
ABSTRACT
Introduction
Mass spectrometry-based single-cell proteomics (scMS) is experiencing rapid evolution due to the increased sensitivity of mass spectrometers as well as advances in multiplexing and sample preparation. To date, researchers have focused on two general approaches to scMS: label-free and isobaric label-based multiplexing. While label-free analysis provides straightforward sample preparation and a clear path to automation, it currently lacks the throughput necessary to practically analyze thousands of single cells. Multiplexed analysis utilizing isobaric labels requires additional sample manipulation but increases throughput such that analyzing thousands of cells is currently achievable. A key feature of multiplexed scMS experiments is a ‘carrier proteome’ – a sample added at 25x-500x, the single-cell sample that increases the number of proteins that can be identified in an MS analysis.
Areas covered
Here, we review early examples of carrier proteomes in quantitative proteomics before summarizing advantages and challenges of using a carrier proteome in scMS experiments.
Expert opinion
We conclude that the addition of carrier proteomes improves depth of identification for scMS, but high levels of carrier proteomes can have adverse effects on quantitative accuracy and precision.
1. Introduction
Cellular heterogeneity is an important factor in disease biology, but only recently have technologies that enable in-depth characterization of single cells at a large scale matured (). These advances have demonstrated that a systemic understanding of cellular variability is crucial to understand both the normal and disease biology. For example, single-cell technologies have been instrumental in teaching us about the processes in drug resistance, viral infection, and cell signaling [Citation1,Citation2]. In fact, scRNAseq technologies have evolved beyond measuring transcript expression to characterizing structural aspects of the genome (e.g. single-cell Hi-C) [Citation3] and DNA accessibility (e.g. scATACSEQ) [Citation4] (). Although we have achieved significant progress in single-cell RNA sequencing, the full understanding of the cellular signaling landscape is incomplete without inclusion of the effector molecules/functional units of the cell, proteins.
Figure 1. (a) Single-cell-based technologies and their technical maturity. Single-cell proteomics (SCP) is the most recent and emerging technique among most single-cell methods. (b) Mass spectrometry-based single-cell proteomics (scMS) workflow that uses a carrier proteome (e.g. SCoPE-MS).
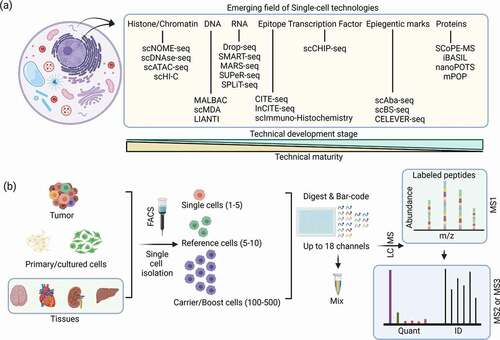
As the product of the transcripts that code them, proteins represent an important readout of cellular state and function. Additionally, multiple studies have documented the poor correlation between RNA and protein levels within cells [Citation5–7]. To date, antibody-based techniques have been the lens through which researchers have viewed proteins at single-cell resolution. These approaches have included sequencing technologies (e.g. CITE-seq) [Citation8], mass cytometry [Citation9], and immunohistochemistry [Citation10] (). As antibody-based detection methods, these techniques require prior knowledge of a protein target as well as compatible antibody-based reagents for the analysis. Unlike antibody approaches, mass spectrometry (MS)-based proteomics can identify and quantify proteins (proteomes) and post-translational modification of proteins (PTMomes) in an unbiased fashion, enabling detection of cell-specific signatures that are not currently known. However, the sensitivity of single-cell mass spectrometry (scMS) had been viewed as a barrier to identifying or quantifying an impactful number of proteins from common cellular systems (e.g. single cells from cell culture or isolated from tissue). Due to the recent introduction of new mass spectrometry technologies and multiplexed labeling reagents, scMS is currently undergoing a drastic increase in interest and adoption.
While scMS is gaining widespread interest, analysis of proteins from single cells is not a new concept. Early work identified proteins from single cells using capillary electrophoresis mass spectrometry (CE-MS) [Citation11,Citation12], a technique that only needs a small amount of material for analysis. Before SCoPE-MS, liquid chromatography mass spectrometry (LC-MS) analysis of single cells typically focused on cellular systems where single cells contain sufficient protein material for analysis (e.g. frog embryos and human oocytes) [Citation13,Citation14]. Since the publication of SCoPE-MS, scMS experienced a rapid increase in interest as this approach demonstrated that the quantification of single cells from common laboratory cell lines was possible [Citation15]. The key attributes of SCoPE-MS were the use of multiplexed isobaric labeling reagents (e.g.TMT) that employ unique mass barcodes to enable multiple samples to be quantified in one analysis and the introduction of a carrier proteome, a sample added at 200x, a single-cell sample that drastically improves the number of identified peptides and proteins [Citation15] ().
The carrier proteome was identified as the enabling feature of SCoPE-MS and serves two main roles increases the signal of intact peptides such that they are identified by the mass spectrometer and selected for fragmentation and 2) provides fragment ion signal that enable identification using a database search (). These features increase the number of peptides and proteins that can be identified in a scMS experiment. However, adding a carrier proteome also introduces a number of challenges relating to data acquisition and quantitative accuracy of results.
Here, we provide a detailed history of the use of carrier proteomes. We first summarize non-single cell methods that utilized similar approaches to the carrier proteome (e.g. Super SILAC, TMTCalibrator, BASIL) before providing an extensive discussion of the use of carrier proteomes in single-cell proteomics (i.e. SCoPE-MS, iBASIL, etc.). Through this process we highlight examples of carrier proteomes improving the number of identified and quantified peptides, the challenges associated with utilizing a carrier proteome, and provide an expert opinion for the current best practices for utilizing a carrier proteome within scMS experiments.
2. Evolution of the carrier proteome approach within multiplexed quantitative proteomics
Multiplexed proteome quantification typically employs a data-dependent acquisition (DDA) approach in which the most intense peptides are selected from an MS1 scan and fragmented for identification in an MS2 analysis. This approach enables the identification of a large number of unique peptides but has limitations related to the stochastic process of selecting peptides for sequencing. In particular, low abundance peptides may not be quantified because they do not meet a threshold signal within an MS1 scan and therefore do not get isolated, fragmented, and subsequently identified or quantified. This challenge was a main driver for the development of techniques that utilize a carrier proteome. In fact, several experimental strategies have been proposed that utilize different forms of carrier proteomes to improve identification as well as quantitation of peptides.
