
Abstract
We observed lack of clarity and consistency in end point definitions of large randomized clinical trials in diffuse large B-cell lymphoma. These inconsistencies are such that trials might, in fact, address different clinical questions. They complicate interpretation of results, including comparisons across studies. Problems arise from different ways to account for events occurring after randomization including absence of improvement in disease status, treatment discontinuation or the initiation of new therapy. We call for more dialogue between stakeholders to define with clarity the questions of interest and corresponding end points. We illustrate that assessing different end point rules across a range of plausible patient journeys can be a powerful tool to facilitate such a discussion and contribute to better understanding of patient-relevant end points.
Plain language summary
What is this article about?
This article talks about the lack of clarity and consistency in the definitions of outcomes used in clinical trials that investigate new treatments for diffuse large B-cell lymphoma. This is mainly due to how these different outcome definitions handle events such as absence of improvement in disease status, treatment discontinuation or initiation of new treatment. The authors discuss how these inconsistencies make it hard to interpret the results of individual clinical trials and to compare results across clinical trials.
Why is it important?
Defining the above events and consequently defining outcomes affects what we can learn from the trials and can lead to different results. Some approaches may not reflect good and bad outcomes for patients appropriately. This makes it challenging for patients, physicians, health authorities and payors to understand the true benefit of treatments under investigation and which one is better.
What are the key take-aways?
This article serves as a call-to-action for more dialogue among all stakeholders involved in drug development and the decision-making process related to drug evaluations. There is an urgent need for clinical trials to be designed with more clarity and consistency on what is being measured so that relevant questions for patients and prescribing physicians are addressed. Understanding patient journeys will be key to successfully understand what truly matters to patients and how to measure the benefit of new treatments. Such discussions will contribute toward more clarity and consistency in the evaluation of new treatments.
TWEETABLE ABSTRACT
Time for more dialogue to harmonize end point definitions in lymphoma! Use patient journeys to bring more clarity, consistency and patient-relevance! Learn about key considerations for the choice of end points and why PFS is not always meaningful!
Background
End points in clinical trials should reflect good and bad outcomes for patients appropriately.
Clarity on end point definitions is critical when interpreting trial results.
Review of phase III trials in diffuse large B-cell lymphoma (DLBCL) highlights the prevailing lack of clarity and consistency in the choice of end points and their definitions.
Overall survival
Combines the effect on survival of both the initial and any subsequent therapies received until death and captures potential effects of initial therapy on the choice, safety and efficacy of subsequent therapies.
Challenging interpretation of clinical trial results in DLBCL with rapidly changing treatment options and multiple potentially curative subsequent treatments.
Progression-free survival
The response of stable disease is a bad outcome for DLBCL patients, and as progression-free survival (PFS) does not count it as event it might not be patient-relevant.
Different approaches to consider the impact of new therapies initiated prior to PFS event or to exclude their impact address different questions.
Good understanding of the reasons for treatment discontinuations and start of new therapy critical to interpret PFS results.
Event-free survival
Event-free survival (EFS) considers not only death and progression as events, but also a type of ‘treatment failure’.
Treatment discontinuation generally not used in confirmatory trials as 'treatment failure’ event as it can be influenced by other factors not related to efficacy.
Start of new therapy often considered as treatment failure event regardless of the reason, i.e., even if it follows discontinuation due to toxicities.
In some trials, start of a new therapy for efficacy-related reasons is defined as an event, showing a focus on the anti-tumour activity of the drug.
Additional considerations required for studies of treatment strategies with multiple interventions such as CAR-T or transplant strategies.
Patient-reported outcomes
Challenging to interpret the results in the absence of clear, well-defined research objectives.
When defining research objectives, it is important to differentiate between short-term and long-term effects and to consider duration of treatment, frequency of patient-reported outcomes (PRO) assessments, the impact of new therapies and deaths and whether quality of life (QoL) at later timepoints reflects only the results in responding patients.
Discussion
Various elements of patient journey, disease setting and therapy type impact the appropriateness of end points.
Dialogue between all stakeholders needed to define more explicitly the questions of interest and corresponding end points in DLBCL and to ensure that relevant data are collected, clinical trials are designed to address these questions and the results are easier to interpret.
1. Background
An improvement in overall survival (OS) has been the ultimate objective in oncology trials. However, in diseases with extended survival duration and multiple therapeutic interventions throughout the patient's life, OS as the primary end point in confirmatory clinical trials has practical limitation that can delay patients' access to efficacious treatment options [Citation1]. Considering these points, the US FDA guidance on end points for cancer trials suggests that end points based on tumor assessments, such as progression-free survival (PFS) and event-free survival (EFS), can be used as primary efficacy end points in confirmatory trials, since tumor measures commonly trigger treatment decisions in clinical practice [Citation2]. A significant improvement in PFS or EFS with an acceptable side effects profile, demonstrates a clinically relevant benefit for many indications. The disappearance, shrinkage or stabilization of a tumor can also be associated with a direct benefit to the patient, for example, based on reduction of tumor-related symptoms and delaying potential subsequent treatments, with their own additional side effects [Citation3]. Additionally, a patient's perception of their disease and treatment is captured in that are typically assessed as secondary end points in clinical trials.
The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) aims to align the development of new medicinal products in Europe, North America and Asia. The ICH guideline on ‘General considerations for clinical studies’ emphasizes that good planning and implementation of a clinical study derive from attention to study design elements such as end points that are well-defined, measurable, clinically meaningful and relevant to patients [Citation4]. Thus, end points and data handling should appropriately reflect good and bad outcomes for patients as the basis for characterizing treatment effects. Well-defined end points and clarity on what is being estimated is critical for trialists and regulatory reviewers when interpreting the reported differences between two treatments or communicating these differences to others in publications or drug labels. With clarity on an appropriate end point, consistency across trials might not be critical per se, but can be helpful when outcomes on different interventions need to be compared by prescribers, patients, payors and regulators (e.g., by European Medicines Agency when determining significant benefit for maintenance of orphan drug designation or major therapeutic advantage in respect of Conditional Marketing Authorization).
