Abstract
Organophosphate poisoning in the context of self-harm is a common medical emergency in Asia. Prolonged muscle weakness is an important but poorly understood cause of morbidity and mortality of the poisoning. This study examined mitochondrial function and its modulation by nitric oxide in muscle weakness of rats exposed to an acute, oral (0.8LD50) dose of monocrotophos. Muscle mitochondrial ATP synthase activity was inhibited in the rat in acute exposure to monocrotophos while respiration per se was not affected. This was accompanied by decreased mitochondrial uptake of calcium and increased levels of nitric oxide. Reactive cysteine groups of ATP synthase subunits were reduced in number, which may contribute to decreased enzyme activity. The decrease in ATP synthase activity and reactive cysteine groups of ATP synthase subunits was prevented by treatment of animals with the nitric oxide synthase inhibitor, L-NG Nitroarginine methyl ester, at 12 mg/kg body weight for 9 days in drinking water, prior to monocrotophos exposure. This indicated a role for nitric oxide in the process. The alterations in mitochondrial calcium uptake may influence cytosolic calcium levels and contribute to muscle weakness of acute organophosphate exposure.
| Abbreviations | ||
| OPP, | = | organophosphate poisoning |
| MCP, | = | Monocrotophos |
| AChE, | = | acetylcholinesterase |
| BuChE, | = | Butyrylcholinesterase |
| L-NAME, | = | L-NG Nitroarginine methyl ester |
| RBC, | = | Red blood cells |
| BN-PAGE, | = | Blue Native Polyacrylamide Gel Electrophoresis |
| OSCP, | = | Oligomycin sensitivity conferring protein |
| NOS, | = | Nitric oxide synthase |
| NO, | = | Nitric Oxide |
Introduction
Organophosphorous pesticide poisoning is a common method of intentional self-harm in agricultural communities across Asia (Eddleston and Phillips Citation2004). Organophosphates inhibit acetylcholinsterase (AChE), leading to an accumulation of acetylcholine at cholinergic synapses in the central nervous system and at neuromuscular junctions, and that results in overstimulation of these systems. In severe acute organophosphate poisoning (OPP) this can result in an autonomic cholinergic storm and lead to prolonged neuromuscular weakness and paralysis. The pathophysiology underlying muscle weakness that occurs in acute OPP is not completely clear, and thus there is no specific treatment for it. Consequently it is a major cause of morbidity and mortality of hospitalized OPP patients (Samuel et al. Citation1995).
Severe, persistent inhibition of AChE is the primary event responsible for muscle weakness that occurs in OPP. However it is unlikely to be the only factor, as cholinergic symptoms are often absent in prolonged muscle weakness and muscle power recovers despite persistent cholinesterase inhibition.
Functional mitochondria are essential in maintaining the integrity of the high energy requiring skeletal muscle. Muscle weakness often occurs subsequent to dysregulation of mitochondrial respiration and energy production, and consequent perturbation of intracellular Ca2+ levels (DiMauro and Gurgel-Giannetti Citation2005; Sarnat and Marin-Garcia Citation2005). It is possible that, during acute OPP, inhibition of mitochondrial function in the muscle also contributes to muscle weakness. This was studied in paralyzed skeletal muscle, in the early phase of poisoning, in rats subjected to severe acute monocrotophos exposure, an organophosphorus pesticide commonly used in India for intentional self-harm.
Acetylcholine, acting through its receptor, is known to increase cytosolic Ca2+ released from the endoplasmic reticulum, resulting in activation of calmodulin that stimulates nitric oxide synthase (NOS) and production of nitric oxide (NO). Increased levels of NO are a natural sequence to the inhibition of AChE by organophosphates. This study also examined nitric oxide signaling, activated in OPP, in modulating muscle mitochondrial function during the early phase of poisoning, as nitric oxide is known to modify mitochondrial proteins and disrupt mitochondrial function, including the ability to control calcium (Dedkova and Blatter Citation2005).
