
Abstract
Background: HIV-1-infected, virologically suppressed adults wanting to simplify or change their non-nucleoside reverse transcriptase inhibitor (NNRTI)-based regimens may benefit from switching to the single-tablet regimen of elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate (E/C/F/TDF).
Objective: We examined differences in the proportion of participants with HIV-1 RNA < 50 copies/mL (Snapshot analysis), change in CD4 cell count, safety, and patient-reported outcomes in participants switching to E/C/F/TDF from an NNRTI + FTC/TDF (TVD) regimen.
Methods: STRATEGY-NNRTI was a 96-week, phase 3b, randomized, open-label, study examining the efficacy, safety, and tolerability of switching to E/C/F/TDF in virologically suppressed individuals (HIV-1 RNA < 50 copies/mL) on an NNRTI + TVD regimen. Participants were randomized to switch or remain on their NNRTI-based regimen (no-switch).
Results: At Week 96, 87% (251/290) of switch and 80% (115/143) of no-switch participants maintained HIV-1 RNA < 50 copies/mL (difference 6.1%; 95% CI −1.3 to 14.2%; p = 0.12) according to the FDA-defined snapshot algorithm. Both groups had similar proportions of subjects with virologic failure (2.8% switch, 1.4% no-switch). Discontinuations resulting from adverse events were infrequent (3% [9/291] switch, 2% [3/143] no-switch). Three switch participants (1%) discontinued due to renal adverse events (2 of the 3 before Week 48). Switch participants reported significant improvements in neuropsychiatric symptoms by as early as Week 4, and which were maintained through Week 96.
Conclusions: E/C/F/TDF is safe and effective and reduces NNRTI-associated neuropsychiatric symptoms for virologically suppressed HIV-positive adults switching from an NNRTI plus FTC/TDF-based regimen.
Introduction
Current treatment guidelines recommend HIV antiretroviral therapy (ART) changes in virologically suppressed patients to simplify treatment, improve tolerability and adherence, and reduce long-term ART-associated toxicities.Citation1,2 A coformulated single-tablet regimen (STR) E/C/F/TDF containing 150 mg elvitegravir (E, an integrase strand transfer inhibitor, INSTI), 150 mg cobicistat (C, a CYP3A4 enzyme inhibitor and pharmacokinetic enhancer), 200 mg emtricitabine (FTC; F, a nucleoside reverse transcriptase inhibitor, NRTI), and 300 mg tenofovir disoproxil fumarate (TDF, an NRTI), is safe, efficacious, and well-tolerated in ART-naïve, HIV-positive adult patients.Citation3–8 E/C/F/TDF is indicated for treatment-naïve and virologically suppressed (HIV-1 RNA < 50 copies/mL) adults on stable antiretroviral therapy for ≥ 6 months and with no history of treatment failure or drug resistance to the components of E/C/F/TDF.Citation9
STRATEGY-NNRTI was a 96-week, open-label study that examined the efficacy, safety, and tolerability of switching to E/C/F/TDF in virologically suppressed, adult patients on a non-nucleoside reverse transcriptase inhibitor (NNRTI)-based regimen, including coformulated emtricitabine/tenofovir DF (TVD). Week 48 data showed non-inferior efficacy of E/C/F/TDF vs. the NNRTI + TVD regimens (93% vs. 88%, respectively; [5.3% difference; 95%CI −0.5, 12.0%]). The switch to E/C/F/TDF was also associated with improved patient-reported outcomes (PROs) related to NNRTI-associated neuropsychiatric side effects and with greater treatment satisfaction score at Week 48.Citation10 Week 96 results of STRATEGY-NNRTI are presented here.
Methods
STRATEGY-NNRTI was a phase 3b, randomized, open-label, active-controlled study conducted at 72 sites in North America, Europe, and Australia. The study design and procedures have been described previously.Citation11 Eligible participants were aged ≥ 18 years, had plasma HIV-1 RNA < 50 copies/mL on a NNRTI + TVD regimen for ≥ 6 consecutive months, and were on their first or second regimen. All subjects were INSTI-naïve, had no prior virologic failure (VF) or genotypic resistance to TVD, and had an estimated glomerular filtration rate (eGFR)≥70 mL/min at screening. This study was conducted in accordance with the Declaration of HelsinkiCitation11; all participants gave written informed consent.
