
ABSTRACT
Introduction
Arginine deimination (citrullination) is a post-translational modification catalyzed by a family of peptidyl arginine deiminase (PAD) enzymes. Cell-based functional studies and animal models have manifested the key role of PADs in various cardiovascular diseases (CVDs).
Area Covered
This review summarizes the past 10 years of knowledge on the role of PADs in CVD pathogenesis. It focuses on the PAD functions and diverse citrullinated proteins in cardiovascular conditions like deep vein thrombosis, ischemia/reperfusion, and atherosclerosis. Identification of PAD isoforms and citrullinated targets are essential for directing diagnosis and clinical intervention. Finally, anti-citrullinated protein antibodies (ACPAs) are addressed as an independent risk factor for cardiovascular events.
Expert Opinion
PAD is an unique family of enzymes that permanently converts amino acid arginine to amino acid citrulline in protein . Overexpression or increased activity of PAD has been observed in various CVDs with acute and chronic inflammation as the background. Importantly, far beyond being simply involved in forming neutrophil extracellular traps (NETs), accumulating evidence indicated PAD activation as a trigger for numerous processes, such as transcriptional regulation, endothelial dysfunction, and thrombus formation. In summary, the findings so far have testified the important role of deimination in cardiovascular biology, while more basic and translational studies are essential for further exploration.
1. Introduction
Citrullination, also known as protein deimination, is a protein post-translational modification (PTM) carried out by the Ca2+-dependent enzyme family peptidylarginine deiminase (PAD) [Citation1,Citation2]. This PTM is an irreversible conversion of the positively charged arginine residues to neutral citrulline residues. The loss of charge may lead to changes in the structure and function of the target protein and has critical physiological and pathological consequences [Citation3]. In human and mouse, five PAD isozymes (PADs 1–4 and 6) have been identified with characteristic tissue-specificity, subcellular localization, and preferred protein targets [Citation4]. In human, all the PAD genes are clustered in a 335 kb region on chromosome 1p36.1 [Citation4,Citation5]. Although in mammals the five isozymes share 50–70% sequence similarity, each isotype has its own specific tissue distribution, functions, and substrates under physiological conditions [Citation5,Citation6]. Importantly, the prerequisites for PAD activation are the high calcium concentration usually found in the extracellular space and redox potential that keeps the catalytic site in a reduced state [Citation7,Citation8]. Considering the importance of the micro to millimolar calcium concentrations range required for PAD activation [Citation6] it is unclear how protein citrullination occurs under physiologically low calcium conditions. Some data have linked changes in intracellular Ca2+ homeostasis to various stages of normal or altered apoptotic signaling cascade [Citation9]. Actually, apoptosis has been reported under inflammatory and stress conditions seen in many autoimmune diseases in which citrullination is implicated [Citation10]. The activation mechanism of PAD and the detailed model of the catalytic conversion of arginine to citrulline were reported by Liu et al. in 2017 [Citation11], and updated recently [Citation12], so we will not discuss it here.
Citrullination has been reported to be related to a variety of cellular processes, such as gene regulation [Citation2,Citation13–15], transcription (by citrullinating splicing factors and RNA-binding proteins) [Citation16,Citation17], apoptosis [Citation18], immune response [Citation19,Citation20], and epidermal terminal differentiation [Citation21]. By inducing structural change in citrullinated proteins, citrullination can alter the function of target proteins like histones [Citation22], cytokines and chemokines [Citation23,Citation24], fibrinogen [Citation25,Citation26], nicotinamide-N-methyl transferase [Citation27] and transforming growth factor-β (TGFβ) [Citation28]. Structural changes in proteins may also generate autoantigenic neoepitopes, as observed in rheumatoid arthritis (RA) and type I diabetes [Citation29,Citation30]. Given that citrullination can change the structure and function of proteins, several citrullinated proteins have been implicated in a wide range of diseases, including cancer [Citation31,Citation32], central nervous system disorders [Citation33,Citation34], inflammatory diseases [Citation35], and immune dysregulation [Citation36,Citation37].
Upregulation of PAD has also been observed in various CVDs, including myocardial infarction [Citation38], atherosclerosis [Citation39,Citation40], cardiac fibrosis [Citation41], heart failure (HF) [Citation42], and venous thrombosis [Citation43]. Although the list of the diseases in which PAD activation has been reported is growing, the full range of citrullinated proteins and the pathways affected remains undetermined, as are their immunological and clinical effects (, details in ).
Table 1. Reported PADs isoforms, citrullinated proteins and mechanisms, in cardiovascular experimental data sets
Figure 1. Overview of the citrullinated proteins in the cardiovascular system. PAD mediated deimination in the field of cardiovascular diseases has been extensively studied in the last decade. It is suggested that citrullinated proteins play a critical role in the acute and the chronic inflammatory process that impair cardiovascular structure and function.
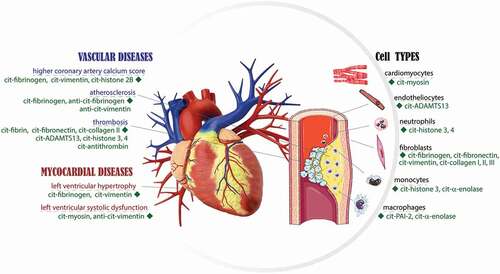
In this review, we highligth the current knowledge of the role of PAD isozymes in cardiovascular events, associated with neutrophils, macrophages, endothelial dysfunction, atherosclerosis, and thrombosis. In addition, we briefly summarized the use of citrullinated proteins as surrogate markers of cardiovascular risk and the correlation between ACPA levels and the outcome of cardiovascular events outcomes [Citation44–46]. By combining the knowledge of PAD function with the biological properties of citrullinated target proteins, we can improve our understanding of the underlying mechanisms of cardiovascular events and pave the way for the development of preventative and therapeutic solutions.
2. Cell-specific expression of PAD isozymes and cardiac disease
Inflammation is a well-known feature of CVDs. Studies have shown that while acute inflammation has a protective effect in isolating damage, removing necrotic cells, and initiating repair, prolonged inflammation can lead to myocardial remodeling, cardiomyocyte apoptosis, and atherosclerosis. Both myelocytes and lymphocytes are implicated in myocardial inflammation. Recent clinical trials have targeted inflammatory cascade as a therapy for CVD with limited success, therefore, a better understanding of the new players and pathways activated in different immune cells and their contribution to the inflammatory response may improve treatment.
2.1. Prevalence of PADs in neutrophils
Five human PAD isoenzymes with distinct tissue specificities are widely expressed throughout the body and are implicated in various physiological processes [Citation1]. PAD1is expressed in epidermis and uterus, PAD3 in epidermis and hair follicles, and PAD6 in ovary, oocytes, and embryo [Citation2,Citation5]. PAD4 expression has been reported in leukocytes, hematopoietic progenitor cells [Citation12,Citation47], various tumors (especially adenocarcinomas) [Citation48–50], oocytes, as well as preimplantation embryos [Citation51,Citation52]. PAD2 is widely expressed in the central nervous system, rheumatoid synovial tissue and mast cells, skeletal muscle, uterus, spleen, pancreas, breast, skin, and cells of the immune system [Citation5,Citation53,Citation54]. Notably, PAD2 and PAD4, are expressed at high levels in some immune cell subsets, primarily neutrophils, monocytes and macrophages [Citation3,Citation55]. NETs are complex webs of chromatin and proteins released from neutrophils during a programmed cell death process termed NETosis [Citation56,Citation57]. Neeli et al. first demonstrated that PAD4-dependent histone deimination occurs in activated neutrophils under inflammatory conditions, and the N terminus of histone H3 must be modified to form NETs [Citation58]. Several other studies confirmed that citrullinated histone H3 (H3Cit) by PAD4 is a marker of ongoing NET formation [Citation59–62].
NETosis can be induced by various pathogens as an antimicrobial defense [Citation61]. NETosis can also be triggered endogenously during noninfectious diseases, such as autoimmune diseases [Citation63], cancer [Citation32,Citation64], and fertility disorders [Citation65]. Additionally, NETosis can be activated by cytokines, chemokines and other physiological stimuli after onset of myocardial infarction (MI) [Citation66,Citation67], vascular dysfunction/thrombosis [Citation68,Citation69], and myocarditis [Citation70–72], where neutrophils infiltrate the damaged tissue and aggravate inflammation that promotes HF [Citation73]. As reported by Bonaventura et al., the increased NET level was correlated with larger myocardial infarct sizes and considered major drivers in adverse CV events [Citation74].
In a study by Vejlstrup et al., the circulating PAD2 levels and enzymatic activity of nearly all patients increased after coronary artery bypass surgery, which suggested that acute inflammation, ischemia, reperfusion, or their combination led to systemic spreading of enzymatically active PAD2 [Citation75]. On the other hand, Du et al. confirmed the contribution of PAD4 to cardiac ischemic injury by exacerbating the inflammatory response and promoting NETosis after MI [Citation66]. In their mouse model of MI with permanent left coronary artery ligation , inhibition of PAD4 by GSK484 reduced MI-induced neutrophil infiltration, histone H3 citrullination (H3Cit), NETs formation, inflammatory cytokine secretion, and cardiomyocyte apoptosis, thereby improved the overall cardiac function.
