
Abstract
Pre-exposure prophylaxis (PrEP) has demonstrated remarkable effectiveness protecting at-risk individuals from HIV-1 infection. Despite this record of effectiveness, concerns persist about the diminished protective effect observed in women compared with men and the influence of adherence and risk behaviors on effectiveness in targeted subpopulations. Furthermore, the high prophylactic efficacy of the first PrEP agent, tenofovir disoproxil fumarate/emtricitabine (TDF/FTC), presents challenges for demonstrating the efficacy of new candidates. Trials of new agents would typically require use of non-inferiority (NI) designs in which acceptable efficacy for an experimental agent is determined using pre-defined margins based on the efficacy of the proven active comparator (i.e. TDF/FTC) in placebo-controlled trials. Setting NI margins is a critical step in designing registrational studies. Under- or over-estimation of the margin can call into question the utility of the study in the registration package. The dependence on previous placebo-controlled trials introduces the same issues as external/historical controls. These issues will need to be addressed using trial design features such as re-estimated NI margins, enrichment strategies, run-in periods, crossover between study arms, and adaptive re-estimation of sample sizes. These measures and other innovations can help to ensure that new PrEP agents are made available to the public using stringent standards of evidence.
Introduction
Pre-exposure prophylaxis (PrEP) against HIV-1 acquisition provides a defense in the fight against the HIV global pandemic. Numerous trials have shown the efficacy of PrEP in providing protection, but substantial work remains to promote access and adherence, understand potential safety issues (particularly long-term side effects), and develop a broader array of PrEP products to meet the diverse needs of people at high risk of HIV-1 infection. Having a broad array of PrEP products, either as new modalities, new technologies, or new agents, would provide important options to individuals seeking protection from HIV-1 infection. In this article, we summarize the current state of knowledge regarding late-stage PrEP study design, discuss specific issues encountered in prior studies, and suggest innovations for smaller trials that retain a level of sensitivity sufficient to detect meaningful effects of preventive interventions.
A substantial and growing body of evidence supports the use of daily, oral tenofovir disoproxil fumarate/emtricitabine (TDF/FTC) to protect against HIV-1. Oral TDF/FTC was approved for use as PrEP by the US Food and Drug Administration (FDA) in 2012,Citation1,2 South Africa’s Medicines Control Council in 2015,Citation3 and the European Medicines Agency in 2016.Citation4 The World Health Organization (WHO) recently revised its antiretroviral guidelines to recommend oral PrEP containing TDF as a prevention option to all people at substantial risk of acquiring HIV-1,Citation5 suggesting that TDF/FTC will become a critical component of the HIV-1 prevention effort.
The efficacy of TDF/FTC is remarkable, with high protection demonstrated in highly adherent populations.Citation6–10 Various lines of evidence support a high degree of protection if the concentration of active drug is sufficiently high when an individual is exposed to HIV-1, especially among men who have sex with men (MSM).Citation6–9 In the IPrEx trial, the relative risk (RR) of HIV-1 acquisition was reduced by an estimated 92% (95% confidence interval [CI], 40–99; p < 0.001) among participants with detectable levels of TDF/FTC compared with participants without detectable levels.Citation6 The regimen resulted in an 86% reduction in HIV-1 acquisition when taken on demand in the IPERGAY study (n = 445)Citation8 and when taken daily in the PROUD study (n = 544).Citation7 Both incidents of post-enrollment HIV-1 infection in the arm of the PROUD study that received TDF/FTC immediately (n = 275) occurred in individuals who seemed to have suboptimal adherence.Citation7 Additionally, no HIV-1 diagnoses were reported during 388 person-years of follow-up (upper limit of 1-sided 97.5% CI, 1.0) in a cohort study in San Francisco, California.Citation9
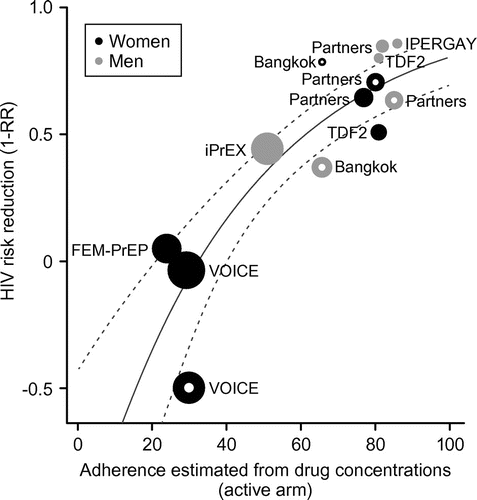
Despite the positive results in these studies in MSM, concerns have been raised about the effectiveness of PrEP in women. Some women-only studies failed to demonstrate significantly reduced risk of HIV-1 infectionCitation11,12 in contrast to positive findings in trials that enrolled both men and women.Citation10,13,14 While there may be biological explanations for this disparity, including the lower concentrations of TDF and FTC metabolites that have been detected in vaginal mucosa compared with rectal mucosa,Citation15 there is a strong correlation between adherence and observed efficacyCitation16 (Figure ). The two major trials that failed to show effectiveness of daily TDF/FTC in women (VOICE and FEM-PrEP)Citation11,12 also identified low levels of adherence (21–30%). However, in trials in which women were more adherent to a daily regimen, a significantly reduced risk of HIV-1 acquisition was demonstrated.Citation10,13 In the Partners PrEP study, which used daily tenofovir, risk of HIV-1 acquisition in women was reduced by 71% versus placebo (p = 0.002),Citation10 and in the TDF2 Study Group trial, the protective efficacy of TDF in the as - treated cohort of women was 75% versus placebo (p = 0.02).Citation13 In the most recent prevention studies in women, a monthly vaginal ring containing dapivirine (DPV) reduced the risk of HIV-1 infection among African women (27% lower than placebo in ASPIRE and 31% lower in the Ring Study), particularly in subgroups with evidence of increased adherence.Citation17,18 When viewed together, the PrEP trial results show a strong association between trial-level adherence and efficacy for both men and women. While it may not account for all the variability, addressing these disparities in adherence and efficacy in PrEP trials for different risk populations remains a challenge for the design of future HIV-1 prevention research. The trial design features discussed in this article offer innovations that can help to ensure that new PrEP agents available to the public adhere to stringent standards of evidence for regulatory authorities and healthcare professionals.