Procedures
Eligible individuals were randomized 2:1 to switch to open-label E/C/F/TDF taken once-daily with food (switch group), or to remain on their current NNRTI + TVD regimen (no-switch group).Citation11 VF for the purposes of identifying subjects for resistance analysis (vs. VF by FDA-defined Snapshot) focused on those subjects with two consecutive visits with plasma HIV-1 RNA ≥ 50 copies/mL. HIV-1 genotyping and phenotyping (protease, reverse transcriptase, and integrase) were then performed specifically in participants with HIV-1 RNA ≥ 400 copies/mL at the confirmation visit, at Week 96, or upon early study discontinuation.
Outcomes
The primary efficacy objective was the proportion of participants who maintained HIV-1 < 50 copies/mL at Week 48 (FDA-defined Snapshot analysis).Citation12 The secondary efficacy objective at Week 96 was identical to the primary objective, plus change in CD4 cell count. Other objectives included safety, tolerability, and PROs.
Statistical analysis
Participants in the full analysis set included those treated with ≥ 1 dose of study drug and with no major protocol violations. Non-inferiority at Week 96 was concluded if the lower bound of a two-sided exact 95% confidence interval (CI) for the difference in proportions of participants (switch minus no-switch group) who maintained HIV-1 viral load < 50 copies/mL was > –12%. If non-inferiority was demonstrated, pre-specified superiority testing (α = 0.05, Fisher’s exact test) was performed. Exploratory analyses of virological success by baseline subgroups were examined, based on age, sex, race, efavirenz [EFV] and non-EFV NNRTI use, and the number of prior ART regimens.
PROs were obtained at all visits and PRO evaluations used the Modified HIV Symptom Index.Citation13,14 The HIV Symptom Index (HIV-SI) is a validated tool to assess 20 commonly experienced symptoms. Modifications of the HIV-SI used in STRATEGY-NNRTI have previously been described, and included addition of symptoms which have been well described in relation to efavirenz-associated neuropsychiatric toxicity.Citation14 PROs were summarized with descriptive statistics (McNemar’s Test for within group comparisons and the Kruskal–Wallis test for comparison between groups). eGFR was estimated via Cockcroft–Gault method.Citation15 Laboratory parameters were assessed using a Wilcoxon Rank Sum test. All statistical analyses were completed using SAS (version 9.2).
Results
Out of 439 participants randomized, 434 were dosed (switch, n = 291; no-switch, n = 143) and 377 completed Week 96 (Figure ). The majority of enrolled participants were men (93%), white (78%), with a mean age of 41 years. At enrollment, 78% were receiving EFV-containing regimens, and the majority of these (74% of enrolled participants) were utilizing the EFV/FTC/TDF STR. The median time since first ART use for all subjects was 3 years (switch, 4 years; IQR: 2–5 years; no-switch, 3 years; IQR: 2–4 years). Most patients were using the EFV/FTC/TDF STR at screening, yet “treatment simplification,” was the most common reason given for study participation (48%). “Simplification” may include dosing with food or dosing at times other than before bedtime, rather than simply reduction in pill number, however, the enrollment questionnaire did not include this level of detail. Concern about current treatment side effects was the second most common reason given for enrollment (31%).
Figure 1 STRATEGY-NNRTI trial profile and disposition of subjects at Week 96
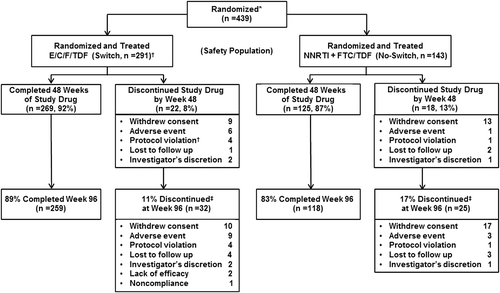
As was observed at Week 48, the switch regimen was non-inferior to the no-switch regimen at Week 96 (Table ). HIV-1 RNA < 50 copies/mL was maintained in 87% of switch and 80% of the no-switch group (6.1% difference; 95% CI: −1.3 to 14.2%, p = 0.12). Although there was a substantial numerical difference in virologic success (6.1%) that favored the switch at Week 96, pre-specified superiority testing did not demonstrate superior efficacy of the switch. The frequencies of VF by the FDA-defined Snapshot algorithm were similar between the two groups (2.8% switch, 1.4% no-switch), whereas a smaller proportion of the switch group had “no virological data in the Week 96 window” (11% vs. 18%; Table ). More discontinuations occurred in the no-switch group (7% switch; 15% no-switch) but in both groups most discontinuations were due to non-virologic reasons (all occurred in patients with HIV- RNA < 50 copies/mL at discontinuation) and included withdrawal of consent, lost to follow-up, or protocol violations. Changes from baseline in mean CD4 cell count were similar between the two groups (switch mean [SD]: 83 [167] cells/mmCitation3; no-switch: 101 [157] cells/mm3, p = 0.16).