Similarly, studies by Helseth et al. strongly supported the role of neutrophil-driven PAD4 activity in CVDs, where the NETs burden was associated with more frequent microvascular obstruction, larger area-at-risk and infarct size, lower myocardial salvage index, and lower left ventricular ejection fraction in patients with ST-elevation MI [Citation76,Citation77]. Others, including Knight et al. and Martinod et al., showed that PAD4-mediated chromatin decondensation in neutrophils is crucial for chronic CVD such as atherosclerosis and vein thrombosis [Citation40,Citation78]. Knight et al. reported that inhibition of PAD4 blocked NET formation, reduced arterial expression of interferon-α (INFα), and recruitment of netting neutrophils and macrophages, consequently delayed time to carotid artery thrombosis in Apolipoprotein-E (Apoe)−/− mouse models of atherosclerosis [Citation40]. In a stenosis mouse model of deep vein thrombosis (DVT), Martinod et al. confirmed that Padi4 knockout (PAD4-/-) mice could not citrullinate histones, and failed to form NETs [Citation78]. Another group reported that NETs acted as a scaffold for thrombus formation through binding of platelets, red blood cells and procoagulant molecules [Citation79]. Kumar et al. showed that citrullinated histones from NETs have a prothrombotic effect by activating platelets [Citation80].
2.2. Prevalence of PADs in macrophages
Macrophages, sentinels of the immune system, are strategically located within all tissues throughout the body. In the early phase of injury, apart from defending against invading microbes, these cells dynamically mediate inflammation and transform it into an anti-inflammatory phenotype. Importantly, macrophages are highly heterogeneous cells that can rapidly change function in response to microenvironmental signals. One of the first confirmed PAD expression in macrophages came from a study by Vossenaar et al. on peripheral blood and synovial fluid cells in patients with RA [Citation81]. The group detected equal amounts of human PAD2 and PAD4 mRNAs in both monocytes and macrophages, but only PAD4 protein in monocytes [Citation81]. PAD expression was also described during the macrophage differentiation, particularly in inflammatory responses, first by Hoko-Nakashima et al. and later by Lai et al. [Citation82,Citation83]. Hoko-Nakashima et al. detected increased PAD2 expression in human THP-1 monocytes at the mRNA and protein levels during TPA-induced differentiation into macrophages, indicating that PAD2 is expressed in THP-1-derived macrophages [Citation82]. Notably, the activation of the PAD2 resulted in the citrullination of the vimentin, and disruption of vimentin filament organization [Citation82].
Similarly, a study by Lai et al. showed that the protein expression of PAD2 and PAD4 increased markedly during the differentiation of monocytes into macrophages, and PAD activation correlated to increased citrullination of certain intracellular proteins, including histone H3 and plasminogen activator inhibitor-2 (PAI-2) [Citation83]. Vice versa, decreased expression of PAD2 and PAD4 was showed to suppress the protein levels of H3Cit and citrullinated PAI-2. PAI-2, a serine protease inhibitor in the serpin superfamily, is a coagulation factor that inhibits tissue plasminogen activator and urokinase plasminogen activator. As one of the most upregulated proteins in activated monocytes and macrophages, it can be considered a stress protein. Notably, the citrullination of PAI-2 reduced its binding ability to proteasome subunit β type-1 (PSMB1) and alternated the function of proteasome. The increase in PAI-2 expression by PAD2 induced the production and secretion of interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) [Citation83]. Noteworthy, the addition of PAD inhibitors significantly suppressed the secretion of cytokines TNF-α, IL-1β, and IL-6 from the differentiated macrophages without affecting cell viability or the expression level of PAD2 or PAD4 [Citation83]. The same group latter showed that in differentiated macrophages, in addition to IL-1β and TNF-α, PAD2 also promoted the production of IL-6, apoptosis through activation of caspase-2, −3, and −9, and enhanced cell adhesion through FAK, paxillin, and p21-activated kinase 1 (PAK1) [Citation84].
The involvement of PADs and citrullinated proteins in macrophage pyroptosis was also described in a recent study by Mishra et al. [Citation85]. Inhibition of PAD2 or knockdown of PADI2 in PADI4-/- macrophages suggested that the PAD2 and PAD4 activity in macrophages is required for NLRP3 inflammasome-dependent IL-1β maturation and release [Citation85]. Interestingly, complementary to Hojo-Nakahima et al.’s results, vimentin was strongly citrullinated in macrophages during inflammasome activation [Citation82]. Both results advocated that the PAD activity may be required for vimentin citrullination and subsequent cleavage, resulting in a fragment that could serve as a scaffolding platform for the assembly of NLRP3 inflammasome [Citation86]. The role of NLRP3 inflammasome activation in macrophages has been recently described in fibrosis process [Citation87]. Although the study did not mention PADs or citrullinated proteins, it could be proposed that PAD activation may contribute to the pathogenesis of fibrosis.
Nevertheless, the role of PADs during macrophage differentiation is an important factor, since macrophages play a decisive role in many pro- and anti-inflammatory processes of atherosclerotic lesions [Citation88]. Macrophages in atherosclerotic lesions actively participate in lipoprotein ingestion and turn into foam cells filled with lipid droplets. The accumulation of foam cells contributes to lipid storage and the growth of atherosclerotic plaques. For more information about macrophages and their role in inflammation and atherosclerosis through citrullination, please see section 3.2., ‘Role of PADs and Citrullination in Atherosclerosis.’ Importantly, according to available literature, PAD targeting could be used as a novel strategy to control different macrophage phenotypes and their roles in the pathogenesis of a wide variety of CVDs.
2.3. Prevalence of PADs in cardiomyocytes/fibroblasts
In addition to being highly expressed in neutrophils and macrophages, PADs were found in cardiac myocytes and fibroblasts. Giles et al. detected PADs in cardiomyocytes (primarily PADs 1 and 3), resident inflammatory cells (primarily PADs 2 and 4), and, to a small extent, endotheliocytes and vascular smooth muscle cells (VSMCs) [Citation89]. Interestingly, there was significantly increased staining of citrullination but no PAD detection in the myocardial interstitium based on immunohistochemical results, and the level of citrullination corresponded with that of myocardial interstitial fibrosis and age in patients with RA [Citation89]. Fert-Bober et al. reported citrullination unique to the cardiac proteome [Citation90]. In contrast to what Giles et al. reported, PADI2 was found to be the main isoform in cardiomyocytes based on the RT-PCR results. Using mass spectrometry,it was found that a number of arginine sites in various proteins were citrullinated in heart samples from patients with HF compared with healthy donors. Fert-Bober et al. showed that citrullination of sarcomeric proteins altered their function by inhibiting the steady-state actin-activated and actin-independent (basal) ATPase cycling and calcium-regulated ATPase activity in permeabilized cardiac myofibrils. According to reports, citrullination of the myosin heavy chain (MHC) reduced ATPase hydrolysis and inhibited the activity of actin-MHC-ATPase. Similarly, inhibition of the activity of actin-MHC-ATPase was also observed after tropomyosin citrullination. Citrullinated tropomyosin showed a stronger affinity for F-actin, which could inhibit the kinetics of rigor actin-binding and of actin-activated phosphate release. More structural studies on intact cardiac muscle are essential to better understand how citrullination may alter myocardial motion. In addition, it will be interesting to determine whether PAD inhibitors reduce myocardial steady-state force, power, and Ca2+ sensitivity that may help develop drug for hyper-constriction. Contrary to the sarcomeric protein hypothesis by Fert-Bober, an in vivo study by Martinod et al. strongly favored age-related interstitial myocardial fibrosis over biochemical alteration of sarcomeric proteins [Citation41]. The group used mice with Padi4-/- neutrophils and ascending aortic constriction mouse model of heart fibrosis to show that the PAD4-medicated release of extracellular chromatin induced heart fibrosis through collagen deposition. Myocardial fibrosis is a major part of cardiac remodeling that leads to HF and death. Since fibroblasts are the principal effector cells of fibrosis, several studies have examined the expression and activity levels of PAD during fibrosis.
Shelef et al. showed how citrullination of fibronectin alters the behavior of synovial fibroblasts and affects adhesion and invasiveness [Citation91]. Similarly, Sun et al. showed that RA-specific ACPAs in response to protein citrullination in RA patients promoted increased migration of synovial fibroblasts [Citation92]. In addition to increased levels of PAD2 and PAD4, the group also detected citrullinated targets using mass spectrometry in most starved synovial fibroblasts from RA patients. Two other examples of regulation of fibroblast migration though citrullination come from the studies by Su et al., and Li et al. [Citation92,Citation93]. Both groups reported that the citrullination of vimentin triggered fibroblast activation and tissue inflammation, resulting in the expression of fibrotic proteins, which in turn promoted pulmonary fibrosis [Citation92].