In this article, we review the definitions of OS, PFS, EFS and PRO end points from large randomized clinical trials in diffuse large B-cell lymphoma (DLBCL), the most common subtype of non-Hodgkin lymphoma and highlight the prevailing lack of clarity and consistency. We discuss which factors in the patient journey might impact the appropriateness of the end point definition and methods of data handling. We call for a dialogue between physicians, patients, regulators, payers and sponsors, which is required to address the challenging aspects of interpretation and the questions related to clinical meaningfulness and relevance to patients. We would hope that such discussion among all stakeholders will ensure more transparency on the outcome being measured, harmonized end point definitions and also a more objective and patient-focused assessment of efficacy for new therapies. This will have a large impact on the design, analysis and interpretation of future phase III trials in DLBCL. This article is inspired by the estimand framework which was introduced by the ICH in 2017 and provides a structured approach to transparently discuss and account for patient journeys in the definition of the research question considering multiple attributes such as population, treatment strategy and post-randomization events that affect the interpretation of clinical outcomes. This structured framework for clinical researchers aims at better alignment of trial objectives, design, conduct, analysis and interpretation, and has been recently adopted by major health authorities [Citation5,Citation6].
summarizes the choice of primary end points, PFS and EFS definitions in completed and ongoing global randomized phase III trials in first- and second-line DLBCL starting with the trial of R-CHOP vs CHOP that changed the standard of care in lymphoma after many decades. For ongoing studies, the information was retrieved from the clinicaltrials.gov website. We note that in contrast to some other oncology indications with a single primary end point consistently used across all randomized trials, PFS, EFS and OS are all frequently used as primary end point in DLBCL trials. In first-line trials both PFS and EFS have been used, although the definitions of both end points differ across trials and sometimes overlap. All three trials in the transplant-eligible second-line population used EFS as the primary end point, each with different definitions. By contrast, six of eight ongoing trials in the transplant-ineligible population do not consider EFS even as a secondary end point. In the following sections we discuss the definitions of end points and their interpretation in the light of the variety of patient journeys that are illustrated in .
Table 1. Primary end points, progression-free survival and event-free survival definitions as primary end points in completed and ongoing randomized phase III diffuse large B-cell lymphoma trials.
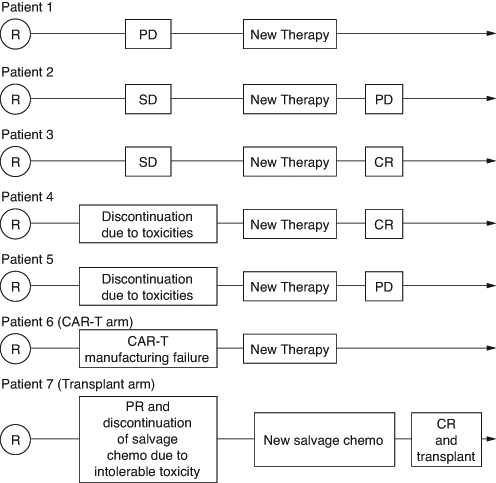
Figure 1. Examples of patient journeys in diffuse large B-cell lymphoma trials.
PD: Progressive disease; R: Randomization; SD: Stable disease; CR: Complete Response.
2. Overall survival
OS is defined across all studies in as time from randomization until death. OS is reported mainly as a secondary end point in randomized trials in DLBCL, however, three ongoing trials in the relapsed or refractory transplant-ineligible population use OS as the primary end point.
We note that the above definition combines the effect on OS of both the initial and any subsequent therapies received until death. This allows the opportunity to capture potential effects of initial therapy on the choice, safety and efficacy of subsequent therapies. However, it can also complicate the interpretation of OS effects, in particular in settings with rapidly changing treatment options such as DLBCL. Furthermore, multiple potentially curative treatments, such as autologous stem cell transplantation or CAR-T can be received by DLBCL patients in second- or later-lines. Therefore, if, for example, Patient 1 discontinues study treatment after disease progression in a clinical trial in first-line, they may subsequently receive curative therapies. In such scenario, OS would include the benefit of a potentially very effective subsequent therapy and not reflect the benefit of the randomized treatment. Statistical methods attempting to exclude the impact of certain subsequent therapies exist, usually relying on complex assumptions which cannot be verified from the data. However, if these subsequent therapies reflect clinical practice such analyses are of limited relevance for prescribers and patients who are interested in OS effects in real life [Citation6,Citation32]. Supplementary Table S1 in the supplementary appendix describes the different OS definitions using the terminology introduced by the estimand framework.
3. Progression-free survival
PFS is traditionally defined in oncology trials as the time from randomization until tumor progression or death, whichever occurs first. Tumor progression in clinical trials enrolling patients with DLBCL is typically determined using Lugano response criteria [Citation33], although one trial with the investigational therapy epcoritamab applied the lymphoma response to immunomodulatory therapy (LYRIC) criteria accounting for the possibility of pseudo-progressions [Citation34]. Pseudo-progression, in other words, initial increase in tumor size followed by decrease, is a recently described phenomenon observed after the introduction of checkpoint inhibitors in approximately 5% of cancer patients receiving these therapies [Citation35]. This demonstrates the need for treatment modality-specific efficacy criteria in order to accurately capture patient's benefit from treatments received.
3.1. The role of stable disease & subsequent new therapies
The above definition of PFS does not consider stable disease as a bad outcome. This can be reasonable from the patient's perspective in many oncology settings including early lines of treatment in indolent follicular lymphoma, another subtype of non-Hodgkin lymphoma. However, clinical guidelines define ‘refractory DLBCL’ based on a best response to previous therapy of stable disease or progressive disease, and recommend to initiate a new therapy for such patients with refractory DLBCL [Citation36,Citation37] This would support considering stable disease as a bad outcome, which is not reflected in the PFS definition. Therefore, despite its wide use as primary or secondary end point in DLBCL trials, it is unclear whether traditionally defined PFS represents a patient-relevant end point in this setting. Moreover, the interpretation of this new PFS definition is complicated if patients with stable disease start a new therapy, for example, Patients 2 and 3 in .
Information on how researchers account for these scenarios is reported only in a few publications across the studies listed in . In the Polarix trial, progression or death after start of a new therapy was counted as an event, in other words, Patient 2's disease progression is a PFS event, but for Patient 3 the response to subsequent therapy contributes to prolongation of PFS. Similarly to OS, the question of interest in this case is the combined effect on PFS of both the initial therapy and any new therapy received before progression or death. On the other hand, disease assessments after start of new therapy were not considered for evaluation of the secondary end point PFS in the Zuma-7 trial, and the statistical translation was that PFS duration for patients starting new therapy without progression were censored in the Kaplan–Meier analysis. Data for Patients 2 and 3 would be censored at the time of SD in such case. However, one of the key assumptions of the Kaplan–Meier method is that censored patients have identical prognosis to patients who did not receive new therapy and continued follow-up for PFS [Citation38]. This is a strong assumption, in particular, in the DLBCL setting as patients with SD with refractory DLBCL have worse prognosis, on average, compared with patients who did not start new therapy (and who are mostly in response). Ignoring progressions and deaths after the start of a new therapy also excludes the impact of subsequent therapies, i.e., the question of interest is not anymore the combined effect on PFS of both the initial therapy and any subsequent therapy, rather, it is the effect on PFS if no alternative therapy existed and patients, hypothetically, would have continued in follow-up without receiving any other therapies. If it is difficult to envisage it happening in real life, which in statistical terms translates into a violation of censoring assumption, other statistical methods can be used to address this question. However, the choice of statistical method is a minor point and only a consequence of the question whose relevance itself remains debated in the community.
