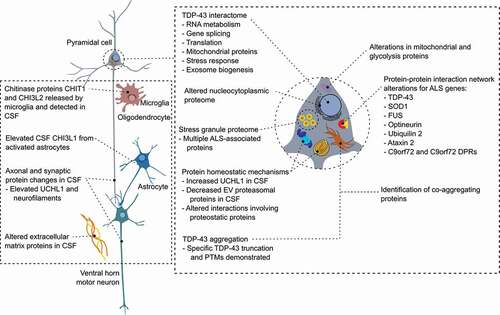
2. Proteomics of human tissue in ALS
Proteomic analysis of post mortem tissue from ALS patients has driven progress in our understanding of underlying disease mechanisms. Studies using proteomic techniques to analyze human ALS tissue are summarized in . Although protein aggregation has long been recognized as a core pathological feature of ALS, it was not until the publication in 2006 of LC-MS/MS analysis of urea soluble, detergent insoluble fractions from neuropathological tissue of patients with ALS and FTD, that TDP-43 was the major aggregate component in over 95% of ALS cases, including ubiquitinated, abnormally truncated, and hyperphosphorylated TDP-43 species [Citation9,Citation11]. Immunoblotting of urea-soluble fractions and immunohistochemistry indicates a pattern of full length and truncated peptides that mostly represent C-terminal peptides at 20–35 kDa in brain tissue from ALS and FTD patients [Citation12]; interestingly, this is a much less consistent finding in spinal cord [Citation13]. Subsequent work has utilized proteomics to further define the nature of TDP-43 in aggregates.
Table 1. Proteomic studies of human tissue samples in ALS. ALS – amyotrophic lateral sclerosis; CSF – cerebrospinal fluid; EV – extracellular vesicle; FT-ICR – Fourier transform ion cyclotron resistance; iTRAQ – isobaric tags for relative and absolute quantitation; LC-MS/MS – liquid chromatography tandem mass spectrometry; MALDI – matrix-assisted laser desorption-ionization; TOF – time-of-flight
Three studies have used proteomic analysis to identify endogenously truncated TDP-43 peptides by identifying semi-tryptic or semi-chymotryptic N- and C-terminal TDP-43 peptides (i.e. TDP-43 specific peptides with one non-enzymatically digested terminus) in urea-soluble fractions from post mortem tissue of patients with ALS or FTD by in-gel digestion of lower molecular weight bands [Citation19–21]. Most endogenous truncation sites were found on the N-terminus of peptides, suggesting the enrichment of C-terminal TDP-43 fragments, but also C-terminal truncations were found, broadening the knowledge of pathological processing of TDP-43 ().
Table 2. Endogenous TDP-43 truncation peptides identified in tissue proteomic studies in ALS. All peptides show the expected trypsin cleavage site after amino acids lysine (K) or arginine (R) or chymotrypsin cleavage site after amino acids phenylalanine (F), tryptophan (W), and tyrosine (Y) and one nonspecific cleavage site suggesting truncation. kDa – kilodaltons
Measurement of the ratio of C- to N-terminal peptides using targeted proteomics appears relatively specific for ALS compared with other neurodegenerative diseases, though abnormal truncation of TDP-43 is also found in Alzheimer’s disease cases accompanied by TDP-43 aggregation [Citation22]. Although alterations in the C:N terminal peptide ratio are not found in ALS spinal cord, concurring with immunoblot findings [Citation20], measurement of C:N terminal ratio appears to be a promising approach for biomarker development. A recent approach using aptamer enrichment prior to quantification to increase the yield of TDP-43 peptides from post mortem tissue might improve the sensitivity of targeted analysis of truncation peptides in biofluids, where such TDP-43 peptides are of much lower abundance [Citation23].
Proteomic analysis of post mortem tissue has also allowed the identification of sites of post-translational modification of proteins involved in ALS, specifically phosphorylation, acetylation, and ubiquitination of TDP-43 [Citation19]. Although a robust finding in tissue samples, it has so far not been possible to reliably recapitulate disease-specific TDP-43 phosphorylation in biofluids, limiting its application as a biomarker. Disease-specific phosphorylation sites in neurofilament heavy chain have also been sought in ALS, though phosphorylation appears similar in ALS patients and controls [Citation24].
Looking beyond disease-associated protein inclusions, unbiased analysis of post mortem brain and spinal cord tissue has been employed to explore broader alterations in the protein network occurring in ALS. Several studies have incorporated analysis of brain tissue from patients with ALS and TDP-43 FTD subtypes. A recent example used LC-MS/MS analysis of brain tissue of patients with ALS (as well as FTD subtypes) identified over 50 proteins enriched in the sarkosyl-insoluble fractions of ALS brain, including a subset of 23 co-aggregating proteins that differentiated ALS from FTD subtypes, such as the presence of Profilin 1, 26S proteasomal subunit D2, and Tubulin alpha 4 A chain, among others [Citation11].
Network analysis of prefrontal cortex of ALS, ALS-FTD, and FTD patients using weighted gene co-correlation network analysis (WGCNA) has been used to identify coordinated changes within the protein network in ALS and FTD [Citation25]. This indicated relatively minor changes in pure ALS cases (as would be expected given the lack of involvement of wider frontal lobe areas in pure ALS), though a protein co-expression module associated with immune system functioning was upregulated in ALS. Further work to define disruptions at the proteome level would benefit from the inclusion of additional CNS tissue regions salient to ALS (such as the brainstem, thalamus and spinal cord) in order to broaden the understanding of regional and mechanistic differences in these overlapping conditions.
A small number of studies have used proteomics to examine spinal cord tissue. Recent studies have identified dysregulation of mitochondrial proteins and those involved in carbohydrate metabolism mRNA splicing and of the neurofilament compartment as well as altered acetylation of glial fibrillary acidic protein (GFAP) [Citation26–28]. Spinal cord lysates from sporadic ALS patients and those carrying a SOD1, FUS, and C9orf72 variants have been used to explore TDP-43 interacting proteins, highlighting the role of TDP-43 in RNA processing and translation, as well as suggesting greater overlap between TDP-43 interactors in FUS and C9orf72 cases and TDP-43 interactors in SOD1 and sporadic ALS cases than between these pairings [Citation29].
A small preliminary study employed matrix-assisted laser desorption-ionization (MALDI) imaging to explore spatial alterations in protein expression; due to technical limitations, only a small number of proteins were identified, though decreased levels of a truncated ubiquitin were observed in the ALS group [Citation30]. Preliminary exploration of the feasibility of using laser capture microdissection of motor neurons from post mortem tissue has been explored as a means to study the human motor neuron-specific proteome, though this has not been applied successfully in comparative study [Citation31].
Another relevant tissue with limited ALS proteomic studies is muscle. While gene expression studies have used muscle samples, only a few studies have focused on proteomics in ALS muscle (). Given the intensified collection of postmortem muscle tissue within ALS biorepository efforts, future studies to explore the proteome in different muscle tissue types are warranted and may provide new mechanistic insights into muscle degeneration that occurs during ALS and potential new blood-based biomarkers released by muscle.
3. Proteomic analysis of disease models in ALS
Proteomic techniques have featured in a vast number of studies of cellular and animal models of ALS, contributing to the major hypotheses of ALS pathogenesis.
3.1. Alterations in protein–protein interactions in ALS
Defining disease-related alterations in protein–protein interaction networks is an essential aspect of understanding the pathophysiological processes that lead to ALS, which has relied heavily upon proteomics. These studies have focused primarily on TDP-43, extending to other ALS-associated gene mutations, using immunoprecipitation coupled with mass spectrometry.