Although, there is no direct link between PADs, cardiac fibroblasts and mechanism of the myocardial fibrosis, many signaling pathways, including glycogen synthase kinase-3β (GSK-3β), β-catenin, and TGF-β1/SMAD-3 networks, have been reported to be regulated by PAD4. Standler et at. first showed that knockdown of PADI4 in breast cancer cells lead to decreased level of nuclear GSK3β protein, E-cadherin expression, and increased TGF-β signaling, which promoted epithelial-to-mesenchymal transition [Citation94]. In addition, the citrullination of TGF-β was reported as early as 2016 by Sipila et al. [Citation28]. First, the group showed that citrullination of integrin αVβ6 altered its binding to β1-latency associated peptide (TGF-β1 LAP). Interaction between integrins and TGF-β1 LAP is a major mechanism for activating the latent TGF-β1 in the extracellular matrix (ECM). Secondly, the citrullination of the active TGF-β further inhibited its binding to recombinant TGF-β receptor II and prevented its ability to activate TGF-β signaling. Importantly, accordingly to the author, the citrullination of both latent and active TGF-β has the potency to regulate the inflammatory process [Citation28]. Although the study did not link citrullination of integrins or TGF-β to CVDs, however, numerous studies have suggested a significant crosstalk between the β-catenin and TGF-β1-SMAD-3 signaling pathways to regulate the organ fibrosis [Citation95,Citation96]. Moreover, Blyszczuk et al. demonstrated the crucial role of crosstalk between β-catenin and TGF-β1 signaling pathwaysin the progression of myofibroblast transformation and myocardial fibrosis [Citation97]. Guo et al. demonstrated that the cardiac fibroblasts-specific GSK-3β conditioned knockout led to hyperactivation of canonical TGF-β1-SMAD-3 signaling, resulting in adverse fibrotic remodeling and cardiac dysfunction in the ischemic heart [Citation98]. A better understanding of the PADs in the regulation of the GSK-3β, β-catenin, and TGF-β1-SMAD-3 signaling network may provide a novel therapeutic target for the management of myocardial fibrosis.
3. Prevalence of PADs in CVDs
3.1. Role of citrullination in angiogenesis related to cardiovascular aging
Angiogenesis is regulated by a complex array of pro- and anti-angiogenic factors [Citation99]. It accompanies cardiac hypertrophy both physiologically and pathologically, and an imbalance between blood vessels and myocardial growth has been considered a key event leading to HF [Citation100–102]. Angiogenesis is also essential for recovery from tissue damage and wound healing, and together with arteriogenesis, it is important for the cardiac and skeletal muscle that recover from ischemia [Citation100,Citation103]. Recent evidence suggested that multiple pathways can drive blood vessel growth and there may be important differences in angiogenesis in different contexts. Furthermore, several groups have reported genetic variability in genes that control angiogenesis, which may affect the susceptibility and progression of angiogenesis-dependent diseases [Citation104]. Recently, the D’Amato group hypothesized that PAD2-mediated citrullination plays a key role in regulating the angiogenic response [Citation105]. First, the genome-wide association indicated that Padi2 gene is the only differentially expressed isoform in a ‘high’ angiogenic mouse strain. Next, the role of PAD2 in angiogenesis was evaluated by in vitro functional assays of human microvascular endothelial cells (ECs) [Citation105]. In the study Padi2−/− and/or inhibition by Cl-Amidine in human microvascular ECs (HMVECs) led to the loss of deiminated proteins and a significant reduction in cell migration. By contrast, Padi2 gene overexpression increased the migration and proliferation of HMVECs, strongly suggesting the ability of Padi2 to be expressed and altered the angiogenic function [Citation105]. This was also supported by functional modeling in zebrafish, where suppression of zebrafish Padi2 expression led to vascular defects in vessels in brain, eye, and internodal vessels in 2-day embryos [Citation105]. Using a 3D microfluidic system, Bai et. al. showed that downregulation of Padi2 correlated with higher Dll4/Notch1 signaling and resulted in N1ICD released, which led to reduced tip cell formation and lower angiogenesis. In contrast, upregulation of Padi2 resulted in lower Dll4/Notch1 signaling with increased tip cell formation and accelerated angiogenesis. Although, future studies are required to show the detailed mechanism of Padi2’s involvement in the Notch signaling pathway, the author suggested that Padi2 may be involved in Notch cleavage that leads to the release of N1ICD and its nuclei translocation [Citation106].
Remarkably, further research is indispensable to examine the new role of PADI2 in regulating the physiological and pathological angiogenesis related to cardiovascular aging.
3.2. Role of PADs and citrullination in atherosclerosis
Atherosclerosis is a chronic inflammatory disease of the arteries, characterized by the accumulation of lipids and fibrous components in the large arteries over time [Citation107,Citation108]. Atherosclerosis is not simply an inevitable degenerative consequence of aging, but a chronic inflammation that can be transformed into an acute clinical event through plaque rupture and thrombosis [Citation109,Citation110]. The event of atherosclerosis can be roughly divided into four main stages: I, inflammation and endothelial dysfunction, II, formation of lipid layer or fatty streak, III, fibrous plaques, and IV, advanced lesions and thrombosis [Citation111]. A detailed outline of the stages of atherosclerosis has been described elsewhere and won’t be discussed here [Citation110,Citation111] . However, we debate the concepts of increased PAD activity as a new risk factor in the stages of lesion formation and complications.
In a coronary artery disease study, Borissoff et al. examined 282 patients with suspected coronary artery disease using tomographic angiography, evaluated cell death and NETosis markers, and plasma markers for activation of coagulation and inflammation [Citation38]. They found the markers of NETosis and of cell death to be independently associated with coronary artery disease, prothrombotic state, and adverse cardiac events [Citation38]. Using a murine surgical model of myocardial ischemia/reperfusion (MI/R) , the same group found that the MI/R injury increased plasma nucleosomes, neutrophil infiltration, and H3Cit at the injury site. They further reported that Padi4−/− mice were protected from MI/R injury, resulting in better postischemic cardiac function [Citation57]. In a more rigorous mouse model by Liu et al. myeloid-specific conditioned Pad4 knockout in Apoe−/− mice diminished NETosis , reduced inflammatory response in aorta, and significantly reduced atherosclerosis [Citation112]. To outline the role of citrullination in the progression of atherosclerotic plaques, we need to look into each stage separately ().
Figure 2. PADs activity interweaves atherosclerosis and thrombosis. NETs with H3Cit and PAD4 are involved in the whole process of atherosclerosis. PAD4 from NETs can stimulate macrophages to oxidize LDL to ox-LDL and turn into foam cells. Hyperlipidemia recruits’ neutrophils in the bone marrow into the circulation by upregulating the expression of granulocyte colony-stimulating factor and downregulating the level of C-X-C motif ligand −12, an important signal for the clearance and recruitment of aged neutrophils to the bone marrow. The macrophages activated by PAD4 driven NETosis undergo METosis. METosis increased pro-inflammatory cytokine production, including IL-1B. IL-1B activates Th17 cells to release IL-17, amplifying the immune cell recruitment to atherosclerotic plaques. NETs promote the expression of VWF and P-selectin on the surface of venous endothelia to entrap platelets and erythrocytes, thereby creating a scaffold for fibrin deposition. Meanwhile, cit-ADAMST13 dramatically inhibits its own activity and reduces the clearance of VWF-platelet strings, thereby accelerating thrombosis.
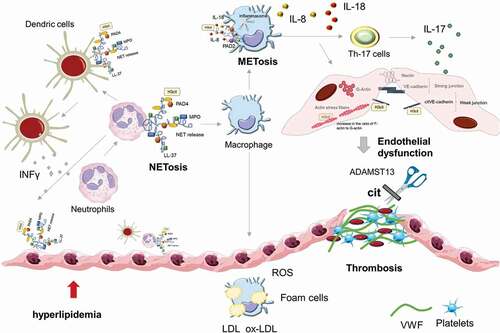
Stage I, Inflammation and endothelial dysfunction. Endothelial dysfunction is characterized by reduced vasodilation and a proinflammatory state with increases in lipid levels and neutrophils priming [Citation113,Citation114]. At the lesion initiation stage, the circulating low-density lipoproteins (LDLs) are retained by the EC matrix, undergo oxidative modification by ROS, and thus possess atherogenic properties [Citation109,Citation115]. The oxidized LDLs (oxLDLs) can trigger the expression of adhesion molecules on the cell surface, especially vascular cell adhesion molecule-1 (VCAM-1/CD106), intercellular adhesion molecule-1 (ICAM-1/CD54), and E-selectin [Citation116,Citation117]. These proteins disturb blood flow and facilitate the intimal retention of monocytes, neutrophils, macrophages, Tcells, and mast cells to the vascular wall [Citation109,Citation116]. The induced ROS can increase the transcription and activation of PAD4, possibly by increasing the intracellular Ca2+ concentration and are essential for PAD4-mediated NETs formation [Citation114,Citation118]. Notably, at this stage, H3Cit released by neutrophils was reported to cause endothelial barrier dysfunction and inflammatory injury by opening adherens junctions and reorganizing the cytoskeleton [Citation119]. Meegan et al. reported that stimulation of human ECs with H3Cit led to rapid thinning of adherens junction protein VE-cadherin at the cell–cell boundary and junction discontinuity. In addition, H3Cit resulted in centralization of filamentous actin (F-actin) and an increased in the ratio of F-actin to globular actin (G-actin), indicating the formation of actin stress fibers [Citation119]. These findings suggested that endothelial permeability induced by citrullination of the histone promotes vascular inflammation and play a key role in the initiation of atherosclerotic lesions.
Stage II, Formation of lipid layer or fatty streak. The characteristics of stage II are the recruitment of leukocytes and formation of foam cells. The recruited monocytes, neutrophils, and macrophages secrete proinflammatory cytokines (e.g., IL-1β and TNF-α), enzymes (e.g. myeloperoxidase), reactive oxygen species (ROS) that promote further retention and modification of LDLs, as well as many other mediators [Citation120,Citation121]. oxLDLs can activate lesional T cells to secrete proinflammatory cytokines TNF-α, TNF-β, IFN-γ, and IL-2 that can activate macrophages and vascular cells to promote inflammation [Citation113,Citation122]. oxLDLs and cytokines like TNF-α and IL-1β can activate nuclear factor-κB (NF-κB) through canonical NF-κB activation pathways, and the upregulation of NF-κB aggravates chronic inflammation throughout the atherogenesis [Citation120,Citation121]. Notably, PAD4 was shown to citrullinate NF-κB p65 subunit in neutrophils and enhance its interaction with importin-α3 to enter the nucleus and led the expression of proinflammatory mediators, including TNF-α and IL-1β, as confirmed in vitro by the inhibition of PAD [Citation123].