Table 1 Analysis of efficacy (HIV-1 RNA < 50 copies/mL) at Week 48 and Week 96 (Snapshot)
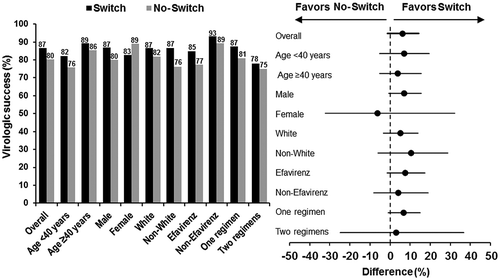
Analyses of response by baseline subgroups were supportive of the overall efficacy result at Week 96. Efficacy of the switch to E/C/F/TDF was numerically favored (but not significantly different (p > 0.05) in all subgroups (Figure ). Three participants (1 [<1%] switch; 2 [1%] no-switch) met criteria for resistance analysis. One participant (no-switch group) developed primary NNRTI resistance mutations (K101K/E, Y181Y/C); viral loads for the other two participants subsequently returned to HIV-1 RNA < 50 copies/mL, while on study drug. No drug resistance, including INSTI resistance, was observed to develop in the switch group.
Figure 2 Analysis of efficacy (HIV-1 RNA < 50 copies/mL, Snapshot) by baseline subgroup
The frequencies of adverse events (AE, any Grade) were comparable between the two groups (85% switch; 80% no-switch) (Table ). At Week 48, the occurrence of headache and nausea differed ≥ 5% between groups (switch vs. no-switch; headache, 10% vs. 3%, respectively; nausea, 8% vs. 3%, respectively)Citation10; by Week 96 the difference was 4% for headache and 6% for nausea (Table ). Consistent with prior studies of E/C/F/TDF, AEs in the switch group were predominantly Grade 1–2, and occurred in the early weeks following the switch to E/C/F/TDF and usually resolved on treatment.Citation10 Among the most common AEs on E/C/F/TDF, the incidence and prevalence of headache, diarrhea, and nausea were examined as these three AEs have been previously observed in other studies of E/C/F/TDF. The incidence of these three AEs was most common in the first 4 weeks of the study, and then declined in the following weeks and remained similar in prevalence to the no-switch group through Week 96 (Supplementary Figure 1). Most occurrences of these three AEs were Grade 1 in severity and did not lead to study drug discontinuation (Supplementary Figure 1). Grade 3–4 AEs occurred with similar frequency (9% switch; 8% no-switch), of which the majority were Grade 3 in both groups. AEs leading to discontinuation were infrequent (switch 3%; no-switch 2%). Three switch participants (1%) discontinued due to renal AEs, two of which were considered related to use of study drug; one subject with evidence of proximal renal tubulopathy disease (PRTD) at baseline developed acquired Fanconi syndrome before Week 48; symptoms resolved upon E/C/F/TDF discontinuation. The two other renal AEs leading to discontinuation were proteinuria (n = 1, considered related to study drug), and elevation in serum creatinine (n = 1, considered not related to study drug). One death (suicide) occurred in the switch group and was considered unrelated to drug. Three subjects had AEs leading to discontinuation in the switch group that were considered related to study drug including one subject with suicidal ideation, depression, and paranoia, and one subject each with dysgeusia, or prurigo. Two additional subjects had AEs leading to discontinuation that were not considered related to study drug including one subject with disturbance in attention, and one subject with arthralgia, coccydynia, paresthesia, muscle atrophy, and hypoesthesia who had a medical history of a fractured head of the right radius. In the no-switch group, three participants (2%) experienced AEs which resulted in discontinuation, including one subject with altered mood, one with anxiety and nightmares, and one with renal cancer. The AEs of altered mood, anxiety, and nightmares occurred in 2 subjects with prior medical history of mood change or depression, both of whom were on EFV plus FTC/TDF at baseline and on study, and these AEs were considered related to study medication. Grade 3–4 laboratory abnormalities also occurred with similar frequency between the two groups, (switch 13% and no-switch 16%). A small increase in serum creatinine (SCr) occurred in the switch group in the first weeks post switch and then remained stable through Week 96. Median changes in SCr from baseline to Week 96 (11.5 μmol/L; IQR: 6.2, 17.7, switch;0 μmol/L; IQR: −5.3, 7.1, no-switch) were similar to that observed in previous studies of E/C/F/TDF.Citation10