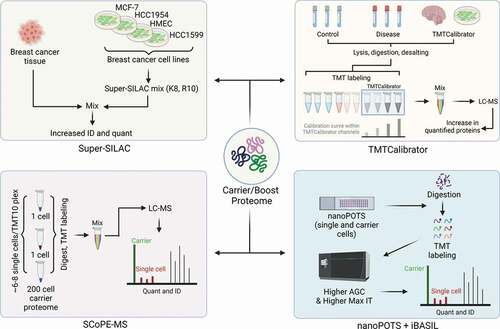
Before the introduction of isobaric labels, stable isotope labeling by amino acids in cell culture (SILAC) was the most common multiplexed quantitative proteomic technique [Citation16]. However, SILAC requires a number of cell doubling events to ensure heavy amino acids are sufficiently incorporated to the cellular proteome and enable quantification. Due to this, SILAC analysis of human tissue was not feasible and SILAC analysis of model systems (e.g. mouse models) was possible – but costly. This led researchers to develop methods to analyze tissue samples that circumvented this challenge. Here, a SILAC mix, termed super-SILAC, was developed as a spike-in internal standard [Citation17] (). A super-SILAC mix is a mixture of two or more labeled cell lines labeled with heavy amino acids that closely represent the experimental proteome being analyzed (). The super-SILAC approach is potentially the first experimental approach to introduce a carrier proteome strategy specifically aimed at enhancing the peptide identification and quantitation of low-level peptides and proteins. More recently, isobaric labels have become the preferred reagents to enable multiplexed quantification due to the increased level of multiplexing compared to SILAC (i.e. 18-plex with isobaric labels vs. 3-plex with SILAC), and the concept of a carrier proteome has evolved within these new multiplexing approaches.
Potentially the first instance of a carrier proteome within an isobaric label-based multiplexed proteome experiment was the TMCcalibrator approach Citation18(). In its initial description, TMTCalibrator samples derived from cell lines or tissues were added to the multiplexed experiment at four different concentrations to construct a calibration curve within the quantitative reporter ion region (). In addition to enabling more accurate quantification, TMTCalibrator channels also improved detection of low-level peptides and proteins. The authors demonstrate that inclusion of TMTCalibrator channels improved the number of low-level proteins quantified from CSF ~23% Citation18. This improvement is attributed to the additional signal provided by the TMTCalibrator channels as they improved the detection of the peptides within MS1 survey scans, leading to subsequent peptide identification and quantification. Importantly, the authors also describe considerations in selecting the TMTCalibrator sample – including ensuring that the TMTCalibrator samples are analogous to the samples you are analyzing and ensuring that the TMTCalibrator samples contain proteins you hope to quantify. This consideration is also true of the carrier proteome within scMS approaches. If proteins are present in experimental single cell-samples but not present in the carrier proteome, then there is no longer an advantage to adding a carrier proteome to your sample. While intuitive, ensuring your carrier proteome reflects the single cells you are attempting to quantify is an essential part of designing an experiment with a carrier proteome.
Figure 2. Historical perspective of experimental approaches that utilize a carrier/boost proteome for multiplexed quantitative proteomics.
2.1. Examples of carrier proteome approaches in non-single cell proteomics experiments
Before discussing the utilization of a carrier proteome within scMS, we want to briefly touch on other quantitative proteomics approaches that have used a carrier proteome in non-single-cell applications. As discussed above, including a carrier proteome improves identification and quantification of low-level samples, and as a result, carrier proteomes have typically been employed when analyzing samples where a limited amount of material is available (e.g. phosphorylated or Human Leukocyte Antigen (HLA)-associated peptides).
Yi et al. introduced boosting to amplify signal with isobaric labeling (BASIL), an experimental design that utilized a carrier proteome, termed ‘boost’ sample, to increase the phosphopeptide identifications from a limited amount of sample [Citation19]. This strategy enhanced the MS1 signal enabling MS2 fragmentation, leading to a higher number of identifications. In one instance, they identified that the phosphopeptide signal intensity was increased by 30-fold by the inclusion of a carrier proteome [Citation19]. Chua et al. also reported the use of a carrier proteome specifically to study phosphotyrosine (pY)-modified peptides [Citation20]. To accomplish this, the authors used a sample treated with pervanadate (PV) and increased the number of identified and quantified low abundance phospho-tyrosine peptides. Notably, the authors propose that a carrier proteome enabled reduction of starting material amount from 20 to 1 mg for phospho tyrosine analysis [Citation20]. This is attractive because a reduction in starting material requirements would make pY analysis more accessible when analyzing low-level samples (e.g. tumor biopsy). In fact, the TMTCalibrator approach was also employed to analyze pY peptides from 1 mg of cells treated with various drug treatments and demonstrated improved proteomic depth when including a carrier proteome for pY peptide analysis [Citation21].
More recently, Stopfer et al. analyzed both pY-modified peptides and HLA-associated peptides in multiplexed experiments that utilized carrier proteomes [Citation22]. Immunopeptidomics and phospho-tyrosine analyses typically require higher amounts of starting material to quantify a meaningful number of peptides. As reported by recent studies, starting material requirements have decreased for peptide-bound major histocompatibility complex (pMHC) studies from billions of cells to millions of cells [Citation22–24]. However, even millions of cells might be a challenging number for the valuable clinical samples. Stofer et al. demonstrated that the protein quantitation measurements were altered by protein carriers, in particular, they noted a significant degree of ratio compression [Citation22]. This was determined by comparing experiments with 0x carrier to experiments where a 20x carrier was used to analyze HLA-associated peptides in response to CDK4/6 inhibitors and 9x-100x was used to analyze phospho-tyrosine changes induced by EGF stimulation. Although overall identifications for pMHC and phospho-tyrosine peptides increased, the quantitative accuracy was poor and ratios were compressed. The study highlighted the limitations associated with the use of a carrier proteome, many of which are also discussed below.