Materials and methods
n-Dodecyl-maltoside, Tricine, aminocaproic acid, Brilliant blue G and R, Cytochrome C (from bovine heart), phosphoenol pyruvate, pyruvate kinase, lactic acid dehydrogenase, oligomycin, sodium succinate dibasic hexahydrate, glutamate (disodium salt), rotenone, Nagarse (proteinase enzyme III), biotin-maleimide, and L-NG Nitroarginine methyl ester (L-NAME) were obtained from Sigma Aldrich Inc. (St Loius, MO). Streptavidin linked secondary antibodies and streptavidin peroxidase were from Bangalore Genei Pvt, Ltd. (Bangalore, India). Mouse monoclonal antibodies to α, β, and oligomycin sensitivity conferring protein (OSCP) subunits of ATP synthase were from Invitrogen (USA). Bovine serum albumin was from National Biochemical Corp. (USA). Monocrotophos (36% SL), containing no other compounds, was from Syngenta India Pvt Ltd. (India). All other chemicals were of the highest purity available.
Animal studies
Wistar rats (female, 150 ± 5 g) were administered monocrotophos by gavage at a dose of 0.8 LD50 in 200 μl water (Monocrotophos LD50 = 18 mg/kg body weight) (Manhande and Chopde Citation1995). Control rats were administered 200 μl water by gavage. Animals were evaluated after exposure to monocrotophos for cholinergic signs (chewing, tremor, salivation, lacrimation, and diarrhea) and for muscle power (Grade 0: normal mobility, Grade 1: ataxic gait, Grade 2: stretch movements after tail stimulation or Grade 3: no voluntary movements after tail stimulation) (De Bleecker et al. Citation1991). Monocrotophos was administered between 8 and 9 am on days of study and animals were sacrificed 2.5 h later when they were paralyzed (no voluntary movements after tail stimulation). Animals were euthanized under ether anesthesia. Blood (2.0 ml) was collected from each rat on sacrifice and RBC and plasma separated and stored at −70°C until assay. For inhibition of nitric oxide synthase (NOS), L-NAME (hydrochloride) was given to rats in drinking water at a dose of 0.1 mg/ml/day (12 mg/kg body weight/day) for 9 days (Thomas et al. Citation2001), following which they were administered monocrotophos as mentioned above. Each parameter was studied in a minimum of six rats. The study was approved by the Institutional Review Board and Institutional Animal Ethics Committee of the Christian Medical College, Vellore.
Skeletal muscle mitochondrial isolation
On sacrifice, upper and lower limb skeletal muscles were immediately dissected from the animal and rinsed with 50 mM Tris HCl pH 7.4 containing 100 mM KCl/2 mM EGTA. Five grams of muscle were finely minced and incubated for 3 min in eight volumes of buffer containing Nagarase (2.1 U/ml), 1 mM ATP, 5 mM MgCl2, 0.5% BSA, homogenized in a Dounce homogenizer and centrifuged at 490 g for 10 min at 4°C to obtain the post-nuclear supernatant. This was filtered through nylon cloth and the filtrate spun at 10,300 g for 10 min at 4°C. The mitochondrial pellet obtained was washed by re-suspension in 50 mM Tris HCl pH 7.4/100 mM KCl (30 ml) and centrifugation at 10,300 g for 10 min at 4°C. Washing was done twice and the final mitochondrial pellet was suspended in 1.0 ml of buffer. Enrichment of the mitochondrial fraction was assessed by measuring the marker enzyme succinate dehydrogenase (Pennington Citation1961). Mitochondria isolated from muscle of non-poisoned rats served as controls.
Assay of cholinesterases
Plasma butyrylcholinesterase and RBC and brain acetylcholinesterase were assayed by the method of Ellman et al. (Citation1961) using butyrylthiocholine iodide or acetylthiocholine iodide, respectively, as substrates. Enzyme units were calculated from the molar extinction co-efficient of 5-mercapto–2-nitro benzoic acid of 13.6 × 103.
Protein was estimated by the method of Lowry et al. (Citation1951) using bovine serum albumin as standard.