Table 2 Treatment emergent adverse events at Week 48 and Week 96
From baseline to Week 96, median changes in some lipid parameters showed small but significant declines (p < 0.05) in the switch group (total cholesterol; −0.13 mmol/L, IQR: −0.52, 0.36; switch; +0.05 mmol/L, IQR: −0.2, 0.54, no-switch; HDL cholesterol; −0.08 mmol/L, IQR −0.23, 0.08, switch; 0.0 mmol/L, IQR: −0.13, 0.15, no-switch). In those who switched from EFV specifically, both of these lipid parameters also decreased significantly (p < 0.05) compared to the no-switch group, and median change in LDL cholesterol also decreased significantly in the switch group vs. the no-switch group (LDL cholesterol; −0.16 mmol/L, IQR: −0.57, 0.21, switch; −0.05 mmol/L, IQR: −0.39, 0.39, no-switch). Median changes in triglycerides at Week 96 were not significantly different (switch vs. no-switch, overall:+0.03 mmol/L, IQR: −0.34, 0.35 vs. +0.01 mmol/L, IQR: −0.29, 0.37, p = 0.83; In those on EFV at baseline: +0.02 mmol/L, IQR: −0.34, 0.34 vs. +0.03 mmol/L, IQR: −0.34, 0.47, p = 0.43). Mean and median changes in total cholesterol/HDL ratios from baseline to Week 96 were also not significantly different (+0.1 for both the mean and median change in both groups, for both the overall population, and for those on EFV at baseline, p > 0.05 in all cases). A similar proportion of patients in both groups started or modified lipid lowering agents through Week 96 (1.7%, switch; 1.4%, no-switch).
Overall, participants who switched from their NNRTI-based regimens to E/C/F/TDF reported that relative to baseline they experienced significant improvements (p < 0.05) in multiple symptoms on the modified HIV Symptom Index. In contrast, within the no-switch group, no symptom demonstrated significant improvement relative to baseline. Symptoms demonstrating significant improvement in the switch group included anxiety, dizziness or light headedness, problems with memory, insomnia, vivid dreams, weird or intense dreams, and diarrhea or loose bowel movements. Symptoms decreased significantly as early as Week 4 following the switch, and this improvement was sustained through Week 96 (p < 0.05, for all noted symptoms). The proportions of participants reporting improvement of several of these symptoms also showed significant differences when comparing between the two groups, including insomnia, dizziness or light headedness, and vivid dreams. Nightmares were also significantly different (p < 0.05) in the between group comparison, but not the within group comparison.
Among those participants that specifically switched from EFV-based regimens, a similar pattern of improvement in neuropsychiatric symptoms relative to baseline was observed in the switch group but not the no-switch group (Supplementary Figures 2(A) and (B)). Neuropsychiatric symptoms that showed significant improvement in the switch group included anxiety, insomnia, dizziness (Supplementary Figure 2(A)) and vivid dreams, weird and intense dreams, and nightmares (Supplementary Figure 2(B)). Other symptoms also showed improvement including fatigue, fevers/chills/sweats, and diarrhea (data not shown). Insomnia, dizziness, fatigue, vivid dreams, weird and intense dreams, and nightmares, also showed significant differences (p < 0.05) favoring the switch when compared between the two groups. Most of these symptoms again showed significant improvement by Week 4. Additionally, in the switch group, four symptoms showed significant (p < 0.05) improvement at all timepoints from Week 4 to Week 96, including dizziness, vivid dreams, weird or intense dreams, and diarrhea or loose bowel movements.