In addition to the aforementioned post mortem tissue study of the TDP-43 interactome [Citation29], tissue culture models have examined the effect of ALS-causing A315T and M337V TDP-43 mutations using primary neuronal cultures [Citation32] and cell lines [Citation33,Citation34] in physiological conditions as well as following oxidative stress, RNA depletion, and DNA damage [Citation35,Citation36]. In addition to highlighting the multifaceted interactions of TDP-43 with splicing and translation machinery, mitochondrial proteins, and proteins involved in the stress response, this work has indicated that the TDP-43 interactome is condition-dependent and altered in the presence of TDP-43 mutations, with effects on the cellular stress response, translation, and exosome biogenesis pathways [Citation32,Citation33]. Analysis of the FUS interactome has highlighted major overlap in the pathway annotations of FUS and TDP-43 interactors around RNA metabolism and splicing, stress granules, exosomes, and mitochondrial proteins [Citation37,Citation38], as well as its involvement in protein degradation pathways [Citation39].b
Proteomic approaches have studied the interactions of other proteins implicated in ALS. Systematic analysis using immunoprecipitation of Ubiquilin 2, FUS, Ataxin 2, C9orf72, TDP-43, and Optineurin in N2a cells demonstrated common interactors and overlapping functional annotations, particularly in relation to DNA and RNA binding, ribosomal proteins, and eukaryotic initiation factors for TDP-43, FUS, and Ataxin 2 interactors and protein homeostatic roles for Ubiquilin 2 and Optineurin interactors [Citation35]. The protein constituents and interactors of SOD1 aggregates have also been probed using proteomic techniques. Cytoskeletal proteins, particularly the intermediate filament protein Vimentin, as well as GFAP and neurofilament proteins, have been consistently identified in native spinal cord detergent-insoluble fractions and whole spinal cord lysate in SOD1 mouse models [Citation40–42]. Other proteins co-aggregating in SOD1 inclusions in the SOD1G93A mouse model include proteins involved in glycolysis and mitochondrial pathways and chaperones [Citation40], pathways overlapping with those of proteins identified as co-aggregating with TDP-43 in ALS brain tissue [Citation11].
3.2. Posttranslational modifications
Abnormal ubiquitination and phosphorylation of aggregated proteins is a core pathological feature of ALS. Proteomic analysis has provided a platform to study different posttranslational modifications in disease models, specifically focusing on TDP-43 and FUS in ALS, illustrating interplay between ubiquitylation, phosphorylation, and acetylation, as well as the importance of posttranslational modifications in the physiological behavior of proteins implicated in ALS [Citation43–45]. It has also revealed a role of less common modifications such as citrullination in maintaining the physiological function of FUS and TDP-43 and inhibit aggregation, eventually through a decrease of binding to proteins relevant for stress granule formation [Citation46].
Posttranslational modification has also been studied using proteomics of SOD1 aggregates from G93A and G37R mouse models, though significant modifications were not identified [Citation41], at odds with earlier and more recent work demonstrating ubiquitination and conjugation of short ubiquitin modifier proteins (SUMOylation) to aggregated SOD1 protein [Citation47,Citation48], likely reflecting the sensitivity of the mass spectrometry techniques employed. More recently, non-enzymatic deamidation of asparagine to aspartic acid within a proteasomally cleaved SOD1 peptide has been identified in the CSF of the SOD1G93A rat model, with a corresponding deamidated peptide detected in the CSF of human carriers (symptomatic and asymptomatic) of ALS-causing SOD1 variants [Citation49]. Such deamidation has been shown to accelerate protein fibrillization [Citation50], providing a potential link to ALS pathogenesis of this common post-translational modification.
3.3. Proteomics in models of C9orf72 ALS
Current leading hypotheses as to how hexanucleotide repeat expansion in an intronic region of C9orf72 leads to neuronal loss and TDP-43 accumulation center on three potentially synergistic mechanisms: loss of function of C9orf72 protein due to haploinsufficiency, toxicity due to sense and antisense repeat RNA transcription products of the GGGGCC repeat region, and toxicity due dipeptide repeat proteins formed through repeat-associated non-AUG translation [Citation51]. Recent work has explored mechanisms of toxicity of C9orf72 hexanucleotide repeat expansion using proteomics, including the interactome of dipeptide repeat proteins, with notably consistent enrichment of ribosomal proteins across studies, as well as RNA splicing and mitochondrial proteins and proteins involved in autophagy and proteasomal systems detected in the interactomes of the toxic arginine-containing dipeptides [Citation52–56]. Protein interactors of repeat RNA transcripts have also been identified using proteomics, with enrichment (perhaps unsurprisingly) of proteins involved in RNA metabolism and containing RNA recognition motifs [Citation57].
Mass spectrometry has also delineated the interactome of C9orf72 protein, demonstrating enrichment for autophagy proteins, cytoskeletal components as well as ubiquilin 2 and heterogeneous ribonucleoprotein A1 and A2/B1, proteins implicated in ALS through rare genetic mutations and roles in proteostasis, RNA processing, and stress granules; and separately enrichment of mitochondrial proteins and chaperones, providing convergence on TDP-43-associated disease alterations [Citation35,Citation58–60].
3.4. Subcellular compartment alterations
Proteomic analysis of subcellular fractions has been applied to cellular and ALS disease models based on SOD1 mutation and overexpression, indicating significant alterations in the proteome of cell lines, spinal cord and brain tissue of rodents overexpressing wild-type and mutant SOD1, relating to multiple pathways including mitochondria, metabolism, and protein degradation and overlapping with proteomic evidence from human tissue in sporadic ALS [Citation26,Citation61–65]. Alterations in the nucleocytoplasmic distribution of TDP-43 are a key histopathological feature of ALS, and nuclear pore complex dysfunction has been observed in ALS models, particularly relating to C9orf72 ALS [Citation66]. Comparative proteomic analysis of nuclear and cytoplasmic fractions from a HEK293 C9orf72 hexanucleotide repeat model indicates a shift in the distribution of proteins involved in RNA metabolism and translation toward the cytoplasm [Citation67]. Alterations in the nucleocytoplasmic distribution of RNA processing and translation proteins are also observed following TDP-43 knock-down [Citation68], while RNA transport pathway alterations have been demonstrated with overexpression of mutant SOD1, though these alterations are opposed to those observed in the C9orf72 model [Citation69].
3.5. Stress granules
Proteomic analysis of stress granule cores – membraneless organelles comprising RNA and protein that form by liquid–liquid phase separation in response to stress [Citation70] – indicates a major role for ALS-associated RNA-binding proteins including TDP-43, FUS, and other heterogeneous ribonucleoproteins (hnRNPs) in stress granule physiology [Citation71]; stress granule cores are proposed to act as a nidus for TDP-43 aggregation [Citation72]; time-series proteomic analysis of stress granule disassembly indicates the importance of altered SUMOylation in delaying stress granule disassembly in a Drosophila C9orf72 ALS model [Citation73]. Accordingly, dipeptide repeat proteins have also separately been found to interact with stress granule proteins [Citation54]. A recent study examining the Caprin-1 proteome in stress granules identified a new hnRNP, SNRNP200, that was also localized to cytoplasmic aggregates in ALS spinal cord [Citation74].
4. Proteomics of biofluids and the search for ALS biomarkers
Biofluid-based biomarkers present several potential opportunities in ALS. Although for the most part, the diagnosis of ALS is not difficult to achieve in the specialist clinic, sensitive and specific biomarkers have long held promise as a means to resolve diagnostically challenging ALS cases or enable earlier nonspecialist diagnosis [Citation75]. Identifying useful biomarkers that fulfil this promise, however, has proven a major challenge. The axonal cytoskeletal neurofilament proteins neurofilament light chain (NFL) and phosphorylated neurofilament heavy chain (pNFH) have long led on this front [Citation76,Citation77]. Although showing promising specificity and sensitivity in retrospective and prospective analysis [Citation78–81], the fact that they are nonspecific markers of axonal degeneration (i.e. are not ALS-specific) and show relatively modest rises in slower-progressing, harder-to-diagnose cases, has hampered their translation into clinical use [Citation82]. ALS has also so far been resistant to combinatorial diagnostic approaches such as CSF Abeta/Tau ratio in Alzheimer’s disease [Citation83] or recently developed protein aggregation-based assays such as RT-QuIC in prion diseases and more recently synucleinopathies [Citation84,Citation85].
A more important role for ALS biomarkers lies in the measurement of underlying disease activity and target engagement in ALS, to support development of novel therapeutics. Drug trials in ALS currently rely on clinical outcome measures, primarily functional decline as measured by the revised ALS functional rating scale (ALSFRS-R), a 48-point score that declines through the course of the disease [Citation86]. Clinical staging systems, decline in respiratory function, muscle strength measures, and survival are also frequently employed [Citation86]. All of these measures accrue slowly over time, and most are confounded by subjectivity or effort and consequently require prolonged follow-up periods with large numbers of participants to measure change. Driven by the desire to detect effects while maintaining relatively small sample size and trial duration, these measures also promote a tendency to limit trial eligibility to those in early disease stages or with aggressive disease, in whom change is detectable over a short timescale; consequently, this may limit the generalizability and eventually access of patients with less aggressive disease to effective treatment [Citation87].
