
Abstract
Patients with early-stage triple-negative breast cancer (TNBC) with residual invasive disease after neoadjuvant therapy have a high risk of recurrence even with neoadjuvant and adjuvant treatment with pembrolizumab. Sacituzumab govitecan, a Trop-2-directed antibody–drug conjugate with a topoisomerase I inhibitor payload, improved progression-free survival (PFS) and overall survival (OS) versus chemotherapy in patients with pre-treated metastatic TNBC. Moreover, preclinical data suggest that topoisomerase I inhibitors may enhance the effects of immune checkpoint inhibitors through activation of the cGAS-STING pathway. Here we describe the international randomized phase III AFT-65/ASCENT-05/OptimICE-RD trial, which evaluates the efficacy and safety of sacituzumab govitecan plus pembrolizumab versus treatment of physician's choice (pembrolizumab ± capecitabine) among patients with early-stage TNBC with residual invasive disease after neoadjuvant therapy.
Clinical Trial Registration: NCT05633654 (ClinicalTrials.gov)
Other Study ID Number(s): Gilead Study ID: GS-US-595-6184
Registration date: 1 December 2022
Study start date: 12 December 2022
Recruitment status: Recruiting
Plain language summary
AFT-65/ASCENT-05/OptimICE-RD is an ongoing clinical trial that is testing a new treatment combination for patients with stage II or III triple-negative breast cancer (TNBC). Stage II–III means the cancer is confined to the breast and/or nearby lymph nodes and can be surgically removed. However, there remains a risk that the cancer could recur after surgery. To reduce this risk, patients with stage II–III TNBC receive anti-cancer medication before and after surgery. For some patients, receipt of anti-cancer medication before surgery produces a pathologic complete response (pCR), meaning there is no observable cancer left behind at surgery. Patients with a pCR have a lower risk of recurrence than patients with residual disease.
The AFT-65/ASCENT-05/OptimICE-RD trial includes people with stage II-III TNBC who have residual cancer after completing their course of pre-surgery anti-cancer medication. All participants have any remaining cancer in their breast and/or lymph nodes removed surgically, after which they are randomly assigned to receive one of two treatments. The experimental therapy consists of pembrolizumab along with a medication called sacituzumab govitecan, which kills cancer cells directly and may strengthen the anti-cancer immune response. Pembrolizumab strengthens the anti-cancer immune response, so the hypothesis of this trial is that the two medications will be more effective together. The control therapy consists of pembrolizumab, alone or in combination with a chemotherapy medication called capecitabine, which is the current standard of care. To study the effectiveness of each treatment, the researchers are following up with all participants to learn if and when their breast cancer returns.
Background
Patients with stage II-III triple-negative breast cancer (TNBC) with residual invasive disease after neoadjuvant therapy have a high risk of recurrence with current standard-of-care adjuvant therapy regimens.
Trop-2 is a transmembrane protein that is highly expressed on epithelial cancers, including TNBC.
Sacituzumab govitecan is an antibody–drug conjugate consisting of an anti-Trop-2 antibody conjugated to SN-38 (the active metabolite of the DNA topoisomerase I inhibitor irinotecan) via a hydrolizable linker. This formulation facilitates release of SN-38 intracellularly as well as outside of cancer cells in the tumor microenvironment, creating a bystander killing effect.
Preclinical evidence suggests that sacituzumab govitecan and anti-PD-1/PD-L1 agents exert synergistic activity. DNA-damaging agents activate cGAS-STING signaling, which results in recruitment of anti-tumor immune cells through type I interferon signaling. Immune cell activity is enhanced by the immune checkpoint inhibitor.
AFT-65/ASCENT-05/OptimICE-RD Trial
The randomized, open-label, phase III AFT-65/ASCENT-05/OptimICE-RD trial investigates the combination of sacituzumab govitecan plus pembrolizumab versus treatment of physician's choice (TPC; pembrolizumab alone or in combination with capecitabine) in patients with stage II-III TNBC with residual invasive disease in the breast or lymph nodes after completion of neoadjuvant therapy.
Patients must be at least 18 years old with histologically confirmed TNBC, clinical stage T1, N1-2 or T2-4, N0-2.
Patients must have received at least 6 cycles of neoadjuvant anthracycline and/or taxane-containing chemotherapy with or without an immune checkpoint inhibitor.
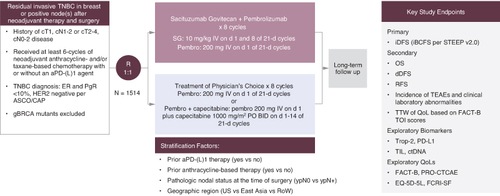
Approximately 1514 patients will be randomized 1:1 to receive sacituzumab govitecan 10 mg/kg IV on day 1 and day 8, every 21 days for 8 cycles, plus pembrolizumab 200 mg IV on day 1 every 21 days for 8 cycles, or TPC, which consists of pembrolizumab 200 mg IV on day 1 every 21 days for 8 cycles with or without capecitabine 1000 mg/m2 orally twice daily on day 1–14 every 21 days for 8 cycles.
The primary end point is iDFS. Secondary end points include overall survival, recurrence-free survival and safety and tolerability.
Exploratory end points include analysis of Trop-2 and PD-L1 expression on tumor tissue and correlation with clinical outcomes.
Patient-reported outcomes (PROs) include the FACT-B, EQ-5D-5L and PRO-CTCAE.
Conclusion
Results from the randomized phase III AFT-65/ASCENT-05/OptimICE-RD trial will provide efficacy and safety data for sacituzumab govitecan plus pembrolizumab versus pembrolizumab (alone or in combination with capecitabine) that could lead to a novel, more effective therapeutic combination to prevent disease recurrence in patients with stage II-III TNBC with residual invasive disease after the completion of neoadjuvant therapy.
1. Background & rationale
Worldwide, breast cancer is the most common type of invasive cancer diagnosed in women as well as the leading cause of cancer-related death [Citation1]. Approximately 10–15% of patients with breast cancer are diagnosed with triple-negative breast cancer (TNBC), which is negative for estrogen receptor (ER), progesterone receptor (PR) and HER2 expression [Citation2–4]. Compared with other breast cancer subtypes, patients with early-stage TNBC are more likely to experience distant recurrence within 5 years of diagnosis [Citation5,Citation6]. Patients with TNBC also have the worst overall survival (OS) and breast cancer-specific survival (BCSS) outcomes [Citation7,Citation8].
Neoadjuvant systemic therapy is a cornerstone of treatment for patients with stage II-III TNBC. The current standard of care consists of neoadjuvant pembrolizumab with multi-agent chemotherapy, followed by adjuvant pembrolizumab [Citation9]. This regimen was investigated in the randomized phase III KEYNOTE-522 trial, in which participants with newly diagnosed stage II-III TNBC were randomized in a 2:1 ratio to receive pembrolizumab or placebo throughout the study. Initially, participants received four cycles of neoadjuvant pembrolizumab or placebo in combination with carboplatin and paclitaxel, followed by four cycles of pembrolizumab or placebo in combination with doxorubicin and cyclophosphamide. After surgery, patients received up to nine cycles of adjuvant pembrolizumab or placebo. The co-primary end points were pathologic complete response (pCR) and event-free survival (EFS) [Citation10]. At the third interim analysis, the pCR rate was 63.0% (95% CI: 59.5–66.4%) in the pembrolizumab group versus 55.6% (95% CI: 50.6–60.6%) in the placebo group [Citation11]. After a median follow-up of 63.1 months, the 5-year EFS was 81.3% (95% CI: 78.4–83.9%) in the pembrolizumab group versus 72.3% (95% CI: 67.5–76.5%) in the placebo group (hazard ratio [HR] = 0.63; 95% CI: 0.49–0.81) [Citation12].