Discussion
The STRATEGY-NNRTI study demonstrated that E/C/F/TDF is an efficacious, non-inferior, and safe option for virologically suppressed patients switching from an NNRTI + TVD regimen (predominantly EFV-containing regimens in this study).Citation10 The switch to E/C/F/TDF was associated with a low frequency of VF and no emergent drug resistance through Week 96. High and similar efficacy occurred in baseline subgroups defined by age, race, gender, EFV or non-EFV NNRTI use, and number of prior ART regimens. No new types of AEs occurred on the switch to E/C/F/TDF compared to Phase 3 trials of E/C/F/TDF in ART-naïve patients.Citation3,6 Renal AEs leading to discontinuation of E/C/F/TDF were infrequent, and consistent with previous studies investigating E/C/F/TDF.Citation3,6 Cobicistat can inhibit the multi-drug and toxin extrusion 1 (MATE-1) transporter, which mediates tubular creatinine secretion.Citation16,17 The resulting small increase in SCr (and corresponding small decrease in eGFR) occurred early on E/C/F/TDF.Citation10 Of three participants who discontinued E/C/F/TDF due to renal AEs, one with pre-existent PRTD developed Fanconi syndrome, which was considered by the investigator to be due to exposure to TDF. As has been observed in multiple clinical trials of TDF-containing regimens, including a study of E/C/F/TDF safety in patients with mild to moderate renal impairment,Citation18 discontinuations due to PRTD and Fanconi syndrome specifically were uncommon.
Limitations of the STRATEGY-NNRTI study included the open-label design, which may have introduced bias in investigator-reported AEs, discontinuations and patient-reported outcomes. Participants in the no-switch group who wanted to change regimens may have been more likely to discontinue treatment, potentially due to the dissatisfaction of not being enrolled in the switch group; this hypothesis is supported by the higher rates of withdrawal of consent in the no-switch group (3% vs. 12%). This difference largely contributed to the noted numerical differences in Week 96 efficacy between the two treatment arms. A double-blind, double-dummy study would remove these biases but would also introduce significant pill burden and prevent examination of the effect of the switch to a single-tablet regimen on treatment satisfaction. The HIV Treatment Satisfaction Questionnaire, a 10-item instrument with five items assessing general treatment satisfaction and five items assessing treatment ease was administered at Week 4 and Week 24 and showed significantly higher satisfaction for the switch group at Week 24.Citation14 The small number of women enrolled in the study also limited interpretation of the efficacy and safety for that gender subset. Other studies have examined the efficacy and safety of E/C/F/TDF in larger numbers of women, but all of these studies were in treatment-naïve patients, including Studies 102/103 and most recently, Study 0128 (WAVES; NCT01705574) which enrolled n = 575 women in a study comparing E/C/F/TDF vs. ATV + RTV + TVD.Citation19 Data on E/C/F/TDF use in women who are virologically suppressed and are switched to the single-tablet regimen remain limited to the current study and its sister study, STRATEGY-PI.Citation20
Nearly one-third of participants reported side effects from their current NNRTI + TVD regimen as a reason for enrollment, consistent with DHHS/EACS guidelines recommendations to improve tolerability by switching ARTs. Almost half of study participants also reported a desire for regimen simplification, which may relate to food requirements or the recommendation for bedtime dosing with EFV/FTC/TDF, although the study enrollment questionnaire did not capture this specific level of detail about reasons for enrollment.Citation1,2 The results of the patient-reported outcomes analysis mirror other studies demonstrating that switching from NNRTIs, and from EFV specifically, to other third agents can result in improvement of a variety of neuropsychiatric symptoms.Citation21–23 Most international HIV treatment guidelines have changed the status of EFV/FTC/TDF from a recommended to an alternative regimen for ART-naïve patients initiating therapy, reflecting EFV’s known neuropsychiatric side effect profile.Citation2,24 STRATEGY-NNRTI demonstrated that switching to E/C/F/TDF reduced multiple NNRTI-associated neuropsychiatric symptoms while maintaining once-daily dosing convenience, durable virologic suppression, and was not associated with the development of drug resistance through Week 96.
Declarations of interests
A. Pozniak reports grants from Gilead Sciences, during the conduct of the study; personal fees from Bristol Myers Squibb, Gilead Sciences, Janssen, Merck, and ViiV Healthcare, outside the submitted work; and is a panel member on EACS and BHIVA ARV Guidelines.
J. Flamm reports other from Gilead, during the conduct of the study and other from ViiV-GSK and Merck, outside the submitted work.
A. Antinori reports grants, personal fees, and non-financial support from Abbvie, Bristol Myers Squibb, Gilead Sciences, and ViiV Healthcare; personal fees from Merck and Janssen-Cilag, outside the submitted work.
M. Bloch reports grants from Gilead Sciences, during the conduct of the study; grants and personal fees from Bristol Myers Squibb, Gilead Sciences, Merck Sharp and Dohme, and ViiV Healthcare; personal fees from Abbvie, outside the submitted work.