Stage III, Fibrous plaques. Fibrous plaques are characterized by a growing mass of extracellular lipid, and accumulation of vascular smooth muscle cells (VSMCs) [Citation124]. In intima, with the accumulation of lipid and inflammatory cells, the overburdened efferocytosis by macrophages results in the development of a necrotic core [Citation125]. In the plaque, cytokines and growth factors secreted by macrophages and T cells are important for VSMC migration, proliferation, and extracellular matrix production. NF-κB leads to the secretion of inflammatory mediators, which can activate VSMCs to undergo vascular remodeling and contribute to plaque formation and vascular stenosis [Citation116]. In intima, the NETs from neutrophils may prime plasmacytoid dendritic cells (pDCs) to produce proinflammatory IFNα, and macrophages proinflammatory cytokines IL-1β and IL-18 [Citation126,Citation127]. Surprisingly, NETs can prime apoptotic macrophages to release macrophage extracellular traps (METosis), similar to NETosis. For example, in an obesity-induced adipose tissue inflammation study, macrophages were detected to surround dead adipocytes at the inflammatory mediator-enriched lesions. TNF-α appeared to stimulate the histone hypercitrullination at H4R3 and DNA release from macrophage (RAW 264.7 cell) nuclei into a MET-like structure, suggesting METosis via-PAD-mediated histone citrullination to be involved in obesity-induced adipose tissue inflammation [Citation128]. The TNF-α induction of ETosis is at least partly due to the nuclear translocation of predominantly cytosolic PAD4, based on a study with a TNF-α mouse model and oligodendrocyte cell models [Citation129]. In a study by Nakazawa et al., PAD4 were detected in the M1 subpopulation of THP-1 macrophages but not unpolarized ones. Importantly, the author suggested that upon interaction with NETs and PAD4, M1 macrophages released decondensed chromatin containing H3Cit [Citation130]. METosis releases additional inflammatory cytokines, PADs, and citrullinated autoantigens into the extracellular milieu, which may perpetuate inflammation and increase thrombogenic potential.
Stage IV, Advanced lesions and thrombosis. Pathological studies suggest that the development of thrombus-mediated acute coronary events depends principally on the composition and vulnerability of a plaque. The cytotoxic histone H4 from the released NETs may perforate and contribute to the eventual lysis of VSMCs that destabilize the plaque [Citation126]. Besides proinflammatory cytokines and ROS, foam cells also secrete metalloproteinases that may degrade the extracellular matrix (ECM) to weaken the fibrous cap, susceptible to rupture and thrombosis [Citation109].
von Willebrand factor (vWF), a glycoprotein produced by ECs, forms multimers and adhere platelets to form thrombogenic long vWF-platelet strings [Citation131]. The wild-type mice treated with vWF antibody significantly reduced the infarct size, and Vwf−/− mice were reported to have significantly reduced infarct size, neutrophil infiltration, and myocyte apoptosis [Citation132]. ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type 1 motif 13) is a plasma protease for vWF multimers. Mild to moderate ADAMTS13 deficiencies are risk factors for myocardial and cerebral infarction, and pre-eclampsia [Citation133]. Adamts13−/− mice showed enlarged infarct size, neutrophil infiltration, and myocyte apoptosis, contrary to the phenotypes of Vwf−/− mice and ADAMTS13-treated wild-type mice [Citation132,Citation134]. Sorvillo et al. reported that citrullinated ADAMTS13, dramatically inhibited its activity in wild-type mice, reduced the clearance of VWF-platelet strings, and thereby accelerated thrombosis after vascular injury [Citation135]. In consistent with this mechanism, they detected elevated citrullination of ADAMTS13 in plasma of patients with sepsis and seniors with comorbidities.
It is noteworthy that the released extracellular traps can provide an important source of PAD4, the role of which has been described before [Citation40] and involved in other arterial diseases, including abdominal aortic aneurysms (AAAs) and peripheral arterial disease [Citation50]. For example, in an angiotensin II (Ang II)-based AAA mouse model, inhibition of the Ang II–induced NETosis with PAD4 inhibitors reduced rupture or prevented aneurysm growth [Citation50,Citation136].
Peripheral artery disease is atherosclerosis of the arteries in the lower extremity [Citation137]. Pan et al. reported high level of PAD4 in the nucleus of endothelial progenitor cells (EPCs) from the peripheral blood of patients with peripheral arterial disease [Citation138]. After injecting PAD4 inhibitor Cl-amidine into lower limb ischemic mice, they observed more functional EPCs with improved proliferation, migration, and tubule-forming ability in vitro, and better blood perfusion, more capillaries, and less apoptosis in gastrocnemius muscle in vivo. From the atherosclerosis story we can see that the impact of protein citrullination is not limited to the initial stage but throughout the atherosclerotic progress.
3.3. Role of citrullination in deep vein thrombosis
In neutrophils, the PAD4-mediated histone citrullination and the consequent NETosis have been reported to be prothrombotic and procoagulant [Citation78,Citation139,Citation140]. In vitro experiments by Fuchs et al. showed that NETs provided scaffolds and stimuli for platelet binding and aggregation, and induced RBC-rich thrombosis [Citation140]. In an inferior vena cava (IVC) stenosis mouse model of deep vein thrombosis (DVT), the same group detected increased extracellular chromatin and H3Cit in proximity to vWF in venous thrombus of wild-type mice [Citation140]. Intriguingly, they found that only less than 10% of Padi4−/− mice thrombosed, compared to 90% of wild-type mice [Citation78]. Since neutrophils from Padi4−/− mice failed to produce H3Cit or NETs after stimulation [Citation78], these observations indicate that histone modification by PAD4 in neutrophils is essential for DVT.
Besides the role of NETosis in thrombosis, citrullinated protein inhibitors have been also involved in thrombosis [Citation43]. Several serine protease inhibitor (SERPIN) superfamily members are pivotal in maintaining an orchestrated balance between clotting (coagulation) and clot dissolution (fibrinolysis). In the coagulation pathway catalyzed by thrombin, antithrombin inhibits thrombin serine protease that converts the soluble plasma fibrinogen into insoluble fibrin meshwork; in the fibrinolysis pathway catalyzed by plasmin, plasminogen activator (PA) inhibitor-1 (PAI-1) (a SERPIN superfamily member), is the main inhibitor of PAs, such as tissue-type PA (tPA) and urokinase-type PA (uPA), which cleave proenzyme plasminogen into active plasmin [Citation141]. α2-antiplasmin (α2AP), another SERPIN superfamily member, is the primary inhibitor of plasmin and a competitor of fibrin to bind plasminogen [Citation142]. Tilvawala et al. found that, in RA patients three SERPIN super-family members, antithrombin, antiplasmin, and PAI-1, were highly citrullinated and could hardly inhibit their cognate proteases [Citation143]. The same group verified in plasma clotting and fibrinolysis in vitro assays that, the citrullination of antithrombin and PAI-1 impaired binding to their cognate proteases, while the citrullination of antiplasmin converted it into a plasmin substrate [Citation43]. Using a mouse model of deep vein thrombosis , they found that the citrullinated antithrombin were increased and citrullinated antiplasmin decreased, which shifted the equilibrium both in the direction of thrombosis [Citation43]. The observation that citrullinated PAI-1 lost inhibitory activity and promoted fibrinolysis both in vitro and ex vivo might imply its physiological role in balancing coagulation and fibrinolysis.
4. Citrullinated autoantibodies as biomarkers
To date, a number of citrullinated protein species have been identified, including vimentin, α-enolase, and fibrin α- and β-chains [Citation144–146]. Accordingly, 100+ citrullination-targeting autoantibody species have been identified in the sera of RA patients [Citation147], and are detected in 70–80% of RA patients and 1–2% of healthy ones. According to reports, ACPAs may present for up to 14 years before manifestation of clinical symptoms, making them good biomarkers for RA [Citation148]. In 2014, Barbarroja et al. demonstrated the direct involvement of ACPAs in an adverse cardiovascular profile in RA patients by revealing strong association of the ACPA level with carotid intima mediathickness, and inflammatory/oxidative stress markers [Citation45] . Interestingly, a pioneering research by El-Barbary et al. reported the anti-modified citrullinated vimentin (anti-MCV) to be associated with subclinical atherosclerosis in early RA [Citation149]. This finding was confirmed by Norouzi et al., who measured the plasma of 135 RA patients and showed a significant inverse correlation between anti-MCV titer (p-value < 0.001) and left ventricular ejection fraction used to evaluate cardiac systolic dysfunction [Citation150]. These data indicate that anti-MCV can be used for screening and early detection of systolic dysfunction in RA patients. Of note, study by El-Barbary et al. showed that, anti-MCV is not only sensitive in diagnosing early RA, but also useful in monitoring subclinical atherosclerosis of early RA [Citation151]. Similar to anti-MCV, anti-citrullinated-fibrinogen was detected in sera from 134 female RA patients and found to be associated with increased aortic plaque burden, a reflection of the progress of atherosclerosis. A study by Hejblum et al. also reported a significant association between anti-citrullinated-fibrinogen and coronary artery disease (CAD) outcomes in RA patients [Citation44]. Geraldino-Pardilla et al. also reported positive association of anti-citrullinated fibrinogen and anti-MCV with higher left ventricular mass index, the most common form of adverse left ventricular remodeling in RA patients [Citation152]. Westerlind et al. further elaborated on the role of ACPAs in a 12-year study to investigate the relationship among ACPAs and incident cardiovascular events in RA patients. The study showed that ACPAs had a clear link to CVD as a comorbid condition in RA individuals, and the autoantibody load increased the risk of heart disease [Citation153].