Figure 1 Relative risk reduction values from the major PrEP trials for men and women according to adherence (measured by plasma level of TDF). The solid line represents the meta-regression fit for all groups combined, and the dashed lines represent the 95% confidence intervals for the regression line. Plot circle size is proportional to the number of events observed in each study. Hollow points show studies (or arms) comparing TDF to placebo, filled points depict TDF/FTC studies. FTC, emtricitabine; PrEP, pre-exposure prophylaxis; RR, relative risk; TDF, tenofovir disoproxil fumarate
General issues for design
Until validated surrogate endpoint(s) for HIV-1 infection or markers of product activity are identified, late-stage clinical trials will continue to use HIV-1 seroconversion as the primary endpoint.Citation19 Given its proven effectiveness and approvals, TDF/FTC is likely to be used as an active control in clinical trials evaluating new agents for PrEP. In an active-controlled study, the trial hypothesis may be a non-inferiority (NI) test, a superiority test, or nested hypotheses, first evaluating NI and then superiority. Superiority studies are appropriate when there is a realistic expectation that the experimental agent will reduce the infection rate below that seen with the active-control agent. Non-inferiority studies are possible once an active control is proven effective and when it could be ethically acceptable to sacrifice some small degree of the efficacy associated with the active control. Despite their complications,Citation20,21 NI designs are likely to be chosen for new PrEP agent studies after careful consideration of three main issues. First, it may not be realistic to expect a new product to reduce the infection rate below that seen with TDF/FTC given its high effectiveness in adherent populations. Second, a new product that offers advantages in either adherence (e.g. long-acting injectable or implantable PrEP) or safety profile would likely be considered acceptable even if it were slightly less effective than oral TDF/FTC. Finally, use of a placebo control may be considered unethical when TDF/FTC (or another agent) has been established to be effective in a risk population. Although future trials will undoubtedly include NI designs, the feasibility challenges of current approaches make it important to consider alternatives that offer innovative solutions.
Non-inferiority margins
The NI margin is the degree to which the experimental intervention can have lower efficacy than the active control without being considered clinically unacceptably worse. At minimum, the NI margin must be set to retain some superiority over no pharmaceutical intervention (NPI) to ensure superiority over a hypothetical placebo arm. The term “NPI” reflects the fact that the assignment is not strictly to placebo but also includes the counseling package for prevention. To make a comparison with an active control, NI trials make an assumption of constancy under which the benefit of an active agent over placebo seen in previous studies applies in the new trial setting. Defining the NI margin requires knowledge of the benefit provided by the active control, preferably based on multiple high-quality controlled trials of the active control versus placebo. The lower bound of that known efficacy is referred to as the M1 margin by FDA guidelines and is estimated based on the lower limit of the 95% CI from a meta-analysis of existing placebo-controlled trials.Citation20–22 This approach provides a conservative estimate of efficacy, acknowledging the uncertainties of sampling variation and the potential that the constancy assumption may not be perfectly satisfied in a new study.
Establishing the NI margin requires an assumption about the “clinically acceptable” degree of inferiorityCitation21 or the proportion of the active comparator drug effect that must be preserved. This is the M2 margin, which is always stricter than M1.Citation23 The M2 margin is typically set to preserve a fixed proportion of M1 because it is believed to be clinically and ethically important that a new prevention modality not just provide minimal efficacy but also preserve a meaningful amount of the active-control effect. One common approach is to set the M2 margin to preserve 50% of the benefit ensured by the M1 margin. In a successful trial, the upper 95% confidence bound on the relative efficacy rate (experimental treatment vs active control) will fall below the pre-specified M2 margin.