D. Ward reports grants from Gilead Sciences, during the conduct of the study; grants and personal fees from Gilead Sciences, Merck, and ViiV Healthcare; personal fees from Bristol Myers Squibb, outside the submitted work.
J. Berenguer reports grants from Gilead, during the conduct of the study; grants and personal fees from Abbvie, Bristol Myers Squibb, Gilead Sciences, Janssen, and ViiV Healthcare; personal fees from Merck Sharp and Dohme, outside the submitted work.
P. Cote has no potential conflict of interest to report.
K. Andreatta, W. Garner, J. Szwarcberg, T. Nguyen-Cleary, D. McColl, and D. Piontkowsky are full time employees and shareholders of Gilead Sciences, Inc.
Funding
This trial was funded by Gilead Sciences [NCT01495702].
Notes on contributors
JS, WG, TN-C, KA, DM, and DP conceived and designed the study, obtained funding and ethics approval, analyzed the data, wrote the article in whole, and revised the article. AP enrolled study subjects, collected and analyzed the data, and reviewed and revised the article. JF enrolled study subjects, collected and analyzed the data, and reviewed and revised the article. AA enrolled study subjects, collected and analyzed the data, and reviewed and revised the article. MB enrolled study subjects, collected and analyzed the data, and reviewed and revised the article. DW enrolled study subjects, collected and analyzed the data, and reviewed and revised the article. JB enrolled study subjects, collected and analyzed the data, and reviewed and revised the article. PC enrolled study subjects, collected and analyzed the data, and reviewed and revised the article.
Supplemental materials
Supplemental data for this article can be accessed https://doi.org/10.1080/15284336.2017.1338844
YHCT_1338844_Supplementary_Material.zip
Download Zip (263.3 KB)Acknowledgments
We thank Raji Swamy and Yu Ning (statistical support), Hoa Chu and Jennifer DeMorin (data organization, review and editing) and the study team including Andrew Plummer, Ramin Ebrahimi, Helen Kuo, Eric Wang, Ting Bai, Sandy Shen, and Desiree Benavente.
Medical writing support was provided by Prescott Medical Communications Group (Chicago, IL, USA).
The authors wish to acknowledge the contributions of study investigator Professor Martin Fisher, from Brighton and Sussex University Hospital, who passed away on April 21st, 2015. He will be sadly missed.
Related Research Data
References
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. Developed by the DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents – A Working Group of the Office of AIDS Research Advisory Council (OARAC). http://aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Last Updated July 14, 2016.
- European AIDS Clinical Society (EACS). Guidelines Version 8.0 English. October 2015.
- Sax P, DeJesus E, Mills A, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet. 2012;379(9835):2439–2448.10.1016/S0140-6736(12)60917-9
- Zolopa A, Sax PE, Dejesus E, et al. A randomized, double-blind comparison of co-formulated elvitegravir/cobicistat/emtricitabine/tenofovir DF versus efavirenz/emtricitabine/tenofovir DF for initial treatment of HIV-1 infection: analysis of week 96 results. J Acquir Immune Defic Syndr. 2013 May 1; 63(1):96–100.
- Wohl DA, Cohen C, Gallant JE, et al. A Randomized, double-blind comparison of single-tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF versus single-tablet regimen efavirenz/emtricitabine/tenofovir DF for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2014;65(3):e118–e120.10.1097/QAI.0000000000000057
- DeJesus E, Rockstroh J, Henry K, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate versus ritonavir-boosted atazanavir plus co-formulated emtricitabine and tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet. 2012;379(9835):2429–2438.10.1016/S0140-6736(12)60918-0
- Rockstroh JK, DeJesus E, Henry K, et al. A randomized, double-blind comparison of co-formulated elvitegravir/cobicistat/emtricitabine/tenofovir versus ritonavir-boosted atazanavir plus co-formulated emtricitabine and tenofovir DF for initial treatment of HIV-1 infection: analysis of week 96 results. J Acquir Immune Defic Syndr. 2013;62(5):483–486.10.1097/QAI.0b013e318286415c
- Clumeck N, Molina J-M, Henry K, et al. A randomized, double-blind comparison of single-tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF vs ritonavir-boosted atazanavir plus emtricitabine/tenofovir DF for initial treatment of HIV-1 infection: analysis of week 144 results. JAIDS. 2014;65(3):e121–e123.