Biomarkers represent an opportunity to provide sensitive, objective, rapidly changing measures that could reduce trial duration and sample sizes, hastening the therapeutic development process while reducing costs and broadening trial inclusion and providing highly valuable information about the underlying frequent failure of preclinically promising drugs in clinical trials [Citation88]. A number of studies have been performed to evaluate specific proteins as pharmacodynamic or prognostic biomarkers in ALS model systems or patient-derived samples [Citation89–93]. Recent ALS clinical trials have explored the use of protein biomarkers as pharmacodynamic biomarkers of treatment effect or as inclusion criteria and then monitoring of treatment effect during the trial [Citation94,Citation95].
The application of proteomic technologies to cerebrospinal fluid (CSF) and blood from ALS patients has been a staple of efforts to identify potential ALS biomarkers over the last two decades. Most proteomic studies have used CSF, due to its proximity to the CNS cells affected by ALS [Citation96]. The relatively low content of highly abundant proteins compared to serum and plasma, reducing the need for depletion methods or separation approaches, and the lower risk of detecting signals of secondary systemic metabolic alterations related to the disease (for example malnutrition due to swallowing difficulties) are additional advantages of studying CSF compared with serum or plasma, though much of the CSF proteome is in fact blood-derived [Citation97]. The obvious disadvantage is, of course, the relatively invasive approach to CSF sampling when compared to blood.
Most frequently, bottom-up shotgun proteomic approaches have been employed, though over this time much of the breadth of proteomic technology has been applied at some point to the study of ALS. Reproducibility has been an issue; despite alterations in over 500 proteins detected over the course of CSF proteomic experiments, only a handful have been demonstrated in two or more studies and even fewer have survived external orthogonal validation techniques [Citation98]. Proteomic studies of human biofluid samples in ALS are summarized in .
Table 3. Proteomic studies of human biofluid samples in ALS. ALS – amyotrophic lateral sclerosis; CSF – cerebrospinal fluid; EV – extracellular vesicle; FT-ICR – Fourier transform ion cyclotron resistance; iTRAQ – isobaric tags for relative and absolute quantitation; LC-MS/MS – liquid chromatography tandem mass spectrometry; MALDI – matrix-assisted laser desorption-ionization; PBMC – peripheral blood mononuclear cells; SELDI – surface-enhanced laser desorption-ionization; SWATH – sequential window acquisition of all theoretical spectra; TMT – tandem mass tag; TOF – time-of-flight
The first mass spectrometric study of CSF in ALS used Fourier transform ion cyclotron resonance (FT-ICR) of tryptically digested CSF samples from a small cohort of ALS patients and healthy controls to produce a classifier based on the resulting spectrograms [Citation107]. Although this early foray into proteomics in ALS identified no individual biomarker candidates, it represents the first mass spectrometric analysis of ALS patient CSF and foretold the later use of multiple proteomic features as the basis for classification algorithms; similar machine learning approaches have been utilized in more recent studies [Citation108].
Subsequent early proteomic biomarker studies in ALS moved toward surface-enhanced laser desorption-ionization TOF (SELDI-TOF) mass spectrometry analysis for top-down proteomics of CSF from ALS patients [Citation109–111]. Between these three studies, some overlap was observed with lower levels of Cystatin-C detected in all three (and validated using CSF immunoblot) as well as decreases in Transthyretin in two studies. As the major constituent of lower motor neuron Bunina body inclusions specific to ALS, Cystatin-C was of particular interest as a biomarker; external validation however has subsequently proved contradictory [Citation112,Citation113]. Additionally, in the most recent SELDI-TOF study [Citation111], incorporating samples from 100 ALS patients and 141 controls, levels of the acute phase protein C-reactive protein (CRP) were found to be elevated in ALS with confirmatory enzyme-linked immunosorbent assay (ELISA); although elevated serum CRP has been associated with worse prognosis in ALS patients, a recent ELISA validation of CSF CRP levels did not confirm this finding [Citation114–116].
Two CSF proteomic studies in ALS incorporated 2D gel electrophoresis to identify differentially abundant proteins in CSF pools from ALS patients and controls [Citation117,Citation118], with subsequent matrix-assisted laser desorption-ionization mass spectrometry or tandem MS identification of differentially abundant protein spots including upregulation of Alpha-1-antitrypsin precursor and Zn-alpha-2-glycoprotein, both demonstrating sometimes contradictory alterations in other proteomic and immunoassay studies [Citation119–121].
More recent studies have employed LC-MS/MS of individual or pooled CSF samples, with preanalytical abundant protein depletion or prefractionation techniques to drive additional proteomic depth and in some cases isobaric labeling to enhance quantitative accuracy [Citation28,Citation108].
A major and consistent feature of recent LC-MS/MS proteomic datasets has been the upregulation of a set of related glial proteins involved in innate immunity, the chitinase proteins, in ALS. The first recognition of coherent alterations in chitinase proteins used LC-MS/MS of pooled CSF samples from ALS patients and controls, identifying a striking upregulation of the active chitinase Chitotriosidase 1 (CHIT1) alongside elevation of the two related inactive chitinase proteins Chitinase 3-like protein 1 (CHI3L1 or YKL-40) and Chitinase 3-like protein 2 (CHI3L2 or YKL-39) [Citation122].
Though the authors were unable to orthogonally validate CHI3L1, subsequent independent proteomic, immunoassay-based, and enzyme activity studies have consistently indicated marked elevation of CHIT1 and, to a lesser extent, CHI3L1 and CHI3L2, in CSF (but not blood) from patients with ALS [Citation28,Citation108,Citation114,Citation123–128].
Emerging literature in ALS indicates that CHIT1 is primarily produced by microglia [Citation98]; intrathecal injection of CHIT1 leads to microglial activation, astrogliosis, and loss of motor neurons in rodents [Citation129]. CHIT1 levels correlate with the rate of functional decline in ALS, a proxy for the aggressiveness of disease as well as neurofilament levels [Citation114,Citation126–128]. CHI3L1, on the other hand, is produced by a subset of activated astrocytes and correlates more closely with the burden of upper motor neuron pathology and cognitive impairment in ALS [Citation114,Citation128]; correspondingly, while CHIT1 levels are markedly increased in ALS but more modestly so in FTD, CHI3L1 levels show more modest elevation in ALS and more pronounced elevation in FTD [Citation123]. CHI3L1 is less closely associated with disease progression, but is similarly correlated with neurofilament levels when compared with CHIT1; both CHIT1 and CHI3L1 have shown inconsistent associations with survival as well as inconsistent small longitudinal increases [Citation114,Citation124,Citation127,Citation128].
Overall, CHIT1 and CHI3L1 represent a recent major success for proteomic biomarker discovery in ALS. Although they do not outperform neurofilaments in terms of prognostic or classifier performance, they represent different dimensions of the underlying disease process – specifically microglial and astrocytic activity – that represent pathways potentially amenable to disease-modifying treatments [Citation114]. Chitinase proteins are therefore well-placed to measure treatment response in these areas, though it should be noted that common CHIT1 and CHI3L1 polymorphisms leading to alterations in expression are recognized (though do not appear to slow the progression of ALS) [Citation123,Citation130].
More recent analyses have used isobaric labeling with prefractionation to improve proteomic depth in CSF to quantification of almost 2000 proteins [Citation28,Citation131], identifying upregulation of the proteins Ubiquitin C-terminal hydrolase L1, Mictrotubule-associated protein 2, and Glycoprotein NMB in ALS patients in addition to neurofilament and chitinase proteins, validated within-cohort using targeted proteomics and subsequently using single molecule array (SIMOA), as well as comparing CSF findings with post mortem tissue [Citation28,Citation131]. The comparison of protein level changes in CSF and tissue contributes to the understanding of the origin of alterations of the CSF proteome. The upregulation of neurofilaments in CSF but lower levels in spinal cord tissue is in agreement with the release of neurofilaments into the extracellular space by degenerating axons. In contrast, neuroinflammatory proteins such as chitinases, Glycoprotein NMB, and Macrophage-capping protein are increased in both, indicating elevated tissue expression during disease [Citation28].
4.1. Extracellular vesicle proteomics – a window on intracellular processes in ALS
Extracellular vesicles (EVs) are 50–200 nm structures, including exosomes and microvesicles, released by virtually all cells, including neurons and glia of the central nervous system [Citation132]. Alterations in EV biogenesis pathways have been identified in cellular models of genetic ALS and implicated as a potential vector for the intercellular spread of toxic oligomers of TDP-43 [Citation32,Citation133]. EVs are also an attractive target for biomarker discovery efforts due to their intracellular origin, potentially opening a window on mechanisms of disease [Citation134].