3.2. Considerations for interpretation of progression-free survival end point
Such differences in end point definitions require various considerations and influence the interpretation of trial results. For example, if PFS is analyzed as it was in the Polarix trial and Patient 4 discontinues randomized treatment due to toxicities and responds to subsequent therapy, the PFS will potentially reflect the benefit of a highly effective subsequent therapy rather than of the initial treatment. On the other hand, the interpretation of PFS results as evaluated in Zuma-7 is also not always straightforward as described in the above section. In that trial 34% of patients in the standard of care arm were censored due to start of new therapy. Therefore, good understanding of the reasons for discontinuations of study treatment and start of new therapy is critical in order to understand the PFS treatment effect. Importantly, the choice of the question of interest and PFS definition also drives data collection and the duration of follow-up. Tumor assessments were not performed in the Zuma-7 trial following the start of new therapy, and, therefore, it is unknown how many of the censored patients responded or progressed after the start of new therapy. In such case, the combined treatment effect on PFS of both initial and any subsequent therapies received prior to progression can only be estimated with models and strong assumptions. The limitation in data collection means that some questions of interest cannot be well answered. Supplementary Table S2 in the supplementary appendix describes the different PFS definitions using the terminology introduced by the estimand framework.
An alternative definition of PFS considering lack of response (i.e. stable disease or progressive disease) as an event is used in the ongoing Polar Bear trial. Another option could be to consider start of new therapy as a PFS event as reported by Coiffier et al. [Citation7,Citation8]. As these approaches have been used more often to define the EFS end point rather than PFS, they will be discussed in the following section dedicated to EFS.
3.3. Determining the date of progression-free survival event
Lastly, the definition of time-to-event end point always requires not only clarity in what constitutes an event, but also in how the date of the event is defined. In the traditional PFS definition the date of death or progression is directly used as the event date. The Polar Bear study represents an exception, in which the PFS event is assigned to the day of randomization for patients who during the study had experienced no better response than progressive disease. Such an approach may be justified from a patient's perspective. For example, if the assigned treatment did not even achieve disease stabilization, only induced toxicities and prevented the use of alternative therapies, then no benefit was derived by the patient and no time post-randomization should be counted as progression free. The decision to start assigned treatment is per se considered a bad outcome. Further dialogue between all stakeholders may be needed to discuss whether backdating observed progression to the day of randomization is a meaningful approach to reflect the lack of benefit post-randomization. Of note, the concept of assigning the day of randomization as the event date has been applied in other settings, e.g., it is proposed in case of induction treatment failure in the FDA guidance for drug development in acute myeloid leukemia [Citation39].
4. Event-free survival
EFS typically refers to time-to-event end points that consider not only death and progression as events, but also a type of ‘treatment failure’. In DLBCL trials EFS has been used as a primary or secondary end point often considering start of new therapy and/or lack of response as treatment failure events.
Preventing or delaying the start of new therapy seems to be a relevant treatment goal for lymphoma treatments, e.g., to prevent or to delay the exposure to new risks and uncertainties associated with a new treatment course, and time to new therapy is a secondary end point in some studies in . Therefore, considering start of new therapy as an event jointly with progression and death can represent a meaningful way to determine the benefit of investigational anticancer treatment. However, is the start of new therapy always informative with regard to the efficacy of the assigned therapy? What if a patient with responding tumor receives new therapy outside of a clinical trial with the intent to consolidate the response? What if a patient is randomized in an open-label study to the standard of care arm and decides to discontinue immediately after randomization in order to receive a promising investigational therapy in another trial? What if a patient with responding tumor discontinues study treatment due to toxicities and starts a new therapy?
4.1. Reasons for start of new therapy & its impact on interpretation of event-free survival
We note that discontinuation of treatment for any reason, including disease progression, toxicity or death, is considered an event in the time to treatment failure end point, but, this end point is generally not accepted from a regulatory point of view as discontinuation can be influenced by other factors not related to efficacy [Citation2,Citation40]. The regulatory position likely explains why time to treatment failure end point has not been used in any of the reviewed DLBCL trials. However, most of the studies using EFS as primary or secondary end point do not distinguish between reasons for starting a new therapy. For example, in the recent Zuma-7 trial, six patients were randomized to standard of care arm, but never received protocol-specified treatment and started a new therapy outside of the trial – these were considered as EFS events [Citation41]. In these cases, the discontinuation and subsequent initiation of new therapies were not related to efficacy and were reported in the regulatory-approved drug label as events in the definition of the primary end point [Citation42].
In contrast, in the Polarix and Transform trials, the researchers considered only the start of a new therapy for efficacy-related reasons as events. Each approach estimates different effects of investigational therapies and address different questions. The former describes the effect of prolonging time to death, progression or start of new therapy for any reason, i.e. also implicitly considering as a bad outcome toxicities resulting in study drug discontinuation and start of a new therapy, or even the perception of a less effective therapeutic option leading patients to refuse the control group treatment and start a new therapy, as likely illustrated by the six patients from the Zuma-7 trial mentioned above. The latter targets the effect of prolonging time to death, progression or start of new therapy for efficacy reasons, i.e., focusing on anti-tumor activity of the drug. However, it also may introduce some subjectivity dependent on how efficacy reasons are defined, collected and reported by investigators. Further discussion among all stakeholders may be warranted to clarify first, in which circumstances one or the other of those different definitions of treatment effect is better for addressing the scientific question targeted by the study. This would facilitate subsequent discussion about precise and consistent definitions of efficacy reasons as well as best practices for the definition of event date. Interestingly, while the start of a new therapy is often directly used to determine the corresponding EFS event dates, other approaches have been recently reported [Citation16,Citation20]. For example, the date of the efficacy reason triggering the start of new therapy was used in the Polarix trial, whereas in the Zuma-7 trial, patients who started new therapy prior to first disease assessment were considered an event on the date of randomization. Supplementary Table S3 in the supplementary appendix describes the different EFS definitions using the terminology introduced by the estimand framework.