Mitochondrial respiration, swelling, and calcium flux
Oxygen uptake was determined polarographically using a Clark type electrode. Mitochondria (300 μg protein) were suspended in 2.0 ml 150 mM sucrose, 1 mM KH2 PO4, 5 mM MgCl2, 20 mM KCl, 10 mM Tris HCl pH 7.4 containing either 5 mM succinate or 5 mM glutamate as respiratory substrates, allowed to stabilize for 3 min and oxygen uptake measured (State 4 respiration). Oxygen uptake stimulated by addition of 0.3 mM ADP was assessed as State 3 respiration. The respiratory control ratio was calculated as the ratio of State 3/State 4 respiration (Madesh et al. Citation1999). Mitochondrial swelling was assessed by following the drop in absorbance at A540 nm as described earlier (Takeyama et al. Citation1993), and calcium flux measurements were carried out by quantifying changes in the absorption spectrum of Arsenazo III at 675/685 nm (Scarpa Citation1979).
Assay of mitochondrial complex IV and V
Complex IV activity was measured by the oxidation of cytochrome c at 550 nm (Darley-Usmar Citation1987). Data is represented as the pseudo first order rate constant (k) divided by the protein concentration. Complex V was assayed by an ATPase coupled reaction measuring oxidation of NADH at 340 nm at 30°C in the presence and absence of oligomycin (Soper and Pedersen Citation1979). Units were defined as μmoles NADH oxidized/min/mg protein.
2D Blue native gel electrophoresis of mitochondrial complexes
Blue Native polyacrylamide gel electrophoresis (BN-PAGE) was performed using the method developed by Schagger and von Jagow (Citation1991) and Schagger et al. (Citation1994), and modified by Brookes et al. (Citation2002). Mitochondrial pellets (0.5 mg of protein) were resuspended in 80 μl of extraction buffer (0.75 M aminocaproic acid, 50 mM BisTris, and 1% n-dodecyl-ß-D-maltoside), incubated on ice for 60 min, centrifuged at 14,000 rpm for 5 min, and the supernatants containing the extracted mitochondrial complexes collected and kept on ice; 2.5 μl of Coomassie Brilliant Blue G-250 (5% w/v suspension in 0.5 M aminocaproic acid) was added to 75 μg of mitochondrial protein and samples stored on ice for no more than 30 min before being subjected to non-denaturing PAGE (5–12% gels) to separate the individual respiratory complexes intact. The identity of the ATP synthase band in mitochondria subjected to BN-PAGE was confirmed by electro-transfer of the gel to PVDF membranes and probing the blots with antibodies to the α-subunit of complex V.
SDS-PAGE, Immunoblotting, and sulfhydryl group labeling of ATP synthase
In further experiments, the band corresponding to ATP synthase in BN-PAGE was excised from the gel and subjected to 2nd dimension SDS-PAGE (10–14% gels) with β-mercaptoethanol to separate individual proteins in the complex. Control and monocrotophos treated samples were run in duplicate so that one gel could be stained with Brilliant blue R for protein visualization and the other transferred to PVDF membranes and probed with antibodies to ATP synthase subunits α, β, and OSCP or analyzed for free sulfhydryl groups. SDS-PAGE and immunoblotting were performed according to standard protocols (Laemmli Citation1970; Towbin et al. Citation1979).
For labeling of free sulfhydryl groups on subunits of ATP synthase, the blots were incubated with biotin-maleimide (12 μg/ml) for 12 h at 4°C, blocked with 2% BSA in PBS Tween 20 (0.02%) for 1 h and incubated with streptavidin-peroxidase (1:5000 diluted) for 45 min. Bands were developed with 0.024% H2O2/3.2 mM diaminobenzidine for 5 min and quantified using densitometric software developed in-house that correlated with Bio-Rad Quantity One Quantitation software version 4 (r = 0.8). Under these conditions, modification of thiol groups is reflected in decreased labeling relative to control.
Estimation of nitrate in mitochondria and plasma
Nitrate in mitochondria (100 μg) and plasma (100 μl) was first reduced to nitrite with copper-cadmium alloy and then assayed by the Griess reaction (Sastry et al. Citation2002).
Statistical analysis
Parameters under study in monocrotophos treated animals were statistically analyzed for differences from controls by the Mann Whitney U-test using the statistical software package SPSS 11.00. A test was considered statistically significant at p < 0.05.