While studies have been mostly focused on low-level proteomes, PTMomes, and peptidomes, carrier channels have also been used outside of these areas. For example, Peck Justice et al. recently reported that the integration of isobaric channels based on affinity purified protein complexes increased the capability to detect the low abundance protein complexes [Citation25]. This approach was called mutant thermal protein profiling (mTPP). While the remainder of the review focuses on carrier proteomes in single-cell proteomics experiments, the principles discussed and lessons learned can be applied to any experiment that utilizes a carrier proteome.
2.2. The use of carrier proteomes in mass spectrometry-based single-cell proteomics experiments
In the initial description of SCoPE-MS, the defining feature of the experimental design was the inclusion of a carrier proteome – a sample added to the multiplexed experiment at 200x, a singlecell channel [Citation15] (). Here, the carrier proteome was created by an equal number of U-937 and Jurkat cells to ensure that it represented the proteomes of the experimental cells being measured [Citation15] (). Even in this early study, the authors took considerations to ensure that data gathered on single cells was not impacted by the presence of isotopic impurities stemming from the carrier proteome. Isobaric labels are constructed using heavy labeled atoms (13C and 15N), and the purity of these isotopes can impact signals measured from quantitative reporter ions. For example, a 126C reporter ion might have isotopic impurities that spill into 127C, or a 131C reporter ion might have isotopic impurities that spill into 130C. These isotopic impurities are typically known and can be corrected using values provided by the reagent’s manufacturer – but the presence of these isotopic impurities is an important consideration when using a carrier proteome in an experiment. In SCoPE-MS, the multiplexed channel occupied by +1 impurities from the carrier proteome reporter ion were left empty – ensuring that these isotopic impurities did not interfere with single cell signal. By using a carrier proteome, the authors identified ~750 proteins and demonstrated that scMS could provide quantitative information for a portion of a single cell’s proteome.
Following the introduction of SCoPE-MS, Dou et al. published a study demonstrating that the NanoPOTS sample preparation workflow could be used for the analysis of single cells [Citation26] (). Here, the NanoPOTS-TMT workflow utilized carrier ratios of 0x, 25x, and 250x to analyze cells from three separate mouse cell lines. The authors demonstrate that protein identifications increase in proteins quantified from ~200 with 0x carrier to ~900 and ~1400 with 25x and 250x carrier, respectively. Interestingly, the authors also demonstrated that cells from specific cell types clustered together well with 0x and 25x carrier proteomes, but clustering was hampered when the carrier level was increased to 250x. This phenomenon was most likely due to undersampling of single cell peptide ions in the 250x sample – a challenge that will be discussed below.
Extending from this work, Tsai et al. introduced an update to the BASIL workflow, termed iBASIL, that specifically focused on single-cell proteomics [Citation27] (). Like SCoPE-MS, the iBASIL method used a carrier proteome, but called it a ‘boost’ channel to align with their previous study. Interestingly, this study was the first published account of analyzing the direct impact of increasing carrier proteome levels as it related to identification rate, ion sampling, and quantitative precision. The authors found that increasing carrier proteome levels increased the number of identified peptides at the expense of increased variability within the data. They demonstrate that increasing ion sampling by requesting more ions and increasing the time allowed to sample them can improve overall data quality within scMS experiments. Note, Cheung et al. made similar observations linking data quality to ion sampling, and these will be discussed further below [Citation28]. Coupled with the improved data quality, this improved version of BASIL was able to detect ~2,000 proteins per multiplexed experiment, representing a major advance for scMS approaches [Citation27].
As an improvement to SCoPE-MS, Specht et al. introduced SCoPE2, an updated version of the SCoPE-MS workflow that quantified ~3,000 proteins across ~1,500 single cells [Citation29]. Both SCoPE-MS and SCoPE2 report using a 200x carrier proteome channel, but SCOPE2 also includes a blank control channel lacking a single cell as well as a reference channel added at 5x, a single-cell channel. Although the reference channel is not meant to serve the purpose of a carrier proteome, it should be considered as part of the carrier proteome level because it still contributes to the challenges of additional non-single cell peptide ions during analysis. However, because a 200x carrier proteome is used, an additional 5x carrier proteome within the reference channel should have a negligible effect. In addition to the reference channel, SCoPE2 also left both the 127 N and 127C TMT channels empty when using a carrier proteome within the 126 channel. This was likely done to avoid unfavorable interactions between the 13C isotope of the carrier proteome reporter ion found in 127C with a single-cell reporter ion within 127 N. If the number of ions within the 127C channel is too large, it can cause erroneous measurement of the number of ions within the 127 N channel; this is a phenomenon known as coalescence and is described further below. With this improved approach, the authors demonstrate that analysis of protein abundance at single-cell levels closely matches bulk proteome measurements, giving confidence to quantitative values determined by SCoPE2.
Following this study, Schoof et al. presented their work studying the differentiation of myeloid cells in the context of leukemia at the single-cell level [Citation30]. Similar to SCoPE2, this approach used a 200x carrier sample but omitted a reference sample in its final iteration. Like previous studies [Citation27,Citation28], the authors present an extensive analysis of the impact of ion sampling and instrument parameters on quantitative precision within scMS. They arrive at a similar conclusion that quantitative accuracy and precision can be improved by sampling more ions through increasing the number of ions requested as well as the time allowed to sample the single-cell peptide ions. Using these methods, the authors demonstrate that their scMS approach yields ~700-1,000 quantified proteins per cell. In addition to the detailed look at optimization of instrument parameters, Schoof et al. also introduced SCepter – a computational workflow that enables analysis of single-cell proteomics data [Citation30]. This is an important contribution because the underlying models of scepter enabled the authors to omit the reference channel within their approach and decrease the amount of non-single cell peptide ions sampled during analysis.
One of the main challenges associated with scMS experiments is performing single-cell sample preparation in a fashion that limits protein losses. Bulk proteome sample preparation includes a number of tube transfers and fractionation steps that cause protein losses before samples are analyzed by the mass spectrometer. While the carrier protome can limit the impact of these losses, specific protocols have been developed to improve the preparation of single cells for scMS analysis, including NanoPOTS [Citation26], mPOP Citation29, and the recently introduced ProteoCHIP [Citation31]. Here the authors describe an all-in-one cell sorting and sample preparation solution specifically focused on multiplexed single-cell proteomics experiments. As part of this study, the samples were analyzed with 0x or 20x carrier proteome in order to determine the importance of the carrier proteome when protein losses were minimized during preparation. They find that omitting a carrier often leads to more single cell signal being detected, likely due to the fact that all ions being injected belong to single cells. Interestingly, they found that the number of quantified proteins only differed slightly between 0x and 20x carrier. In fact, they demonstrated that while there is a benefit to the carrier proteome, analysis of ~1,000 proteins per cell was possible when the carrier proteome was absent. Further work will be needed to replicate this finding as it currently represents an outlier within the field as other studies have demonstrated an increase in identified and quantified proteins when a carrier proteome is utilized [Citation26,Citation27].
