
Abstract
Lead (Pb) is recognized as the first heavy metal of the top six toxic air pollutants threatening human health and the second hazardous substance. Pb exposure is associated with lung impairment and high incidences of lung cancer. Nuclear factor kappa B (NF-κB) and aryl hydrocarbon receptor (AhR) signaling pathways are known to be expressed and play an important role in the lung. However, the link between Pb lung toxicity and NF-κB and/or AhR pathways remains unclear. This study was established to explore the role of NF-κB and AhR modulation in Pb-induced lung toxicity in human lung cancer A549 cells. In the current study, treatment of A549 cells with Pb significantly induced cell apoptosis as evidenced by increasing a) the percentage of cells underwent apoptosis determined by flow cytometry and b) p53 mRNA level. Pb treatment induced oxidative stress by a) increasing the formation of reactive oxygen species and b) decreasing GSTA1 mRNA levels. The toxic effects of Pb on the lung was associated with significant increases in NF-κB and AhR levels which was accompanied with increases in downstream targets genes, iNOS and CYP1A1, respectively. Inhibition of NF-κB or AhR either chemically using resveratrol or genetically using small interfering RNA (siRNA) significantly rescued A549 cells from Pb-mediated lung toxicity. The results clearly indicate that Pb-mediated lung toxicities are NF-κB and AhR-dependent mechanism.
Keywords:
1. Introduction
Lung cancer is the second most widespread cancer and accounts for the highest number of cancer-related deaths worldwide. Air pollution is associated with 45% increased risk of lung cancer and is responsible for 36% of deaths from lung cancer (Mu et al. Citation2013; Jemal and Torre Citation2018). Heavy metals are the most important air pollutants because of their toxicities and higher stability (Jarup Citation2003). Among heavy metals, lead (Pb) is recognized as the first among the top six toxic air pollutants threatening human health and has been identified as a major pollutant of concern worldwide (Csavina et al. Citation2012).
Pb is very toxic even in very small traces and thus ranked the second most commonly encountered toxic substance according to the Agency for Toxic Substances and Disease Registry (ATSDR Citation2017). Since it is widely used in industrial and mining activities, Pb-exposure remains a major concern in several countries and many people worldwide are still at high risk of Pb-exposure particularly in polluted areas. In this context, previous studies have reported that long-term exposure to Pb is associated with immune dysfunction and increased susceptibility to various diseases and serious toxicities to various organs, such as the kidneys, heart, liver, brain, and lung (Anttila et al. Citation1995; Jarup Citation2003).
The lung is a critical organ composed of different types of cells with various metabolic and immune functions. Lung is the primary organ exposed to air pollutants, including heavy metals. Pb-exposure has been shown to induce oxidative stress and altered expression of genes related to inflammation. Previous studies have reported increased incidences of lung cancer among Pb smelters and workers at Pb battery plants (Kumar et al. Citation1991; Wong and Harris Citation2000; Liu et al. Citation2012). In addition, studies on occupational hazards have demonstrated the increased risk of lung cancer in workers exposed to Pb (Anttila et al. Citation1995; Lundstrom et al. Citation1997).
In vivo and in vitro studies demonstrated that Pb-exposure induces the modulation of several signaling pathways and transcriptional factors. Among which, the nuclear factor kappa B (NF-κB) and the aryl hydrocarbon receptor (AhR) are targeted by heavy metals and have been shown to play a role in their toxicities (Korashy and El-Kadi Citation2005; Korashy and El-Kadi Citation2008; Ansari et al. Citation2013). The cascades between AhR and NF-κB on the toxic effect of heavy metals are not clear. In that, AhR activator 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces several inflammatory responsive genes such as Interleukin 1 (IL-1), IL-6, and IL-8 through the activation of NF-κB in non-small cell lung cancer patients (Kobayashi et al. Citation2008; Kimura et al. Citation2009; Vogel and Matsumura Citation2009; Chen et al. Citation2012; Vogel et al. Citation2014). On the other hand, Vogel et al. have shown that inhibition of NF-κB activity using PDTC blocked the expression of AhR and subsequently its downstream target cytochrome P450 1A1 (CYP1A1) activity. A previous work from our laboratory in human hepatocellular carcinoma HepG2 cells have reported that activation of NF-κB using PMA significantly blocked the activation of AhR/CYP1A1 by Pb, whereas NF-κB inhibitor PDTC potentiated Pb-induced CYP1A1 (Korashy and El-Kadi Citation2008).