However, the low number of EVs in CSF, combined with the contribution of multiple CNS cell types and the use of MS-incompatible isolation techniques (such as those involving polyethylene glycol based precipitation), poses major challenges to the application of proteomic approaches [Citation134]. Research in this field applying mass spectrometry approaches to CSF EVs for biomarker discovery in ALS has so far been very limited, including one targeted proteomic study measuring relative exosomal TDP-43 levels, which did not differ between ALS patients and controls [Citation135], and two small shotgun proteomic studies, which identified decreased proteasomal and proteasome-like proteins in ALS [Citation136], a pathway previously implicated through post mortem analysis of spinal cord tissue; and increased levels of the nucleolar protein Novel INHAT repressor [Citation137], both of which await external verification.
An alluring means to simplify access to CNS biomarkers has emerged through the analysis of CNS-derived EVs extracted from serum by immunoprecipitation of EVs carrying the neuronal lineage marker L1CAM [Citation138]. This method has been used in targeted biomarker development approaches in Alzheimer’s disease and Parkinson’s disease [Citation139,Citation140]. Whether EVs isolated in this way truly represent a pool of CNS origin is hotly debated, in part due to expression of L1CAM beyond the CNS and some evidence indicating that most serum L1CAM is a cleaved ectodomain [Citation141]. Further work to delineate their origin and identify more robust means of extracting a relevant EV population using proteomics would be highly valuable. Ultimately, a combination of proteomics and transcriptomics of EVs may provide the optimal ALS-specific biomarker.
4.2. CSF proteomics in the presymptomatic period
The identification of highly penetrant ALS-causing genetic variants, particularly C9orf72 hexanucleotide repeat expansion and mutations in SOD1, in upwards of 10% of ALS patients has spawned a cohort of first degree relatives of ALS gene carriers with known high risk of carrying a developing ALS [Citation5]. Evidence from neurophysiological studies and measurement of neurofilament and chitinase proteins in asymptomatic gene carriers suggests that significant neurodegeneration and microglial activation is detectable only months before symptom onset [Citation125,Citation142–144]. Studying gene carriers during the period before onset of neurodegeneration therefore offers an opportunity to define early events preceding neurodegeneration, identify biomarkers that might predict the onset of symptoms, or measure presymptomatic therapeutic response, thereby enabling treatment prior to the onset of symptoms [Citation145].
To date, only one proteomic study has addressed this, comparing 14 asymptomatic carriers of SOD1 and C9orf72 mutations with controls and ALS patients using isobaric tag labeled, prefractionated LC-MS/MS approach [Citation28]. Despite quantifying 1929 proteins, no proteins with significantly differing levels between gene carriers and non-carriers were identified, perhaps attributable to the relatively small asymptomatic carrier group and the mixture of underlying gene mutations reflecting multiple pathway alterations upstream of motor neuron degeneration [Citation28].
4.3. Blood proteomics
Relatively few studies have examined the serum, plasma, or peripheral blood mononuclear cell (PBMC) proteome in ALS. Recent studies have used isobaric labeling of brain tissue alongside plasma samples in order to improve the relevance of the identified proteome and circumvent the problems of highly abundant proteins suppressing signal from more low abundance, potentially more interesting, proteins [Citation146,Citation147]. Consistent themes indicate alterations in complement proteins and apolipoproteins, though reproducibility of individual findings has been lacking [Citation146–152].
4.4. Proteomics transcending biofluid, pathology and disease model boundaries
Modern bioinformatic and proteomic techniques offer the capability to link alterations in the tissue, model, and biofluid proteomes. Proteomic biofluid studies in ALS have generally detected changes presumed to reflect downstream consequences of neurodegeneration, such as the leakage of neurofilament proteins from damaged neurons, activation of glial cells, the effects of synaptic loss, or altered extracellular matrix regulation [Citation28,Citation108,Citation124]. A handful of studies have attempted to bridge this gap using pathway analysis [Citation108], network analysis [Citation153], or direct comparison of post mortem tissue and biofluid proteomic changes [Citation28,Citation154]. These have identified a degree of overlap between network-level changes in the CSF proteome with RNA processing, cellular stress, and metabolic pathways identified in ALS models. Detecting clear perturbations of these pathways in the biofluid proteome that could find clinical use has not yet occurred.
5. Expert opinion
Proteomic analysis has cut across the field of ALS research. It has redefined our understanding of the molecular histopathological hallmarks of ALS and diverted the course of scientific study accordingly [Citation9]. As outlined herein, proteomics has also highlighted pathophysiological mechanisms of ALS, including alterations in protein–protein interaction networks brought about by ALS-associated genetic variants, the importance of proteins implicated in ALS in the cellular stress response, and widespread changes in nuclear and cytoplasmic proteomes. Recent proteomic studies have identified major biomarkers capable of quantifying different dimensions of the disease and linked findings from disease models and post mortem tissue with alterations in the protein network in patient CSF.
Many techniques have been utilized, including a range of preanalytical methods, ionization and separation methods, mass spectrometers, and bioinformatic approaches [Citation155]. Within the field of mechanistic study, proteomics has provided highly valuable insights into the consequences of ALS gene mutations and pathways involved in ALS, though interpretation is necessarily tempered by the types of model used, particularly in relation to overexpression models and the use of SOD1 mutation-based models, which, given the pathological differences between SOD1 and other familial and sporadic ALS forms, may not be a faithful reflection of upstream biological differences leading to sporadic ALS [Citation156]. Given alterations in gene transcription, translation, and metabolic pathways in ALS, it would be well-suited to multi-omic analysis, integrating proteomic, transcriptomic and metabolomic datasets together, which has been thus far limited in ALS [Citation157,Citation158].
Proteomics of pathological tissue and disease models stands to benefit from spatial proteomic techniques such as MALDI imaging, which have so far found limited use in ALS research [Citation30,Citation159,Citation160], in order to resolve compartmentalized aspects of ALS pathology. Techniques to separate tissues and enhance the purity of in vitro models, such as laser capture micro-dissection and fluorescence-activated cell sorting (FACS), offer additional means to decipher changes occurring within individual cell types and their relative contribution to the disease process [Citation161], which could in future be further enhanced by nascent single cell proteomics [Citation162]; newer techniques such as MALDI-2 mass spectrometry promise subcellular compartment resolution [Citation159]. Newer technologies that use multiplex immunofluorescence microscopy data from up to 40 different proteins could also enable spatial resolution of many proteins within the same tissue sample [Citation163].
Within the biomarker field [,], reproducibility of proteomic discoveries has been a major problem, driven in part by the issues of inconsistent preanalytical sample handling, the stochastic nature of data dependent acquisition (DDA) proteomic pipelines and the heterogeneity of the disease [Citation164,Citation165]. In the last decade, however, consistent signals in the chitinase proteins have been demonstrated initially in proteomic and subsequently immunoassay studies [Citation114,Citation122–124,Citation126]. Elevated levels of neurofilament proteins, initially identified in candidate-driven immunoassay studies, have also been identified with increasing consistency in recent proteomic studies, particularly Neurofilament medium polypeptide, which has so far been neglected by target-driven studies [Citation28,Citation108,Citation131]. Chitinase proteins, particularly Chitotriosidase 1, represent a major success for proteomic biomarker development and are now front-running ALS biomarkers; though they have not improved upon the classifier or prognostic performance of neurofilament proteins, they encapsulate alternative dimensions of the disease process so might find use in drug trials targeting glial mechanisms or through the eventual advent of personalized treatment of ALS. Identifying ALS-specific biomarkers, such as those based on disease-associated truncated or posttranslationally modified forms of TDP-43, remains a major challenge to which targeted proteomic methodologies could offer solutions in future [Citation12].
The multiplex nature and absolute quantitative capabilities of targeted mass spectrometry, protein arrays, and aptamer-based proteomics would also be highly suitable for a panel approach to biochemical diagnosis of ALS, though a suitable set of proteins remains elusive [Citation23,Citation166,Citation167].