4.2. Event-free survival in studies with treatment strategies
The start of a new therapy requires additional considerations when evaluating the effect of treatment strategies, where multiple interventions combined constitute the therapeutic approach of interest. Three second-line trials compared autologous CAR-T treatment strategies with the standard of care treatment strategy, consisting of salvage chemotherapy followed by high-dose chemotherapy and autologous stem cell transplantation in responding patients (). Autologous CAR-T therapies are not readily available at the time of randomization and require personalized manufacturing. Time to CAR-T infusion is impacted by manufacturing time and various logistical factors such as shipment of apheresis material prior to manufacturing and of the product after manufacturing. Bridging chemotherapy may be administered during this time, until lymphodepleting chemotherapy is provided before the CAR-T infusion. While real-world data suggests that bridging chemotherapy is used for the majority of patients during the manufacturing period regardless of the CAR-T product [Citation43–45], different bridging strategies were studied in these three clinical trials. In the Zuma-7 trial, only corticosteroid bridging therapy was allowed per investigator discretion. In contrast, immunochemotherapy bridging was allowed in Transform (one cycle) and Belinda (at least one cycle).
Figure 2. Scheme of CAR-T and transplant treatment strategies.
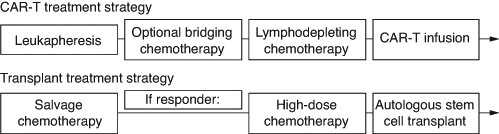
The study design and interpretation of the trial results in this setting require considerations related to the variety of patient journeys and the reasons for starting new therapies. The researchers need to consider the role of administered therapies in the planned treatment arms prior to observed progressions. The intent of new therapies may also differ between and within treatment arms, which makes it challenging to define a general rule. For example, Patient 6 in receives new therapy following manufacturing failure. From a patient perspective, manufacturing failure for CAR-T therapy is undoubtedly preventing the patient from receiving the main benefit of a CAR-T based treatment strategy, and in that sense, represents a bad outcome. On the other hand, non-protocol specified bridging therapy may not be a detrimental event for a patient if in the end it allows the patient to ultimately receive CAR-T infusion. For the standard of care treatment strategy, patients may receive new therapy in the absence of disease progression after observing insufficient response to transplant, or due to toxicities of initial salvage chemotherapy. Patient 7 experiences PR after the first cycle of salvage chemotherapy, accompanied by intolerable toxicity and then changes the salvage regimen and proceeds to transplant after achieving CR. The start of new therapy in such cases does not represent failure of assigned treatment strategy and considering it as an event will not reflect the real benefit that the patient receives from it (namely to be able to receive the intended and possibly curative therapeutic intervention of transplant). However, in a similar situation after the first cycle with intolerable toxicity, it could also be decided that the patient will tolerate neither salvage chemotherapy nor any other therapy. In such a scenario, an EFS event would only be reported if disease progression or death is observed at a later timepoint and not after the first cycle of salvage chemotherapy when it was determined that the patient will not be able to continue receiving the assigned treatment strategy. This would also not be an accurate reflection of the clinical status of the patient at the time it is determined.
This raises a number of questions in settings with complex treatment strategies, and highlights the importance to carefully review a wide range of possible patient journey scenarios to define the relevant end point characteristics. What is the most meaningful way to reflect an unfavorable outcome from a patient perspective, and still rely on objective assessments to do so? How should failure to receive the potentially curative component of the treatment strategy, CAR-T infusion or transplant, be taken into account when defining the primary question of interest? And what if patients require new therapies, but in the end can proceed to CAR-T infusion or transplant?
4.3. Short-term versus long-term effects & EFS definition
Another important question for understanding the EFS end point is the timeframe to be considered before declaring a treatment failure and an EFS event. Should potential delay in CAR-T infusion or transplant be reflected in the end point, if it is eventually administered? For example, in the Belinda trial, all patients with SD or PD at week 12 were considered EFS events, although six of them responded later due to delayed CAR-T infusion. By contrast, such patients were not considered as events in the Zuma-7 trial as long as their response was observed prior to day 150. This illustrates that more than one definition of the end point may be needed to capture all regulatory and patient-relevant aspects. Both pharmacological effects (short term) and overall outcome (long-term) can be relevant for different stakeholders dependent on their question of interest. This also exemplifies the challenges in interpreting different treatment effects across different trials.
5. Patient-reported outcomes
Patient-reported outcomes (PRO) aim to capture the patient's own perception of their health condition and are often included as a secondary objective in randomized clinical trials. Typically, quality of life (QoL) questionnaires such as the European Organization for Research and Treatment of Cancer Quality of Life Core 30 (EORTC QLQ-C30) or the Functional Assessment of Cancer Therapy – Lymphoma (FACT-Lym) are used for this purpose [Citation46,Citation47] In a recent paper, FDA authors acknowledged that it has been challenging to appropriately interpret the results, primarily because of the absence of clear, well defined PRO research objectives in protocols and statistical analysis plans [Citation48]. Indeed, the same questions raised in previous sections are relevant for PRO end points. For example, it is important to differentiate between short-term and long-term effects on QoL. Patients' QoL may deteriorate in the first weeks of treatment due to newly experienced toxicities. Such deterioration would be captured in a time to first deterioration end point or in change from baseline at an early timepoint. However, as acute toxicities resolve or patients learn to cope with adverse event and concomitant medications, and the anti-tumor effect of the therapy starts to improve the underlying disease, their QoL may improve over time. Generally, while it is important to understand the trajectory of QoL and short-term effects, long-term improvements represent the ultimate goal for new therapies. Time to definitive deterioration end points consider such improvements as benefit for a patient and only include definitive worsening in QoL without further improvement as events. Alternatively, such long-term effects could be reflected in changes from baseline at later timepoints.
5.1. Role of subsequent therapy in the interpretation of quality of life
When defining the questions of interest for the PRO end points clarity is needed whether the researchers are interested in a combined effect of the assigned therapy and any subsequent new therapies on QoL, or only in the effect of the assigned therapy until its discontinuation or the start of new therapy. For example, for Patients 1–5, considering assessments after disease progression, stable disease or treatment discontinuation due to toxicities may allow capturing potential further worsening of QoL prior to starting a new therapy. However, any disease assessment after the start of a new therapy would reflect QoL of both the assigned therapy and the new therapy. The new therapy could have a greater impact on QoL and either further worsen or improve it.