Although NF-κB and AhR are known to be expressed in lung and play critical roles in lung development, maintenance, survival, and inflammation as well as apoptosis (Ghosh et al. Citation1998; Beamer and Shepherd Citation2013), there is very limited data on the impact and mechanism of NF-κB and AhR in Pb-modulated pulmonary immunotoxicity are not fully understood. Therefore, the main objectives of the current study are (a) to investigate the pulmonary toxic effects of Pb exposure on the immune system; (b) to explore the role of NF-κB and AhR pathways in vitro A549 human lung cell line.
2. Materials and methods
2.1. Materials
Lead (II) nitrate was obtained from Sigma-Aldrich (St. Louis, MO). TRIzol was purchased from Invitrogen Co. (Grand Island, NY). High Capacity cDNA Reverse Transcription kit and SYBR Green PCR Master Mix were obtained from Applied Biosystems (Foster City, CA). Apoptosis detection kit was obtained from Millipore (Muse® Cell Analyzer, Millipore, Billerica, MA). The Western blot detection Enhanced Chemiluminescence ECL kit was obtained from EMD Millipore Co. (Billerica, MA). Acrylamide, bromophenol blue, glycine, β-mercaptoethanol, N'N'-bis-methylene-acrylamide, ammonium persulfate, sodium dodecyl sulfate (SDS), N,N,N',N'-tetramethylethylenediamine (TEMED), and nitrocellulose membrane (0.45 μm), were purchased from Bio-Rad Laboratories (Hercules, CA). Primary and secondary antibodies against target proteins, small interfering RNA (siRNA) against NF-κB p65, and AhR, and transfection reagents and kits were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). All other chemicals were purchased from Fisher Scientific Co. (Toronto, ON).
2.2. Cell culture and treatment
Human adenocarcinoma alveolar type II cells (A549), obtained from American Type Culture Collection (Rockville, MD), were maintained in DMEM with phenol red supplemented with fetal bovine serum (10%) and 100× Antibiotic-Antimycotic (1%) in 75-cm2 tissue culture flasks in a humidified 5% CO2 environment at 37 °C. For Pb-exposure studies, fresh Pb solutions were prepared for each experiment by dissolving lead nitrate in distilled water and DMSO. An equal volume of DMSO (0.05%) was added to the control. The medium was changed every other day and the cells were sub-cultured every 3 days at a 3:1 ratio.
2.3. Determination of apoptosis
The percentage of cells undergoing apoptosis/necrosis was determined by Muse® Annexin V and Dead Cell assay kit (Merck Millipore, Darmstadt, Germany) according to manufacturer's instructions and as described previously (Abdullah et al. Citation2018). The A549 cells were treated with increasing concentrations of Pb for 24 h. The cells were then washed with phosphate buffer saline (PBS), detached by trypsin 0.25%, collected by centrifugation at 300 × g for 5 min, and then resuspended in 0.5 ml PBS before incubated with Annexin V and 7-amino actinomycin D (7-AAD) in the dark for 20 min at 25 °C. The apoptotic and necrotic populations were analyzed using Muse Cell Analyzer (Merck Millipore, Billerica, MA).
2.4. Determination ROS generation
Intracellular ROS generation was measured using dihydroethidium (DHE), a well-characterized reagent that has been used to detect superoxide radicals in cellular populations. DHE is cell permeable and it reacts with superoxide anions and undergoes oxidation to form the DNA-binding fluorophore ethidium bromide which intercalates with DNA and emits red fluorescence. The percentage of intracellular ROS production in A549 cells exposed to Pb was determined by flow cytometry using DHE reagent following the manufacturer’s instructions. Briefly, A549 cells were treated with Pb for 24 h. The cells were then harvested, washed with PBS and 10 μL A549 cell suspension was added to 190 μL DHE reagent, and incubated for 30 min at 37 °C. Cellular fluorescence was analyzed using a Muse Cell Analyzer (Merck Millipore, Billerica, MA) and the percentage change in ROS generation was calculated by comparing with the control untreated cells.