A link between ACPAs and coronary heart disease (CHD) in the absence of RA has also been reported by Hermans et al. [Citation154]. The author reported that 11% of RA-negative ST-elevation MI patients were ACPA positive . Noteworthy, the long-term mortality (p-value = 0.01) as well as combined endpoint of re-infarction and death rate (p-value = 0.01) in this group were associated with ACPA positivity. The author further suggested that the ACPAs detected in CHD patients might act as an independent pro-atherogenic factor. However, more studies are needed to better elucidate the association of ACPAs with risk of CHD. To investigate whether ACPA is related to CVD independent of RA, Cambridge et al. showed that 10.4% of CHD cases were ACPA positive compared with 3.8% from controls (p-value = 0.008) [Citation155]. Interestingly, the predominance of IgG in CHD patients suggested that the ACPA response may be related to previous exposure to citrullinated proteins and affected by different immunogenetics [Citation155]. Fantus et al. noted that the risk of CVD increased with ACPA levels, although the results were not statistically significant [Citation156]. A similar case study by Hasbani et al. reported significantly elevated titers of ACPA in acute coronary syndrome [Citation157], adding to the existing body of literature that ACPA positivity should be consider an independent risk factor for cardiovascular events.
Considering the evidence discussed above, it can be hypothesized that selected citrullinated cardiac proteins or ACPAs could be used as biomarkers for diagnosis and identification of high-risk patients for myocardial injury and CVD in both RA and non-RA patients.
5. Citrullinated histone H3 as a novel prognostic marker and therapeutic target
Many studies have suggested enhanced histone citrullination to be a potential diagnostic or prognostic biomarker for diseases such as cancer [Citation22], septic shock [Citation158], and CVDs [Citation57,Citation119,Citation136].
In a clinical investigation, Eilenberg et al. observed that high levels of H3Cit in plasma (p-value = 0.004) and aortic tissue (p-value < 0.001) showed the value of diagnostic markers in the pathogenesis of AAAs. The author believed that H3Cit may be a promising AAA biomarker, because in their longitudinal observation study, the plasma levels of H3Cit were able to predict aneurysm growth (77% sensitivity and 64% specificity) [Citation136]. In addition, the plasma level of H3Cit decreased to the level of healthy controls after surgical intervention. As a proof of concept, they used Ang II–induced mouse model of AAA to inhibit PAD and blocked AAA progression [Citation136].
Another study with a similar mouse model showed that a PAD4 inhibitor markedly reduced Ang II–induced AAA rupture, as revealed by the reduction in aortic diameter, apoptosis of VSMCs, and elastin degradation [Citation159]. While the results from both studies showed strong evidence of H3Cit as a potential therapeutic target, further longitudinal studies are needed to definitively quantify H3Cit levels and link them to adverse disease outcomes.
Besides AAA, NETs, and H3Cit have also been found in other arterial pathological conditions, including myocardial infarction and thrombosis that cause ischemic stroke [Citation160]. Likewise, Ducroux et al. recently reported the presence of NETs and citrullinated H4 in 34 human acute ischemic stroke thrombi [Citation161].
Considering these and other results, it can be suggested that circulating citrullinated histones may be used as a prognostic tool for patients with conditions along with a higher rate of NETs formation. However, more research is needed to determine whether to use circulating NETs component markers alone or in conjunction with established clinical parameters of CVD.
6. Expert opinion
In recent years, solid evidence has revealed that PAD activation is involved in acute and chronic cardiovascular events, including atherosclerosis, ischemia, venous thrombosis, and cardiac fibrosis. However, the exact regulatory mechanisms and biological significance of protein citrullination in these and other cardiovascular events are still unclear, and additional efforts are called to uncover the real impact of PADs.
The biggest challenge in this field is the lack of specific tools to detect and identify citrullinated proteins in complex biological system. Mass spectrometry approaches have been developed to locate accurate citrullination sites at the peptide level. However, quantitative and high-throughput methods for detecting both known and uncharacterized sites of disease-relevant citrullination will truly benefit the field. Specifically, the identification of citrullinated candidates and their networks will help understand the repertoire of the biological functions mediated by citrullination, which is more diverse than we think.
Notably the presence of serum ACPAs has contributed to the clinical diagnosis and assessment of histopathology and disease development, such as in RA and type 1 diabetes. Despite the breadth of evidence, there is little consensus on the specific functions of ACPAs in various forms of CVD. Do ACPAs act as advocates of cardiac damage, or rather bystanders and indicators in the disease process? Hopefully, the identification of citrullination markers and understanding of their sources will help clarify these issues in the future.
Considering the pathophysiological relevance of PADs, plenty of work attempted to develop PAD4 inhibitors [Citation31,Citation162]. For example, the inhibition of PAD4 has been proved effective in different conditions, such as MI [Citation159], stroke [Citation163], and atherosclerosis [Citation164]. Although progress has been made and several compounds are currently available, including F- and Cl-amidine, YW3-56 (a Cl-amidine analog) [Citation165], GSK199, and GSK484 [Citation166], the development of isotype-specific PAD inhibitors requires more research on the structure, function, and regulation of PAD enzymes. Today, no PAD inhibitors are currently available for clinical use.
Article highlights
Upregulation of PAD2 and PAD4 have been observed in various CVDs, for example, myocardial infarction, acute inflammation induced by coronary artery bypass surgery, fibrinogen clog formation, and fibrosis.
Acute inflammation, ischemia/reperfusion, or a combination thereof leads to the systemic spreading of enzymatically active PAD that may citrullinate protein substrates, alter their functions, or/and generate citrullinated self-antigens.
Studies have shown that inhibiting PAD can reduce the burden of atherosclerosis and the formation of atherothrombosis by blocking the neutrophil extracellular trapping network and regulating the innate immune response.
ACPAs in patients with and without RA might act as an independent risk factor for the development of cardiovascular events.
Declaration of interests
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Amin B, Voelter W. Human deiminases: isoforms, substrate specificities, kinetics, and detection. Prog Chem Org Nat Prod. 2017;106:203–240.
- Wang S, Wang Y. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim Biophys Acta. 2013;1829(10):1126–1135.
- Mondal S, Thompson PR. Protein Arginine Deiminases (PADs): biochemistry and chemical biology of protein citrullination. Acc Chem Res. 2019;52(3):818–832.
- Chavanas S, Méchin MC, Takahara H, et al. Comparative analysis of the mouse and human peptidylarginine deiminase gene clusters reveals highly conserved non-coding segments and a new human gene, PADI6. Gene. 2004;330:19–27.
- Vossenaar ER, Zendman AJ, van Venrooij WJ, et al. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays. 2003;25(11):1106–1118.
- Knuckley B, Causey CP, Jones JE, et al. Substrate specificity and kinetic studies of PADs 1, 3, and 4 identify potent and selective inhibitors of protein arginine deiminase 3. Biochemistry. 2010;49(23):4852–4863.
- Damgaard D, Senolt L, Nielsen MF, et al. Demonstration of extracellular peptidylarginine deiminase (PAD) activity in synovial fluid of patients with rheumatoid arthritis using a novel assay for citrullination of fibrinogen. Arthritis Res Ther. 2014;16(6):498–506.
- Damgaard D, Bjørn ME, Jensen P, et al. Reactive oxygen species inhibit catalytic activity of peptidylarginine deiminase. J Enzyme Inhib Med Chem. 2017;32(1):1203–1208.
- Rizzuto R, Pinton P, Ferrari D, et al. Calcium and apoptosis: facts and hypotheses. Oncogene. 2003;22(53):8619–8627.
- Darrah E, Andrade F. Rheumatoid arthritis and citrullination. Curr Opin Rheumatol. 2018;30(1):72–78.
- Liu YL, Lee CY, Huang YN, et al. Probing the roles of calcium-binding sites during the folding of human peptidylarginine deiminase 4. Sci Rep. 2017;7(1):2429.
- Liu X, Arfman T, Wichapong K, et al. PAD4 takes charge during neutrophil activation: impact of PAD4 mediated NET formation on immune-mediated disease. J Thromb Haemost. 2021;19(7):1607–1617.
- Anzilotti C, Pratesi F, Tommasi C, et al. Peptidylarginine deiminase 4 and citrullination in health and disease. Autoimmun Rev. 2010;9(3):158–160.
- Zhai Q, Wang L, Zhao P, et al. Role of citrullination modification catalyzed by peptidylarginine deiminase 4 in gene transcriptional regulation. Acta Biochim Biophys Sin (Shanghai). 2017;49(7):567–572.
- Li P, Wang D, Yao H, et al. Coordination of PAD4 and HDAC2 in the regulation of p53-target gene expression. Oncogene. 2010;29(21):3153–3162.
- Guo Q, Fast W. Citrullination of inhibitor of growth 4 (ING4) by peptidylarginine deminase 4 (PAD4) disrupts the interaction between ING4 and p53. J Biol Chem. 2011;286(19):17069–17078.