5.2. Time to deterioration end point of quality of life & death
QoL assessments cannot be collected after patient death. Different approaches to account for death are possible and result in different questions of interest [Citation48,Citation49] In the Belinda trial, the time to definitive deterioration end point considered death as an event, i.e., the researchers were interested in the effect of prolonging of time to deterioration of QoL or death. Alternatively, censoring deaths in Kaplan–Meier analysis rather than counting them as events has been used in other oncology trials [Citation48]. This approach can be used to evaluate the effect on QoL while alive, however, Kaplan–Meier estimates in such situations are biased and an alternative statistical methodology is more appropriate [Citation50]. Furthermore, the results of an analysis in which deaths are censored can also be interpreted as the effect on QoL in the hypothetical scenario if deaths had not occurred, however, its relevance for clinicians, patients and regulators is debatable. We note that time to deterioration is also reported as a secondary end point in other trials in [Citation9,Citation16,Citation29,Citation30], but without describing how they account for deaths.
5.3. Change in quality of life from baseline at a pre-specified timepoint
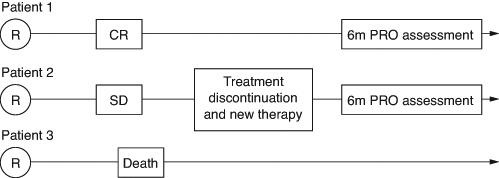
The change in QoL from baseline is another secondary end point used in completed and ongoing DLBCL trials [Citation10,Citation19,Citation22,Citation29]. This end point requires careful selection of timepoints for communication of PRO effects. As illustrates, QoL at e.g., 6 months after randomization can have a very different meaning dependent on how researchers account for patient journeys. For example, if the researchers are interested in QoL until treatment discontinuation, the 6-month assessment of Patient 2 (i.e., after start of new therapy) will not contribute to the evaluation. In fact, as DLBCL patients in SD or PD are expected to start new therapy, the results of such analysis will likely implicitly reflect only the QoL of responding patients at 6 months and should be interpreted accordingly. If the researchers consider assessments after start of a new therapy, Patient 2's 6-month data will be included in the analysis and the results will also reflect the effect of subsequent therapies. In such cases, it is relevant to know the number of discontinuations and new therapies for the interpretation of reported results. We note that, as discussed above, assessment of QoL is complicated by the occurrence of deaths. Therefore, it is also important to consider how many patients are expected to be alive when selecting a clinically meaningful timepoint for QoL assessment.
Figure 3. Patient journeys and assessment of quality of life at 6 months.
CR: Complete Response; SD: Stable Disease; PRO: Patient-reported outcomes.
Duration of treatment and the frequency of PRO assessments are also critical factors that need to be considered when defining the PRO end point and interpreting the results. For example, results at different timepoints can reflect QoL in different treatment periods (on-treatment vs post-treatment), possibly associated with different toxicities. Moreover, complex CAR-T and transplant treatment strategies require additional considerations. For example, in the Transform trial, QoL was compared between these strategies on day 29, day 64, day 126 and month 6 [Citation51]. In the CAR-T arm, Day 29 reflects the pre-infusion period capturing the effect of possible bridging and lymphodepleting chemotherapies and day 64 likely reflects the first month post-infusion with possible hospitalizations. On the other hand, for the transplant treatment strategy, day 29 and day 64 reflect the effect of salvage chemotherapy prior to transplant. Assessments after day 126 mainly reflect QoL of responding patients after CAR-T infusion and successful transplant in both arms if data for non-responding patients is not collected or used after the start of new therapies. As discussed in the EFS section above, the complexity in this setting raises a number of questions when defining the most meaningful way to reflect patients' benefit and warrants a discussion with all stakeholders. In addition, new disease-specific and treatment-specific PRO instruments may be required to assess the impact of novel therapies on QoL.
6. Discussion
In our review we observed lack of clarity and consistency in end point definitions of large randomized clinical trials in DLBCL. Clarity in end point definitions is of paramount importance to understand what is being estimated and how to interpret the treatment effects. The diversity of patient journeys raises fundamental questions regarding the evaluation of drug effects in clinical trials to inform clinical practice [Citation6]. When defining the treatment effect of interest and the corresponding end point in a trial, the researcher needs to account for events occurring after randomization, such as treatment discontinuation or the start of a new therapy, before observing progression or death or QoL deterioration. Different ways to account for such events actually address different clinical questions and result in different estimated effect sizes for summary measures such as hazard ratio, even if the overall conclusion concerning the superiority of investigational therapy may not change. Therefore, when communicating trial results, it is critical to provide not only what constitutes an event for time-to-event end point such as PFS, but also whether and how e.g., disease assessments after treatment discontinuation and start of new therapy were considered for its evaluation. Moreover, inconsistent definitions of PFS and EFS further complicate the interpretation of results across trials for patients, prescribing physicians, regulators and payors. Importantly, it is unclear why there are different ways to define end points and rules for dates of event and censoring. For instance, some studies consider all new therapies as events in the EFS or PFS definition, other studies only consider new therapies started for efficacy reasons and yet other trials do not consider new therapies as events. Some elements in end point definitions are debatable and may introduce bias in efficacy assessments. More clarity on the scientific rationale for the chosen approach, which would clarify the reasons for the inconsistencies would help in the design of future trials, and also facilitate a discussion among all stakeholders that could lead to more harmonization and reduce the risk of bias. Such discussion should consider the type of subsequent therapy, e.g., in the Zuma-7 trial, patients receiving new therapy were considered an event, except if they received HSCT after CAR-T infusion.
The ICH E8 guideline emphasizes that end points should not only be well-defined, but also be clinically meaningful and relevant to patients, appropriately reflecting good and bad outcomes for patients. This raises important questions - how do we define patient-relevant end points in DLBCL trials and what should be reflected in the label to inform prescribers? For example, clinical guidelines for DLBCL recommend starting a new therapy for patients with stable disease as these patients have similarly poor prognosis to progressing patients. Therefore, the response of stable disease to assigned treatment is expected to represent a bad outcome for patients and yet the PFS end point not counting stable disease as an event has been used as primary or secondary end point in almost all DLBCL trials and has been reported in drug labels. While patients with stable disease are expected to progress quickly, their progressions will not be captured by a traditional PFS end point if observed after starting of new therapies and thus censored. On the other hand, clinical trial protocols that discourage initiating new therapy for patients with stable disease until observed progression raise ethical issues in this setting.