While the papers listed above are examples of the carrier proteome used to analyze single-cell samples, two studies have been published specifically addressing quantitative accuracy and precision within scMS approaches that utilize a carrier proteome. Cheung et al. utilized a number of controlled mixtures containing a range of carrier proteome levels (up to 2,000x) to define the carrier proteome limit for single-cell proteomics experiments [Citation28]. A number of these experiments and conclusions are discussed in the following section, but briefly the authors demonstrate that increasing the carrier proteome level within a sample requires more ions to be sampled to maintain quantitative accuracy. Furthermore, they demonstrated that even when sufficient ions are sampled, single-cell signals can be truncated when the carrier proteome level is too high. Ultimately, the authors provide experimental conditions that improve quantification of samples at single-cell levels as compared to the same samples measured at bulk proteome levels. They also introduced SCPCompanion – a computational tool that enables researchers to better understand the level of quantitative signal within their experiments as well as optimize method parameters around ion sampling.
In a separate study, Ctortecka et al. compared both the SCoPE-MS and SCoPE2 methods to their own approaches that used 0x, 5x, 10x, 20x carrier proteomes [Citation32]. These analyses included experimental designs that utilized two 5x or 10x carrier channels, noting that when two carrier channels are used, the carrier proteome level should be considered as equal to sum of both. Based on these analyses, the authors found that two carrier channels did not improve analysis over one equally abundant carrier proteome. They also agreed with Cheung et al. that carrier proteome levels should be limited, for example to 20x, as higher carrier levels (e.g. 200x) increase quantitative variability of measurements [Citation28]. Interestingly, they introduced an offset carrier proteome approach that utilized TMTzero – an isobaric label that has the same chemical structure but unique mass as compared to TMT. This imparted a mass shift that separated the TMTzero-labeled carrier proteome from the same isotopic cluster as TMT. They found that the increased complexity led to fewer proteins quantified in ‘single cell’ channels as the amount of carrier proteome increased. However, it is possible that this approach would benefit from higher carrier proteome levels, which would make MS1 scans less complex as the single-cell signals become a minority of the signal. This alteration could make this approach more attractive but would likely need to be paired with an intelligent data acquisition approach that enables offset mass triggering on a proteome scale.
2.3. Impact of the carrier proteome on proteomic depth of scMS
As summarized above, scMS experiments can quantify ~1,000 proteins per cell and ~3,000 proteins across thousands of single cells. The carrier proteome is an important aspect in achieving this depth because it adds a consistent signal that is matched across all individual multiplexes experiments. The constant background provided by the carrier proteome also ensures that proteins that are variably expressed across single cells have the opportunity to be detected. A similar effect was seen when a common ‘bridge’ sample comprising multiple cell lines was used in the analysis of proteomes from 375 cell lines from the cancer cell line encyclopedia [Citation33]. Ultimately, the number of proteins quantified will correlate with quantification of lower copy-per-cell proteins because quantification by scMS is mainly determined by protein abundance and length. Due to this, increasing the number of peptides and proteins quantified will also increase quantification of lower level, functionally relevant proteins.
Early scMS studies have demonstrated sufficient proteomics depth to separate cells into unique populations according to their cell type or differentiation state. However, proteomic analysis is often employed to understand the function of proteins that perform biochemical reactions within the cell (e.g. receptors, kinases, or other signaling molecules), and many of these proteins exist at low copies per cell. Currently, scMS can quantify proteins as low as ~50,000 copies per cell, and the detection of a protein of interest will depend on its abundance in the cell being analyzed [Citation34].
Even at the current proteomic depth, applications have emerged that demonstrate the potential of scMS to enable biological understanding or therapeutic development. Schoof and colleagues demonstrated how scMS can be used to monitor the differentiation of cell types as they enter different cellular lineages [Citation30]. This approach could be used to monitor the diversity of the proteome for iPSC-derived cell models as they differentiate toward more biologically relevant states. Slavov and colleagues demonstrated that p53 protein levels correlate more closely with up- or downregulated targets as compared to p53 RNA levels [Citation29]. This is an example of how monitoring protein levels of pathway regulators could be used to understand how certain cell populations respond to therapeutic intervention or even identify cell populations resistant to a particular drug treatment. Lastly, Zhu and colleagues presented the identification of cell surface proteins that were not detected in RNAseq analysis [Citation35]. The identification of new, cell-type-specific surface markers could leverage the development of therapeutics that are targeted to specific cell populations through molecules such as bispecific antibodies. Carrier proteome-based scMS methods continue to improve toward a goal of quantifying ~5,000 proteins across single cells, a proteomic depth that would improve the quantification of functionally relevant proteins [Citation34].
3. Analytical considerations when utilizing a carrier proteome
Before the publication of SCoPE-MS, carrier proteomes had previously been used for unbiased proteome quantification (e.g. TMTCalibrator); however, the description of using a carrier proteome to enable single-cell proteomics prompted much discussion in the proteomics community. Early discussions of the carrier proteome in scMS focused on the dynamic range of the mass analyzer most commonly used for mass analysis of TMT reporter ions, the Orbitrap. However, this was just one of a number of considerations that have arisen as the use of carrier proteomes has become more widespread. Here we discuss challenges associated with the carrier proteome including sampling of single-cell peptide ions, the dynamic range of the mass analyzer, effective transfer of single-cell ions within the mass spectrometer, and quantitative reporter ion signal stemming from isotopic impurities of the isobaric label ().
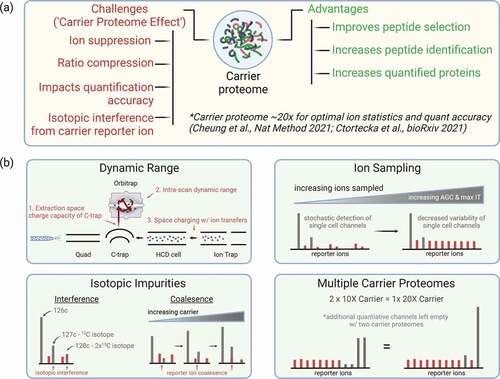
Figure 3. (a) Advantages and disadvantages of carrier proteome in SCP experiments and suggested range of carrier proteome amount based on the recent studies. (b) Experimental considerations and instrumental challenges for scMS experiments that use a carrier proteome.