1.1. The importance of pCR & the challenge of residual invasive disease
Patients with early-stage TNBC who achieve a pCR to neoadjuvant therapy have significantly better survival outcomes than patients with residual invasive disease at surgery [Citation13,Citation14]. Unfortunately, a substantial percentage of patients with residual invasive disease experience recurrence despite standard-of-care adjuvant regimens. This was demonstrated by a prespecified exploratory analysis of KEYNOTE-522, which evaluated EFS outcomes based on whether participants achieved a pCR. Among participants who achieved a pCR, the 5-year EFS was 92.2% in the pembrolizumab group and 88.2% in the placebo group (HR = 0.65; 95% CI: 0.39–1.08). EFS events were far more frequent in participants with residual invasive disease, in whom the 5-year EFS was 62.6% in the pembrolizumab group and 52.3% in the placebo group (HR = 0.72; 95% CI: 0.54–0.96) [Citation12].
Currently, the US FDA has approved adjuvant pembrolizumab only for patients with stage II-III TNBC who received neoadjuvant pembrolizumab with multi-agent chemotherapy irrespective of pCR [Citation15]. Patients with residual invasive disease after receipt of neoadjuvant chemotherapy (without an immune checkpoint inhibitor) may receive adjuvant capecitabine. This regimen was established by the randomized phase III CREATE-X trial, which enrolled patients with early-stage HER2-negative breast cancer with residual invasive disease after the completion of neoadjuvant chemotherapy (containing an anthracycline, taxane, or both), before pembrolizumab was approved. Participants were randomized 1:1 to receive six to eight cycles of adjuvant capecitabine or no further systemic therapy (aside from adjuvant endocrine therapy in patients with hormone receptor-positive disease). Among the subgroup of 286 participants with TNBC, the 5-year disease-free survival (DFS) was 69.8% in the capecitabine group versus 56.1% in the control group (HR = 0.58; 95% CI: 0.39–0.87), and the 5-year OS was 78.8% in the capecitabine group versus 70.3% in the control group (HR = 0.52; 95% CI: 0.30–0.90) [Citation16]. We do not yet know if capecitabine improves outcomes using more modern regimens including pembrolizumab as capecitabine was not used in KEYNOTE-522. A limited sensitivity analysis of patients who took adjuvant capecitabine on KEYNOTE-522 did not demonstrate a benefit, but these data are not conclusive [Citation17].
Altogether, findings from prior studies demonstrate that roughly a third of patients with stage II-III TNBC with residual invasive disease after neoadjuvant systemic therapy will experience disease recurrence within 5 years despite receipt of adjuvant capecitabine or pembrolizumab. Escalation of adjuvant therapy may help to improve recurrence and survival outcomes in this patient population.
Aside from capecitabine and pembrolizumab, the PARP inhibitor olaparib is available for a subset of early-stage TNBC patients. Pathogenic germline variants in BRCA1 or BRCA2 are observed in approximately 15% of TNBC patients [Citation18–21]. Adjuvant olaparib is available for early-stage TNBC patients with a germline BRCA1 or BRCA2 mutation and residual invasive disease after the completion of neoadjuvant chemotherapy. FDA approval was based on the results of the randomized phase III OlympiA trial, which enrolled patients with high-risk early-stage HER2-negative breast cancer and a pathogenic germline variant in BRCA1 or BRCA2 and who had completed definitive local therapy and neoadjuvant or adjuvant chemotherapy. Participants were randomized 1:1 to receive adjuvant olaparib or placebo for 1 year. After a median follow-up of 2.5 years, the 3-year invasive disease-free survival (iDFS) was 85.9% in the olaparib group versus 77.1% in the placebo group (HR = 0.58; 95% CI: 0.41–0.82; p < 0.001) [Citation22]. After a median follow-up of 3.5 years, the median OS was significantly improved in the olaparib versus the placebo group (HR = 0.68; 98.5% CI: 0.47–0.97; p = 0.009) [Citation23].
1.2. Sacituzumab govitecan
Trop-2 is a transmembrane glycoprotein that promotes cell proliferation, migration and invasion [Citation24,Citation25]. It is overexpressed in many solid tumor types, including breast cancer, where its expression correlates with poorer prognosis [Citation26,Citation27]. Sacituzumab govitecan is an antibody–drug conjugate (ADC) consisting of an anti-Trop-2 antibody conjugated via the cleavable CLA2 linker to SN-38, the active metabolite of the topoisomerase I inhibitor irinotecan. Upon binding to Trop-2-overexpressing cancer cells, the ADC is internalized and trafficked to lysosomes, where SN-38 is released by hydrolysis of the linker. The free SN-38 inhibits DNA topoisomerase I, leading to DNA damage and cancer cell apoptosis [Citation28–30]. Sacituzumab govitecan also exhibits bystander killing, both through diffusion of free SN-38 through the cell membrane after lysosomal degradation of the linker, and through hydrolysis of the linker outside of cancer cells within the tumor microenvironment [Citation31–33].
Sacituzumab govitecan is FDA approved for the treatment of patients with unresectable locally advanced or metastatic TNBC who have received two or more prior systemic therapies, with at least one in the metastatic setting [Citation34]. Approval was based on the results of the randomized phase III ASCENT trial, in which patients with metastatic TNBC who had received two or more prior lines of systemic therapy in the metastatic setting were randomized 1:1 to receive sacituzumab govitecan or single-agent chemotherapy of physician's choice (capecitabine, eribulin, vinorelbine, or gemcitabine). At the primary analysis, the median progression-free survival (PFS) was 5.6 months (95% CI: 4.3–6.3 months) with sacituzumab govitecan versus 1.7 months (95% CI: 1.5–2.6 months) with chemotherapy (HR = 0.41; 95% CI: 0.32–0.52; p < 0.0001). The median overall survival (OS) was 12.1 months (95% CI: 10.7–14.0 months) with sacituzumab govitecan versus 6.7 months (95% CI: 5.8–7.7 months) with chemotherapy (HR = 0.48; 95% CI: 0.38–0.59; p < 0.0001) [Citation35]. In an exploratory analysis, patients derived benefit from sacituzumab govitecan regardless of Trop-2 expression level [Citation36].
1.3. Rationale for combining sacituzumab govitecan with pembrolizumab
An accumulating body of preclinical evidence suggests that sacituzumab govitecan may enhance anti-tumor immunity. DNA-damaging agents like topoisomerase I inhibitors cause the release of genomic DNA fragments into the cytosol of cancer cells. Cytosolic DNA is detected by cGAS, which results in activation of the cGAS-STING pathway and concomitant upregulation of anti-tumor immunity through type I interferon (IFN) signaling [Citation37–39].