Results
Monocrotophos Treatment of Rats
In rats administered monocrotophos (0.8LD50) RBC AChE was inhibited 97% and plasma BuChE 91.5% within 2.5 h of poisoning. RBC AChE levels in control rats were 8.96 ± 0.456 units/mg protein that decreased to 0.253 ± 0.218 units/mg protein in monocrotophos treated animals (mean ± SD, n = 6). The values for plasma BuChE levels were 313.23 ± 34.72 units/liter and 26.69 ± 8.98 units/liter in control and monocrotophos treated animals, respectively.
All rats treated with monocrotophos exhibited cholinergic symptoms and muscle weakness. Cholinergic symptoms began at 12.92 ± 5.58 min and continued until 41 ± 12.21 min after acute monocrotophos poisoning. Muscle weakness developed at 37.5 ± 26.94 min and progressed until 57.5 ± 23.67 min (mean ± SD, n = 6). Muscle paralysis occurred within 2 h of poisoning.
Skeletal muscle mitochondrial integrity and respiration in monocrotophos exposure
Mitochondrial swelling, respiration rates, respiratory control ratios (RCR) using substrates for complex I and II, and the activity of complex IV were not altered significantly in rats exposed to monocrotophos.
Enzyme activity of muscle mitochondrial complex V in rats exposed to monocrotophos
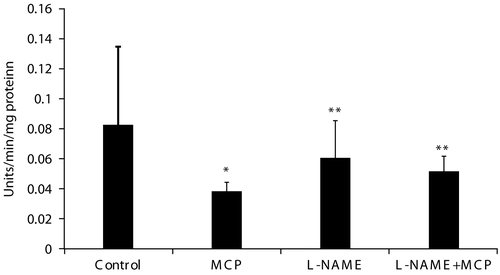
Complex V (ATP synthase) was significantly inhibited by 55% in muscle mitochondria isolated from rats treated with 0.8LD50 dose of monocrotophos compared to control animals ().
Figure 1. Muscle mitochondrial complex V activity of control, monocrotophos (MCP), L-NAME, and L-NAME + MCP treated rats. Muscle mitochondria were isolated from control, MCP, L-NAME, and L-NAME + MCP treated rats and complex V activity measured. Values are mean ± SD of nine control and six rats in all other groups. * p < 0.05 compared to Control. ** p < 0.05 compared to MCP.
Mitochondrial calcium uptake in rats exposed to monocrotophos
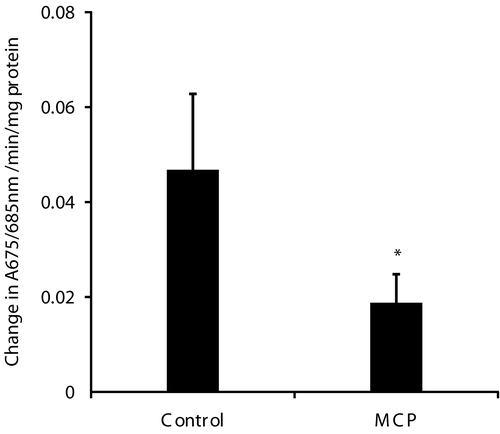
The uptake of calcium by muscle mitochondria from monocrotophos treated animals was inhibited 60% compared to control animals ().
Figure 2. Calcium uptake by muscle mitochondria isolated from control and monocrotophos treated rats. Muscle mitochondria isolated from control animals and after monocrotophos induced paralysis were used for measurement of calcium uptake using the dye Arsenazo III and following absorbance at 675/685 nm. Values are mean ± SD of six rats. *p< 0.05 compared to Control.
Nitric oxide levels in plasma and mitochondria of rats treated with monocrotophos
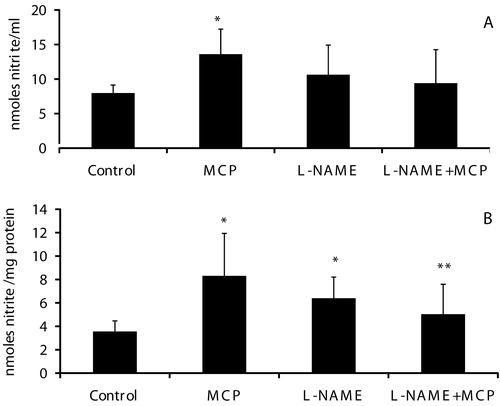
A significant increase in nitrate (measured as nitrite), the stable end product of NO, was noted in plasma and skeletal muscle mitochondria of paralyzed rats during the early phase of acute monocrotophos exposure (0.8LD50 dose) ( and , respectively).