Defining the biochemical landscape in the preclinical period in ALS gene carriers is a major challenge that looms large [Citation145]. Antisense oligonucleotide therapies targeting the common gene mutations in ALS have reached the clinical trial arena in symptomatic patients [Citation94]. Asymptomatic gene carriers probably have the most potential to benefit from these treatments, but the unpredictable age of onset, even in genetic cases, high costs, and the invasiveness of intrathecal treatment are major barriers to use in this group [Citation145]. Proteomics is ideally placed to identify biomarkers of treatment response or predict symptom onset that could help to remove this barrier by enabling better timing and monitoring of treatment. Detecting subtle proteomic changes in this group, though, will require major improvements in proteomic depth, quantitative accuracy, and large longitudinal cohorts. Some of this may occur by improving the relevance of the proteome of study, for example using analysis of CSF or neuronal-derived serum EVs, or through advances in proteomic approaches such as data independent acquisition methodologies, which have been seldom used in the ALS field to-date.
The goals of ALS biomarker studies are to provide insights into disease mechanisms and biomarkers that are useful in drug development and clinical trials. Continued studies that incorporate biomarkers in ALS drug development programs and clinical trials will generate the data necessary for regulatory agencies to accept biomarkers in their decision-making processes regarding new treatments for ALS.
Article highlights
Proteomics has contributed across the field of ALS in study of post mortem tissue, disease models and identification of biomarkers
Identification of TDP-43 as the major aggregate component in 95% of cases of ALS was brought about by proteomics as well as identifying disease-specific truncation and posttranslational modification of TDP-43
Proteomics has helped delineate the effects of ALS-causing gene mutations on protein interactions and in cellular and animal models
Major advances in ALS biomarkers have come about through the identification of chitinase protein alterations in cerebrospinal fluid of ALS patients
Future advances are likely to include spatial proteomic methods, improvements in proteomic depth through data independent acquisition and through the study of ‘at-risk’ populations to understand early events in ALS
Declaration of Interests
AGT is supported by the Medical Research Council and Motor Neurone Disease Association (MNDA) Lady Edith Wolfson Clinician Scientist Fellowship MR/T006927/1. PO receives research support from the Michael J. Fox Foundation for Parkinson´s Research (Grant ID: MJFF-010349) and Alzheimer Forschung Initiative e.V. (20059CB). RB receives research support from NIH/NINDS grant NS116385, Target ALS, the Fein Foundation, and the Barrow Neurological Foundation. RB is a founder of Iron Horse Diagnostics, a company focused on biomarker assays and therapeutics for ALS and other neurologic diseases. RF and BK were supported by the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Science (CIFMS), China (grant number: 2018-I2M-2-002). MO is funded by ALSA. The authors report no other conflicts of interest. MRT receives support from the MNDA.
References
- Hardiman O, Al-Chalabi A, and Chio A, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;5 (3):7071.
- Renton AE, Chiò A, and Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. . 2014;17 (1):17–23.
- Al-Chalabi A, Fang F, and Hanby MF, et al. An estimate of amyotrophic lateral sclerosis heritability using twin data. J Neurol Neurosurg Psychiatry . 2010;81(12):1324–1326.
- van Blitterswijk M, van Es MA, and Hennekam EA, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet . 2012;21(17):3776–3784.
- Zou ZY, Zhou ZR, and Che CH, et al. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry . 2017;88(7):540–549.
- Taylor JP, Brown RHJ, and Cleveland DW. Decoding ALS: from genes to mechanism. Nature. . 2016;539(7628):197–206.
- Talbot K, Feneberg E, Scaber J, et al. Amyotrophic lateral sclerosis: the complex path to precision medicine. J Neurol. 2018;265(10):2454–2462.
- Al-Chalabi A, Calvo A, and Chio A, et al. Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol . 2014;13(11):1108–1113.
- Neumann M, Sampathu DM, and Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science . 2006;314(5796):130–133.
- Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537(7620):347–355.
- Laferrière F, Maniecka Z, Pérez-Berlanga M, et al. TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat Neurosci. 2019;22(1):65–77.
- Feneberg E, Gray E, and Ansorge O, et al. Towards a TDP-43-Based Biomarker for ALS and FTLD. Mol Neurobiol . 2018; . ;55(10):7789–7801.
- Igaz LM, Kwong LK, Xu Y, et al. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol. 2008;173(1):182–194.
- Alonso R, Pisa D, Marina AI, et al. Evidence for fungal infection in cerebrospinal fluid and brain tissue from patients with amyotrophic lateral sclerosis. Int J Biol Sci. 2015;11(5):546–558.
- Vassileff N, Vella LJ, Rajapaksha H, et al. Revealing the proteome of motor cortex derived extracellular vesicles isolated from amyotrophic lateral sclerosis Human Postmortem Tissues. Cells. 2020;9(7):1709.
- Iridoy MO, Zubiri I, and Zelaya MV , et al. Neuroanatomical quantitative proteomics reveals common pathogenic biological routes between amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Int J Mol Sci. 2019; 20(1):4 .
- Conti A, Riva N, Pesca M, et al. Increased expression of Myosin binding protein H in the skeletal muscle of amyotrophic lateral sclerosis patients. Biochim Biophys Acta - Mol Basis Dis. 2014;1842(1):99–106.
- Elf K, Shevchenko G, Nygren I, et al. Alterations in muscle proteome of patients diagnosed with amyotrophic lateral sclerosis. J Proteomics. 2014;108:55–64.
- Kametani F, Obi T, and Shishido T, et al. Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci Rep. 2016;6(1).
- Feneberg E, Charles PD, and Finelli MJ, et al. Detection and quantification of novel C-terminal TDP-43 fragments in ALS-TDP. Brain Pathol. 2020; 31(4):e129 ;
- Nonaka T, Kametani F, Arai T, et al. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum Mol Genet. 2009;18(18):3353–3364.
- Tomé SO, Vandenberghe R, Ospitalieri S, et al. Distinct molecular patterns of TDP-43 pathology in Alzheimer’s disease: relationship with clinical phenotypes. Acta Neuropathol Commun. 2020;8(1). DOI:https://doi.org/10.1186/s40478-020-00934-5.
- Pobran TD, Yang D, Mackenzie IRA, et al. Aptamer-based enrichment of TDP-43 from human cells and tissues with quantification by HPLC-MS/MS. J Neurosci Methods. 2021;363:109344.
- Strong MJ, Strong WL, Jaffe H, et al. Phosphorylation state of the native high-molecular-weight neurofilament subunit protein from cervical spinal cord in sporadic amyotrophic lateral sclerosis. J Neurochem. 2001;76(5):1315–1325.
- Umoh ME, Dammer EB, and Dai J, et al. A proteomic network approach across the ALS-FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain. EMBO Mol Med. 2018;10(1):48–62.
- Engelen-Lee J, Blokhuis AM, Spliet WGM, et al. Proteomic profiling of the spinal cord in ALS: decreased ATP5D levels suggest synaptic dysfunction in ALS pathogenesis. Amyotroph Lateral Scler Front Degener. 2017;18(3–4):210–220.
- Liu D, Liu C, and Li J, et al. Proteomic analysis reveals differentially regulated protein acetylation in human amyotrophic lateral sclerosis spinal cord. PLoS One. 2013; 8(12):e779
- Oeckl P, Weydt P, and Thal DR, et al. Proteomics in cerebrospinal fluid and spinal cord suggests UCHL1, MAP2 and GPNMB as biomarkers and underpins importance of transcriptional pathways in amyotrophic lateral sclerosis. Acta Neuropathol. 2020;139(1):119–134.
- Volkening K, Keller BA, and Leystra-Lantz C, et al. RNA and Protein Interactors with TDP-43 in Human Spinal-Cord Lysates in Amyotrophic Lateral Sclerosis. J Proteome Res. 2018 17(4) 1712–29 ;
- Hanrieder J, Ekegren T, Andersson M, et al. MALDI imaging of post-mortem human spinal cord in amyotrophic lateral sclerosis. J Neurochem. 2013;124(5):695–707.
- Ekegren T, Hanrieder J, Aquilonius SM, et al. Focused proteomics in post-mortem human spinal cord. J Proteome Res. 2006;5(9):2364–2371.
- Feneberg E, Gordon D, Thompson AG, et al. An ALS-linked mutation in TDP-43 disrupts normal protein interactions in the motor neuron response to oxidative stress. Neurobiol Dis. 2020;144:105050.
- Freibaum BD, Chitta RK, and High AA, et al. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res. 2010;9(2):1104–1120.
- Davis SA, Itaman S, Khalid-Janney CM, et al. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci Lett. 2018;678:8–15.