In mouse models of solid human tumors, the combination of irinotecan with an anti-PD-L1 antibody resulted in greater tumor control than either agent alone [Citation40,Citation41]. Mice treated with the combination had higher numbers of intra-tumoral CD8+ T cells and lower numbers of regulatory T cells (Tregs) compared with mice treated with either agent alone. Moreover, irinotecan increased MHC class I expression on tumor cells and PD-L1 expression on tumor cells and tumor-infiltrating immune cells [Citation41]. Altogether, these findings suggest that sacituzumab govitecan plus pembrolizumab may produce synergistic activity in breast cancer patients. The ongoing randomized phase II Saci-IO TNBC trial (NCT04468061) is investigating the safety and efficacy of this combination versus sacituzumab govitecan alone in patients with metastatic TNBC [Citation42].
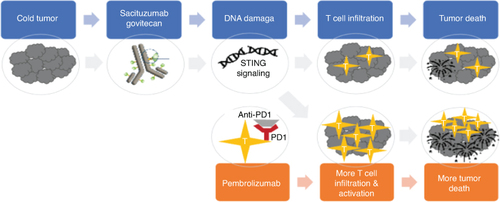
The trial described in this article, AFT-65/ASCENT-05/OptimICE-RD, will test the hypothesis that sacituzumab govitecan plus pembrolizumab will produce synergistic activity through increased induction of anti-tumor immunity (), and will thus be more effective than pembrolizumab (alone or with capecitabine) in patients with stage II-III TNBC with residual invasive disease after neoadjuvant therapy [Citation43].
Figure 1. Primary hypothesis: Sacituzumab govitecan induces DNA damage, which results in STING activation and increased anti-tumor immunity. Efficacy will be synergistically enhanced through combination with pembrolizumab.
2. Methods
2.1. Study design
AFT-65/ASCENT-05/OptimICE-RD is an open-label, international, multicenter, randomized phase III trial evaluating the efficacy and safety of sacituzumab govitecan plus pembrolizumab versus treatment of physician's choice (TPC) consisting of pembrolizumab alone or in combination with capecitabine in patients with early-stage TNBC with residual invasive disease after at least six cycles of anthracycline and/or taxane-based neoadjuvant chemotherapy with or without an immune checkpoint inhibitor. The primary objective is to compare invasive disease-free survival (iDFS) between patients who receive sacituzumab govitecan plus pembrolizumab versus TPC (). Approximately 1514 participants will be enrolled at 300 academic and community sites across the USA (including 65 AFT/Alliance sites; a full list of study sites is available on clinicaltrials.gov). Participants are stratified according to prior anti-PD-1 or anti-PD-L1 therapy (yes/no), prior anthracycline-based therapy (yes/no), pathologic nodal status at the time of surgery (ypN0/ypN+) and geographic region (USA/East Asia/Rest of World).
Figure 2. Study schema.
BID: Twice daily; d: Day; dDFS: Distant disease-free survival; ER: Estrogen receptor; iDFS: Invasive disease-free survival; IV: Intravenous; OS: Overall survival; PO: Orally; PR: Progesterone receptor; QoL: Quality of life; RFS: Recurrence-free survival; ROW: Rest of world; TEAEs: Treatment-emergent adverse events; TIL: Tumor-infiltrating lymphocytes; TNBC: Triple-negative breast cancer; TOI: Trial outcome index; TTW: Time to worsening.
2.2. Inclusion criteria
To be eligible for the study, participants must have a history of clinical stage T1, N1-2 or T2-4, N0-2 histologically confirmed TNBC, defined as ER and PR <10% and HER2-negative per ASCO/CAP 2018 guidelines [Citation44]. Patients must have received at least 6 cycles of neoadjuvant anthracycline and/or taxane-containing chemotherapy with or without an immune checkpoint inhibitor, with any amount of residual invasive disease in the breast or lymph nodes after completion of neoadjuvant therapy. For patients who have received pembrolizumab in the neoadjuvant setting, up to 3 cycles of adjuvant pembrolizumab administered with or without radiotherapy is allowed prior to study entry. Participants must have adequate excision and surgical removal of all clinically evident disease in the breast and/or lymph nodes, followed by appropriate radiation therapy. The presence of metastatic disease must be ruled out.
Participants must have recovered from surgery and any adverse events (AEs) due to previously administered neoadjuvant treatment or radiation therapy to grade ≤1 or baseline at the time of study entry, with the exception of any grade alopecia, grade ≤2 neuropathy, or grade ≤2 immune-mediated endocrinopathies due to neoadjuvant immune checkpoint inhibitor therapy (for this last criterion, participants must have been stable on endocrine therapy and able to continue immune checkpoint inhibitor in the neoadjuvant setting). Other prior immune-related AEs (irAEs) are allowed if they resolved, and the participant tolerated subsequent immune checkpoint inhibitor therapy without requiring chronic steroid therapy for irAE.
Prior to starting therapy, participants must have adequate hematologic counts without transfusion or growth factor support (hemoglobin ≥9 g/dl, absolute neutrophil count [ANC] ≥1500/mm3 and platelets ≥100,000/μl). Patients must also have adequate hepatic function (bilirubin ≤1.5 x upper limit of normal [ULN], aspartate aminotransferase [AST] and alanine aminotransferase [ALT] ≤2.5 x ULN, and serum albumin >3 g/dl) and creatinine clearance ≥30 ml/min as assessed by the Cockroft-Gault equation. Participants must have an international normalized ratio (INR)/prothrombin (PT) or activated partial thromboplastin time (aPTT) ≥1.5 x ULN unless the patient is receiving anticoagulation therapy.
Male and female participants of childbearing potential who engage in heterosexual intercourse must agree to use protocol-specified method(s) of contraception. Patients with HIV infection are eligible if they are on antiretroviral therapy and have a well-controlled HIV infection. Patients must be at least 18 years old with an Eastern Cooperative Oncology Group (ECOG) performance score of 0 or 1, and able to understand and provide written informed consent.
2.3. Exclusion criteria
Participants with stage IV (metastatic) breast cancer or any history of prior ipsilateral or contralateral invasive breast cancer are not eligible. Patients with prior ductal carcinoma in situ (DCIS) are eligible if they meet other inclusion criteria. Participants who received any prior neoadjuvant HER2-directed therapy, topoisomerase I inhibitors, ADCs containing a topoisomerase I inhibitor, or prior therapy with an agent directed to another stimulatory or coinhibitory T cell receptor (e.g., CTLA-4, OX-40, CD137) are not eligible. In addition, participants must not have known or severe (grade ≥3) hypersensitivity or allergy to sacituzumab govitecan, pembrolizumab, their metabolites or formulation excipients.
Additional exclusionary criteria include germline BRCA1/2 mutations (these patients should receive adjuvant olaparib as standard of care), evidence of recurrent disease following neoadjuvant therapy and surgery, or history of other malignancy within the last 5 years, except for appropriately treated carcinoma in situ of the cervix, non-melanoma skin carcinoma, stage I uterine cancer, or other non-breast malignancies with an outcome similar to those mentioned above. Participants must not have participated in a study with an investigational agent or device within 4 weeks prior to randomization, though participation in observational studies is allowed. Participants must not have had a myocardial infarction or unstable angina pectoris within 6 months prior to enrollment or a history of serious ventricular arrhythmia or left ventricular ejection fraction (LVEF) <50%. Participants must not have an active serious infection requiring anti-microbial therapy. Female participants with a positive serum pregnancy test or who are breastfeeding are not eligible.