The above considerations cast doubt on the relevance of the conventional definition of PFS for patients and prescribers in DLBCL and highlight the need for indication-specific discussions about end points, as for example, stable disease can represent a good outcome in other oncology indications. Moreover, even within one indication, the relevance of the end point may depend on the exact setting, as the meaning of stable disease for patients may change depending on the line of therapy as the disease becomes more aggressive (e.g. as with follicular lymphoma). Different considerations may also be required for one-time treatments compared with therapies with fixed durations or treatment until progression as e.g., discontinuations due to toxicities are not possible for the former. Furthermore, with some therapies a delayed response after initial progression may be observed either because of pseudo-progression due to immunomodulating effects, or, for CAR-T treatments because of delayed infusions. This suggests that different treatment-specific end points may be needed to fully describe short-term and long-term benefits for patients, which makes selection of an end point to compare two very different treatments even more challenging. We note that some payors such as the Federal Joint Committee in Germany do not consider PFS as a patient-relevant end point in many oncology settings as it is based on imaging assessment and not related to patient symptoms, although this end point is accepted by regulators [Citation3]. Regulators, payors, clinicians and patients may have different perspectives on how to define treatment benefit in the context of their decision-making and considering different effects on tumor size, alleviation of symptoms, reduction of toxicities and the choice and efficacy of subsequent therapies.
Patients with DLBCL and treating physicians deserve more transparency and clarity in end point definitions so that relevant data are collected, clinical trials are designed to address their questions and the results are easier to interpret. Similar inconsistencies exist in other oncology settings. For example, Punt et al. found that the definition of end points in studies of adjuvant treatments in colon cancer varied widely and a panel of experts then reached consensus on the definition of each end point (see Supplementary Table S4) [Citation52]. A similar overview could be a starting point for the DLBCL community and should also reflect the different approaches related to specific treatment strategies and patient journeys while ensuring the objectivity of the end point.
More recently, the value of the estimand framework to facilitate the dialogue in such situations has been highlighted for the disease-free survival end point for adjuvant therapies in renal cell carcinoma [Citation53]. The DLBCL community would also benefit from a dialogue between patients, physicians, regulators, payors and sponsors to define more explicitly the scientific questions of interest. Such discussion could take place, for example, in the form of several workshops co-sponsored by medical societies, regulators and other stakeholders. This would create opportunities to define better and more transparent end point definitions to address these questions. Additionally, we encourage all stakeholders to better justify their choices for preferred end point definitions at every step of drug development, in particular, when discussing clinical trial protocols. As illustrated herein, assessing different end point rules through a systematic review of complex plausible patient journeys can be a powerful tool to facilitate such a discussion and to explain the rationale for end point definitions. Regulatory guidelines for development of new therapies in DLBCL could contribute to harmonization and better understanding of patient-relevant end points.
7. Future perspective
The last few years have seen major advances in the management of DLBCL with the introduction of CAR-T therapies and bi-specific antibodies. We applaud the researchers for these scientific breakthroughs and bringing these therapies to patients with DLBCL. However, we also recognize that different end points have been chosen for clinical trials without clear rationale and justification of their relevance to patients. Importantly, differences are observed not only in the choice of primary and secondary end points, but also in the definitions of the same end point (e.g., PFS) used across different trials. These subtle differences originate in the different ways to take account of various elements of the patient journey, such as the start of new therapy prior to progression or death, implying that these trials address different scientific questions.
Currently, more than ten large randomized phase III trials in first and second-line in DLBCL are ongoing. In the absence of greater clarity and consistency, the interpretation of these trials risks being compromised and the impact will not be subtle. This can complicate approval and reimbursement decisions and lead to uninformative prescribing information without clear descriptions of the studied questions and corresponding end points. Cross-trial comparisons between these trials will be especially challenging and further complicate the selection of treatments by physicians and patients. It also decreases the scientific value of these trials, giving little return on the participants' investment in contributing to the trial.
We advocate that all stakeholders recognize the need for change and pick up the challenge to work together to develop key questions of interest from regulatory, payor, physician and patient perspective. Engaging in such dialogue can assist in establishing consensus on the rationale and value of existing end points. This can also lead to collaboration on the development of novel end points measuring outcomes of high importance for patients (e.g. disease-burden and QoL impairments, symptom-related burden on healthcare services). Finding a common language between all stakeholders is the key to the success of these discussions, although it has been historically challenging. The review of patient journeys and the use of estimand thinking to better understand favorable and unfavorable outcomes for patients and to define the questions of interest for clinical trials can become an important and regularly used tool to facilitate such discussions.
Author contributions
All authors contributed to the concept of this work, interpretations, conclusions and critical revisions.
Financial disclosure
E Degtyarev, A Buchbinder, M Fuchs, A Masood, S Newsome, N Yateman and E Zuber are employees and stock owners of Novartis. C Saxton is a co-principal investigator on a study that was funded by Bristol Myers Squibb and Blue Note Therapeutics. The authors who represent Cancer Support Community and Lymphoma Coalition did not receive any compensation for their work on this manuscript. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Supplementary Materials
Download Zip (53.9 KB)Acknowledgments
We would like to thank Pal A and Kasala ER for providing editorial and submission support.