Figure 3. Nitric oxide levels in plasma and muscle mitochondria of control, monocrotophos (MCP), L-NAME, and L-NAME + MCP treated rats. Nitrate, the stable end product of nitric oxide was measured (as nitrite) in plasma (a) and mitochondrial samples (b) from control, MCP treated, L-NAME, and L-NAME + MCP treated animals. Values are mean ± SD of nine control and six rats in all other groups. *p < 0.05 compared to Control. **p < 0.05 compared to MCP.
Modification of reactive cysteines on ATP synthase subunits from rats treated acutely with monocrotophos
Protein levels of ATP synthase on 1D BN-PAGE () and of ATP synthase subunits in the second dimension SDS PAGE, from gels stained for total protein, did not differ between control and monocrotophos treated rats ().
Figure 4. Sulfhydryl group reactivity of muscle mitochondrial ATP synthase from control, monocrotophos (MCP), L-NAME, and L-NAME + MCP treated rats. Muscle mitochondria isolated from control and monocrotophos poisoned rats were subjected to 2D BN-PAGE, immunoblotting and sulfhydryl labeling. (a) First dimension blue native gel after staining with coomassie blue (Complex V (CV) indicated with arrow) and immune blot after probing with the α-subunit of ATP synthase. (b) Second dimension SDS-PAGE gel of ATP synthase identified and excised from the 1st dimension gel, stained with coomassie blue. (c) Immune blot of the 2nd dimension SDS-PAGE gel with separated ATP synthase subunits after probing with the anti-α-subunit, anti-β-subunit, and anti-OSCP subunit of ATP synthase. (d) Sulfhydryl group labeling on the 2nd dimension SDS-PAGE gel with separated ATP synthase subunits using biotin maleimide in controls and monocrotophos poisoned animals in the presence and absence of L-NAME, an NOS inhibitor. (a–d) are representative of one control and one monocrotophos treated rat. (e) The intensity of sulfhydryl labeled bands in (d) were determined and plotted as fold change from control. Values are mean ± SD of three rats. *p < 0.05 compared to Control. #p < 0.05 compared to MCP treated rats.
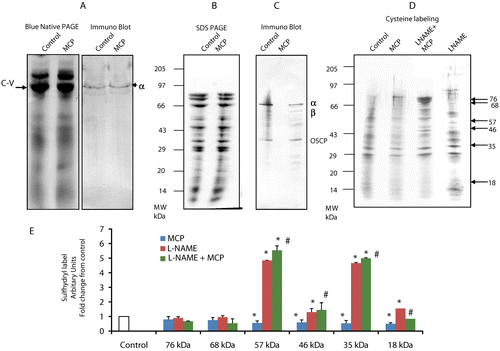
Blots probed for α, β, and OSCP subunits of ATP synthase resulted in detection of proteins at approximately 58, 52, and 27 kD ().
Sulfhydryl labeling indicated that a number of ATP synthase subunits were modified in acute monocrotophos exposure. Six subunits with reactive cysteines were labeled under these conditions. The one at 57 kD showed approximately 43% decrease in labeling, while the one at 18 kD showed a 50% reduction in labeling compared to controls ( and ). The fact that the molecular weights of these proteins correlated closely with those obtained on the immunoblots for the α, β, and OSCP subunits suggested that these subunits were modified. In addition a 46 kD and 35 kD band also showed 40% and 50% decrease in labeling, respectively. Sulfhydryl group modification appeared to be specific (to certain subunits) and not a universal phenomenon in monocrotophos poisoning, since the two bands at 76 kD and 68 kD did not show any change in labeling after poisoning ( and ).
Inhibition of NOS and ATP synthase activity after monocrotophos exposure
L-NAME, a general NOS inhibitor, treatment of animals increased plasma and muscle mitochondrial levels of nitrate compared to control animals, although the increase was not significant ( and ). L-NAME, however, prevented the increase in NO levels seen during monocrotophos exposure in plasma and significantly in muscle mitochondria (p < 0.05) ( and , respectively).