2.4. Informed consent
The investigator is responsible for obtaining written informed consent from each individual participating in this study after adequate explanation of the rationale, methods, objectives and potential hazards of the study before undertaking any study-related procedures. The informed consent form (Appendix A) will inform participants about genomic testing and/or planned sample retention, as well as optional procedures relating to study-specific research.
In addition to the study-specific informed consent document, participants will be required to sign additional informed consent documents to provide additional samples for optional genomic and/or future research and to provide permission to use the remainder of their already collected biomarker and/or pharmacokinetic specimens for optional future research. The specimens collected for optional future and/or genomic research and/or residual biologic samples consented for optional future use will be destroyed no later than 15 years after the end of the study or per country requirements.
2.5. Screening & randomization procedures
Potential participants are screened within 28 days before enrollment in the study. Patients who meet all the inclusion criteria and none of the exclusion criteria return to the clinic within 28 days for randomization into the study. All participants who are randomized must receive their first dose of study treatment within 5 days of randomization.
Randomization is conducted centrally using interactive response technology (IRT) to minimize bias in the assignment of participants to treatment arms. For each participant, the IRT implements a randomization scheme and assigns a unique patient number. Randomized patients are stratified by receipt of prior anti-PD-(L)1 therapy (yes versus no), prior anthracycline-based therapy (yes versus no), pathologic nodal status at time of surgery (ypN0 versus npN+) and geographic region (USA versus East Asia versus Rest of World).
This is an open-label study, which means the investigators and participants are not blinded to treatment assignment. For the sponsor study management team, until the analysis of in-house study unblinding, the access to the treatment assignment is limited and well controlled, depending on the functional or study roles.
2.6. Interventions
Participants are randomized 1:1 to receive sacituzumab govitecan 10 mg/kg IV on day 1 and day 8, every 21 days for 8 cycles, plus pembrolizumab 200 mg IV on day 1 every 21 days for 8 cycles, or treatment of physician's choice (TPC), which consists of pembrolizumab 200 mg IV on day 1 every 21 days for 8 cycles. Treating investigators have the option of adding capecitabine 1000 mg/m2 orally twice daily on day 1–14 every 21 days for 8 cycles to pembrolizumab in the control arm only. Participants on both arms are treated until a maximum of 8 cycles, local or distant disease recurrence, unacceptable toxicity, investigator or treating physician decision, consent withdrawal or death.
Tumor tissue and blood biospecimens are collected for research purposes. Participants must provide representative formalin-fixed paraffin-embedded (FFPE) tumor specimen in tumor tissue blocks (preferred) or freshly sectioned unstained slides from both pretreatment diagnostic biopsy and from resected surgical specimen. Participants will also provide blood and plasma samples for research purposes at pre-dose on Cycle 1 Day 1, Cycle 3 Day 1, Cycle 5 Day 1, at the end of treatment and once per year thereafter if the participant had not experienced disease recurrence.
2.7. Concomitant medications
Premedication and prophylaxis are permitted while on study. Supportive medications, such as pain medications, entiemetics, antidiarrheal medications, transfusions and growth factor support are allowed at the investigator's discretion. Concomitant administration of inhibitors or inducers of UGT1A1 should be used with caution because of the potential to either increase (inhibitors) or decrease (inducers) the exposure to SN-38, the active metabolite of sacituzumab govitecan. Patients are prohibited from receiving live vaccines within 30 days of planned treatment start and while receiving study treatment. High-dose corticosteroids (≥10 mg prednisone or equivalent) are not allowed within 14 days prior to randomization.
2.8. Efficacy outcomes
The intention-to-treat (ITT) analysis set includes all randomized participants. This is the primary analysis set for all efficacy analyses, demographic characteristics, baseline assessments and quality of life assessments. The primary end point is iDFS, which is defined as the time from the date of randomization to invasive local, regional or distant recurrence, invasive contralateral breast cancer or death from any cause. This definition is called invasive breast cancer-free survival (IBCFS) in the updated STEEP 2.0 criteria [Citation45]. Secondary end points include OS, defined as the time from the date of randomization to death from any cause; distant disease-free survival (dDFS), defined as the time from the date of randomization to distant recurrence, second primary invasive cancer, or death from any cause; recurrence-free survival (RFS), defined as the time from the date of randomization to invasive local, regional, or distant recurrence, or death from any cause.
Disease status evaluations are performed every 3 months during the first 2 years after randomization, every 6 months during Years 3 and 4 after randomization and annually starting in Year 5 and beyond. Disease status evaluations will be based on all available clinical assessments per local standards that may include (but are not limited to) physical examination, radio-imaging procedures (bone scan, computed tomography [CT], magnetic resonance imaging [MRI], positron emission tomography [PET]) and biopsy. Clinically suspected recurrent disease must be confirmed pathologically, if possible. If pathological confirmation is not feasible due to location of the disease, then evidence of recurrence must be proven radiographically. In the event that a participant passes away without confirmation of disease recurrence, and in whom disease recurrence is suspected, then efforts should be made to obtain an autopsy report.
Participants will be followed up for survival status as part of their disease status evaluations. A participant who experiences a confirmed disease recurrence event will no longer follow the disease status evaluation schedule and will instead be followed annually or more frequently for OS, subsequent anti-cancer therapy, and any new relapse or disease progression events, unless the participant explicitly indicates their desire to forego survival follow-up in writing to their study investigator. Participants will be followed until death or withdrawal of consent, whichever occurs first. For participants who are lost to follow-up, study staff may use public records (i.e., public health records) to obtain information about survival status where allowable by local regulation.
2.9. Adverse events
The safety analysis set includes all participants who received at least one dose of any study treatment. The treatment-emergent period is defined as the time from the first dose of study treatment to the earlier of 30 days following the last dose of study treatment or the initiation of subsequent anti-cancer therapy. All safety data during the treatment-emergent period will be summarized by treatment arm. The safety variables to be analyzed include exposure to study treatment, AEs, deaths, clinical laboratory test results (hematology and chemistry), physical examination and vital sign measurements. AEs will be assessed in accordance with the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE), version 5.0.
Clinical and laboratory AEs will be coded using the current version of the Medical Dictionary for Regulatory Activities. System organ class, high-level group term, high-level term, preferred term and lower-level term will be attached to the clinical database. For each AE, the number and percentage of patients who experience at least one occurrence of the event will be summarized. The number and percent of participants with treatment-emergent adverse events (TEASs) will be summarized according to severity grade and drug relationship, as well as categorized by system organ class and preferred term. Summaries, listings, datasets, or patient narratives may be provided, as appropriate, for those patients who die, discontinue treatment due to an AE, or who experience a severe AE or serious AE (SAE).