Supplementary material
Supplemental data for this article can be accessed at https://doi.org/10.1080/14796694.2024.2357537
Competing interests disclosure
The authors have no competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- Richardson NC, Kasamon Y, Pazdur R, et al. The saga of PI3K inhibitors in haematological malignancies: survival is the ultimate safety endpoint. Lancet Oncol. 2022;23(5):563–566. doi:10.1016/S1470-2045(22)00200-5
- FDA Guidance for Industry. Clinical trial endpoints for the approval of cancer drugs and biologics. December 2018. Available from: www.fda.gov/media/71195/download
- Dabisch I, Dethling J, Dintsios CM, et al. Patient relevant endpoints in oncology: current issues in the context of early benefit assessment in Germany. Health Econ Rev. 2014;4(1):2. doi:10.1186/2191-1991-4-2
- ICH Harmonised Guideline. E8(R1) General considerations for clinical studies. [ updated 2021 Oct 06]. Available from: www.ema.europa.eu/en/documents/regulatory-procedural-guideline/ich-guideline-e8-r1-general-considerations-clinical-studies_en.pdf
- ICH Harmonised Guideline. E9(R1) Addendum on estimands and sensitivity analysis in clinical trials. [ updated 2019 Nov 20]. Available from: https://database.ich.org/sites/default/files/E9-R1_Step4_Guideline_2019_1203.pdf
- Degtyarev E, Zhang Y, Sen K, et al. Estimands and the patient journey: addressing the right question in oncology clinical trials. JCO Precis Oncol. 2019;3:1–10. doi:10.1200/PO.18.00381
- Coiffier B, Lepage E, Briere J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(4):235–242. doi:10.1056/NEJMoa011795
- Coiffier B, Thieblemont C, Van Den Neste E, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d'Etudes des Lymphomes de l'Adulte. Blood. 2010;116(12):2040–2045. doi:10.1182/blood-2010-03-276246
- Younes A, Sehn LH, Johnson P, et al. Randomized Phase III trial of ibrutinib and rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in non-germinal center B-cell diffuse large B-cell lymphoma. J Clin Oncol. 2019;37(15):1285–1295. doi:10.1200/JCO.18.02403
- Bartlett NL, Wilson WH, Jung SH, et al. Dose-adjusted EPOCH-R compared with R-CHOP as frontline therapy for diffuse large B-cell lymphoma: clinical outcomes of the Phase III intergroup trial Alliance/CALGB 50303. J Clin Oncol. 2019;37(21):1790–1799. doi:10.1200/JCO.18.01994
- ClinicalTrials.gov. Study to compare axicabtagene ciloleucel with standard of care therapy as first-line treatment in participants with high-risk large B-cell lymphoma (ZUMA-23). [ Last updated: 2024 Feb 22; accessed: 2024 Feb 23]. Available from: https://clinicaltrials.gov/ct2/show/NCT05605899
- Seymour JF, Pfreundschuh M, Trněný M. R-CHOP with or without bevacizumab in patients with previously untreated diffuse large B-cell lymphoma: final MAIN study outcomes. Haematologica. 2014;99(8):1343–1349. doi:10.3324/haematol.2013.100818
- ClinicalTrials.gov. A study to evaluate change in disease activity of subcutaneous (sc) epcoritamab combined with intravenous and oral rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine, and prednisone (R-CHOP) or R-CHOP in adult participants with newly diagnosed diffuse large B-cell lymphoma (DLBCL) (EPCORE DLBCL-2). [ Last updated: 2024 Feb 23; accessed: 2024 Feb 23]. Available from: https://clinicaltrials.gov/study/NCT05578976
- Vitolo U, Trněný M, Belada D, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol. 2017;35(31):3529–3537. doi:10.1200/JCO.2017.73.3402
- Nowakowski GS, Chiappella A, Gascoyne RD, et al. ROBUST: a Phase III study of lenalidomide plus R-CHOP versus placebo plus R-CHOP in previously untreated patients with ABC-type diffuse large B-cell lymphoma. J Clin Oncol. 2021;39(12):1317–1328. doi:10.1200/JCO.20.01366
- Tilly H, Morschhauser F, Sehn LH, et al. Polatuzumab vedotin in previously untreated diffuse large B-cell lymphoma. N Engl J Med. 2022;386(4):351–363. doi:10.1056/NEJMoa2115304
- ClinicalTrials.gov. A randomized, multicenter, Phase III trial comparing treatment with R-mini-CHOP with R-mini-CHP + polatuzumab vedotin in patients with diffuse large cell B cell lymphoma (POLAR BEAR). [ Last updated: 2024 Feb 13; accessed: 2024 Feb 23]. Available from: https://clinicaltrials.gov/study/NCT04332822
- ClinicalTrials.gov. Tafasitamab + lenalidomide + R-CHOP versus R-CHOP in newly diagnosed high-intermediate and high risk DLBCL patients (frontMIND). [ Last updated: 2023 Nov 29; accessed: 2024 Feb 23]. Available from: www.clinicaltrials.gov/ct2/show/NCT04824092
- Bishop MR, Dickinson M, Purtill D, et al. Second-line tisagenlecleucel or standard care in aggressive B-cell lymphoma. N Engl J Med. 2022;386(7):629–639. doi:10.1056/NEJMoa2116596
- Locke FL, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. N Engl J Med. 2022;386(7):640–654. doi:10.1056/NEJMoa2116133
- Kamdar M, Solomon SR, Arnason JE, et al. Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second-line treatment in patients with relapsed or refractory large B-cell lymphoma (TRANSFORM): results from an interim analysis of an open-label, randomised, Phase III trial. Lancet. 2022;399(10343):2294–2308. doi:10.1016/S0140-6736(22)00662-6
- Belada D, Georgiev P, Dakhil S, et al. Pixantrone-rituximab versus gemcitabine-rituximab in relapsed/refractory aggressive non-hodgkin lymphoma. Future Oncol. 2016;12(15):1759–1768. doi:10.2217/fon-2016-0137
- ClinicalTrials.gov. Study to evaluate loncastuximab tesirine with rituximab versus immunochemotherapy in participants with relapsed or refractory diffuse large B-cell lymphoma (LOTIS 5). [ Last updated: 2024 Jan 24; accessed: 2024 Feb 23]. Available from: https://clinicaltrials.gov/ct2/show/NCT04384484
- ClinicalTrials.gov. A study evaluating efficacy and safety of mosunetuzumab in combination with polatuzumab vedotin compared to rituximab in combination with gemcitabine plus oxaliplatin in participants with relapsed or refractory aggressive B-cell non-hodgkin's lymphoma (SUNMO). [ Last updated: 2024 Jan 31; accessed: 2024 Feb 23]. Available from: https://clinicaltrials.gov/ct2/show/NCT05171647
- ClinicalTrials.gov. A study of rituximab-gemcitabine-dexamethasone-platinum (R-GDP) with or without selinexor in patients with relapsed/refractory diffuse large B-cell lymphoma. [ Last updated: 2023 Oct 03; accessed: 2024 Feb 23]. Available from: https://clinicaltrials.gov/ct2/show/NCT04442022