Complex V activities (ATP synthase) did not differ significantly between control, L-NAME, and L-NAME + monocrotophos treated animals (). This indicated that L-NAME inhibition of NOS prevented inhibition of ATP synthase induced by monocrotophos exposure, which suggested that NO had a role to play in this process.
Prevention of NO synthesis by L-NAME significantly increased sulfhydryl labeling of ATP synthase subunits 57, 46, 35, 18 kda by 4.5-, 1.3-, 4.7-, and 1.6-fold, respectively, compared to control animals (p < 0.05) (). This significant increase in sulfhydryl label of these ATP synthase subunits was preserved in animals treated with L-NAME prior to monocrotophos treatment compared to those treated only with monocrotophos (p < 0.05) (). This data further supports the role of NO in the modification of sulfhydryl groups on ATP synthase in monocrotophos exposure.
Discussion
OPP results in rapid toxicity due to efficient absorption and high affinity of binding to AChE of these compounds. The nature and rapidity of the toxicity may explain why cellular events that occur in the early phase of poisoning initiate prolonged muscle pathology. Muscle weakness of OPP patients is associated with muscle damage that occurs early in the poisoning (John et al. Citation2003). This study was to elucidate intracellular mechanisms in muscle that occur soon after acute OPP that may contribute to muscle injury and focused on the mitochondria.
It was found that acute exposure to monocrotophos did not affect skeletal muscle mitochondrial respiration or coupling to oxidative phosphorylation which need to be tightly coupled in order to generate ATP for cellular energy. The activity of ATP synthase was, however, decreased by 55%.
This would result in levels of ATP which may be insufficient to maintain the integrity of the cell (Wilkinson and Robinson Citation1974) and therefore contribute to muscle weakness and injury that occurs in acute OPP.
Mitochondria also contribute to maintaining calcium homeostasis in the muscle by calcium uptake through a uniporter, a process that requires ATP (Moreau et al. Citation2006). The inhibition of energy-dependent mitochondrial Ca2+ influx observed in this study may lead to elevated sarcoplasmic Ca2+ that can injure the muscle. Muscle damage induced within 2 h of exposure to organophosphates has been shown earlier by Leonard and Salpeter (Citation1979) to be a myopathy mediated by calcium.
Interventions that prevent or reduce ATP synthase inhibition in OPP may decrease the resulting muscle injury. Increased NO levels are known to disrupt muscle contraction, cause muscle weakness and fatigue, and to underlie muscle degeneration and toxicity of organophosphate poisoned rats (Jeyarasasingam et al. Citation2000; Gupta et al. Citation2002; Smith and Reid Citation2006). The data in this study also showed increased systemic levels of NO in acute monocrotophos exposure that correlated to the inhibition of ATP synthase, as inhibition of NOS and reduction of NO levels resulted in partial recovery of ATP synthase activity. These results support the studies of Gupta et al. (Citation2002) that show a marked decrease in rat muscle and brain ATP levels, within an hour of treatment with the organophosphate compound diisopropylphosphorofluoridate. This decrease was accompanied by an increase in NO levels. Nitric oxide is also implicated in toxicity to the brain in organophosphate pesticide and nerve agent poisoned rats (Chang et al. Citation2001; Damodaran et al. Citation2006). In the brain, organophosphates disrupt respiration by inhibition of Complex I/IV (Yen et al. Citation2004). This clearly differs from the situation in organophosphate poisoned paralyzed muscle where NO targets Complex V (ATP synthase).
NO is known to modify reactive cysteines on proteins through post-translational modification and initiate signaling (Jaffrey et al. Citation2001). To explore the possibility of such modifications on mitochondrial ATP synthase, we used a proteomics approach coupled to labeling reactive cysteines with biotinylated maleimide. The labeling of four subunits of ATP synthase was substantially decreased in monocrotophos exposure. This suggested that certain subunits of ATP synthase are susceptible to thiol modification subsequent to acute monocrotophos exposure. NO may play a role in this modification as it was prevented by L-NAME treatment.