2.5. RNA isolation, cDNA synthesis, and RT-PCR
Total RNA from A549 cells was isolated using the TRIzol method (Korashy et al. Citation2016a). The RNA quantity and quality were determined using a NanoDrop® 8000 (Thermo Scientific, Waltham, MA). The optical density (OD) at 260 nm and 280 nm were measured and an OD 260/280 ratio ∼ 2.0 was considered as indicator of pure RNA. cDNA synthesis was performed and the changes in the mRNA levels of NF-κB, AhR, CYP1A1, glutathione Transferase A1 (GSTA1), inducible nitric oxide synthase (iNOS), and p53 () in response to Pb treatment was quantified by Real-Time PCR System (qRT-PCR) (Applied Biosystems®, Foster City, CA) using SYBR Green Master mix as described previously (Korashy et al. Citation2016a). The qRT-PCR data were analyzed using the relative gene expression (ΔΔ CT) method and the data is presented as the fold change in gene expression normalized to the endogenous reference gene β-actin.
Table 1. Primers sequences used for teal-time polymerase chain (RT-PCR) reactions.
2.6. Protein extraction and Western blot analysis
The total protein from lung A549 cell lysate was extracted as reported previously (Abrams et al. Citation2003) and the protein concentrations were determined using a direct-detect infrared spectrophotometer (Millipore, Billerica, MA). The expressions of NF-κB and AhR proteins was measured by Western blot analysis using specific primary and secondary antibodies and the results were normalized to GAPDH as described previously (Korashy et al. Citation2017). Briefly, 30–35 µg of protein was separated in 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred to nitrocellulose membrane by electrophoresis. Protein blots were blocked overnight by blocking solution at 4 °C, and then washed several times with TBST (Tris-buffered saline, 0.1% Tween 20) before further incubated for 2 h at room temperature with primary antibodies against NF-κB and AhR in TBS solution. Protein blots were then incubated with secondary antibodies in blocking solution for 2 h at room temperature. The quantification of western blots was performed by C-DiGit® Blot Scanner (LI-COR Biosciences, Franklin Lakes, NJ) using the enhanced chemiluminescence method according to the manufacturer’s instructions (Merck Millipore, Billerica, MA).
2.7. Gene silencing using small interfering RNA (siRNA)
Silencing of NF-κB and AhR genes was determined by transfecting the cells with either NF-κB or AhR siRNA according to manufacture instruction (Santa Cruz Biotechnology, Inc., Santa Curz, CA) and as described previously (Korashy et al. Citation2016b). Briefly, A549 cells (70% confluent) were transfected using Lipofectamine® 2000 (Santa Cruz, CA) for 24 h with either AhR or NF-κB siRNA. Thereafter, the cells were washed and then incubated with Pb for additional 24 h. NF-κB and AhR mRNAs were then quantified by RT-PCR, to ensure significant (50–70%) knockdown, and the effect of NF-κB or AhR knockdown on apoptotic and oxidative stress markers were determined.
2.8. Statistical analyses
Data are presented as mean ± SEM. All statistical analyses were performed using Sigma Plot for Windows (Systat Software, Inc, San Jose, CA). Statistical significance was determined by either Student’s t-test or one-way analysis of variance (ANOVA) followed by a Student–Newman–Keuls test. A p-value < 0.05 was considered statistically significant.