- ClinicalTrials.gov. A trial to evaluate the efficacy and safety of tafasitamab with bendamustine (BEN) versus rituximab (RTX) with BEN in adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) (B-MIND). [ Last updated: 2022 Aug 02; accessed: 2024 Feb 23] https://clinicaltrials.gov/ct2/show/NCT02763319
- Dang NH, Ogura M, Castaigne S, et al. Randomized, Phase III trial of inotuzumab ozogamicin plus rituximab versus chemotherapy plus rituximab for relapsed/refractory aggressive B-cell non-hodgkin lymphoma. Br J Haematol. 2018;182(4):583–586. doi:10.1111/bjh.14820
- ClinicalTrials.gov. A Phase III trial of epcoritamab vs investigator's choice chemotherapy in R/R DLBCL (EPCORE DLBCL-1). [ Last updated: 2024 Feb 06; accessed: 2024 Feb 23]. Available from: https://clinicaltrials.gov/ct2/show/NCT04628494
- ClinicalTrials.gov. A study to evaluate the safety and efficacy of polatuzumab vedotin in combination with rituximab, gemcitabine and oxaliplatin compared to rituximab, gemcitabine and oxaliplatin alone in participants with relapsed or refractory diffuse large B-cell lymphoma (POLARGO). [ Last updated: 2023 Dec 14; accessed: 2024 Feb 23]. Available from: https://clinicaltrials.gov/ct2/show/NCT04182204
- ClinicalTrials.gov. A Phase III study evaluating glofitamab in combination with gemcitabine + oxaliplatin vs rituximab in combination with gemcitabine + oxaliplatin in participants with relapsed/refractory diffuse large B-cell lymphoma. [ Last updated: 2024 Feb 13; accessed: 2024 Feb 23]. Available from: www.clinicaltrials.gov/ct2/show/NCT04408638
- ClinicalTrials.gov. Polatuzumab vedotin plus rituximab, ifosfamide, carboplatin and etoposide (Pola-R-ICE) versus R-ICE alone in second line treatment of diffuse large B-cell lymphoma (DLBCL). [ Last updated: 2023 Oct 12; accessed: 2024 Feb 23]. Available from: www.clinicaltrials.gov/ct2/show/NCT04833114
- Manitz J, Kan-Dobrosky N, Buchner H, et al. Estimands for overall survival in clinical trials with treatment switching in oncology. Pharm Stat. 2022;21(1):150–162. doi:10.1200/pst.2158
- Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of hodgkin and non-Hodgkin lymphoma: the lugano classification. J Clin Oncol. 2014;32(27):3059–3068. doi:10.1200/JCO.2013.54.8800
- Cheson BD, Ansell S, Schwartz L, et al. Refinement of the lugano classification lymphoma response criteria in the era of immunomodulatory therapy. Blood. 2016;128(21):2489–2496. doi:10.1182/blood-2016-05-718528
- Park HJ, Kim KW, Pyo J, et al. Incidence of pseudoprogression during immune checkpoint inhibitor therapy for solid tumors: a systematic review and meta-analysis. Radiology. 2020;297(1):87–96. doi:10.1148/radiol.2020200443
- Barth M, Xavier AC, Armenian S, et al. Pediatric aggressive mature B-cell lymphomas, version 3.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2022;20(11):1267–1275. doi:10.6004/jnccn.2022.0057
- Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130(16):1800–1808. doi:10.1182/blood-2017-03-769620
- Fojo T, Simon RM. Inappropriate censoring in Kaplan-Meier analyses [comment]. Lancet Oncol. 2021;22(10):1358–1360. doi:10.1016/S1470-2045(21)00473-3
- FDA Guidance for Industry. Acute Myeloid Leukemia: Developing Drugs and Biological Products for Treatment. October 2022. Available from: www.fda.gov/media/162362/download
- Pazdur R. Endpoints for assessing drug activity in clinical trials. The Oncol. 2008;13(S2):19–21. doi:10.1634/theoncologist.13-S2-19
- Clinical Review and Evaluation – YESCARTA. 2022 Apr 01. Available from: www.fda.gov/media/157687/download
- US Package Insert – YESCARTA. [ revised: Dec 2023]. Available from: www.fda.gov/media/108377/download
- Riedell PA, Brower J, Nastoupil LJ, et al. A multicenter retrospective analysis of outcomes and toxicities with commercial axicabtagene ciloleucel and tisagenlecleucel for relapsed/refractory aggressive B-cell lymphomas. Oral presentation 52. Presented at: Orlando, FL: 2020 TCT; February 19–23, 2020.
- Kwon M, Iacoboni G, Reguera JL, et al. Axicabtagene ciloleucel compared to tisagenlecleucel for the treatment of relapsed or refractory large B-cell lymphoma in the real world setting in Spain. Blood. 2021;138(Suppl. 1):1742. doi:10.1182/blood-2021-147369
- Chiappella A, Guidetti A, Dodero A, et al. Real-life CAR-T cell treatment in large B-cell lymphomas indicates that axi-cel and tisa-cel have similar outcomes, but long-term cytopenia is an emerging problem. Blood. 2021;138(Suppl. 1):3867. doi:10.1182/blood-2021-154037
- Quality of Life of Cancer Patients - EORTC QLQ-C30. Available from: https://qol.eortc.org/questionnaire/eortc-qlq-c30/
- FACT-Lym (facit.org). Functional Assessment of Cancer Therapy – Lymphoma. Available from: www.facit.org/measures/FACT-Lym
- Fiero MH, Roydhouse JK, Bhatnagar V, et al. Time to deterioration of symptoms or function using patient-reported outcomes in cancer trials. Lancet Oncol. 2022;23(5):e229–e234. doi:10.1016/S1470-2045(22)00021-3
- Lawrance R, Degtyarev E, Griffiths P, et al. What is an estimand & how does it relate to quantifying the effect of treatment on patient-reported quality of life outcomes in clinical trials? J Patient RepOutcomes. 2020;4(1):68. doi:10.1186/s41687-020-00218-5
- Rufibach K. Treatment effect quantification for time-to-event endpoints – Estimands, analysis strategies, and beyond. Pharm Stat. 2019;18:144–164. doi:10.1002/pst.1917
- Abramson JS, Solomon SR, Arnason JE, et al. Improved quality of life (QOL) with lisocabtagene maraleucel (liso-cel), a CD19-directed chimeric antigen receptor (CAR) T cell therapy, compared with standard of Care (SOC) as second-line (2L) treatment in patients (Pts) with relapsed or refractory (R/R) large B-cell lymphoma (LBCL): results from the Phase III Transform study. Blood. 2021;138(Suppl. 1):3845. doi:10.1182/blood-2021–151611
- Punt CJ, Buyse M, Köhne CH, et al. Endpoints in adjuvant treatment trials: a systematic review of the literature in colon cancer and proposed definitions for future trials. J Natl Cancer Inst. 2007;99(13):998–1003. doi:10.1093/jnci/djm024
- Casey M, Degtyarev E, Lechuga MJ, et al. Estimand framework: are we asking the right questions? a case study in the solid tumor setting. Pharm Stat. 2021;20:324–334. doi:10.1002/pst.2079