L-NAME treatment was expected to reduce NO levels to less than those in controls. Paradoxically, plasma and mitochondrial NO levels were higher, although not significantly, in the L-NAME treated animals compared to controls. Chronic low dose L-NAME treatment of rats is known to activate NO synthesis through negative feedback regulation of NOS by NO and may also account for the observations in this study (Bernatova et al. Citation2007). The lower NO levels in L-NAME + monocrotophos treated animals compared to those treated only with L-NAME lend support to negative feedback regulation of NOS by NO in animals on the treatment schedule of this study.
L-NAME inhibition of NO synthesis is associated with increased levels of glutathione (GSH) in animals (Schild et al. Citation2003). Under these conditions NO induced by negative feedback inhibition of NOS may be rapidly oxidized by GSH to GSNO, the transport and storage form of NO. This sequestration of NO may explain the increased free sulfyhdryl groups on ATP synthase subunits observed in L-NAME and L-NAME + monocrotophos treated rats compared to control animals during the initial phase of poisoning.
In conclusion, the studies demonstrate that muscle mitochondrial ATP synthase is inhibited in paralyzed muscle during the early phase of acute monocrotophos exposure, an effect possibly mediated through NO. The inhibition of ATP synthase may be responsible for reduced calcium uptake by the mitochondria, which can affect cellular calcium homeostasis and lead to muscle damage. As inhibition of NOS appears to protect against modification and inhibition of ATP synthase, scavenging NO early in OPP may help maintain myocyte integrity and prevent muscle weakness. This is currently under study.
Acknowledgement
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Bernatova, I., Kopincova, J., Puzserova, A., Janega, P., and Babal, P. 2007. Chronic low dose L-NAME treatment increases nitric oxide production and vasorelaxation in normotensive rats. Physiol. Res. 56(Suppl 2):S17–S24.
- Brookes, P. S., Pinner, A., Ramachandran, A., Coward, L., Barnes, S., Kim, H., and Darley-Usmar, V. M. 2002. High throughput two-dimensional blue-native electrophoresis: a tool for functional proteomics of mitochondria and signaling complexes. Proteomics 2:969–977.
- Chang, A. Y., Chan, J. Y., Kao, F. J., Huang, C. M., and Chan, S. H. 2001. Engagement of inducible nitric oxide synthase at the rostral ventrolateral medulla during mevinphos intoxication in the rat. J. Biomed. Sci. 8:475–483.
- Damodaran, T. V., Patel, A. G., Greenfield, S. T., Dressman, H. K., Lin, S. M., and Abou-Donia, M. B. 2006. Gene expression profiles of the rat brain both immediately and 3 months following acute sarin exposure. Biochem. Pharmacol. 7:497–520.
- Darley-Usmar, V. 1987. The molecular aetiology of human mitochondrial myopathies. Biochem. Soc. Trans. 1:102–103.
- De Bleecker, J., Willems, J., De Reuck, J., Santens, P., and Lison, D. 1991. Histological and histochemical study of paraoxon myopathy in the rat. Acta Neurol. Belg. 9:255–270.
- Dedkova, E. N., and Blatter, L. A. 2005. Modulation of mitochondrial Ca2+ by nitric oxide in cultured bovine vascular endothelial cells. Am. J. Physiol. Cell Physiol. 289:C836–C845.
- DiMauro, S., and Gurgel-Giannetti, J. 2005. The expanding phenotype of mitochondrial myopathy. Curr. Opin. Neurol. 18:538–542.
- Eddleston, M., and Phillips, M. R. 2004. Self poisoning with pesticides. Br. Med. J. 328:42–44.
- Ellman, G. L., Courtney, K. D., AndresJr., V. and Feather-Stone, and R. M. 1961. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7:88–95.
- Gupta, R. C., Milatovic, D., and Dettbarn, W. D. 2002. Involvement of nitric oxide in myotoxicity produced by diisopropylphosphorofluoridate (DFP)-induced muscle hyperactivity. Arch. Toxicol. 76:715–726.
- Jaffrey, S. R., Erdjument-Bromage, H., Ferris, C. D., Tempst, P., and Snyder, S. H. 2001. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nature Cell Biol. 3:193–197.