3. Results
3.1. Effect of Pb treatment on induction of apoptosis in A549 cells
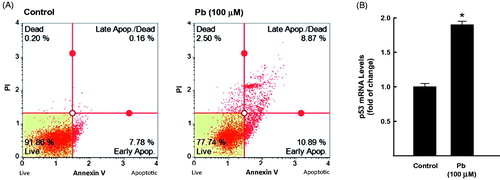
The ability of Pb to induce apoptosis in human lung A549 cells was assessed by (a) measuring the percentage of cells underwent apoptosis/necrosis using flow cytometry assay and (b) quantifying the mRNA expression of p53, a well-known apoptotic marker. shows that treatment of A549 cells with 100 μM Pb increased the percentage of both early and late apoptotic cells to approximately 20% compared to healthy untreated cells (8%). In addition, Pb treatment increased the mRNA expression levels of p53 by 90% (.
Figure 1. Effect of Pb treatment on apoptosis levels in A549 cells. The cells were treated for 24 h with Pb (100 µM). (A) the percentage of cells underwent apoptosis was determined by flow cytometry using annexin V/PI. (B) p53 mRNA level was quantified by RT-PCR and normalized to β-actin housekeeping gene. The values represent mean of fold change ± SEM (n = 6, triplicate). *p < 0.05 compared to control.
3.2. Effect of Pb treatment on oxidative stress markers in A549 cells
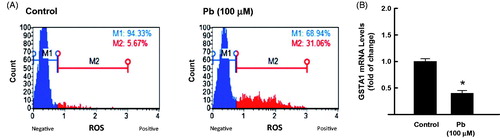
The ability of Pb to induce oxidative stress markers in human lung A549 cells was assessed by two approaches: (1) determining the ROS formation in response to Pb and (2) quantifying the mRNA expression of GSTA1, a known oxidative stress marker. shows that Pb 100 μM significantly increases the formation of ROS by approximately 6-folds (from 6% in control to 31% in Pb-treated cells). Furthermore, Pb treatment significantly inhibited the mRNA expression of GSTA1 by approximately 60% (.
Figure 2. Effect of Pb treatment on oxidative stress levels in A549 cells. The cells were treated for 24 h with Pb (100 µM). (A) the percentage of oxidative cells was determined by flow cytometry using DCF as a substrate. (B) GSTA1 mRNA level was quantified by RT-PCR and normalized to β-actin housekeeping gene. The values represent mean of fold change ± SEM (n = 6, triplicate). *p < 0.05 compared to control.
3.3. Effect of Pb treatment on the expression levels of NF-κB and AhR in A549 cells
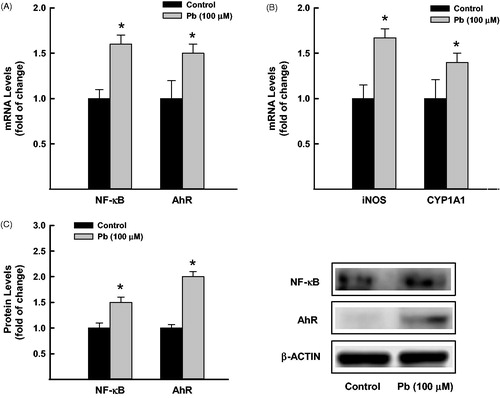
To determine the effect of Pb treatment on the expression of NF-κB and AhR, we measured the mRNA and protein expression levels of NF-κB and AhR in A549 cells exposed to Pb for 24 h. Our results showed that treatment of A549 cells with Pb 100 μM significantly increased the mRNA levels of NF-κB and AhR by approximately 60% and 55%, respectively compared to control cells (. To further confirm that induction of NF-κB and AhR by Pb was associated with increase in their downstream targets, we quantified the mRNA expression of iNOS and CYP1A1, respectively. shows that Pb treatment significantly increased the iNOS and CYP1A1 mRNA levels by approximately 75% and 45%, respectively. At the protein level, similarly, Pb treatment for 24 h increased the protein expression levels of NF-κB and AhR by approximately 50% and 100%, respectively (.