3.1. Dynamic range and detection of single-cell ions
When SCoPE-MS was first introduced, concerns were raised surrounding the dynamic range of the Orbitrap mass analyzer used for analysis of the quantitative reporter ion signals. The Orbitrap mass analyzer was an important advancement because it made high mass accuracy FTMS analyzers more accessible than traditional FT-ICR mass analyzers. In fact, the Orbitrap is a key aspect of increased multiplexing due to its ability to differentiate peptides made heavier by addition of 13C from those made heavier by addition of 15N (i.e. mass defect). Although powerful, Obritraps have challenges related to intra-scan dynamic range [Citation36,Citation37] and accuracy of measuring isotopic ratios [Citation38], and these historical challenges were the reason for concern upon the introduction of SCoPE-MS (). However, earlier iterations of the Orbitrap have a documented intrascan dynamic range of 1:5000 and would seem to be capable of measuring ions even when they are mixed with 100x carrier proteome. Nonetheless, early single-cell data demonstrated highly variable single-cell quantification that could either be attributed to cellular heterogeneity or variability due to limitations of the instrumentation.
While completely isolating the limitations of the mass analyzer from other components in the mass spectrometer is difficult, Cheung et al. attempted to understand how measurement dynamic range was impacted by the presence of the carrier proteome [Citation28]. To do this, an instrument method was constructed that fragmented peptides to produce reporter ions, which were then sent to the ion trap where they were isolated either including or excluding the carrier reporter ion. By doing this, reporter ions belonging to quantitative ‘single cell’ samples could be measured in the presence or absence of the carrier reporter ion (). These methods enabled characterization of both the Orbitrap instra-scan dynamic range as well as the instrument’s ability to get ‘single cell’ ions from the ion trap to the Orbitrap when the carrier ions were or were not present.
Interestingly, these experiments demonstrated that the ‘single cell’ signal was truncated in a manner dependent on the carrier proteome level. This suggested that when enough ions were sampled, the presence of the carrier protome impacted the instrument’s ability to measure the single-cell ions. The most likely causes for this findings are 1) the Orbitrap mass analyzer has difficulty analyzing ‘single cell’ ions in the presence of the carrier reporter ions (i.e. limited intra-scan dynamic range) or2) there is space charging occurring during the operation of this instrument method (). Space charging is a phenomenon that occurs when the number of ions traversing the mass spectrometer becomes too large and the ions stop behaving as expected as they navigate the electric fields within the instrument. For the Orbitrap instrument used in these experiments, the most likely places for space charging would be during ion trap isolation of the reporter ions or extraction of ions from the C-trap into the Orbitrap ( b). At this time, any of these explanations could be possible and further experiments would be needed to determine the cause of ‘single cell’ signal truncation.
These findings are particularly interesting in the context of a recent study by Stopfer et al., which provided a careful look at the quantitative impact of the carrier proteome within the analysis of pY- and MHC-associated peptides [Citation22,Citation23]. Here they found that quantitative ratios were compressed when the carrier proteome was present, as compared to when it was absent. This finding could be related to the truncation of the single-cell signal detected by Cheung et. al. [Citation28], and implementation of the ‘exclude’ method with the samples prepared by Stopfer et al. [Citation22] could shed light on this further.
While these data might seem to explain high variability of early single cell proteomics data, these findings come with the caveat that the truncation in ‘single cell’ signal demonstrated by Cheung et al. was detected only when a large number of ions were injected into the mass spectrometer. In fact, most scMS experiments would struggle to inject the number of ions necessary for this phenomenon to occur. Due to this, ion sampling was considered as the root cause of challenges associated with data precision and accuracy within single-cell proteomics experiments.
3.2. Carrier proteome effect on sampling of single-cell ions
Mass spectrometry methods that utilize data-dependent acquisition (DDA) have been honed over the years to maximize the number of identified or quantified peptides and proteins within a given analysis. This is typically done by sampling only enough peptide ions to enable identification and/or quantification. If too few ions are selected, peptides cannot be identified, and if too many ions are selected, then time is wasted and the total number of identifications will decrease. When researchers first started to explore single-cell proteomics, many started with MS methods akin to those used on bulk proteome samples. These methods typically sampled a relatively small number of ions and ensured that any peptide was not analyzed for too long (i.e. lower maximum injection time). Applying these methods to samples with a high carrier level (e.g. 200x) will lead to a deficit in ion sampling of single-cell ions and ultimately data exhibiting high variability and low precision ().
As discussed in Cheung et al., ion sampling is a major consideration in the optimization of any technique that utilizes a carrier proteome (including SCoPE-MS). For a given number of ions, any increase in the amount of carrier proteome will decrease the number of single cell ions sampled, leading to higher measurement variability and lower accuracy. These effects can be compensated for by increasing the number of ions sampled by increasing the number of ions requested and the time the instrument is allowed to accumulate them (). Interestingly, Ctortecka et al. used an offset mass carrier proteome to demonstrate that the carrier proteome also causes ion suppression of single-cell peptide ions [Citation32]. In these experiments, even when a carrier proteome is not within the same isotopic cluster, ion sampling will need to be optimized in order to obtain sufficient single cell signal.
Based on several reports, it is now generally accepted that ion sampling is likely the major component of obtaining quantitative data with high degrees of accuracy and precision [Citation27,Citation28,Citation30]. Making matters a bit more complicated, each mass spectrometer might have a slightly different sensitivity as compared to a separate mass spectrometer of the same model. Due to this, Cheung at al. introduced SCPCompanion, a program that analyzes single-cell proteomics data to suggest instrument parameters such as maximum injection time based on the data collected on a particular mass spectrometer [Citation28]. Note, increasing ion sampling will increase data quantity but will likely also decrease the number of peptides and proteins that are quantified. Ultimately, there needs to be a balance between the level of carrier proteome, the number of ions requested, and the amount of time allowed to sample ions in order to optimize quantified peptides and proteins.