- STRIBILD®, Gilead Sciences Inc. STRIBILD® (elvitegravir, cobicistat, emtricitabine, tenofovir disoproxil fumarate) tablets, for oral use Revised ed. Foster City, CA: US Prescribing Information; April 2017.
- Pozniak A, Markowitz M, Mills A, et al. Switching to coformulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus continuation of non-nucleoside reverse transcriptase inhibitor with emtricitabine and tenofovir in virologically suppressed adults with HIV (STRATEGY-NNRTI): 48 week results of a randomised, open-label, phase 3b non-inferiority trial. Lancet Infect Dis. 2014;14:590–599.10.1016/S1473-3099(14)70796-0
- Dixon JR. The international conference on harmonization good clinical practice guideline. Qual Assur Good Pract Regul Law. 1999;6(2):65–74.
- U. S. Department of Health and Human Services, Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER). Human Immunodeficiency Virus-1 Infection: Developing Antiretroviral Drugs for Treatment. Guidance for Industry. Silver Spring, MD: Office of Communications, Division of Drug Information Center for Drug Evaluation and Research Food and Drug Administration. November 2015. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
- Justice AC, Holmes W, Gifford AL, et al. Development and validation of a self-completed HIV symptom index. J Clin Epidemiol. 2001;54(12):S77–S90.10.1016/S0895-4356(01)00449-8
- Mills A, Garner W, Pozniak A, et al. Patient-reported symptoms over 48 weeks in a randomized, open-label, phase IIIb non-inferiority trial of adults with HIV switching to co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir DF versus continuation of non-nucleoside reverse transcriptase inhibitor with emtricitabine and tenofovir DF. Patient. 2015;8:359–371.10.1007/s40271-015-0129-9
- Cockcroft D, Gault Henry. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41.10.1159/000180580
- Stray K, Bam R, Birkus G, et al. Evaluation of the effect of cobicistat on the in vitro renal transport and cytotoxicity potential of tenofovir. Antimicrob Agents Chemother. 2013;57(10):4982–4989.10.1128/AAC.00712-13
- German P, Liu HC, Szwarcberg J, et al. Effect of cobicistat on glomerular filtration rate in subjects with normal and impaired renal function. J Acquir Immune Defic Syndr. 2012;61(1):32–40.10.1097/QAI.0b013e3182645648
- Post FA, Winston J, Andrade-Villanueva JF, et al. Elvitegravir/cobicistat/emtricitabine/tenofovir DF in HIV-infected patients with mild-to-moderate renal impairment. J Acquir Immune Defic Syndr. 2015;68(3):310–313.10.1097/QAI.0000000000000476
- Squires K, Kityo C, Hodder S, et al. Integrase inhibitor versus protease inhibitor based regimen for HIV-1 infected women (WAVES): a randomised, controlled, double-blind, phase 3 study. Lancet HIV. 2016;3:e410–e420.10.1016/S2352-3018(16)30016-9
- Arribas JR, Pialoux G, Gathe J, et al. Simplification to coformulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus continuation of ritonavir-boosted protease inhibitor with emtricitabine and tenofovir in adults with virologically suppressed HIV (STRATEGY-PI): 48 week results of a randomised, open-label, phase 3b, non-inferiority trial. Lancet Infect Dis. 2014;14(7):581–589.10.1016/S1473-3099(14)70782-0
- Nguyen A, Calmy A, Delhumeau C, et al. A randomized cross-over study to compare raltegravir and efavirenz (SWITCH-ER study). AIDS. 2011;25(12):1481–1487.10.1097/QAD.0b013e328348dab0
- Nelson M, Winston A, Waters L, et al. Multicentre open-label study of switching from atripla to eviplera for possible efavirenz associated CNS toxicity. Paper presented at: The 53rd ICAAC Interscience Conference on Antimicrobial Agents and Chemotherapy. Denver, CO; 2013. Abstract H-672b.
- Waters L, Fisher M, Winston A, et al. A phase IV, double-blind, multicentre, randomized, placebo-controlled, pilot study to assess the feasibility of switching individuals receiving efavirenz with continuing central nervous system adverse events to etravirine. AIDS. 2011;25(1):65–71.10.1097/QAD.0b013e328341685b
- British HIV Association. British HIV Association guidelines for the treatment of HIV-1-positive adults with antiretroviral therapy 2015 (2016 interim update). 2016.