Exploratory biomarker analyses include testing for Trop-2 and PD-L1 expression on tumor tissue from the pretreatment diagnostic core biopsy or surgical specimen. In the KEYNOTE-355 trial, the combination of pembrolizumab with chemotherapy resulted in significantly longer OS than chemotherapy alone only among patients with metastatic TNBC with a PD-L1 combined positive score (CPS) ≥10 [Citation46], though PD-L1 was not a biomarker predictor of outcome in the KEYNOTE-522 trial. Tumor samples in the study will also be evaluated for the percentage of tumor-infiltrating lymphocytes (TILs). Increased TILs have been associated with better survival outcomes among patients with early-stage TNBC with residual invasive disease after neoadjuvant chemotherapy [Citation47]. Blood samples will be evaluated for circulating tumor DNA (ctDNA), which has been associated with distant recurrence in patients with early-stage breast cancer [Citation48–50].
The study also includes several patient-reported outcome (PRO) instruments, including the Functional Assessment of Cancer Therapy-Breast (FACT-B), EuroQol (EQ)-5D-5L and the Patient-Reported Outcome-Common Terminology Criteria for Adverse Events (PRO-CTCAE). The EQ-5D-5L is a six-item generic patient-reported preference-based instrument designed to assess the health status of patients using a descriptive system and visual analog scale [Citation51]. The FACT-B is a 37-item instrument that assesses the symptoms and impacts associated with breast cancer from the patient perspective, across five domains: physical, social/family, emotional and functional well-being, along with a breast cancer-specific subscale (BCS) [Citation52]. The PRO-CTCAE is a patient-reported instrument that evaluates the symptom attributes of frequency, severity, interference and presence or absence of AEs [Citation53].
2.10. Study discontinuation & poststudy care
If a participant discontinues study treatment (e.g., due to an AE), then every attempt should be made to keep the participant in the study and continue to perform the required study-related follow-up and procedures. If this is not possible or acceptable to the participant or investigator, the participant may be withdrawn from the study. A participant who discontinues study treatment will be asked to return to the investigational site within 30 (± 7 days) of stopping study treatment to attend an end-of-treatment visit. A participant who discontinues from the study will have the reason of discontinuation captured.
Following study treatment completion or early discontinuation, participants may receive other breast cancer-related care under the discretion of their primary treating physician. Subsequent breast cancer therapies should be recorded on the subsequent anti-cancer therapy electronic case report form.
2.11. Statistical methods
The study will enroll 1514 participants. Strategies to achieve this enrollment target include regular investigator meetings, newsletters sent to participating sites, discussion of the trial at AFT meetings with study investigators and discussion at monthly Alliance Breast Steering Committee meetings.
Analysis of the primary end point of iDFS for comparing the efficacy of sacituzumab govitecan plus pembrolizumab versus TPC will be performed using the log-rank test stratified by randomization stratification factors. The study is planned to have one superiority interim analysis of iDFS performed on the ITT population when approximately a total of 254 events (75% information fraction) have occurred; at this point, the primary hypothesis will be tested at a one-sided significance level of 0.96%. The interim analysis is projected to occur approximately 40 months after the first participant is randomized.
For the primary end point of iDFS, 339 events are needed to detect an HR of 0.72 with 85% power at a one-sided significance level of 2.5% using a log-rank test, given one interim efficacy analysis at 75% information fraction. Assuming a 3-year iDFS of 72% for the TPC arm with a total of 1514 participants, and a negligible annual dropout rate of 2%, it is estimated that the final iDFS analysis will occur approximately 49 months after the first participant is randomized.
The HR will be presented with corresponding 95% CIs using a Cox proportional hazard regression model stratified by randomization stratification factors. iDFS will be summarized by treatment arm using Kaplan–Meier estimates, which include median and the proportion of event-free patients at benchmark time points, such as 3 and 5 years. The key secondary end point OS will be formally tested in the hierarchical setting, only when/if the analysis of the primary end point iDFS is significant. Analyses of OS, dDFS and RFS will be performed using a similar methodology as for the analysis of iDFS described above. If the null hypothesis with respect to the key secondary end point is not rejected at any of the interim analyses, the final analysis of OS will be conducted on the ITT population when approximately 244 deaths have occurred across both treatment arms (~91 months after the first participant is randomized).
For safety assessments, continuous variables will be summarized using descriptive statistics (n, mean, median, standard deviation, standard error and range). Categorical variables will be summarized using frequencies and percentages. No formal statistical testing is planned.
For each biomarker assessed in tumor tissue and blood (Trop-2, PD-L1, ctDNA), the baseline level, absolute level and change from baseline level over time will be summarized using descriptive statistics for each biomarker at the sample collection timepoint by treatment arm, as appropriate. Exploratory analyses may be performed to evaluate the association of each biomarker or combination of biomarkers with clinical outcomes in the ITT analysis set.
3. Ethical considerations
The study will be performed in accordance with the ethical principles of the Declaration of Helsinki and conducted in adherence to the study protocol. All trial participants must provide written informed consent to their study investigator before receiving any study medications or interventions.
3.1. Institutional review board/independent ethics committee approval
The investigator at each site (or Gilead, as appropriate according to local regulations) must submit the study protocol, informed consent form, and any accompanying material to be provided to each trial participant (such as advertisements, patient information sheets, or descriptions of the study used to obtain informed consent) to an institutional review board (IRB) or independent ethics committee (IEC). The investigator will not begin any study patient activities until approval from the IRB/IEC has been documented and provided as a letter to the investigator. Before implementation, the investigator will submit to and receive documented approval from the IRB/IEC for any modifications made to the protocol or any accompanying material to be provided to the patient after initial IRB/IEC approval, with the exception of those necessary to reduce immediate risk to study participants.
The trial was approved by the IRB at each of the 111 currently participating sites in USA. These sites are listed on ClinicalTrials.gov and the Gilead website. Gilead is currently opening additional sites outside the U.S. IRB/IEC approval will be obtained prior to trial initiation at each site.
3.2. Data monitoring committee
A multidisciplinary data monitoring committee (DMC) consisting of non-Gilead personnel will review the progress of the study, perform reviews of safety data at intervals of approximately 6 months after the first patient has enrolled (initial safety evaluation will be performed after the first 60 patients have enrolled or 6 months after the first patient has enrolled, whichever occurs later), perform review of the iDFS efficacy interim analysis, and provide recommendation to Gilead whether the nature, frequency and severity of AEs associated with study treatment warrant the early termination of the study in the best interests of the patient, whether the study should continue as planned, or whether the study should continue with modifications. The DMC may also provide recommendations as needed regarding study design. The membership, conduct, role and responsibilities and meeting schedule of the DMC will be defined by a mutually agreed charter.
While the DMC will be asked to advise Gilead regarding future conduct of the study (including possible early study termination), Gilead retains final decision-making authority on all aspects of the study. If the DMC recommends stopping the study, a Gilead Oversight Committee will be unblinded to confirm the DMC recommendation.
3.3. Participant confidentiality
The investigator is responsible for maintaining the anonymity of each patient and ensuring that their identity is protected from unauthorized parties. Only an identification code and any other unique identifier(s) as allowed by local law (such as year of birth) will be recorded on any form or biological sample submitted to Gilead, the IRB, IEC, or any laboratory. Laboratory specimens will be labeled in such a way as to protect patient identity while allowing the results to be linked to the proper patient. The investigator is responsible for maintaining a screening log with details for all patients screened and enrolled in the study, in accordance with site procedures and regulations. Patient data will be processed in accordance with all applicable regulations.