3.3. Influence of isotopic impurities from stemming from the carrier proteome
Multiplexed isobaric labeling reagents are a core feature of scMS approaches such as SCoPE-MS as they drastically increase the number of samples that can be analyzed in a single analysis. However, one main consideration when using isobaric labeling reagents should be the isotopic impurities present within the tags themselves (). In the initial description of SCoPE-MS, the authors wisely left the light −1 reporter ion channel (12C impurity) empty to avoid any contamination of the light isotopic impurity with a single cell channel [Citation15]. Interestingly, Cheung et al. demonstrated that at higher carrier levels, impurities can be detected in the +1 as well as +2 reporter ion channels [Citation28] (). This had not previously been demonstrated and has implications for many single cell approaches that use carrier levels above 200x, particularly those that use multiple carrier proteome channels because there are now multiple sources of impurity interference. This challenge can be easily addressed by omitting quantitative channels where impurities may occur, but this does reduce multiplexing capacity of an isobaric label-based experiment and ultimately will lead to longer data collection duration or the analysis of fewer cells.
In addition to overlapping directly with single-cell reporter ions, isotopes from the carrier proteome ion can also impact single-cell reporter ions in neighboring channels (). As noted above, the multiplexing capacity of TMT labels was increased through introduction of similar tags differing only by the inclusion of a 13C or 15N. These mass barcodes differ by 0.0063 Da, a difference that can be detected by resolutions at or above 50,000 in tribrid Orbitrap instruments (e.g. Orbitrap Fusion, Lumos, or Eclipse) under normal conditions. However, the accurate detection of these unique mass barcodes can become compromised if one of the two ion populations separated by 0.0063 Da becomes much larger than the other [Citation39]. Cheung et al. demonstrate that when using a large amount of carrier proteome in channel 126C, the large population of ions from the 13C isotope in channel 127C negatively affects detection of the single-cell signal in channel 127N [Citation28]. This effect was dependent on the signal within the 127C channel and was likely due to ion coalescence, a phenomenon where one larger ion population can merge with another smaller ion population, leading to the detection of one peak instead of two (). This would likely only occur when a large amount of carrier proteome is used and when a large number of ions were analyzed. Due to this, it is unlikely this would occur in scMS, but one way in which this can be monitored is to measure the mass error of the isobaric label reporter ions. If the masses of two neighboring reporter ions begin to drift toward one another, it is likely a sign that coalescence is occurring.
3.4. Use of multiple carrier proteome channels within a multiplexed experiment
Multiple groups have proposed utilizing multiple carrier proteome channels as a way to improve scMS experiments. Note that splitting a larger carrier, for example 500x, into two separate channels of 250x does not alter the physical demands of the carrier proteome (). This is an important consideration because the ion sample demands and the caution around space charge effects will be dependent on the sum of all carrier channels within a sample. Due to this, two 250x channels should be treated the same as one 500x channel. In fact, SCPCompanion will consider all channels labeled as ‘Carrier’ as part of the calculation for the estimated carrier ratio, enabling researchers to determine the effective carrier ratio within their samples.
4. Conclusion
Mass spectrometry-based single-cell proteomics is undergoing a rapid evolution as it matures from a concept that was thought to be out of reach to something that is now obtainable. The introduction of the SCoPE-MS approach and the utilization of a carrier proteome was the key development that spurred on the current excitement around scMS development. In the few years since the introduction of SCoPE-MS, biologically important findings have been demonstrated around the identification of cell-specific receptors [Citation35] and understanding cellular hierarchies in a leukemia model system [Citation30].
While the carrier proteome was an enabling feature of SCoPE-MS, it has taken multiple groups to understand the current best way to employ a carrier proteome within the bounds of a single-cell proteomics experiment. Carrier proteome have been used at levels as high as 500x the single cell channels, but studies by multiple groups suggest that carrier levels be limited to 20x to avoid negative effects on quantitative accuracy and precision [Citation22,Citation23,Citation28]. However, Stofer et al. describe that even a carrier level of 20x could lead to a truncated dynamic range when measuring differences between experimental samples [Citation22]. While the main concern of a carrier proteome was initially the dynamic range limitations of the Orbitrap mass analyzer, Cheung et al. found that truncation of single cell signal only occurs when a large number of ions are analyzed [Citation28]. In single-cell proteomics experiments, this large number of ions is not likely to be reached and the most likely factor associated with increased quantitative variability and decreased accuracy is the number of peptide ions sampled during MS analysis. Multiple groups have demonstrated that sampling more single-cell peptide ions can increase data quality but at the expense of peptide and protein identification events. Lastly, the presence of high levels of carrier proteome can introduce high levels of isotopic impurities stemming from the carrier proteome reporter ion. If the carrier proteome is added at a high level, these signals can impact single-cell signals in the +2 reporter ion channel and lead to erroneous quantitative information.
Ultimately, utilizing a carrier proteome requires a balancing act between the level of carrier proteome used, design of the multiplexed experiment, and configuration of the MS method to ensure optimal precision and accuracy of the data. Looking into the future, the carrier proteome has multiple paths within single-cell proteomics workflows 1) continued to be used as is, 2) implemented and utilized in a new fashion, and 3) omitted from the experiment entirely.
As described above, a carrier proteome is a simple addition to a scMS experiment that will increase the number of peptides and proteins that are identified. However, the inclusion of a high level of carrier proteome can pose challenges to collecting data that has a high level of precision and accuracy. If researchers continue to use a carrier proteome, the current best practice will be to use it at a relatively low level (e.g. 20x) to limit the negative effects of the carrier proteome on ion sampling and dynamic range (). If a higher level is used, great care should be taken to optimize mass spectrometry data acquisition parameters to ensure sufficient ions are sampled and quantitative signals reflect the biology of the samples being analyzed.