To begin to determine the NI margin for the likely comparator for many future studies of PrEP, we conducted a meta-regression of data from FEM-PrEP,Citation12 VOICE,Citation11 iPrEX,Citation6 Bangkok,Citation14 Partners PrEP,Citation10 TDF2 (Botswana),Citation13 and IPERGAY.Citation8 The PROUD study resultsCitation7 were not included in the model due to lack of a parallel adherence measure. Adherence was assessed by measuring plasma concentrations of tenofovir; however, the threshold for defining adherence was not the same in all trials. Threshold values ranged from 0.1 to 10 ng/mL, but most trials used a threshold of 0.31 ng/mL. Results demonstrated a clear and consistent association between trial-level adherence and TDF/FTC efficacy (Figure ). The meta-analysis allows the estimation of the observed (RR estimate) and demonstrated (RR upper bound) effect of TDF/FTC, conditional on sex and a given level of adherence.
Table provides estimates of the demonstrated effect for men and women assuming 45, 65, and 85% adherence rates, as well as potential NI margins. For adherence of 45%, TDF/FTC exhibits a modest but significant improvement compared with NPI (demonstrated effects of 0.98 and 0.96 in men and women, respectively). As adherence increases so does the demonstrated effect of TDF/FTC. Table also shows the consequent M2 margins derived from these estimated effects. With the lowest levels of adherence and similarity among treatment efficacies, the impracticality of conducting an NI study is obvious because it could require more than 100,000 HIV infections. Yet, as the estimated effect of TDF increases, the NI margin becomes wider (from 1.02 to 1.42 for women). Similar estimates could be generated for any trial based on the projected population and level of adherence.
Table 1 Meta-regression of data from FEM-PrEP,Citation12 VOICE,Citation11 iPrEX,Citation6 Bangkok,Citation14 Partners PrEP,Citation10 TDF2 (Botswana),Citation13 and IPERGAYCitation8: Sex-specific margins based on combined model
Sample size
PrEP trials have traditionally assessed the relative reduction of HIV-1 infection between arms during the trial period by monitoring the occurrence and timing of HIV-1 infections. For these trials, in addition to alpha (the probability of a type 1 error) and power, sample size depends on two factors: the signal (i.e. treatment difference) that the trial must detect and the incidence rate in the population to be studied.Citation20 The former determines the number of events required and the latter determines how many person-years are required to observe those events. In a superiority study, the treatment difference is the expected reduction, or perhaps clinically meaningful reduction, in the infection rate in the experimental arm compared with the control arm. In an NI study, the treatment difference is the potential acceptable loss of efficacy or M2.Citation20 Represented by the hazard ratio (HR), the H0 for a superiority test would typically be that HR ≥ 1 (no difference or worse) and for an NI test that HR ≥ M2 (difference as bad as or worse than M2). The H1 for a superiority test would be that HR is, for example, 0.8 (experimental is 20% better than control) and for an NI test that HR < 1 (no difference or better than control). Table shows sample size considerations for NI and superiority hypotheses under various assumptions for men and women using results from the meta-regression described in Table . For NI hypotheses, the demonstrated effect of TDF/FTC and the width of the NI margin correlate directly with adherence. Thus, the number of events required to demonstrate NI decreases as adherence increases. For superiority hypotheses, the assumed effectiveness of an experimental agent compared with control decreases as adherence rises and, consequently, the sample size required to show superiority increases.
Table 2 Sample size considerations for NI and superiority hypotheses under various assumptions for men and women using results from the meta-regression
Blinding
Whether PrEP trials should be blinded or unblinded was heavily debated in the microbicide field. Arguments for having an unblinded condom-only or no-gel arm in addition to a gel-placebo arm were made by Fleming and RichardsonCitation24 in 2004 and debated in subsequent correspondence.Citation25–29 The main argument at that time in favor of an unblinded control group was doubt as to whether the placebo was truly inert or did provide some protection against HIV-1 infection through, for example, increased lubrication or dilution of semen.Citation28 It was also argued that having a condom-only control group permits measurement of real-world effectiveness and accounts for behavior change, which may be associated with knowledge of PrEP use.Citation25
These debates were partially informed by the HPTN035 study that included both a gel-placebo and a condom-only control armCitation30 and demonstrated no difference between the two control arms in HIV-1 risk behavior, pregnancy rates, or HIV-1 or other sexually transmitted infection rates. This suggested that sexual behavior was not affected by lack of blinding, but it provided no insight on whether adherence was affected. For trials that measure efficacy without a need to evaluate patient preference, it may be preferable to include a blinded comparison group, particularly when the routes of administration are similar.
However, debates continue as to whether treatment blinding is necessary or not when administration routes substantially differ (e.g. injectable vs oral treatment). An open-label design would enable the evaluation of patient preference for the different modes of drug delivery, with adherence not being impacted by the double-dummy requirements for a blinded comparison. In addition, the conduct of the study would not be encumbered by the complexity of administering double-dummy products (e.g. sham injections). Guarding against the introduction of bias would be an important consideration, although that would be somewhat mitigated because the endpoint of seroconversion is objective rather than subjective.
Base-case non-inferiority design and sample size
The meta-analysis of historical studies previously described yields estimates of the efficacy of TDF/FTC over placebo for a given level of adherence. Table outlines considerations for trial designs in different populations. TDF/FTC will likely be included as the active control in future PrEP trials among MSM populations. For this analysis, we assumed that adherence to TDF/FTC would be 65%, leading to an NI margin of 1.3 among MSM (per Table ).