- Blokhuis AM, Koppers M, and Groen EJN, et al. Comparative interactomics analysis of different ALS-associated proteins identifies converging molecular pathways. Acta Neuropathol. 2016;132(2):175–196.
- Kawaguchi T, Rollins MG, Moinpour M, et al. Changes to the TDP-43 and FUS Interactomes Induced by DNA Damage. J Proteome Res. 2020;19(1):360–370.
- Kamelgarn M, Chen J, Kuang L, et al. Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim Biophys Acta - Mol Basis Dis. 2016;1862(10):2004–2014.
- Sun S, Ling SC, and Qiu J, et al. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat Commun. 2015; 6:6171 .
- Wang T, Jiang X, Chen G, et al. Interaction of amyotrophic lateral sclerosis/frontotemporal lobar degeneration-associated fused-in-sarcoma with proteins involved in metabolic and protein degradation pathways. Neurobiol Aging. 2015;36(1):527–535.
- Basso M, Samengo G, Nardo G, et al. Characterization of detergent-insoluble proteins in ALS indicates a causal link between nitrative stress and aggregation in pathogenesis. PLoS One. 2009;4(12):e8130.
- Shaw BF, Lelie HL, Durazo A, et al. Detergent-insoluble aggregates associated with amyotrophic lateral sclerosis in transgenic mice contain primarily full-length, unmodified superoxide dismutase-1. J Biol Chem. 2008;283(13):8340–8350.
- Une M, Yamakawa M, Watanabe Y, et al. SOD1-interacting proteins: roles of aggregation cores and protein degradation systems. Neurosci Res. 2021;170:295–305.
- Hans F, Eckert M, Von Zweydorf F, et al. Identification and characterization of ubiquitinylation sites in TAR DNA-binding protein of 43 kDa (TDP-43). J Biol Chem. 2018;293(41):16083–16099.
- Monahan Z, Ryan VH, Janke AM, et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017;36(20):2951–2967.
- Cohen TJ, Hwang AW, and Restrepo CR, et al. An acetylation switch controls TDP-43 function and aggregation propensity. Nat Commun. 2015;6.
- Tanikawa C, Ueda K, Suzuki A, et al. Citrullination of RGG Motifs in FET Proteins by PAD4 Regulates Protein Aggregation and ALS Susceptibility. Cell Rep. 2018;22(6):1473–1483.
- Niikura T, Kita Y, Abe Y. SUMO3 modification accelerates the aggregation of ALS-Linked SOD1 mutants. PLoS One. 2014;9(6):e101080.
- Bruijn LI, Becher MW, Lee MK, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18(2):327–338.
- Gertsman I, Wuu J, and McAlonis-Downes M, et al. An endogenous peptide marker differentiates SOD1 stability and facilitates pharmacodynamic monitoring in SOD1 amyotrophic lateral sclerosis. JCI Insight. 2019;4(10). .
- Shi Y, Rhodes NR, Abdolvahabi A, et al. Deamidation of asparagine to aspartate destabilizes Cu, Zn superoxide dismutase, accelerates fibrillization, and mirrors ALS-linked mutations. J Am Chem Soc. 2013;135(42):15897–15908.
- Hutten S, Dormann D. RAN translation down. Nat Neurosci. 2019;22(9):1379–1380.
- Radwan M, Ang CS, Ormsby AR, et al. Arginine in C9ORF72 dipolypeptides mediates promiscuous proteome binding and multiple modes of toxicity. Mol Cell Proteomics. 2020;19(4):640–654.
- Moens TG, Niccoli T, Wilson KM, et al. C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol. 2019;137(3):487–500.
- Hartmann H, Hornburg D, Czuppa M, et al. Proteomics and C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Sci Alliance. 2018;1(2):e201800070.
- Kanekura K, Yagi T, Cammack AJ, et al. Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum Mol Genet. 2016;25(9):1803–1813.
- May S, Hornburg D, Schludi MH, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014;128(4):485–503.
- Cooper-Knock J, Walsh MJ, and Higginbottom A, et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain . 2014;137(7):2040–2051. .
- Sivadasan R, Hornburg D, Drepper C, et al. C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat Neurosci. 2016;19(12):1610–1618.
- Ho WY, Tai YK, and Chang JC, et al. . In: Autophagy. 2019; 15(5):827–42 .
- Farg MA, Sundaramoorthy V, Sultana JM, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014;23(13):3579–3595.
- Li Q, Vande VC, Israelson A, et al. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc Natl Acad Sci U S A. 2010;107(49):21146–21151.
- Fukada K, Zhang F, Vien A, et al. Mitochondrial proteomic analysis of a cell line model of familial amyotrophic lateral sclerosis. Mol Cell Proteomics. 2004;3(12):1211–1223.
- Allen S, Heath PR, Kirby J, et al. Analysis of the cytosolic proteome in a cell culture model of familial amyotrophic lateral sclerosis reveals alterations to the proteasome, antioxidant defenses, and nitric oxide synthetic pathways. J Biol Chem. 2003;278(8):6371–6383.
- Di Poto C, Iadarola P, Bardoni AM, et al. 2-DE and MALDI-TOF-MS for a comparative analysis of proteins expressed in different cellular models of amyotrophical lateral sclerosis. Electrophoresis. 2007;28(23):4320–4329.
- Shin JH, London J, Le Pecheur M, et al. Proteome analysis in hippocampus of mice overexpressing human Cu/Zn-superoxide dismutase 1. Neurochem Int. 2005;46(8):641–653.
- Zhang K, Donnelly CJ, Haeusler AR, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525(7567):56–61.
- Ortega JA, Daley EL, Kour S, et al. Nucleocytoplasmic Proteomic Analysis Uncovers eRF1 and Nonsense-Mediated Decay as Modifiers of ALS/FTD C9orf72 Toxicity. Neuron. 2020;106(1):90–107.e13.
- Štalekar M, Yin X, Rebolj K, et al. Proteomic analyses reveal that loss of TDP-43 affects RNA processing and intracellular transport. Neuroscience. 2015;293:157–170.
- Kim JE, Hong YH, and Kim JY, et al. Altered nucleocytoplasmic proteome and transcriptome distributions in an in vitro model of amyotrophic lateral sclerosis. PLoS One. 2017; 28(12):e0176462 .
- Wolozin B, Ivanov P. Stress granules and neurodegeneration. Nat Rev Neurosci. 2019;20(11):649–666.
- Jain S, Wheeler JR, and Walters RW, et al. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell . 2016;164(3):487–498.
- Fernandes N, Nero L, Lyons SM, et al. Stress granule assembly can facilitate but is not required for TDP‐43 cytoplasmic aggregation. Biomolecules. 2020;10(10):1367.
- Marmor-Kollet H, Siany A, Kedersha N, et al. Spatiotemporal Proteomic Analysis of Stress Granule Disassembly Using APEX Reveals Regulation by SUMOylation and Links to ALS Pathogenesis. Mol Cell. 2020;80(5):876–891.e6.
- Vu L, Ghosh A, Tran C, et al. Defining the Caprin-1 Interactome in Unstressed and Stressed Conditions. J Proteome Res. 2021;20(6):3165–3178.
- Turner MR. Progress and new frontiers in biomarkers for amyotrophic lateral sclerosis. Biomark Med. 2018;12(7):693–696.
- Poesen K, and Van Damme P. Diagnostic and prognostic performance of neurofilaments in ALS. Front. Neurol. 2019; 9 1167 .
- Figlewicz DA, Krizus A, Martinoli MG, et al. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum Mol Genet. 1994;3(10):1757–1761.
- Steinacker P, Feneberg E, and Weishaupt J, et al. Neurofilaments in the diagnosis of motoneuron diseases: a prospective study on 455 patients. J Neurol Neurosurg Psychiatry . 2016;87(1):12–20.
- Feneberg E, Oeckl P, and Steinacker P, et al. Multicenter evaluation of neurofilaments in early symptom onset amyotrophic lateral sclerosis. Neurology . 2018;90(1):e22–e30.
- Lu CH, Macdonald-Wallis C, and Gray E, et al. Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis. Neurology . 2015;84(22):2247–2257.
- Ganesalingam J, An J, Shaw CE, et al. Combination of neurofilament heavy chain and complement C3 as CSF biomarkers for ALS. J Neurochem. 2011;117(3):528–537.
- Turner MR, and Gray E. Are neurofilaments heading for the ALS clinic? J Neurol Neurosurg Psychiatry. . 2016;87(1):3–4.