- Jeyarasasingam, G., Yeluashvili, M., and Quik, M. 2000. Nitric oxide is involved in acetylcholinesterase inhibitor-induced myopathy in rats. J. Pharmacol. Exp. Ther. 295:314–320.
- John, M., Oommen, A., and Zachariah, A. 2003. Muscle injury in organophosphorous poisoning and its role in the development of intermediate syndrome. Neurotoxicology 24:43–53.
- Laemmli, U. 1970. Cleavage of structural proteins during the assembly of the head of bacteriphage T4. Nature 227:680–685.
- Leonard, J. P., and Salpeter, M. M. 1979. Agonist induced myopathy at the neuromuscular junction is mediated by calcium. J. Cell Biol. 82:811–819.
- Lowry, O. H., Rosenbrough, N. H., Farr, A. L., and Randall, R. J. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275.
- Madesh, M., Ramachandran, A., and Balasubramanian, K. A. 1999. Nitric oxide prevents anoxia-induced apoptosis in colonic HT29 cells. Arch. Biochem. Biophys. 366:240–248.
- Manhande, S. N., and Chopde, C. 1995. Neurobehavioral effect of acute monocrotophos administration in rats and mice. Indian J. Pharmacol. 27:245–249.
- Moreau, B., Nelson, C., and Parekh, A. B. 2006. Biphasic regulation of mitochondrial Ca2+ uptake by cytosolic Ca2+ concentration. Curr. Biol. 16:1672–1677.
- Pennington, R. J. 1961. Biochemistry of dystrophic muscle. Mitochondrial succinate-tetrazolium reductase and adenosine triphosphatase. Biochem. J. 80:649–654.
- Samuel, J., Thomas, K., Jeyaseelan, L., Peter, J. V., and Cherian, A. M. 1995. Incidence of intermediate syndrome in organophosphorus poisoning. J. Ass. Physicians India 43:321–323.
- Sarnat, H. B., and Marin-Garcia, J. 2005. Pathology of mitochondrial encephalomyopathies. Can. J. Neurol. Sci. 32:152–166.
- Sastry, K. V., Moudgal, R. P., Mohan, J., Tyagi, J. S., and Rao, G. S. 2002. Spectrophotometric determination of serum nitrite and nitrate by copper-cadmium alloy. Anal. Biochem. 306:79–82.
- Scarpa, A. 1979. Measurements of cation transport with metallochromic indicators. Methods Enzymol. 56:301–338.
- Schagger, H., and von Jagow, G. 1991. Blue Native Electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 199:223–231.
- Schagger, H., Cramer, W. A., and von Jagow, G. 1994. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 217:220–230.
- Schild, L., Reinheckel, T., Reiser, M., Horn, T. F. W., Wolf, G., and Augustin, W. 2003. Nitric oxide produced in rat liver mitochondria causes oxidative stress and impairment of respiration after transient hypoxia. FASEB J. 17:2194–2201.
- Smith, M. A., and Reid, M. B. 2006. Redox modulation of contractile function in respiratory and limb skeletal muscle. Respir. Physiol. Neurobiol. 151:229–241.
- Soper, J. W., and Pedersen, P. L. 1979. Isolation of an oligomycin-sensitive ATPase complex from rat liver mitochondria. Methods Enzymol. 55:328–333.
- Takeyama, N., Matsuo, N., and Tanaka, T. 1993. Oxidative damage to mitochondria is mediated by the Ca (2+)-dependent inner-membrane permeability transition. Biochem. J. 294:719–725.
- Thomas, S., Ramachandran, A., Susama, P., and Balasubramanian, K. A. 2001. Nitric oxide prevents intestinal mitochondrial dysfunction induced by surgical stress. Br. J. Surgery 88:393–399.
- Towbin, H., Staehelin, T., and Gordon, J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 76:4350–4354.
- Wilkinson, J. H., and Robinson, J. M. 1974. Effect of ATP on release of intracellular enzymes from damaged cells. Nature 249:662–663.
- Yen, D. H., Chan, J. Y., Tseng, H. P., Huang, C. I., Lee, C. H., Chan, S. H., and Chang, A. Y. 2004. Depression of mitochondrial respiratory enzyme activity in rostral ventrolateral medulla during acute mevinphos intoxication in the rat. Shock 21:358–363.