Table 3 Summary of potential trial designs for different populationsTable Footnotea
The anticipated reduction in infection rate is dependent on the investigational agent. For studies of oral agents or new dosing regimens for TDF/FTC in men, there is little reason to expect an improvement in efficacy. These studies are therefore classic NI designs with 611 events potentially required.
Long-acting formulations or vaccines may address the adherence challenges for daily oral PrEP. Such an experimental intervention could overcome the challenge of uncertain adherence in other settings, because exposure would be directly observed in these cases and, thus, known. If such an intervention were expected to be 80% effective compared with NPI, making the incidence on this intervention roughly one-half that seen on TDF/FTC, 72 events would be required to test a superiority hypothesis.
The anticipated reduction in infection rate is also dependent on some amount of nonadherence to TDF/FTC. If adherence to TDF/FTC is 85% (instead of 65%) and its efficacy relative to NPI is 72%, the effectiveness of a vaccine/long-acting agent with 80% efficacy relative to NPI is only slightly superior to that of TDF/FTC (Table ). Thus, a larger sample size would be required for adequate power to demonstrate superiority (n = 372 events).
Current WHO guidelines recommend offering oral PrEP containing TDF as part of the prevention package to all people at substantial risk of HIV infection.Citation5 This implies that the control arm in prevention trials among women will likely provide participants with TDF, raising the possibility of employing an NI design. Without improved adherence, however, it is not possible to define an NI margin for the use of TDF/FTC in women because it has not reliably demonstrated improvement over placebo.
It is possible to define a margin for DPV rings as a comparator, albeit one that is so narrow (NI margin, 1.02 at 45% adherence) that an NI study would require a prohibitive number of events (n = 110,028). The base-case sample size is only feasible for agents with a reasonable possibility of superiority to the comparator. We assumed an adherence rate of 45%, the upper end of that seen in studies of women (excepting serodiscordant couples). With assumed effectiveness of an experimental agent over a control of 71%, a superiority study in this setting would require 24 events. In contrast, some of the sample sizes described in Table are prohibitive. The power of a study is often dependent on the rate of adherence to the active control in the trial, yet this cannot be predicted reliably when a study is being planned. Given the relationship between adherence and efficacy, and the growing body of evidence supporting advances in PrEP, it is conceivable that adherence rates in women might improve. It is therefore worth considering innovations that could reduce sample sizes or lead to more reliable inferences about the relative benefits of treatment options.
Potential design innovations
Combined non-inferiority/superiority designs
Concerns over sample size can sometimes be managed by combining NI and superiority endpoints in a trial design with an active control. In a superiority study among MSM with an assumed 65% adherence rate to TDF/FTC, H0 is no difference and H1 is a relative difference of at least 54% (HR = 0.46). In this setting, the signal is a difference of 54% (Table ). If a degree of clinical inferiority, such as HR = 1.3, is acceptable, then H0 is HR = 1.3 and H1 is HR = 0.46, making the signal a relative difference of 65% and requiring 40 events instead of 611. Similarly, a standard NI study among women using DPV rings as a comparator requires more than 100,000 events. An agent with a reasonable expectation of 74% efficacy over DPV could be studied in an NI/superiority design with 23 events.
Such a bare-minimum sample size has risks. The first example has 90% power to show NI (to beat a worst-case scenario of HR = 1.3) but not to show superiority (beating a no-difference scenario of HR = 1). If the true benefit of the investigational intervention does not match its assumed value (or adherence to TDF/FTC is greater than expected), there may not even be 90% power to show NI. The target populations for superiority and NI trials differ, whereas NI studies require conditions of moderate-to-high adherence to justify the constancy assumption.
Pre-specified re-estimation of non-inferiority margins
Adherence is not reliably predictable, especially with participant-controlled dosing. The iPrEX study found moderate adherence, moderate efficacy (50% reduction in infection rates), and a 2–3% per annum rate of infection for patients on TDF/FTC.Citation6 The IPERGAY and PROUD studies demonstrated greater adherence, greater efficacy (~85% reduction in infection rates), and a lower infection rate.Citation7,8 If adherence to the active control in the new trial is lower than in previous trials, its effect (relative to placebo) in the new trial will be lower than expected and the pre-defined NI margin too generous. This could lead to acceptance of an experimental drug that does not provide benefit. Alternatively, adherence rates may be higher than in prior trials, making the pre-specified M2 margin too stringent, leading to the inappropriate rejection of a new agent.
By using an objective laboratory measure of drug adherence, together with a model for the relationship between drug concentrations and reduced HIV-1 incidence, it may be possible to pre-specify adjusting the NI marginCitation16 to a margin that corresponds to the observed active-control arm adherence in the trial. For instance, the adherence/efficacy association can be quantified using meta-analysis (Figure ) and adherence measured in the active-control arm in the new trial (using the same plasma-level concentration of the control arm study drug). These adherence measures can be used to estimate the effect of the active control compared with a hypothetical placebo arm (M1). The NI margin used to assess the new therapy can then be re-computed based on the estimated M1 margin, including corrections that preserve an appropriate pre-specified level of benefit relative to placebo.
There is tension between the need to state an a priori standard for establishing NI and the desire to choose a margin that will correctly characterize the efficacy of the active control in the NI trial. Careful study of the statistical and operational implications of re-estimating the margin is needed. The precise formula and algorithm to be used for margin re-estimation would need to be pre-specified in the protocol.