Figure 3. Effect of Pb treatment on NF-κB and AhR expression in A549 cells. The cells were treated for 24 h with Pb (100 µM). (A) NF-κB and AhR, and (B) CYP1A1, and iNOS mRNA levels were determined by RT-PCR. The values represent mean of fold change ± SEM (n = 6, triplicate). (C) NF-κB and AhR protein levels were measured by Western blot analysis. The intensity of protein bands was quantified relative to the signals obtained for β-actin protein, using C-DiGit® blot scanner, LI-COR Biotechnology (Lincoln, NE). The values represent mean of fold change ± SEM (n = 3, triplicate). *p < 0.05 compared to control.
3.4. Effect of NF-κB and AhR inhibition on Pb-induced lung toxicities
To further explore the role of AhR and NF-κB in Pb-induced lung toxicities, we tested whether inhibition of NF-κB or AhR either chemically or genetically would prevent Pb to induce lung apoptosis and oxidative stress. For this purpose, two independent experiments were conducted as follows.
3.4.1. Effect of NF-κB and AhR chemical inhibitor resveratrol on Pb-induced lung apoptosis and oxidative stress
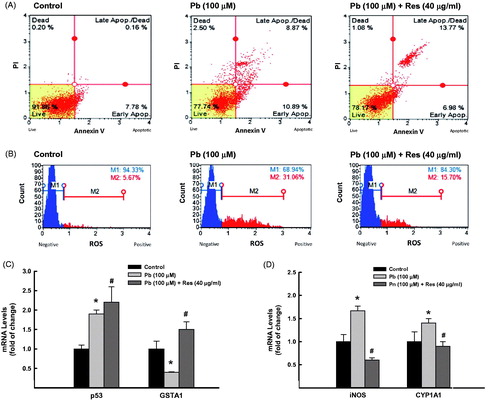
A549 cells were pretreated for 2 h with 40 μg/ml resveratrol (Res), a well-known inhibitor for AhR and NF-κB (Ren et al. Citation2013), before incubated with Pb 100 μM for additional 24 h. Thereafter, apoptotic and oxidative stress markers were determined. shows that Res treatment did not neither reduce the percentage of apoptotic cells () nor downregulate p53 mRNA expression () induced by Pb. On the other hand, Res treatment successfully blocked Pb-induced oxidative stress. This was evidenced by (1) the ability of Res to reduce the ROS formation by Pb by 50% (from 31% to 16%) with significant increase in the percentage of healthy cells from 69% to 84% and (2) by restoring Pb-induced GSTA1 mRNA inhibition by three-fold (. Furthermore, the effects of Res on Pb-induced lung toxicities were associated with significant inhibition of NF-κB and AhR downstream genes, iNOS and CYP1A1 (.
Figure 4. Effect of Res on Pb-induced lung toxicities. A549 cells were treated for 24 h with Pb (100 µM) in the presence and absence of Res (40 μg/ml). (A) the percentage of cells underwent apoptosis was determined by flow cytometry using annexin V/Propidium iodide (PI). (B) The formation of ROS was determined using DCF as a substrate. (C) GSTA1, p53 and (D) iNOS, CYP1A1 mRNA levels were quantified by RT-PCR normalized to β-actin housekeeping gene. The values represent mean of fold change ± SEM (n = 6, triplicate). *p < 0.05 compared to control; #p < 0.05 compared to Pb treatment.
3.4.2. Effect of NF-κB and AhR silencing on Pb-induced lung apoptosis and oxidative stress
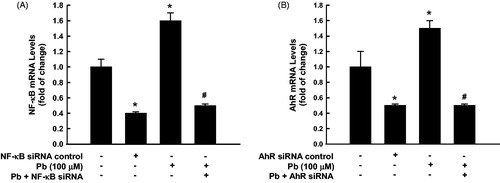
To further confirm the exact role of NF-κB and AhR in Pb-induced-lung toxicities, we examined the effect of knock down of either NF-κB or AhR on Pb-induced apoptosis and oxidative stress. Initially, transfection of the untreated cells with siRNA against either NF-κB or AhR caused approximately 50% knockdown of the constitutive gene expression of both NF-κB () and AhR (). Importantly, induction of NF-κB and AhR mRNA by Pb was inhibited by 70% () and 65% (), respectively, by gene silencing ().