- Lewczuk P, Riederer P, and O’Bryant SE, et al. Cerebrospinal fluid and blood biomarkers for neurodegenerative dementias: an update of the consensus of the task force on biological markers in psychiatry of the world federation of societies of biological psychiatry. World J Biol Psychiatry . 2018;19(4):244–328.
- Fairfoul G, McGuire LI, Pal S, et al. Alpha-synuclein RT-Qu IC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol. 2016;3(10):812–818.
- Atarashi R, Sano K, Satoh K, et al. Real-time quaking-induced conversion: a highly sensitive assay for prion detection. Prion. 2011;5(3):150–153.
- Paganoni S, Cudkowicz M, and Berry JD. Outcome measures in amyotrophic lateral sclerosis clinical trials Clin Investig (Lond) . . 2014;4(7):605–618
- Van Eijk RPA, Westeneng HJ, Nikolakopoulos S, et al. Refining eligibility criteria for amyotrophic lateral sclerosis clinical trials. Neurology. 2019;92(5):e451–e460.
- Turner MR, and Benatar M. Ensuring continued progress in biomarkers for amyotrophic lateral sclerosis. Muscle Nerve. . 2015;51(1):14–18.
- Winer L, Srinivasan D, and Chun S, et al. SOD1 in cerebral spinal fluid as a pharmacodynamic marker for antisense oligonucleotide therapy. JAMA Neurol . 2013;70(2):201–207.
- Gendron TF, Chew J, and Jn S, et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci Transl Med. 2017; 9(38: eaai7866
- Shepheard SR, Wuu J, Cardoso M, et al. Urinary p75 ECD. Neurology. 2017;88(12):1137–1143.
- Gendron TF, Daughrity LM, Heckman MG, et al. Phosphorylated neurofilament heavy chain: a biomarker of survival for C9ORF 72 -associated amyotrophic lateral sclerosis. Ann Neurol. 2017;82(1):139–146.
- Benatar M, Zhang L, Wang L, et al. Validation of serum neurofilaments as prognostic and potential pharmacodynamic biomarkers for ALS. Neurology. 2020;95(1):e59–e69.
- Miller T, Cudkowicz M, Shaw PJ, et al. Phase 1–2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. 2020;383(2):109–119.
- Milligan C, Atassi N, Babu S, et al. Tocilizumab is safe and tolerable and reduces C-reactive protein concentrations in the plasma and cerebrospinal fluid of ALS patients. Muscle Nerve. 2021;64(3):309–320.
- Guldbrandsen A, Farag Y, Kroksveen AC, et al. CSF-PR 2.0: an interactive literature guide to quantitative cerebrospinal fluid mass spectrometry data from neurodegenerative disorders. Mol Cell Proteomics. 2017;16(2):300–309.
- Dayon L, Cominetti O, Wojcik J, et al. Proteomes of Paired Human Cerebrospinal Fluid and Plasma: relation to Blood-Brain Barrier Permeability in Older Adults. J Proteome Res. 2019;18(3):1162–1174.
- Barschke P, Oeckl P, and Steinacker P, et al. Proteomic studies in the discovery of cerebrospinal fluid biomarkers for amyotrophic lateral sclerosis. Expert Rev Proteomics . 2017;14(9):769–777.
- Ranganathan S, Nicholl GC, and Henry S, et al. Comparative proteomic profiling of cerebrospinal fluid between living and post mortem ALS and control subjects. Amyotroph Lateral Scler . 2007;8(6):373–379.
- Brettschneider J, Lehmensiek V, Mogel H, et al. Proteome analysis reveals candidate markers of disease progression in amyotrophic lateral sclerosis (ALS). Neurosci Lett. 2010;468(1):23–27.
- Collins M, Riascos D, Kovalik T, et al. The RNA-binding motif 45 (RBM45) protein accumulates in inclusion bodies in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) patients. Acta Neuropathol. 2012;124(5):717–732.
- Bereman MS, Beri J, and Enders JR, et al. Machine Learning Reveals Protein Signatures in CSF and Plasma Fluids of Clinical Value for ALS. Sci Rep. 2018;8(1).
- Zhu S, Wuolikainen A, Wu J, et al. Targeted Multiple Reaction Monitoring Analysis of CSF Identifies UCHL1 and GPNMB as Candidate Biomarkers for ALS. J Mol Neurosci. 2019;69(4):643–657.
- Mellinger AL, Griffith EH, Bereman MS. Peptide variability and signatures associated with disease progression in CSF collected longitudinally from ALS patients. Anal Bioanal Chem. 2020;412(22):5465–5475.
- Palma AS, De Carvalho M, Grammel N, et al. Proteomic analysis of plasma from Portuguese patients with familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2008;9(6):339–349.
- Häggmark A, Mikus M, Mohsenchian A, et al. Plasma profiling reveals three proteins associated to amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2014;1(8):544–553.
- Ramstrom M, Ivonin I, and Johansson A, et al. Cerebrospinal fluid protein patterns in neurodegenerative disease revealed by liquid chromatography-Fourier transform ion cyclotron resonance mass spectrometry. Proteomics . 2004;4(12)::4010–4018.
- Collins MA, An J, and Hood BL, et al. Label-Free LC-MS/MS Proteomic Analysis of Cerebrospinal Fluid Identifies Protein/Pathway Alterations and Candidate Biomarkers for Amyotrophic Lateral Sclerosis. J Proteome Res . 2015;14(11):4486–4501.
- Ranganathan S, Williams E, and Ganchev P, et al. Proteomic profiling of cerebrospinal fluid identifies biomarkers for amyotrophic lateral sclerosis. J Neurochem . 2005;95(5):1461–1471.
- Pasinetti GM, Ungar LH, and Lange DJ, et al. Identification of potential CSF biomarkers in ALS. Neurology . 2006;66(8):1218–1222.
- Ryberg H, An J, and Darko S, et al. Discovery and verification of amyotrophic lateral sclerosis biomarkers by proteomics. Muscle Nerve . 2010;42(1):104–111.
- Sako W, Ishimoto S. Can cystatin C in cerebrospinal fluid be a biomarker for amyotrophic lateral sclerosis? A lesson from previous studies. Neurol Clin Neurosci. 2014;2(3):72–75.
- Tsuji-Akimoto S, Yabe I, Niino M, et al. Cystatin C in cerebrospinal fluid as a biomarker of ALS. Neurosci Lett. 2009;452(1):52–55.
- Thompson AG, Gray E, Bampton A, et al. CSF chitinase proteins in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2019;90(11):1215–1220.
- Lunetta C, Lizio A, Maestri E, et al. Serum C-reactive protein as a prognostic biomarker in amyotrophic lateral sclerosis. JAMA Neurol. 2017;74(6):660.
- Beers DR, Zhao W, and Neal DW, et al. Elevated acute phase proteins reflect peripheral inflammation and disease severity in patients with amyotrophic lateral sclerosis. Sci Rep. 2020;10(1).
- Brettschneider J, Mogel H, and Lehmensiek V, et al. Proteome analysis of cerebrospinal fluid in amyotrophic lateral sclerosis (ALS). Neurochem Res . 2008;33(11):2358–2363.
- Mendonca DM, Pizzati L, and Mostacada K, et al. Neuroproteomics: an insight into ALS. Neurol Res . 2012;34(10):937–943.
- Wormser U, Mandrioli J, and Vinceti M, et al. Reduced levels of alpha-1-antitrypsin in cerebrospinal fluid of amyotrophic lateral sclerosis patients: a novel approach for a potential treatment. J Neuroinflammation. 2016;13(1).
- Von Neuhoff N, Oumeraci T, Wolf T, et al. Monitoring CSF proteome alterations in amyotrophic lateral sclerosis: obstacles and perspectives in translating a novel marker panel to the clinic. PLoS One [Internet]. 2012;7(9):e44401.
- Chen Y, Liu XH, Wu JJ, et al. Proteomic analysis of cerebrospinal fluid in amyotrophic lateral sclerosis. Exp Ther Med [Internet]. 2016;11(6):2095–2106.
- Varghese AM, Sharma A, Mishra P, et al. Chitotriosidase - a putative biomarker for sporadic amyotrophic lateral sclerosis. Clin Proteomics [Internet]. 2013;10(1):19.
- Oeckl P, Weydt P, Steinacker P, et al. Different neuroinflammatory profile in amyotrophic lateral sclerosis and frontotemporal dementia is linked to the clinical phase. J Neurol Neurosurg Psychiatry. 2019;90(1):4–10.