Enrichment approaches to trial enrollment
Enrichment refers to preferential enrollment of certain participants in a study. A biomarker present at randomization can be used to determine whether individuals belong to a subgroup with characteristics that might offer specific advantages to trial outcomes. Adaptive enrichment is a variation in which interim analyses are conducted on observed efficacy in subgroups to determine which types of individuals to continue enrolling, with eligibility criteria updated adaptively. These designs preserve type 1 error and may provide an increase in power.
Selection of study participants and settings is important and guided by current ethics guidelines. The likelihood of seeing an effect of a preventive product is increased by enrolling a population at higher risk of HIV-1 infection (prognostic enrichment). Another type of enrichment would be to choose those likely to respond to the preventive drug, or those likely to use the experimental agent while less likely to adhere to the active-control agent (predictive enrichment).Citation31 Successful outcomes are favored by low heterogeneity of a population, decreasing nondrug-related variability primarily by improving rates of adherence. If we could rely on participant characteristics observed in previous trials that correlate with high rates of adherence to the experimental intervention or high risk of HIV-1 infection, we could use pre-randomization characteristics of the current trial to continue preferentially enrolling subjects who are likely to be highly adherent to the experimental agent or likely to be at high risk or both.
Run-in designs
A run-in period is the time before randomization in a clinical trial during which no treatment is given but specific characteristics are evaluated (e.g. adherence to an inactive but measurable compound). Data from this stage of the trial are used as a baseline stratification factor or to characterize noncompliant participants. The run-in period is an example of an enrichment strategy and can be used to encourage adherence by making participants aware of the conditions and demands of the trial.Citation23
The duration of the run-in period should be carefully considered. A short run-in period may not provide realistic estimates of the adherence rates expected during a long study. A long run-in period increases the cost of the study without providing data addressing the primary and secondary objectives.
At the end of the run-in period, an assessment of adherence could be used to identify levels for a stratified randomization or to cap the number of participants with low adherence (for an NI study) or with high-adherence (for a superiority study) levels. If adopted, the run-in period will increase the overall study duration and the number of individuals required at screening to enroll participants who meet enrichment criteria. Therefore, this approach may not lead consistently to cost reductions, and it can be expected to produce benefits for the trial only if adherence can be measured reliably at the end of the run-in period.
Crossover designs
In the crossover family of designs, trial participants are randomly assigned to a new agent or a control drug, assessed for a defined period of time, and then switched to the opposite treatment arm and reassessed.Citation32 Although they were once thought to be inappropriate for absorbing endpoints such as HIV-1 infection, crossover designs have been shown to be statistically valid and efficient under certain circumstances.Citation32–34 For a superiority study, a crossover design has the same efficiency as a parallel design in the absence of heterogeneity. The crossover design gains potentially substantial efficiency as heterogeneity increases. An advantage of crossover designs is that they do not require measurement of heterogeneity (both in infection risk and treatment adherence) to control for it. However, if heterogeneity can be measured and controlled by an approach such as stratification, the advantage of the crossover design may be diminished. There are operational challenges to a crossover design, including the time needed to observe trial participants for two time periods rather than one, the issue of seroconversion in period one, the potential for carryover effects, and a probable increase in discontinuation rates. This innovation is not appropriate for vaccines or agents with long half-lives due to carryover. Therefore, it would be most useful for oral agents for which NI designs are the norm. However, methodological research and regulatory scrutiny of this design should be conducted to enable assessment of its potential for future studies.
Adaptive re-estimation of sample size
During a study, the overall event rate (pooled from both arms) can be compared with the assumptions used in planning. If the data are examined in a blinded analysis, statistical bias is not a concern, and the sample size can be adapted with no statistical adjustments required. In contrast, a change in study sample size related to an unblinded data analysis (using the observed treatment effect or infection rate in one arm) can increase the type 1 error rate. However, regulatory guidance provides established methods for making these adjustments.Citation21,35
The uncertainty about adherence to protocol medication schedules or the infection rate in a given population during a trial make PrEP studies natural candidates for ongoing monitoring of each of these factors with clear guidelines for adaptations to trial characteristics (curtailment or changes in sample size) in the event of significant differences between observed and planned trial characteristics.
Addressing an anticipated result of low incidence(s)
In a successful NI study, low incidence rates might be observed in both arms in the new trial. Whether or not the new agent is effective is not obvious because the observation could be explained by two possible scenarios. In Scenario 1, the new trial may have been conducted in a population with a low underlying risk of HIV-1 infection with various levels of adherence to PrEP, and the trial simply has insufficient data to establish effectiveness. In Scenario 2, the trial may have been conducted in a population with a high underlying risk of HIV-1 infection with high levels of adherence in both study arms. The efficacy of a new intervention as a PrEP agent relative to the standard of care can only be demonstrated in Scenario 2. To separate these explanations, the key issue is establishing the underlying HIV-1 infection risk without pharmaceutical intervention in the study population. Knowing the outcomes of placebo would provide a useful context for interpreting a treatment effect. However, a rigorous estimate of the placebo effect is difficult in practical terms. An idealized trial design would incorporate a contemporaneous control group, such as a randomized no-treatment arm, but this is ethically unacceptable in many contexts. Hence, the NPI risk of a trial population must be estimated by other means.