- Snijders AP, Hautbergue GM, Bloom A, et al. Arginine methylation and citrullination of splicing factor proline- and glutamine-rich (SFPQ/PSF) regulates its association with mRNA. Rna. 2015;21(3):347–359.
- Fan L, Wang Q, Liu R, et al. Citrullinated fibronectin inhibits apoptosis and promotes the secretion of pro-inflammatory cytokines in fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther. 2012;14(6):R266–R266.
- van Venrooij WJ, Pruijn GJ. Citrullination: a small change for a protein with great consequences for rheumatoid arthritis. Arthritis Res. 2000;2(4):249–251.
- Wiik AS. The immune response to citrullinated proteins in patients with rheumatoid arthritis. Clin Rev Allergy Immunol. 2007;32(1):13–21.
- Méchin M-C, Takahara H, Simon M. Deimination and peptidylarginine deiminases in skin physiology and diseases. Int J Mol Sci. 2020;21(2):566.
- Zhu D, Zhang Y, Wang S. Histone citrullination: a new target for tumors. Mol Cancer. 2021;20(1):90.
- Proost P, Loos T, Mortier A, et al. Citrullination of CXCL8 by peptidylarginine deiminase alters receptor usage, prevents proteolysis, and dampens tissue inflammation. J Exp Med. 2008;205(9):2085–2097.
- Moelants EA, Mortier A, Grauwen K, et al. Citrullination of TNF-α by peptidylarginine deiminases reduces its capacity to stimulate the production of inflammatory chemokines. Cytokine. 2013;61(1):161–167.
- Nakayama-Hamada M, Suzuki A, Furukawa H, et al. Citrullinated fibrinogen inhibits thrombin-catalysed fibrin polymerization. J Biochem. 2008;144(3):393–398.
- Bezuidenhout JA, Venter C, Roberts TJ, et al. Detection of citrullinated fibrin in plasma clots of rheumatoid arthritis patients and its relation to altered structural clot properties, disease-related inflammation and prothrombotic tendency. Front Immunol. 2020;11:577523–577535.
- Nemmara VV, Tilvawala R, Salinger AJ, et al. Citrullination Inactivates Nicotinamide- N-methyltransferase. ACS Chem Biol. 2018;13(9):2663–2672.
- Sipilä KH, Ranga V, Rappu P, et al. Extracellular citrullination inhibits the function of matrix associated TGF-β. Matrix Biol. 2016;55:77–89.
- Trela M, Perera S, Sheeran T, et al. Citrullination facilitates cross-reactivity of rheumatoid factor with non-IgG1 Fc epitopes in rheumatoid arthritis. Sci Rep. 2019;9(1):12068–12075.
- Rondas D, Crèvecoeur I, D’Hertog W, et al. Citrullinated glucose-regulated protein 78 is an autoantigen in type 1 diabetes. Diabetes. 2015;64(2):573–586.
- Uysal-Onganer P, D’Alessio S, Mortoglou M, et al. Peptidylarginine deiminase inhibitor application, using Cl-Amidine, PAD2, PAD3 and PAD4 isozyme-specific inhibitors in pancreatic cancer cells, reveals roles for PAD2 and PAD3 in cancer invasion and modulation of extracellular vesicle signatures. Int J Mol Sci. 2021;22(3):1396–1417.
- Moshkovich N, Ochoa HJ, Tang B, et al. Peptidylarginine deiminase IV regulates breast cancer stem cells via a novel tumor cell–autonomous suppressor role. Cancer Res. 2020;80(11):2125–2137.
- Jang B, Jeon YC, Shin HY, et al. Myelin basic protein citrullination, a hallmark of central nervous system demyelination, assessed by novel monoclonal antibodies in prion diseases. Mol Neurobiol. 2018;55(4):3172–3184.
- Musse AA, Li Z, Ackerley Ca, et al. Peptidylarginine deiminase 2 (PAD2) overexpression in transgenic mice leads to myelin loss in the central nervous system. Dis Model Mech. 2008;1(4–5):229–240.
- Makrygiannakis D, Af Klint E, Lundberg IE, et al. Citrullination is an inflammation-dependent process. Ann Rheum Dis. 2006;65(9):1219–1222.
- Alghamdi M, Alasmari D, Assiri A, et al. An overview of the intrinsic role of citrullination in autoimmune disorders. J Immunol Res. 2019;2019:7592851.
- Dragoni G, De Hertogh G, Vermeire S. The role of citrullination in inflammatory bowel disease: a neglected player in triggering inflammation and fibrosis? Inflamm Bowel Dis. 2020;27(1):134–144.
- Borissoff JI, Joosen IA, Versteylen MO, et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol. 2013;33(8):2032–2040.
- Varjú I,N, Sorvillo D, Cherpokova Z, et al. Citrullinated fibrinogen renders clots mechanically less stable, but lysis-resistant. Circ Res. 2021;129(2):342–344.
- Knight JS, Luo W, O’Dell AA, et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res. 2014;114(6):947–956.
- Martinod K, Witsch T, Erpenbeck L, et al. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J Exp Med. 2016;214(2):439–458.
- Ahlers MJ, Lowery BD, Farber-Eger E, et al. Heart failure risk associated with rheumatoid arthritis-related chronic inflammation. J Am Heart Assoc. 2020;9(10):e014661.
- Tilvawala R, Nemmara VV, Reyes AC, et al. The role of SERPIN citrullination in thrombosis. Cell Chem Biol. 2021;28(12):1728–1739 .
- Hejblum BP, Cui J, Lahey LJ, et al. Association between anti–citrullinated fibrinogen antibodies and coronary artery disease in rheumatoid arthritis. Arthritis Care Res (Hoboken). 2018;70(7):1113–1117.
- Barbarroja N, Pérez-Sanchez C, Ruiz-Limon P, et al. Anticyclic citrullinated protein antibodies are implicated in the development of cardiovascular disease in rheumatoid arthritis. Arterioscler Thromb Vasc Biol. 2014;34(12):2706–2716.
- Chirivi RGS, van Rosmalen JWG, van der Linden M, et al. Therapeutic ACPA inhibits NET formation: a potential therapy for neutrophil-mediated inflammatory diseases. Cell Mol Immunol. 2021;18(6):1528–1544.
- Rohrbach AS, Slade DJ, Thompson PR, et al. Activation of PAD4 in NET formation. Front Immunol. 2012;3:360–374.
- Chang X, Han J. Expression of peptidylarginine deiminase type 4 (PAD4) in various tumors. Mol Carcinog. 2006;45(3):183–196.
- Chen H, Luo M, Wang X, et al. Inhibition of PAD4 enhances radiosensitivity and inhibits aggressive phenotypes of nasopharyngeal carcinoma cells. Cell Mol Biol Lett. 2021;26(1):9–19.
- Wei L, Wang X, Luo M, et al. The PAD4 inhibitor GSK484 enhances the radiosensitivity of triple-negative breast cancer. Hum Exp Toxicol. 2021;40(7):1074–1083.
- Brahmajosyula M, Miyake M. Localization and expression of peptidylarginine deiminase 4 (PAD4) in mammalian oocytes and preimplantation embryos. Zygote. 2013;21(4):314–324.
- Brahmajosyula M, Miyake M. Role of peptidylarginine deiminase 4 (PAD4) in pig parthenogenetic preimplantation embryonic development. Zygote. 2013;21(4):385–393.
- Arandjelovic S, McKenney KR, Leming SS, et al. ATP induces protein arginine deiminase 2-dependent citrullination in mast cells through the P2X7 purinergic receptor. J Immunol (Baltimore, Md: 1950). 2012;189(8):4112–4122.
- Beato M, Sharma P. Peptidyl Arginine Deiminase 2 (PADI2)-mediated arginine citrullination modulates transcription in cancer. Int J Mol Sci. 2020;21(4):1351–1367.
- Witalison EE, Thompson PR, Hofseth LJ. Protein arginine deiminases and associated citrullination: physiological functions and diseases associated with dysregulation. Curr Drug Targets. 2015;16(7):700–710.
- Koushik S, Joshi N, Nagaraju S, et al. PAD4: pathophysiology, current therapeutics and future perspective in rheumatoid arthritis. Expert Opin Ther Targets. 2017;21(4):433–447.
- Savchenko AS, Borissoff JI, Martinod K, et al. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood. 2014;123(1):141–148.
- Neeli I, Khan SN, Radic M. Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol. 2008;180(3):1895–1902.
- Wang Y, Li M, Stadler S, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184(2):205–213.
- Thiam HR, Wong SL, Qiu R, et al. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc Natl Acad Sci U S A. 2020;117(13):7326–7337.
- Li P, Li M, Lindberg MR, et al. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207(9):1853–1862.
- Saha P, Yeoh BS, Xiao X, et al. PAD4-dependent NETs generation are indispensable for intestinal clearance of Citrobacter rodentium. Mucosal Immunol. 2019;12(3):761–771.
- Lood C, Blanco LP, Purmalek MM, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22(2):146–153.
- SenGupta S, Hein LE, Parent CA. The recruitment of neutrophils to the tumor microenvironment is regulated by multiple mediators. Front Immunol. 2021;12:3719–3734.
- Niedźwiedzka-Rystwej P, Repka W, Tokarz-Deptuła B, et al. “In sickness and in health” – how neutrophil extracellular trap (NET) works in infections, selected diseases and pregnancy. J Inflam. 2019;16(15):1–13.