- Thompson AG, Gray E, Thézénas ML, et al. Cerebrospinal fluid macrophage biomarkers in amyotrophic lateral sclerosis. Ann Neurol. 2018;83(2):258–268.
- Gray E, Thompson AG, Wuu J, et al. CSF chitinases before and after symptom onset in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2020;7(8):1296–1306.
- Steinacker P, Verde F, Fang L, et al. Chitotriosidase (CHIT1) is increased in microglia and macrophages in spinal cord of amyotrophic lateral sclerosis and cerebrospinal fluid levels correlate with disease severity and progression. J Neurol Neurosurg Psychiatry [Internet]. 2018;89(3):239–247.
- Gille B, De Schaepdryver M, and Dedeene L, et al. Inflammatory markers in cerebrospinal fluid: independent prognostic biomarkers in amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatry. 2019;1338–46. .
- Vu L, An J, Kovalik T, et al. Cross-sectional and longitudinal measures of chitinase proteins in amyotrophic lateral sclerosis and expression of CHI3L1 in activated astrocytes. J Neurol Neurosurg Psychiatry. 2020;91(4):350–358.
- Varghese AM, Ghosh M, and Bhagat SK, et al. Chitotriosidase, a biomarker of amyotrophic lateral sclerosis, accentuates neurodegeneration in spinal motor neurons through neuroinflammation. J Neuroinflammation. 2020;17(1).
- Gaur N, Perner C, and Witte OW, et al. The Chitinases as Biomarkers for Amyotrophic Lateral Sclerosis: signals From the CNS and Beyond. Front Neurol. 2020;11.
- Barschke P, Oeckl P, Steinacker P, et al. Different CSF protein profiles in amyotrophic lateral sclerosis and frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. J Neurol Neurosurg Psychiatry. 2020;91(5):503–511.
- Thompson AG, Gray E, and Heman-Ackah SM, et al. Extracellular vesicles in neurodegenerative disease - pathogenesis to biomarkers. Nat Rev Neurol . 2016;12(6):346–357.
- Aoki Y, Manzano R, Lee Y, et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain. 2017;140(4):887–897.
- Thompson AG, Gray E, Mager I, et al. UFLC-Derived CSF Extracellular Vesicle Origin and Proteome. Proteomics. 2018;18:1800257.
- Feneberg E, Steinacker P, and Lehnert S, et al. Limited role of free TDP-43 as a diagnostic tool in neurodegenerative diseases. Amyotroph Lateral Scler Front Degener . 2014;15:351–356. (
- Thompson AG, Gray E, and Mäger I, et al. CSF extracellular vesicle proteomics demonstrates altered protein homeostasis in amyotrophic lateral sclerosis. Clin Proteomics. 2020;17(1).
- Hayashi N, Doi H, Kurata Y, et al. Proteomic analysis of exosome-enriched fractions derived from cerebrospinal fluid of amyotrophic lateral sclerosis patients. Neurosci Res. 2019;160:43–49.
- Mustapic M, Eitan E, and Werner JK, et al. Plasma extracellular vesicles enriched for neuronal origin: a potential window into brain pathologic processes. Front Neurosci. 2017;11.
- Goetzl EJ, Boxer A, Schwartz JB, et al. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology. 2015;85(1):40–47.
- Jiang C, Hopfner F, Hopfner F, et al. Serum neuronal exosomes predict and differentiate Parkinson’s disease from atypical parkinsonism. J Neurol Neurosurg Psychiatry. 2020;91(7):720–729.
- Norman M, Ter-Ovanesyan D, Trieu W, et al. L1CAM is not associated with extracellular vesicles in human cerebrospinal fluid or plasma. Nat Methods. 2021;18(6):631–634.
- Benatar M, Wuu J, Andersen PM, et al. Neurofilament light: a candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann Neurol. 2018;84(1):130–139.
- Vucic S, Nicholson GA, and Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain. 2008;131(6):1540–1550.
- Weydt P, Oeckl P, Huss A, et al. Neurofilament levels as biomarkers in asymptomatic and symptomatic familial amyotrophic lateral sclerosis. Ann Neurol. 2016;79(1):152–158.
- Benatar M, Wuu J. Presymptomatic studies in ALS: rationale, challenges, and approach. Neurology. 2012;79(16):1732–1739.
- Leoni E, Bremang M, and Mitra V, et al. Combined Tissue-Fluid Proteomics to Unravel Phenotypic Variability in Amyotrophic Lateral Sclerosis. Sci Rep. 2019;9(1).
- Zubiri I, Lombardi V, and Bremang M, et al. Tissue-enhanced plasma proteomic analysis for disease stratification in amyotrophic lateral sclerosis. Mol Neurodegener. 2018;13(1).
- Conraux L, Pech C, Guerraoui H, et al. Plasma peptide biomarker discovery for amyotrophic lateral sclerosis by MALDI-TOF mass spectrometry profiling. PLoS One. 2013;8(11):e79733.
- Xu Z, Lee A, Nouwens A, et al. Mass spectrometry analysis of plasma from amyotrophic lateral sclerosis and control subjects. Amyotroph Lateral Scler Front Degener. 2018;19(5–6):362–376.
- Katzeff JS, Bright F, and Lo K, et al. Altered serum protein levels in frontotemporal dementia and amyotrophic lateral sclerosis indicate calcium and immunity dysregulation. Sci Rep. 2020;10(1) .
- Nardo G, Pozzi S, Mantovani S, et al. Nitroproteomics of peripheral blood mononuclear cells from patients and a rat model of ALS. Antioxid Redox Signal. 2009;11(7):1559–1567.
- Nardo G, Pozzi S, Pignataro M, et al. Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS One. 2011;6(10):e25545.
- Thompson AG, Gray E, and Charles PD, et al. Network Analysis of the CSF Proteome Characterizes Convergent Pathways of Cellular Dysfunction in ALS. Front Neurosci. 2021;15.
- Higginbotham L, Ping L, Dammer EB, et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Sci Adv. 2020;6(43):eaaz9360.
- Aslam B, Basit M, Nisar MA, et al. Proteomics: technologies and their applications. J Chromatogr Sci. 2017;55(2):182–196.
- Van Damme P, Robberecht W, Van Den Bosch L. Modelling amyotrophic lateral sclerosis: progress and possibilities. DMM Dis Model Mech. 2017;10(5):537–549.
- Wong CO, Venkatachalam K. Motor neurons from ALS patients with mutations in C9ORF72 and SOD1 exhibit distinct transcriptional landscapes. Hum Mol Genet. 2019;28(16):2799–2810.
- Morello G, Salomone S, and D’Agata V, et al. From Multi-Omics Approaches to Precision Medicine in Amyotrophic Lateral Sclerosis. Front Neurosci. 2020;14.
- Niehaus M, Soltwisch J, Belov ME, et al. Transmission-mode MALDI-2 mass spectrometry imaging of cells and tissues at subcellular resolution. Nat Methods. 2019;16(9):925–931.
- Acquadro E, Caron I, Tortarolo M, et al. Human SOD1-G93A specific distribution evidenced in murine brain of a transgenic model for amyotrophic lateral sclerosis by MALDI imaging mass spectrometry. J Proteome Res. 2014;13(4):1800–1809.
- Xu BJ. Combining laser capture microdissection and proteomics: methodologies and clinical applications. Proteomics - Clin Appl. 2010;4(2):116–123.
- Schoof EM, Furtwängler B, and Üresin N, et al. Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat Commun. 2021;12(1).
- Gut G, Herrmann MD, and Pelkmans L. Multiplexed protein maps link subcellular organization to cellular states. Science. 2018;361(6401).
- Li KW, Gonzalez-Lozano MA, and Koopmans F, et al. Recent Developments in Data Independent Acquisition (DIA) Mass Spectrometry: application of Quantitative Analysis of the Brain Proteome. Front Mol Neurosci. 2020;13.
- Halvey P, Farutin V, and Koppes L, et al. Variable blood processing procedures contribute to plasma proteomic variability. Clin Proteomics. 2021;18(1).
- Robelin L, Gonzalez De Aguilar JL. Blood biomarkers for amyotrophic lateral sclerosis: myth or reality? BioMed Res Int. 2014;2014:1–11.
- Carlyle BC, Trombetta BA, Arnold SE. Proteomic approaches for the discovery of biofluid biomarkers of neurodegenerative dementias. Proteomes. 2018;6(3):32.