There are certain populations (e.g. perinatal transmission, serodiscordant couples) for whom the risk of HIV transmission is from a known source and thus ongoing and well characterized. Predictions based on the observed rates of infection in one population can be adjusted to account for different distributions of baseline characteristics.Citation36 A compelling reduction from a projected risk to an observed risk can add indirect evidence to the case for Scenario 2 rather than Scenario 1 previously discussed.
Using external historical controls (including participants from the preparedness phase when a clinical trial is planned) is an inferior option because of the concern that HIV-1 infection rates may be based on a group who no longer resembles the trial population. However, in light of the ethical considerations and current WHO guidelines, as well as the challenges of planning and conducting extremely large complicated NI trials, if it is clear that the risk of HIV-1 exposure remains consistent and the resulting HIV-1 reduction is compelling, such an alternative design may warrant careful consideration.
A change in perspective: additive and relative scales in hypothesis testing
One formidable challenge that confronts investigators in active-controlled trials of PrEP interventions is the heterogeneity of effect sizes in the intent-to-treat (ITT) populations across trials, which is probably driven by variable adherence levels across populations. This variation makes defining NI margins challenging, particularly on a multiplicative scale.Citation37 To illustrate, consider a scenario with a new agent that is 70% as effective as TDF/FTC with an ideal level of adherence. If implemented in a population for whom adherence to TDF/FTC yields an ITT effectiveness of 90%, the net effectiveness of the new agent is 63% in this population – a substantial level of protection. A regulatory agency would evaluate the new agent on the strength of this evidence. However, in a population with lower adherence levels in which the ITT effectiveness of TDF/FTC is 50%, the 70% effectiveness relative to TDF/FTC would yield a net effectiveness of 35%. Hence, it is difficult to specify a single multiplicative margin that would be interpreted in the same way for these diverse scenarios. This is the major motivation for the discussion mentioned previously regarding the pre-specified approach to re-estimating the NI margin based on the observed adherence level in the trial relative to the assumed adherence level that was used for planning purposes. However, the additive scale may be worth considering, namely the rate difference rather than the ratio of rates. Both of these previously mentioned scenarios assume the new agent produces an RR reduction that is 70% of the reduction produced by TDF/FTC. With a background HIV infection rate of 3 per 100 person-years for a cohort of 10,000 individuals followed for 1 year, 300 infections would be expected for NPI compared with 30 infections for active-control treatment and 111 for new treatment (Scenario 1). With a background HIV infection rate of 8 per 100 person-years for a cohort of 10,000 individuals followed for 1 year, 800 infections would be expected for NPI compared with 80 infections for active-control treatment and 296 infections for new treatment (Scenario 2). The rate difference in Scenario 1 is 81 additional infections on the test treatment compared with Scenario 2 with 216 additional infections on the test treatment. These considerations can also be applied to the justification of NI margins. A margin of 1.22 requires 1062 events, and a margin of 1.3 requires 611 events. The difference between these margins may seem substantial on the relative scale. If the background rate of infection is 6 per 100 person-years under NPI, this difference in margins could correspond to assumed infection rates on control of 2.88% versus 2.58% (Table ). An intervention approved under the broader margin would allow for an extra 30 infections in a cohort of 10,000 people followed for 1 year. This information could be helpful in the evaluation of the clinical acceptability of different NI margins.
Combining historical controls and the additive scale
An innovative solution would be to consider a process that first tests for non-inferiority between the experimental agent and the control on the additive scale (i.e. the rate difference) and then demonstrates a compelling relative reduction from the projected risk per the background incidence to the observed risk through a single-arm approach using historical controls as described above.
Table shows the effect of rate differences using different NI margins on power. Lower incidence rates in the treated groups and numbers of incident infections are associated with greater power, which is the opposite of inference on a rate-ratio scale. The increased power in the lower incidence rate cases derives from an assumption of a much higher RR margin. The definition of an acceptable NI margin (both scale and size) is a challenging issue. For example, should this be a function of the estimated underlying incidence of HIV-1 infection in the study population or of the incidence of HIV-1 infection anticipated in the TDF/FTC arm? To illustrate, excluding a rate difference of 0.5 events per 100 person-years requires a very large trial, whereas excluding a rate difference of 2.0 events per 100 person-years may be achievable with a trial of several hundred participants (Table ). A decision could be made based on clinical judgment depending on the environment surrounding the trial itself, the treatments involved in the trial, the uptake of PrEP in the local setting, and reaching consensus on the largest clinically acceptable difference.
Table 4 Power based on rate difference sample size assumptionsTable Footnotea
It is important to re-emphasize that supplementary evidence of a high underlying risk of HIV-1 infection in the study population is essential for the trial to be interpretable. The data from historical controls previously described could be used in projecting what that underlying risk would be.
Conclusion
Important advances have been made in developing effective agents to prevent HIV-1 infection, particularly in men. While these developments provide tremendous benefits for individuals interested in taking PrEP, they also impose considerable hurdles for the development of new PrEP agents. In the context of low incidence of HIV-1 infection and high-adherence rates, traditionally designed non-inferiority trials may require unrealistically large sample sizes.