It is possible that new approaches could enable utilization of a carrier proteome in a new fashion. Ctrotecka et al. introduced the concept of an offset mass carrier protome that does not occupy the same precursor ion cluster as the single cell samples [Citation32]. This approach has been utilized in a technique called TOMAHAQ, a targeted mass spectrometry approach that combines sample and peptide multiplexing to enable accurate quantification of peptides within multiplexed samples [Citation40]. While Ctortecka saw little benefit from the offset mass carrier proteome, it is possible that increasing the level of carrier proteome in this approach combined with intelligent data acquisition could improve the depth and quality of proteome quantification. Recently, there has been an increase in the number of intelligent data acquisition strategies within mass spectrometry-based proteomics. The most commonly used strategies within isobaric label-based multiplexing experiments are those that use real-time search to identify peptides while data is being collected [Citation41–43]. This approach improves the accuracy and depth of bulk proteomics and has recently been applied to the analysis of single-cell proteomics as well [Citation44]. If new approaches such as an offset-mass carrier proteome are to be used, it will require an approach that at a minimum includes a real-time database search capable of identifying a peptide from the carrier and triggering subsequent scans on offset endogenous peptides.
Perhaps, the most likely path for scMS experiments will be moving away from carrier proteomes entirely. Carrier proteomes are currently used because the sensitivity of mass spectrometers remains on the cusp of routine quantification of thousands of proteins from single cells. Improvements to the sensitivity of mass spectrometers or the increased levels of multiplexing for isobaric label reagents are both likely developments that could negate the need for carrier proteomes within scMS. In fact, alternative tag chemistries have been developed that enable a larger number of samples to be multiplexed [Citation45]. Increased number of multiplexing channels will increase the combined signal of a multiplexed sample, potentially to the level of current 20x carrier proteome levels and beyond. While sensitivity of mass spectrometers has increased dramatically in recent years, it remains possible that additional improvements in this area might also remove the need for carrier proteomes. But until these developments are realized, carrier proteomes are likely to be utilized, and while they remain a staple of multiplexed scMS experiments, the considerations outlined within this review will remain extremely important.
5. Expert opinion
Carrier proteomes – samples added to multiplexed quantitative proteomics experiments at 20–500x the experimental samples – have the potential to increase the number of identification and quantification events from low-abundance samples (e.g. single cells, HLA-associated peptides, pY IP). However, the addition of carrier proteomes at high levels challenges the ability to sample quantitative ions of peptides originating from single cells as well as the ability of mass spectrometers to effectively transfer ions throughout the instrument. If not addressed, these challenges could lead to under-sampling of quantitative single-cell reporter ions that leads to more variable single cell measurements or a truncation of single-cell signal and ratio compression of measurements between experimental conditions, respectively. These challenges can be addressed by alterations to the experimental design as well as the operation of the mass spectrometer. To improve sampling of single cell ions, the instrument can be instructed to select more ions (i.e. increase AGC target) for a longer time (i.e. longer maximum injection time). While these changes will limit the number of quantified proteins, they will improve the precision and accuracy of data that are collected. Additionally, limiting the amount of carrier proteome can limit the effects stemming from inadequate transfer of single cell ions within the mass spectrometer. Multiple publications have now demonstrated that a carrier proteome of ~20x works well within a SCoPE-MS experimental design as it limits the truncation of single cell signal within the mass spectrometer [Citation28,Citation32]. The ultimate solution may be that increased multiplexing enables deep scMS analysis without the use of a carrier proteome; however, this is dependent on the production of new multiplexing reagents that enable a higher level of multiplexing in one experiment. It is likely that most scMS experiments will continue to use a carrier proteome as it is a simple addition that enables deeper proteome quantification, but if employing a carrier proteome, it is important to monitor the amount of signal collected within a scMS experiment and adjust mass spectrometer parameters accordingly. One could do this through a program like SCPCompanion that analyzes the amount of single-cell signal detected in an experiment and provides mass spectrometer method parameters to improve data quality based on the specific experimental design and instrument characteristics.
Article highlights
Summarization of carrier proteome use in quantitative multiplexed proteomics
Carrier proteomes within single-cell proteomics
Experimental advantages and challenges of using a carrier proteome
Analytical considerations when utilizing a carrier proteome
Conflict of Interest
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
The figures were prepared using Biorender.com.
References
- Miller AJ, Yu Q, Czerwinski M, et al. In vitro and in vivo development of the human airway at single-cell resolution. Dev Cell. 2020 Apr 6;53(1):117–128.e6.
- Izar B, Tirosh I, Stover EH, et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Met. 2020;26. 1271–1279.
- Ramani V, Deng X, Qiu R, et al. Massively multiplex single-cell Hi-C. Nature Methods. 2017;14(3):263–266.
- Buenrostro J, Wu B, Chang H, et al. ATAC-seq:Amethod for assaying chromatin accessibility genome wide. Curr Protoc Mol Biol. 2015;209:21.29.1–21.29.9.
- Foutnier ML, Paulson A, Pavelka N, et al. Delayed correlation of mRNA and protein expression in rapamycin-treated cells and a role for Ggc1 in cellular sensitivity to rapamycin. Mol. Cell. Prot. 2010Feb; 92: 271–284.
- Gygi SP, Rochon Y, Franza BR, et al. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999 Mar;19(3):1720–1730.
- Liu Y, Beyer A, Aebersold R. On the dependency of cellular protein levels on mRNA abundance. Cell. 2016 Apr 21;165(3):535–550.
- Stoeckius M, Hafemeister C, Stephenson W, et al. Simultaneous epitope and transcriptome measurements in single cells. Nat. Met. 2017;14. 865–868.
- Spitzer MH, Nolan GP. Mass cytometry: single cell, many features. Cell. 2016 May 5;165(4):780–791.
- Ren X, Zhong G, Zhang Q, et al. Reconstruction of cell spatial organization from single cell RNA sequencing data based on ligand-receptor mediated self assembly. Cell Res. 2020;30(9):763–778.
- Dovichi NJ, Hu S, and Michels D, et al. Single Cell Proteomics. In: Veenstra TD, and Yates JR, editors. Proteomics for Biological Discovery. Hoboken, NJ, USA: John Wiley & Sons, Inc; 2009. p. 225–245.
- Rubakhin SS, Lanni EJ, Sweedler J. Progress towards single cell metabolomics. Curr Opin Biotech. 2013 Feb;24(1):95–104.
- Wagner M, Yoshihara M, Douagi I, et al. Single-cell analysis of human ovarian cortex identifies distinct cell populations but no oogonial stem cells. Nat Comm. 2020;11(1):1147.
- Sun L, Dubiak KM, Peuchen EH, et al. Single cell proteomics using frog (Xenopus laevis) blastomeres isolated from early stage embryos, which form a geometric progression in the protein content. Anal Chem. 2016 Jul 5;88(13):6653–6657.