- Du M, Yang W, Schmull S, et al. Inhibition of peptidyl arginine deiminase-4 protects against myocardial infarction induced cardiac dysfunction. Int Immunopharmacol. 2020;78:106055.
- Fousert E, Toes R, Desai J. Neutrophil Extracellular Traps (NETs) take the central stage in driving autoimmune responses. Cells. 2020;9:4.
- Qi H, Yang S, Zhang L. Neutrophil extracellular traps and endothelial dysfunction in atherosclerosis and thrombosis. Front Immunol. 2017;8(928):1–12.
- Damiana T, Damgaard D, Sidelmann JJ, et al. Citrullination of fibrinogen by peptidylarginine deiminase 2 impairs fibrin clot structure. Clin Chim Acta. 2020;501:6–11.
- De Meyer SF, Suidan GL, Fuchs TA, et al. Extracellular chromatin is an important mediator of ischemic stroke in mice. Arterioscler Thromb Vasc Biol. 2012;32(8):1884–1891.
- Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175.
- Döring Y, Soehnlein O, Weber C. Neutrophil extracellular traps in atherosclerosis and atherothrombosis. Circ Res. 2017;120(4):736–743.
- Ling S, Xu J-W. NETosis as a pathogenic factor for heart failure. Oxid Med Cell Longev. 2021;2021:6687096.
- Bonaventura A, Vecchié A, Abbate A, et al. Neutrophil extracellular traps and cardiovascular diseases: an update. Cells. 2020;9:1.
- Vejlstrup A, Møller AM, Nielsen CH, et al. Release of active peptidylarginine deiminase into the circulation during acute inflammation induced by coronary artery bypass surgery. J Inflamm Res. 2019;12:137–144.
- Helseth R, Knudsen EC, Eritsland J, et al. Glucose associated NETosis in patients with ST-elevation myocardial infarction: an observational study. BMC Cardiovasc Disord. 2019;19(1):221.
- Helseth R, Shetelig C, Andersen G, et al. Neutrophil extracellular trap components associate with infarct size, ventricular function, and clinical outcome in STEMI. Mediators Inflamm. 2019;2019:7816491.
- Martinod K, Demers M, Fuchs TA, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A. 2013;110(21):8674–8679 .
- Thålin C, Hisada Y, Lundström S, et al. Neutrophil extracellular traps: villains and targets in arterial, venous, and cancer-associated thrombosis. Arterioscler Thromb Vasc Biol. 2019;39(9):1724–1738.
- Kumar SV, Kulkarni OP, Mulay SR, et al. Neutrophil extracellular trap-related extracellular histones cause vascular necrosis in severe GN. J Am Soc Nephrol. 2015;26(10):2399–2413.
- Vossenaar ER, Radstake TRD, van der Heijden A, et al. Expression and activity of citrullinating peptidylarginine deiminase enzymes in monocytes and macrophages. Ann Rheum Dis. 2004;63(4):373–381.
- Hojo-Nakashima I, Sato R, Nakashima K, et al. Dynamic expression of peptidylarginine deiminase 2 in human monocytic leukaemia THP-1 cells during macrophage differentiation. J Biochem. 2009;146(4):471–479.
- Lai N-S, Yu H-C, Tung C-H, et al. Increased peptidylarginine deiminases expression during the macrophage differentiation and participated inflammatory responses. Arthritis Res Ther. 2019;21(1):108.
- Yu H-C, Tung C-H, Huang K-Y, et al. The essential role of peptidylarginine deiminases 2 for cytokines secretion, apoptosis, and cell adhesion in macrophage. Int J Mol Sci. 2020;21(16):5720.
- Mishra N, Schwerdtner L, Sams K, et al. Cutting edge: protein arginine deiminase 2 and 4 regulate NLRP3 inflammasome-dependent IL-1β maturation and ASC speck formation in macrophages. J Immunol. 2019;203(4):795–800.
- Dos Santos G, Rogel MR, Baker MA, et al. Vimentin regulates activation of the NLRP3 inflammasome. Nat Commun. 2015;6(1):6574–6590.
- Alyaseer AAA, de Lima MHS, Braga TT. The role of NLRP3 inflammasome activation in the epithelial to mesenchymal transition process during the fibrosis. Front Immunol. 2020;11:883–903.
- Bobryshev YV, Ivanova EA, Chistiakov DA, et al. Macrophages and their role in atherosclerosis: pathophysiology and transcriptome analysis. Biomed Res Int. 2016;2016:9582430.
- Giles JT, Fert-Bober J, Park JK, et al. Myocardial citrullination in rheumatoid arthritis: a correlative histopathologic study. Arthritis Res Ther. 2012;14(1):R39.
- Fert-Bober J, Giles JT, Holewinski RJ, et al. Citrullination of myofilament proteins in heart failure. Cardiovasc Res. 2015;108(2):232–242.
- Shelef MA, Bennin DA, Mosher DF, et al. Citrullination of fibronectin modulates synovial fibroblast behavior. Arthritis Res Ther. 2012;14(6):R240.
- Sun M, Rethi B, Krishnamurthy A, et al. Anticitrullinated protein antibodies facilitate migration of synovial tissue-derived fibroblasts. Ann Rheum Dis. 2019;78(12):1621–1631.
- Li FJ, Surolia R, Li H, et al. Citrullinated vimentin mediates development and progression of lung fibrosis. Sci Transl Med. 2021;13:585.
- Stadler SC, Vincent CT, Fedorov VD, et al. Dysregulation of PAD4-mediated citrullination of nuclear GSK3β activates TGF-β signaling and induces epithelial-to-mesenchymal transition in breast cancer cells. Proc Nat Acad Sci. 2013;110(29):11851–11856.
- Akhmetshina A, Palumbo K, Dees C, et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun. 2012;3(1):735.
- Yousefi F, Shabaninejad Z, Vakili S, et al. TGF-β and WNT signaling pathways in cardiac fibrosis: non-coding RNAs come into focus. Cell Commun Signaling. 2020;18(1):87.
- Blyszczuk P, Müller-Edenborn B, Valenta T, et al. Transforming growth factor-β-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur Heart J. 2017;38(18):1413–1425.
- Guo Y, Gupte M, Umbarkar P, et al. Entanglement of GSK-3β, β-catenin and TGF-β1 signaling network to regulate myocardial fibrosis. J Mol Cell Cardiol. 2017;110:109–120.
- Cai W, Schaper W. Mechanisms of arteriogenesis. Acta Biochim Biophys Sin (Shanghai). 2008;40(8):681–692.
- Lähteenvuo J, Rosenzweig A, Sinclair D. Effects of aging on angiogenesis. Circ Res. 2012;110(9):1252–1264.
- D’Alessio A, Moccia F, Li J-H, et al. Angiogenesis and vasculogenesis in health and disease. Biomed Res Int. 2015;2015:126582–126584.
- Khurana R, Simons M, Martin JF, et al. Role of angiogenesis in cardiovascular disease. Circulation. 2005;112(12):1813–1824.
- Honnegowda TM, Kumar P, Udupa EGP, et al. Role of angiogenesis and angiogenic factors in acute and chronic wound healing. Plast Aesthetic Res. 2015;2(5):243–249.
- Galan A, Ferlin A, Caretti L, et al. Association of age-related macular degeneration with polymorphisms in vascular endothelial growth factor and its receptor. Ophthalmology. 2010;117(9):1769–1774.
- Khajavi M, Zhou Y, Birsner AE, et al. Identification of Padi2 as a novel angiogenesis-regulating gene by genome association studies in mice. PLoS Genet. 2017;13(6):e1006848.
- Bai J, Khajavi M, Sui L, et al. Angiogenic responses in a 3D micro-engineered environment of primary endothelial cells and pericytes. Angiogenesis. 2021;24(1):111–127.
- Ala-Korpela M. The culprit is the carrier, not the loads: cholesterol, triglycerides and apolipoprotein B in atherosclerosis and coronary heart disease. Int J Epidemiol. 2019;48(5):1389–1392.
- Whelton SP, Deal JA, Zikusoka M, et al. Associations between lipids and subclinical coronary atherosclerosis. Aids. 2019;33(6):1053–1061.
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874.
- Bentzon JF, Otsuka F, Virmani R, et al. Mechanisms of plaque formation and rupture. Circ Res. 2014;114(12):1852–1866.
- Lusis AJ. Atherosclerosis. Nature. 2000;407(6801):233–241.
- Liu Y, Carmona-Rivera C, Moore E, et al. Myeloid-specific deletion of peptidylarginine deiminase 4 mitigates atherosclerosis. Front Immunol. 2018;9:1680–1691.
- Szmitko PE, Wang CH, Weisel RD, et al. New markers of inflammation and endothelial cell activation: part I. Circulation. 2003;108(16):1917–1923.
- Shah MS, Brownlee M. Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circ Res. 2016;118(11):1808–1829.
- Borén J, Chapman MJ, Krauss RM, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European atherosclerosis society consensus panel. Eur Heart J. 2020;41(24):2313–2330.
- Mussbacher M, Salzmann M, Brostjan C, et al. Cell type-specific roles of NF-κB linking inflammation and thrombosis. Front Immunol. 2019;10:85.
- Döring Y, Soehnlein O, Weber C. Neutrophil extracellular traps in atherosclerosis and atherothrombosis. Circ Res. 2017;120(4):736–743.