Even feasible non-inferiority studies face further challenges: the difficulty of attributing uniformly low infection rates to the successful interventions and the difficulty of predicting adherence (and any consequent expectations of superiority or non-inferiority margins) in the participants who enter the study.
We propose several innovations to address these challenges, each of which may be suitable in a different intervention or trial setting. The interventions have the potential to reduce the sample size needed to achieve acceptable power. For example, for studies exploring a long-acting agent with expectations of better adherence than TDF/FTC, a trial could incorporate a run-in period during which adherence measures for a non-active drug are used to stratify the population into rates of low- and high-adherence groups with a primary assessment of superiority taking place in the low-adherence group, with sample-size re-estimation used to adjust the sample size to match the infection rate in that randomized subset.
In an NI setting, a run-in period could potentially be used to estimate the incidence rate of infection among all enrolled participants, with a re-estimated NI margin pre-specified in the protocol allowing the final analysis to use a margin relevant to the population recruited in the study.
Innovative solutions are needed to ensure that new PrEP agents can be made available to the public while upholding appropriate standards of evidence for regulatory authorities and healthcare professionals and maintaining realistic trial sizes.
Declaration of interest
AC and RLC are the employees of ViiV Healthcare and stockholders in GlaxoSmithKline. DD reports grants from the National Institutes of Health during preparation of the submitted work and from the Bill and Melinda Gates Foundation outside the submitted work. DTD was supported by the UK Medical Research Council (MR_UU_12023/23) during preparation of and outside the submitted work. DVG reports personal fees from ViiV Healthcare outside the submitted work. BSS reports personal fees from GSK and ViiV Healthcare during preparation of and outside the submitted work. RDM and RW are the employees of Pfizer. AG and BH report no declarations of interest.
Funding
Funding for this work was provided by ViiV Healthcare, including editorial assistance under the direction of the authors. All listed authors meet the criteria for authorship set forth by the International Committee of Medical Journal Editors.
Contributors
AC, DD, DTD, DVG, AG, BH, BSS, RDM, RW, and RLC jointly conceived and designed, wrote, and reviewed and revised this manuscript.
Acknowledgments
The authors wish to acknowledge the following individuals for editorial assistance during the development of this manuscript: Anthony Hutchinson and Diane Neer at MedThink SciCom, Cary, NC.
References
- Truvada [package insert]. Foster City, CA: Gilead Sciences, Inc; 2013.
- US Food and Drug Administration. FDA approves first drug for reducing the risk of sexually acquired HIV infection [press release]. https://aidsinfo.nih.gov/news/1254/fda-approves-first-drug-for-reducing-the-risk-of-sexually-acquired-hiv-infection. Accessed September 22, 2017.
- Medicines Control Council (South Africa). Medicines Control Council approves fixed-dose combination of tenofovir disoproxyl fumarate and emtricitabine for pre-exposure prophylaxis of HIV [press release]. http://www.mccza.com/documents/2e4b3a5310.11_Media_release_ARV_FDC_PrEP_Nov15_v1.pdf. Accessed March 15, 2017.
- European Commission grants marketing authorization for Gilead’s once-daily Truvada® for reducing the risk of sexually acquired HIV-1 [press release]. https://www.gilead.com/news/press-releases/2016/8/european-commission-grants-marketing-authorization-for-gileads-oncedaily-truvada-for-reducing-the-risk-of-sexually-acquired-hiv1. Accessed March 15, 2017.
- World Health Organization. Guideline on when to start antiretroviral therapy and on pre-exposure prophylaxis for HIV. http://www.who.int/hiv/pub/guidelines/earlyrelease-arv/en/. Accessed March 15, 2017.
- Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363(27):2587–2599.10.1056/NEJMoa1011205
- McCormack S, Dunn DT, Desai M, et al. Pre-exposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open-label randomised trial. Lancet. 2016;387(10013):53–60.10.1016/S0140-6736(15)00056-2
- Molina JM, Capitant C, Spire B, et al. On-demand preexposure prophylaxis in men at high risk for HIV-1 infection. N Engl J Med. 2015;373(23):2237–2246.10.1056/NEJMoa1506273
- Volk JE, Marcus JL, Phengrasamy T, et al. No new HIV infections with increasing use of HIV preexposure prophylaxis in a clinical practice setting. Clin Infect Dis. 2015;61(10):1601–1603.10.1093/cid/civ778
- Baeten JM, Donnell D, Ndase P, et al. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med. 2012;367(5):399–410.10.1056/NEJMoa1108524
- Marrazzo JM, Ramjee G, Richardson BA, et al. Tenofovir-based preexposure prophylaxis for HIV infection among African women. N Engl J Med. 2015;372(6):509–518.10.1056/NEJMoa1402269
- Van Damme L, Corneli A, Ahmed K, et al. Preexposure prophylaxis for HIV infection among African women. N Engl J Med. 2012;367(5):411–422.10.1056/NEJMoa1202614
- Thigpen MC, Kebaabetswe PM, Paxton LA, et al. Antiretroviral preexposure prophylaxis for heterosexual HIV transmission in Botswana. N Engl J Med. 2012;367(5):423–434.10.1056/NEJMoa1110711
- Choopanya K, Martin M, Suntharasamai P, et al. Antiretroviral prophylaxis for HIV infection in injecting drug users in Bangkok, Thailand (the Bangkok Tenofovir Study): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2013;381(9883):2083–2090.10.1016/S0140-6736(13)61127-7
- Patterson KB, Prince HA, Kraft E, et al. Penetration of tenofovir and emtricitabine in mucosal tissues: implications for prevention of HIV-1 transmission. Sci Transl Med. 2011;3(112):112re114.