3.4. Data integrity
An electronic case report form (eCRF) casebook will be completed by an authorized study personnel member whose training for this function is completed in the electronic data capture system unless otherwise directed. The eCRF casebook will only capture the data required per the protocol schedule of events and procedures, unless collected by a non-electronic data capture vendor system (e.g., central laboratory). Data entry must be performed in accordance with the CRF Completion Guidelines provided by Gilead. Subsequent to data entry, a study monitor may perform source data verification to identify potential data discrepancies. The site investigator, site coordinator or other designee is responsible for responding to queries from Gilead personnel regarding potential data discrepancies in a timely manner, within the system, either by confirming the data as correct or updating the original entry, and providing the reason for the update. Original entries as well as any changes to data fields are stored in the audit trail of the system.
3.5. Data storage
The investigator must maintain a complete record of clinical study documents for at least 2 years or according to local laws (whichever is longer) after the last approval of a marketing application in an International Council for Harmonization (ICH) region and until there are no planned or pending marketing application in an ICH region. If no application is filed or if the application is not approved for such indication, the investigator must maintain a record for 2 years after the investigation is discontinued and regulatory authorities have been notified. The investigator may be required to retain documents longer than 2 years if specified by regulatory requirements, local regulations, or by an agreement with Gilead. The investigator must notify Gilead before destroying any clinical study records.
Trial data are being collected by Gilead in a central database. Gilead will have access to the final dataset. The members of the steering committee will have access to the final data analyses.
3.6. Inspections/audits
Representatives of regulatory authorities or Gilead may conduct inspections or audits of the study. The investigator agrees to provide to representatives of a regulatory agency or Gilead access to records, facilities and personnel for the effective conduct of any inspection or audit.
4. Study report & publications
A clinical study report will be prepared and provided to the regulatory agency within 1 year of the last visit date for the last patient enrolled. Gilead will ensure that the report meets the standards established in the ICH Guideline for Structure and Content of Clinical Study Reports. Investigators in this study may communicate, orally present or publish the results of this study in scientific journals or other scholarly media only after the following conditions have been met: the results of the study in their entirety have been publicly disclosed by or with the consent of Gilead in an abstract, manuscript or presentation, or the study has been completed at all investigational sites for at least 2 years; the investigator will submit to Gilead any proposed publication or presentation along with the respective scientific journal or presentation forum at least 30 days before submission of the publication or presentation; no such communication, presentation, or publication will include Gilead's confidential information; the investigator will comply with Gilead's request to delete references to its confidential information (other than the study results) in any paper or presentation and agrees to withhold publication or presentation for an additional 60 days to obtain patent protection if deemed necessary. Individual patient data will not be shared.
5. Study modifications
Modifications to the study protocol, except those intended to reduce immediate risk to study participants, may be made only by Gilead. The investigator must submit all protocol modifications of the IRB/IEC in accordance with local requirements and receive documented IRB/IEC approval before modifications can be implemented.
6. Study design & oversight
The trial was designed by the principal investigator (Sara M Tolaney) in coordination with the Alliance Foundation Trials (AFT) leadership team, the trial steering committee and the trial sponsor (Gilead). Within the USA, the trial is run collaboratively by AFT and Gilead along with the steering committee (Sara M Tolaney, Simonetta Mocci, Ann H Partridge, Lisa A Carey, Angela DeMichele, Toshimi Takano, William Sikov, See Phan, Filipa Lynce, Heather Anne Parsons, Cesar Augusto Santa-Maria, Clinton Yam, Gabrielle Betty Rocque, and Hope S Rugo). Outside the USA, the trial is overseen by Gilead and the steering committee.
7. Intended use of professional writers
Publications resulting from this trial may be prepared with the assistance of Timothy K Erick, PhD, a full-time medical writer employed by Dana-Farber Cancer Institute. Editing and submission assistance may be provided by Kaitlyn T Bifolck and/or Valerie H Goldstein, full-time editors employed by Dana-Farber Cancer Institute.
8. Conclusion
For patients with stage II-III TNBC with residual invasive disease after neoadjuvant therapy, the risk of recurrence and death remains high even with pembrolizumab. In addition to its direct anti-tumor effects, sacituzumab govitecan may potentiate the activity of pembrolizumab by enhancing anti-tumor immunity through activation of the cGAS-STING pathway. The open-label, international, multicenter, randomized phase III AFT-65/ASCENT-05/OptimICE-RD trial described in this article will determine whether the combination of sacituzumab govitecan and pembrolizumab can improve iDFS compared with pembrolizumab, alone or in combination with capecitabine, in patients with stage II-III TNBC with residual invasive disease after neoadjuvant therapy. Positive results may lead to a new treatment option that improves recurrence and survival outcomes in these patients, and thus addresses an important unmet need.
Author contributions
Conceptualization: SM Tolaney, LA Carey, AH Partridge, S Mocci and SW Sun. Funding acquisition: SM Tolaney, S Mocci and SW Sun. Investigation: SM Tolaney, A DeMichele, T Takano, HS Rugo, C Perou, F Lynce, HA Parsons, CA Santa-Maria, GB Rocque, AH Partridge and LA Carey. Methodology: SM Tolaney, S Mocci, SW Sun. Project administration: SM Tolaney, S Mocci, SW Sun, F Lynce and W Yao. Supervision: SM Tolaney, A DeMichele, T Takano, HS Rugo, C Perou, F Lynce, HA Parsons, CA Santa-Maria, GB Rocque, AH Partridge, LA Carey, S Mocci, SW Sun, W Yao.
Financial disclosure
The trial is funded by Gilead Sciences. Contact information: Gilead Clinical Study Information Center, 1-833-445-3230; [email protected]
SM Tolaney reports consulting or advisory role Novartis, Pfizer (SeaGen), Merck, Eli Lilly, AstraZeneca, Genentech/Roche, Eisai, Sanofi, Bristol Myers Squibb, CytomX Therapeutics, Daiichi Sankyo, Gilead, Zymeworks, Zentalis, Blueprint Medicines, Reveal Genomics, Sumitovant Biopharma, Umoja Biopharma, Artios Pharma, Menarini/Stemline, Aadi Bio, Bayer, Incyte Corp, Jazz Pharmaceuticals, Natera, Tango Therapeutics, Systimmune, eFFECTOR, Hengrui USA, Cullinan Oncology, Circle Pharma, Arvinas, BioNTech, Johnson&Johnson, research funding from Genentech/Roche, Merck, Exelixis, Pfizer, Lilly, Novartis, Bristol Myers Squibb, Eisai, AstraZeneca, Gilead, NanoString Technologies, Seattle Genetics, OncoPep, Jazz Pharmaceuticals; and travel support from Eli Lilly, Sanofi, Gilead, Jazz Pharmaceuticals, Pfizer. FL reports participation on a steering committee for Gilead; and research funding from Merck and Gilead. GBR reports research support from Genentech and Pfizer; and consulting for Pfizer, Gilead and Armada. AH Partridge reports royalties from Wolters Kluwer (UpToDate). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing disclosure
This manuscript was prepared with the assistance of TK Erick and KT Bifolck. Both are full-time employees of Dana-Farber Cancer Institute, with expertise in medical writing and editing/publishing, respectively.