- Budnik B, Levy E, Harmange G, et al. SCoPE-MS: mass spectrometry of single mammalian cells quantifies protome heterogeneity during cell differentiation. Genome Biol. 2018;19(1):161.
- Ong SE, Blagoev B, Kratchmarova I, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Prot. 2002 May;1(5):376–386.
- Griger T, Cox J, Ostasiewicz P, et al. Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat. Met. 2010;7(5):383–385.
- Russell C L, Heslegrave A, Mitra V, Zetterberg H, Pocock J M, Ward M A and Pike I. (2017). Combined tissue and fluid proteomics with Tandem Mass Tags to identify low-abundance protein biomarkers of disease in peripheral body fluid: An Alzheimer's Disease case study. Rapid Commun. Mass Spectrom., 31(2), 153–159. https://doi.org/10.1002/rcm.7777
- Yi L, Tsai CF, Dirice E, et al., Boosting to Amplify Signal with Isobaric Labeling (BASIL) strategy for comprehensive quantitative phosphoproteomic characterization of small populations of cells. Anal Chem. 2019;91(9): 5794–5801.
- Chua XY, Mensah T, Aballo T, et al. Tandem mass tags approach utilizing pervanadate BOOST channels delivers deeper quantitative characterization of the tyrosine phosphoproteome. Mol. Cell. Prot. 2020;19(4):730–743.
- Fang B, Izumi V, Remsing Rix LL, et al., Lowering sample requirement to study phospho tyrosine kinase signaling using phosphoproteomics with the TMT calibrator approach. Proteomics. 2020;20(24): e2000116.
- Stofer LE, Conage-Pough JE, White FM. Quantitative consequences of protein carriers in immunopeptidomics and tyrosine phosphorylation MS2 analyses. Mol. Cell. Prot. 2021;20:100104.
- Stofer LE, Mesfin JM, Joughin BA, et al. Multiplexed relative and absolute quantitative immunopeptidomics reveals MHC I repertoire alterations induced by CDK4/6 inhibition. Nat Comm. 2020;11:1–14.
- Chong C, Marino F, Pak HS, et al. High-throughput and sensitive immunopeptidomics platform reveals profound interferon-mediated remodeling of the human leukocyte antigen (HLA) ligandome. Mol. Cell. Prot. 2018;17(3):533–548.
- Peck Justice SA, McCracken NA, Victorino JF, et al. Boosting detection of low-abundance proteins in thermal proteome profiling experiments by addition of an isobaric trigger channel to TMT multiplexes. Anal Chem. 2021;93(11):7000–7010.
- Dou M, Clair G, Tsai CF, et al. High-throughput single cell proteomics enabled by multiplex isobaric labeling in a nanodroplet sample preparation platform. Anal Chem. 2019;91(20):13119–13127.
- Tsai C-F, Zhao R, Williams SM, et al., An Improved Boosting to Amplify Signal with Isobaric Labeling (iBASIL) strategy for precise quantitative single-cell proteomics. Molecular & Cellular Proteomics. 2020;19(5): 828–838.
- Cheung TK, Lee C-Y, Bayer FP, et al., Defining the carrier proteome limit for single-cell proteomics. Nature Methods. 2021;18(1): 76–83.
- Specht H, Emmott E, Petelski AA, et al., Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 2021;22(1): 50.
- Schoof EM, Furtwangler B, Uresin N, et al., Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nature Communications. 2021;12(1): 3341.
- Hartlmayr D, Ctortecka C, Seth A, et al. An automated workflow for label-free and multiplex single-cell proteomics sample preparation at unprecedented sensitivity. bioRxiv. 2021;04:14.439828.
- Ctortecka C, Krssakova G, Stejskal K, et al., Comparative proteome signatures of trace samples by multiplexed data-independent acquisition. Molecular & Cellular Proteomics. 2022;21(1): 100177.
- Nusinow DP, Szpyt J, Ghandii M, et al. Quantitative proteomics of the cancer cell line encyclopedia. Cell. 2020;180(2):387–402.e16.
- Slavov N. Unpicking the proteome in single cells: Single cell mass spectrometry will help reveal mechanisms that underpin health and disease. Science. 2020;367(6477):512–513.
- Woo J, Williams SM, Markillie LM, et al. High-throughput and high-efficiency sample preparation for single-cell proteomics using nested nanowell chips. Nat Comm. 2021;12:6246.
- Kaufmann A, Walker S. Comparison of linear intrascan and interscan dynamic ranges of orbitrap and ion-mobility time-of-flight mass spectrometers. Rapid Comm. Mass Spectrom. 2017;31(22):1915–1926.
- Kaufmann A, Walker S. Extension of the Q orbitrap intrascan dynamic range by using a dedicated customized scan. Rapid Commun Mass Spectrom. 2016;30(8):1087–1095.
- Zubarev RA, Makarov A. Orbitrap mass spectrometry. Anal Chem. 2013;85(11):5288–5296.
- Werner T, Sweetman G, Savitski MF, et al. Ion coalescence of neutron encoded TMT 10-plex reporter ions. Anal Chem. 2014;86:3594–3601.
- Erickson BK, Rose CM, Braun CR, et al. A strategy to combine sample multiplexing with targeted proteomics assays for high-throughput protein signature characterization. Mol Cell. 2017;65:361–370.
- Bailey DJ. Instant spectral assignment for advanced decision tree-driven mass spectrometry. Pnas. 2012;109(22):8411–8416.
- Graumann J, Scheltema RA, Zhang Y, et al. A framework for intelligent data acquisition and real-time database searching for shotgun proteomics. Mol. Cell. Prot. 2012;11(3): M111.013185.
- Hendriks IA, Akimov V, Blagoev B, et al. MaxQuant.Live enables enhanced selectivity and identification of peptides modified by endogenous SUMO and ubiquitin. J. Prot. Res. 2021;20(4): 2042–2055.
- Furtwangler B, Uresin N, Motamedchaboki K, et al. Real-time search assisted acquisition on a tribrid mass spectrometer improves coverage in a multiplexed single cell proteomics. bioRxiv. 2021;08(16):456445.
- Braun CR, Bird GH, Wuhr M, et al. Generation of multiple reporter ions from a single isobaric reagent increases multiplexing capacity for quantitative proteomics. Anal Chem. 2016;87(19):9855–9863.