- Hanscom B, Janes HE, Guarino PD, et al. Brief report: preventing HIV-1 infection in women using oral preexposure prophylaxis: a meta-analysis of current evidence. J Acquir Immune Defic Syndr. 2016;73(5):606–608.10.1097/QAI.0000000000001160
- Baeten JM, Palanee-Phillips T, Brown ER, et al. Use of a vaginal ring containing dapivirine for HIV-1 prevention in women. N Engl J Med. 2016;375(22):2121–2132.10.1056/NEJMoa1506110
- Nel A, Saidi K, Bekker L-G, et al. Abstract presented at: Conference on Retroviruses and Opportunistic Infections 2016; February 22–25, 2016; Boston, MA.
- Institute of Medicine. Methodological Challenges in Biomedical HIV Prevention Trials. Washington, DC: The National Academies Press; 2008.
- Donnell D, Hughes JP, Wang L, Chen YQ, Fleming TR. Study design considerations for evaluating efficacy of systemic preexposure prophylaxis interventions. J Acquir Immune Defic Syndr. 2013;63(suppl 2):S130–S134.10.1097/QAI.0b013e3182986fac
- US Food and Drug Administration. Human immunodeficiency virus-1 infection: developing antiretroviral drugs for treatment: guidance for industry, revision 1. Silver Spring, MD: US Dept of Health and Human Services; US Food and Drug Administration; Center for Drug Evaluation and Research; 2015.
- Hernandez AV, Pasupuleti V, Deshpande A, Thota P, Collins JA, Vidal JE. Deficient reporting and interpretation of non-inferiority randomized clinical trials in HIV patients: a systematic review. PlS One. 2013;8(5):e63272.10.1371/journal.pone.0063272
- Spilker B. Guide to Clinical Trials. New York, NY: Raven Press; 1991.
- Fleming TR, Richardson BA. Some design issues in trials of microbicides for the prevention of HIV infection. J Infect Dis. 2004;190(4):666–674.10.1086/jid.2004.190.issue-4
- Padian NS. Evidence‐based prevention: increasing the efficiency of HIV intervention trials. J Infect Dis. 2004;190(4):663–665.10.1086/jid.2004.190.issue-4
- Stein ZA, Susser MW. Control groups in microbicide trials: in defense of orthodoxy. J Infect Dis. 2005;191(8):1377–1378.10.1086/jid.2005.191.issue-8
- Skoler S, Govender S, Altini L, et al. Risks in the use of an unblinded‐control group. J Infect Dis. 2005;191(8):1378–1379.10.1086/jid.2005.191.issue-8
- Fleming TR. Reply to Skoler et al. and to Stein and Susser. J Infect Dis. 2005;191(8):1379–1380.10.1086/jid.2005.191.issue-8
- Padian NS. Reply to Stein and Susser. J Infect Dis. 2005;191(8):1380–1381.10.1086/jid.2005.191.issue-8
- Richardson BA, Kelly C, Ramjee G, et al. Appropriateness of hydroxyethylcellulose gel as a placebo control in vaginal microbicide trials: a comparison of the two control arms of HPTN 035. J Acquir Immune Defic Syndr. 2013;63(1):120–125.10.1097/QAI.0b013e31828607c5
- US Food and Drug Administration. Guidance for industry: adaptive design clinical trials for drugs and biologics. https://www.fda.gov/downloads/drugs/guidances/ucm201790.pdf. Accessed March 15, 2017.
- Nason M, Follmann D. Design and analysis of crossover trials for absorbing binary endpoints. Biometrics. 2010;66(3):958–965.10.1111/j.1541-0420.2009.01358.x
- Auvert B, Sitta R, Zarca K, Mahiane SG, Pretorius C, Lissouba P. The effect of heterogeneity on HIV prevention trials. Clin Trials. 2011;8(2):144–154.10.1177/1740774511398923
- Makubate B, Senn S. Planning and analysis of cross-over trials in infertility. Stat Med. 2010;29(30):3203–3210.10.1002/sim.3981
- European Medicines Agency. Reflection paper on methodological issues in confirmatory clinical trials planned with an adaptive design. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003616.pdf. Accessed March 15, 2017.
- Baeten JM, Heffron R, Kidoguchi L, et al. Integrated delivery of antiretroviral treatment and pre-exposure prophylaxis to HIV-1-serodiscordant couples: a prospective implementation study in Kenya and Uganda. PLoS Med. 2016;13(8):e1002099.10.1371/journal.pmed.1002099
- Dunn DT, Glidden DV. Statistical issues in trials of preexposure prophylaxis. Curr Opin HIV AIDS. 2016;11(1):116–121.10.1097/COH.0000000000000218