- Stoiber W, Obermayer A, Steinbacher P, et al. The Role of Reactive Oxygen Species (ROS) in the formation of Extracellular Traps (ETs) in humans. Biomolecules. 2015;5(2):702–723.
- Meegan JE, Yang X, Beard RS Jr., et al. Citrullinated histone 3 causes endothelial barrier dysfunction. Biochem Biophys Res Commun. 2018;503(3):1498–1502.
- de Winther MP, Kanters E, Kraal G, et al. Nuclear factor kappaB signaling in atherogenesis. Arterioscler Thromb Vasc Biol. 2005;25(5):904–914.
- Yu XH, Zheng XL, Tang CK. Nuclear factor-κB activation as a pathological mechanism of lipid metabolism and atherosclerosis. Adv Clin Chem. 2015;70:1–30.
- Hansson GK, Libby P, Schönbeck U, et al. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. 2002;91(4):281–291.
- Sun B, Dwivedi N, Bechtel TJ, et al. Citrullination of NF-κB p65 promotes its nuclear localization and TLR-induced expression of IL-1β and TNFα. Sci Immunol. 2017;2(12):12.
- Libby P, Pasterkamp G, Crea F, et al. Reassessing the mechanisms of acute coronary syndromes. Circ Res. 2019;124(1):150–160.
- Schrijvers DM, De Meyer GR, Herman AG, et al. Phagocytosis in atherosclerosis: molecular mechanisms and implications for plaque progression and stability. Cardiovasc Res. 2007;73(3):470–480.
- Silvestre-Roig C, Braster Q, Ortega-Gomez A, et al. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol. 2020;17(6):327–340.
- Warnatsch A, Ioannou M, Wang Q, et al. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349(6245):316–320.
- Mohanan S, Horibata S, McElwee JL, et al. Identification of macrophage extracellular trap-like structures in mammary gland adipose tissue: a preliminary study. Front Immunol. 2013;4:67–75.
- Mastronardi FG, Wood DD, Mei J, et al. Increased citrullination of histone H3 in multiple sclerosis brain and animal models of demyelination: a role for tumor necrosis factor-induced peptidylarginine deiminase 4 translocation. J Neurosci. 2006;26(44):11387–11396.
- Nakazawa D, Shida H, Kusunoki Y, et al. The responses of macrophages in interaction with neutrophils that undergo NETosis. J Autoimmun. 2016;67:19–28.
- Smeets MWJ, Mourik MJ, Niessen HWM, et al. Stasis Promotes Erythrocyte Adhesion to von Willebrand Factor. Arterioscler Thromb Vasc Biol. 2017;37(9):1618–1627.
- Gandhi C, Motto DG, Jensen M, et al. ADAMTS13 deficiency exacerbates VWF-dependent acute myocardial ischemia/reperfusion injury in mice. Blood. 2012;120(26):5224–5230.
- Zheng XL. Structure-function and regulation of ADAMTS-13 protease. J Thromb Haemost. 2013;Suppl 11(1):11–23.
- De Meyer SF, Savchenko AS, Haas MS, et al. Protective anti-inflammatory effect of ADAMTS13 on myocardial ischemia/reperfusion injury in mice. Blood. 2012;120(26):5217–5223.
- Sorvillo N, Mizurini DM, Coxon C, et al. Plasma peptidylarginine deiminase IV promotes VWF-platelet string formation and accelerates thrombosis after vessel injury. Circ Res. 2019;125(5):507–519 .
- Eilenberg W, Zagrapan B, Bleichert S, et al. Histone citrullination as a novel biomarker and target to inhibit progression of abdominal aortic aneurysms. Transl Res. 2021;233:32–46.
- Morley RL, Sharma A, Horsch AD, et al. Peripheral artery disease. Bmj. 2018;360:j5842.
- Pan J, Liu W, Chen Y, et al. Effect of peptidylarginine deiminase 4 on endothelial progenitor cell function in peripheral arterial disease. Evid Based Complement Alternat Med. 2021;2021:7550693.
- Brill A, Fuchs TA, Savchenko AS, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012;10(1):136–144.
- Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107(36):15880–15885.
- Yasar Yildiz S, Kuru P, Toksoy Oner E, et al. Functional stability of plasminogen activator inhibitor-1. ScientificWorldJournal. 2014;2014:858293.
- Carpenter SL, Mathew P. Alpha2-antiplasmin and its deficiency: fibrinolysis out of balance. Haemophilia. 2008;14(6):1250–1254.
- Tilvawala R, Nguyen SH, Maurais AJ, et al. The rheumatoid arthritis-associated citrullinome. Cell Chem Biol. 2018;25(6):691–704.
- Raptopoulou A, Sidiropoulos P, Katsouraki M, et al. Anti-citrulline antibodies in the diagnosis and prognosis of rheumatoid arthritis: evolving concepts. Crit Rev Clin Lab Sci. 2007;44(4):339–363.
- Van Steendam K, Tilleman K, Deforce D. The relevance of citrullinated vimentin in the production of antibodies against citrullinated proteins and the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford). 2011;50(5):830–837.
- Wegner N, Lundberg K, Kinloch A, et al. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol Rev. 2010;233(1):34–54.
- Yu H-C, Lu M-C. The roles of anti-citrullinated protein antibodies in the immunopathogenesis of rheumatoid arthritis. Ci Ji Yi Xue Za Zhi. 2019;31(1):5–10.
- van de Stadt LA, de Koning MH, van de Stadt RJ, et al. Development of the anti-citrullinated protein antibody repertoire prior to the onset of rheumatoid arthritis. Arthritis Rheum. 2011;63(11):3226–3233.
- El-Barbary AM, Kassem EM, El-Sergany MA, et al. Association of anti-modified citrullinated vimentin with subclinical atherosclerosis in early rheumatoid arthritis compared with anti-cyclic citrullinated peptide. J Rheumatol. 2011;38(5):828–834.
- Norouzi S, Javinani A, Aminorroaya A, et al. Anti-modified citrullinated vimentin antibody: a novel biomarker associated with cardiac systolic dysfunction in patients with rheumatoid arthritis. BMC Cardiovasc Disord. 2020;20(1):390–398.
- Sokolove J, Brennan MJ, Sharpe O, et al. Brief report: citrullination within the atherosclerotic plaque: a potential target for the anti–citrullinated protein antibody response in rheumatoid arthritis. Arthritis Rheumatism. 2013;65(7):1719–1724.
- Geraldino-Pardilla L, Russo C, Sokolove J, et al. Association of anti-citrullinated protein or peptide antibodies with left ventricular structure and function in rheumatoid arthritis. Rheumatology (Oxford). 2017;56(4):534–540.
- Westerlind H, Rönnelid J, Hansson M, et al. Anti-citrullinated protein antibody specificities, rheumatoid factor isotypes, and incident cardiovascular events in patients with rheumatoid arthritis. Arthritis Rheumatol. 2020;72(10):1658–1667.
- Hermans MPJ, van der Velden D, Montero Cabezas JM, et al. Long-term mortality in patients with ST-segment elevation myocardial infarction is associated with anti-citrullinated protein antibodies. Int J Cardiol. 2017;240:20–24.
- Cambridge G, Acharya J, Cooper JA, et al. Antibodies to citrullinated peptides and risk of coronary heart disease. Atherosclerosis. 2013;228(1):243–246.
- Fantus S, Heisler A, Briones M, et al. Association of elevated anti-cyclic citrullinated peptide antibody titer with increased cardiovascular risk. J Rheumatol Arthritis Res. 2018;1.
- Hasbani GE, Ghazzal Z, Dakik H, et al. Acute coronary syndrome in a male with elevated anti-cyclic citrullinated peptide and no evidence of longstanding rheumatoid arthritis. Mediterranean journal of rheumatology. 2020;31(3):362–365.
- Pan B, Alam HB, Chong W, et al. CitH3: a reliable blood biomarker for diagnosis and treatment of endotoxic shock. Sci Rep. 2017;7(1):8972–8981.
- Wei M, Wang X, Song Y, et al. Inhibition of peptidyl arginine deiminase 4-dependent neutrophil extracellular trap formation reduces angiotensin II-Induced abdominal aortic aneurysm rupture in mice. Front Cardiovasc Med. 2021;8:805–816.
- Laridan E, Denorme F, Desender L, et al. Neutrophil extracellular traps in ischemic stroke thrombi. Ann Neurol. 2017;82(2):223–232.
- Ducroux C, Di Meglio L, Loyau S, et al. Thrombus neutrophil extracellular traps content impair tPA-induced thrombolysis in acute ischemic stroke. Stroke. 2018;49(3):754–757.
- Aliko A, Kamińska M, Falkowski K, et al. Discovery of novel potential reversible peptidyl arginine deiminase inhibitor. Int J Mol Sci. 2019;20(9):2174–2186.
- Lange S, Rocha-Ferreira E, Thei L, et al. Peptidylarginine deiminases: novel drug targets for prevention of neuronal damage following hypoxic ischemic insult (HI) in neonates. J Neurochem. 2014;130(4):555–562.
- Wong SL, Wagner DD. Peptidylarginine deiminase 4: a nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J. 2018;32(12):6258–6370.
- Wang Y, Li P, Wang S, et al. Anticancer peptidylarginine deiminase (PAD) inhibitors regulate the autophagy flux and the mammalian target of rapamycin complex 1 activity. J Biol Chem. 2012;287(31):25941–25953.
- Lewis HD, Liddle J, Coote JE, et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol. 2015;11(3):189–191.