Ethical conduct of research
The protocol received approval from the appropriate institutional review board. The study will be performed in accordance with the ethical principles of the Declaration of Helsinki and conducted in adherence to the study protocol. All trial participants will provide written informed consent before receiving any study medications or interventions.
Acknowledgments
We thank the trial participants and their caregivers for helping us realize the possibilities of this research. We are also grateful to the dedicated clinical trial investigators and their devoted team members for participating in the AFT-65/ASCENT-05/OptimICE-RD trial. We thank Alliance Foundation Trials (AFT) for their collaboration on this project.
Competing interests disclosure
The authors have no competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
- Desantis CE, Fedewa SA, Goding Sauer A, et al. Breast cancer statistics, 2015: convergence of incidence rates between black and white women. CA Cancer J Clin. 2016;66(1):31–42. doi:10.3322/caac.21320
- Kohler BA, Sherman RL, Howlader N, et al. Annual Report to the Nation on the Status of Cancer, 1975–2011, Featuring Incidence of Breast Cancer Subtypes by Race/Ethnicity, Poverty, and State. J Natl Cancer Inst. 2015;107(6):djv048. doi:10.1093/jnci/djv048
- Plasilova ML, Hayse B, Killelea BK, et al. Features of triple-negative breast cancer: analysis of 38,813 cases from the national cancer database. Medicine (Baltimore). 2016;95(35):e4614. doi:10.1097/MD.0000000000004614
- Lin NU, Vanderplas A, Hughes ME, et al. Clinicopathologic features, patterns of recurrence, and survival among women with triple-negative breast cancer in the National Comprehensive Cancer Network. Cancer 2012;118(22):5463–5472. doi:10.1002/cncr.27581
- Dent R, Trudeau M, Pritchard KI, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429–4434. doi:10.1158/1078-0432.CCR-06-3045
- Li X, Yang J, Peng L, et al. Triple-negative breast cancer has worse overall survival and cause-specific survival than non-triple-negative breast cancer. Breast Cancer Res Treat. 2017;161(2):279–287. doi:10.1007/s10549-016-4059-6
- Howlader N, Cronin KA, Kurian AW, Andridge R. Differences in breast cancer survival by molecular subtypes in the United States. Cancer Epidemiol Biomarkers Prev. 2018;27(6):619–626. doi:10.1158/1055-9965.EPI-17-0627
- Korde LA, Somerfield MR, Hershman DL, et al. Use of immune checkpoint inhibitor pembrolizumab in the treatment of high-risk, early-stage triple-negative breast cancer: ASCO Guideline Rapid Recommendation Update. J Clin Oncol. 2022;40(15):1696–1698. doi:10.1200/JCO.22.00503
- Schmid P, Cortes J, Pusztai L, et al. Pembrolizumab for early triple-negative breast cancer. N Engl J Med. 2020;382(9):810–821. doi:10.1056/NEJMoa1910549
- Oncologic Drugs Advisory Committee. BLA 125514 Supplement-089 Drug name: Pembrolizumab. Applicant: Merck Sharp & Dohme Corp. Combined FDA and Applicant ODAC Briefing Document. 2021.
- Schmid P, Cortés J, Dent RA, et al. LBA18 Pembrolizumab or placebo plus chemotherapy followed by pembrolizumab or placebo for early-stage TNBC: updated EFS results from the Phase III KEYNOTE-522 study. Ann Oncol. 2023;34:S1257. doi:10.1016/j.annonc.2023.10.008
- Cortazar P, Zhang L, Untch M, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 2014;384(9938):164–172. doi:10.1016/S0140-6736(13)62422-8
- Shepherd JH, Ballman K, Polley MC, et al. CALGB 40603 (Alliance): long-term outcomes and genomic correlates of response and survival after neoadjuvant chemotherapy with or without carboplatin and bevacizumab in triple-negative breast cancer. J Clin Oncol. 2022;40(12):1323–1334. doi:10.1200/JCO.21.01506
- U.S. Food and Drug Administration. FDA approves pembrolizumab for high-risk early-stage triple-negative breast cancer. 2021. www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pembrolizumab-high-risk-early-stage-triple-negative-breast-cancer
- Masuda N, Lee SJ, Ohtani S, et al. Adjuvant capecitabine for breast cancer after preoperative chemotherapy. N Engl J Med. 2017;376(22):2147–2159. doi:10.1056/NEJMoa1612645
- Schmid P CJ, Dent R, Pusztai L, et al. GS1-01 KEYNOTE-522 study of neoadjuvant pembrolizumab + chemotherapy vs placebo + chemotherapy, followed by adjuvant pembrolizumab vs placebo for early-stage TNBC: event-free survival sensitivity and subgroup analyses. Cancer Res. 2022;82(Suppl. 4):GS1–01.
- Engel C, Rhiem K, Hahnen E, et al. Prevalence of pathogenic BRCA1/2 germline mutations among 802 women with unilateral triple-negative breast cancer without family cancer history. BMC Cancer. 2018;18(1):265. doi:10.1186/s12885-018-4029-y
- Rhiem K, Zachariae S, Waha A, et al. Prevalence of pathogenic germline variants in women with non-familial unilateral triple-negative breast cancer. Breast Care (Basel). 2023;18(2):106–112. doi:10.1159/000528972
- Tung N, Lin NU, Kidd J, et al. Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J Clin Oncol. 2016;34(13):1460–1468. doi:10.1200/JCO.2015.65.0747
- Sharma P, Klemp JR, Kimler BF, et al. Germline BRCA mutation evaluation in a prospective triple-negative breast cancer registry: implications for hereditary breast and/or ovarian cancer syndrome testing. Breast Cancer Res Treat. 2014;145(3):707–714. doi:10.1007/s10549-014-2980-0
- Tutt ANJ, Garber JE, Kaufman B, et al. Adjuvant olaparib for patients with BRCA1- or BRCA2-mutated breast cancer. N Engl J Med. 2021;384(25):2394–2405. doi:10.1056/NEJMoa2105215
- Geyer CE Jr, Garber JE, Gelber RD, et al. Overall survival in the OlympiA Phase III trial of adjuvant olaparib in patients with germline pathogenic variants in BRCA1/2 and high-risk, early breast cancer. Ann Oncol. 2022;33(12):1250–1268. doi:10.1016/j.annonc.2022.09.159
- Shvartsur A, Bonavida B. Trop2 and its overexpression in cancers: regulation and clinical/therapeutic implications. Genes Cancer. 2015;6(3–4):84–105. doi:10.18632/genesandcancer.40
- Goldenberg DM, Stein R, Sharkey RM. The emergence of trophoblast cell-surface antigen 2 (TROP-2) as a novel cancer target. Oncotarget. 2018;9(48):28989–29006. doi:10.18632/oncotarget.25615
- Ambrogi F, Fornili M, Boracchi P, et al. Trop-2 is a determinant of breast cancer survival. PLOS ONE. 2014;9(5):e96993. doi:10.1371/journal.pone.0096993
- Lin H, Huang J-F, Qiu J-R, et al. Significantly upregulated TACSTD2 and Cyclin D1 correlate with poor prognosis of invasive ductal breast cancer. Exp Mol Pathol. 2013;94(1):73–78. doi:10.1016/j.yexmp.2012.08.004
- Starodub AN, Ocean AJ, Shah MA, et al. First-in-human trial of a novel anti-trop-2 antibody-SN-38 conjugate, sacituzumab govitecan, for the treatment of diverse metastatic solid tumors. Clin Cancer Res. 2015;21(17):3870–3878. doi:10.1158/1078-0432.CCR-14-3321
- Goldenberg DM, Cardillo TM, Govindan SV, et al. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody–drug conjugate (ADC). Oncotarget. 2015;6(26):22496–22512. doi:10.18632/oncotarget.4318
- Sharkey RM, Mcbride WJ, Cardillo TM, et al. Enhanced delivery of SN-38 to human tumor xenografts with an anti-Trop-2-SN-38 antibody conjugate (sacituzumab govitecan). Clin Cancer Res. 2015;21(22):5131–5138. doi:10.1158/1078-0432.CCR-15-0670
- Perrone E, Manara P, Lopez S, et al. Sacituzumab govitecan, an antibody–drug conjugate targeting trophoblast cell-surface antigen 2, shows cytotoxic activity against poorly differentiated endometrial adenocarcinomas in vitro and in vivo. Mol Oncol. 2020;14(3):645–656. doi:10.1002/1878-0261.12627
- Goldenberg DM, Sharkey RM. antibody–drug conjugates targeting TROP-2 and incorporating SN-38: a case study of anti-TROP-2 sacituzumab govitecan. MAbs. 2019;11(6):987–995. doi:10.1080/19420862.2019.1632115
- Kopp A, Hofsess S, Cardillo TM, et al. Antibody–drug conjugate sacituzumab govitecan drives efficient tissue penetration and rapid intracellular drug release. Mol Cancer Ther. 2023;22(1):102–111. doi:10.1158/1535-7163.MCT-22-0375
- U.S. Food and Drug Administration. FDA grants regular approval to sacituzumab govitecan for triple-negative breast cancer. 2021. www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-regular-approval-sacituzumab-govitecan-triple-negative-breast-cancer
- Bardia A, Hurvitz SA, Tolaney SM, et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. N Engl J Med. 2021;384(16):1529–1541. doi:10.1056/NEJMoa2028485
- Hurvitz S, Tolaney SM, Punie K, et al. Abstract GS3-06: Biomarker evaluation in the phase 3 ASCENT study of sacituzumab govitecan versus chemotherapy in patients with metastatic triple-negative breast cancer. Cancer Res. 2021;81(Suppl. 4):GS3–GS6. doi:10.1158/1538-7445.SABCS20-GS3-06
- Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215(5):1287–1299. doi:10.1084/jem.20180139
- Berger G, Marloye M, Lawler SE. Pharmacological modulation of the STING pathway for cancer immunotherapy. Trends Mol Med. 2019;25(5):412–427. doi:10.1016/j.molmed.2019.02.007
- Zhao J, Ma S, Xu Y, et al. In situ activation of STING pathway with polymeric SN38 for cancer chemoimmunotherapy. Biomaterials. 2021;268:120542. doi:10.1016/j.biomaterials.2020.120542
- Mckenzie JA, Mbofung RM, Malu S, et al. The effect of topoisomerase I inhibitors on the efficacy of T-cell-based cancer immunotherapy. J Natl Cancer Inst. 2018;110(7):777–786. doi:10.1093/jnci/djx257
- Iwai T, Sugimoto M, Wakita D, et al. Topoisomerase I inhibitor, irinotecan, depletes regulatory T cells and up-regulates MHC class I and PD-L1 expression, resulting in a supra-additive antitumor effect when combined with anti-PD-L1 antibodies. Oncotarget. 2018;9(59):31411–31421. doi:10.18632/oncotarget.25830
- Garrido-Castro AC, Keenan TE, Li T, et al. Saci-IO TNBC: randomized Phase II trial of sacituzumab govitecan (SG) +/- pembrolizumab in PD-L1– metastatic triple-negative breast cancer (mTNBC). J Clin Oncol. 2021;39(Suppl. 15):TPS1106. doi:10.1200/JCO.2021.39.15_suppl.TPS1106
- Tolaney SM, Demichele A, Takano T, et al. ASCENT-05/OptimICE-RD (AFT-65): Phase III, randomized, open-label study of adjuvant sacituzumab govitecan (SG) + pembrolizumab (pembro) vs pembro± capecitabine (cape) in patients (pts) with triple-negative breast cancer (TNBC) and residual disease after neoadjuvant therapy (NAT) and surgery. J Clin Oncol. 2023;41(Suppl. 16):TPS619. doi:10.1200/JCO.2023.41.16_suppl.TPS619
- Wolff AC, Hammond MEH, Allison KH, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J Clin Oncol. 2018;36(20):2105–2122. doi:10.1200/JCO.2018.77.8738
- Tolaney SM, Garrett-Mayer E, White J, et al. Updated Standardized Definitions for Efficacy End Points (STEEP) in adjuvant breast cancer clinical trials: STEEP Version 2.0. J Clin Oncol. 2021;39(24):2720–2731. doi:10.1200/JCO.20.03613
- Cortes J, Rugo HS, Cescon DW, et al. Pembrolizumab plus chemotherapy in advanced triple-negative breast cancer. N Engl J Med. 2022;387(3):217–226. doi:10.1056/NEJMoa2202809
- Luen SJ, Salgado R, Dieci MV, et al. Prognostic implications of residual disease tumor-infiltrating lymphocytes and residual cancer burden in triple-negative breast cancer patients after neoadjuvant chemotherapy. Ann Oncol. 2019;30(2):236–242. doi:10.1093/annonc/mdy547
- Radovich M, Jiang G, Hancock BA, et al. Association of circulating tumor DNA and circulating tumor cells after neoadjuvant chemotherapy with disease recurrence in patients with triple-negative breast cancer: preplanned secondary analysis of the BRE12-158 randomized clinical trial. JAMA Oncol. 2020;6(9):1410–1415. doi:10.1001/jamaoncol.2020.2295
- Magbanua MJM, Swigart LB, Wu HT, et al. Circulating tumor DNA in neoadjuvant-treated breast cancer reflects response and survival. Ann Oncol. 2021;32(2):229–239. doi:10.1016/j.annonc.2020.11.007
- Lipsyc-Sharf M, De Bruin EC, Santos K, et al. Circulating tumor DNA and late recurrence in high-risk hormone receptor-positive, human epidermal growth factor receptor 2-negative breast cancer. J Clin Oncol. 2022;40(22):2408–2419. doi:10.1200/JCO.22.00908
- Herdman M, Gudex C, Lloyd A, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10):1727–1736. doi:10.1007/s11136-011-9903-x
- Brady MJ, Cella DF, Mo F, et al. Reliability and validity of the Functional Assessment of Cancer Therapy-Breast quality-of-life instrument. J Clin Oncol. 1997;15(3):974–986. doi:10.1200/JCO.1997.15.3.974
- Basch E, Reeve BB, Mitchell SA, et al. Development of the National Cancer Institute's patient-reported outcomes version of the common terminology criteria for adverse events (PRO-CTCAE). J Natl Cancer Inst. 2014;106(9):dju244. doi:10.1093/jnci/dju244