Figure 5. NF-κB and AhR knockdown. A549 cells were transfected with either NF-κB or AhR siRNA before treated for 24 h with Pb (100 µM). Thereafter, NF-κB (A) and AhR (B) mRNA expression levels were quantified by RT-PCR normalized to β-actin housekeeping gene. The values represent mean of fold change ± SEM (n = 6, triplicate). *p < 0.05 compared to control; #p < 0.05 compared to Pb treatment.
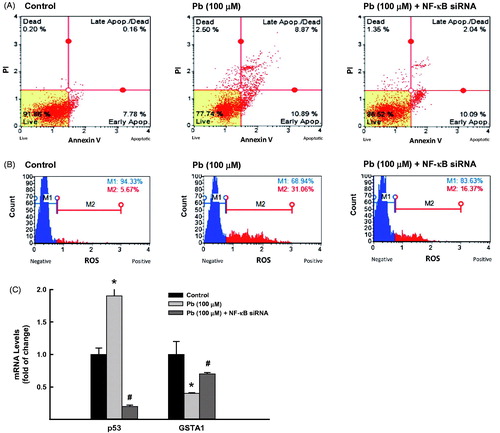
To further explore the role of NF-κB in Pb-induced lung toxicities, we tested the effect of NF-κB silencing using siRNA on Pb-induced lung toxicities. For this purpose, A549 cells were transfected with NF-κB siRNA in the presence and absence of Pb 100 μM. Knockdown of NF-κB significantly decreased the Pb-increased percentage of apoptotic cells from 19% to 12% (37% inhibition) while increased the percentage of healthy cells from 77% to 86% (. This was accompanied with approximately 80% reduction in Pb-induced apoptotic marker, p53 (. Similarly, knockdown of NF-κB significantly decreased the ROS formation in A549 cells treated with Pb from 31% to 16.4% () and restored the inhibition of GSTA1 by Pb from 40% to 70% (.
Figure 6. Effect of NF-κB knockdown on Pb-induced lung toxicities. A549 cells were transfected with NF-κB siRNA before treated for 24 h with Pb (100 µM). (A) the percentage of cells underwent apoptosis was determined by flow cytometry using annexin V/PI. (B) The formation of ROS was determined using DCF as a substrate. (C) GSTA1 and p53 mRNA expression levels were quantified by RT-PCR normalized to β-actin housekeeping gene. The values represent mean of fold change ± SEM (n = 6, triplicate). *p < 0.05 compared to control; #p < 0.05 compared to Pb treatment.
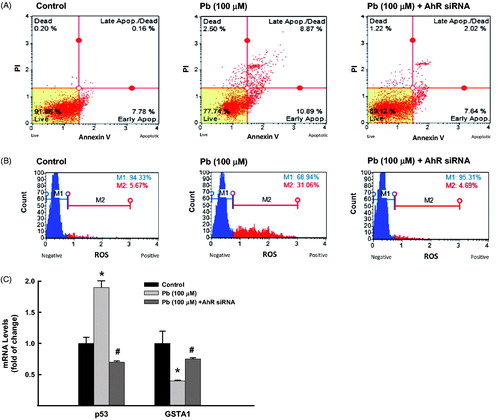
With regard to the role of AhR, we tested the effect of AhR silencing on Pb-induced lung toxicities. Knockdown of AhR significantly decreased the Pb-increased percentage of apoptotic cells from 19% to 10% (47% inhibition) while increased the percentage of healthy cells from 77% to 89% (. This was accompanied with approximately 60% reduction in Pb-induced apoptotic marker, p53 (. Similarly, knockdown of AhR significantly decreased the ROS formation in A549 cells treated with Pb from 31% to 4.7% () and restored the inhibition of GSTA1 by Pb from 40% to 70% (.
Figure 7. Effect of AhR knockdown on Pb-induced lung toxicities. A549 cells were transfected with AhR siRNA before treated for 24 h with Pb (100 µM). (A) the percentage of cells underwent apoptosis was determined by flow cytometry using annexin V/PI. (B) The formation of ROS was determined using DCF as a substrate. (C) GSTA1 and p53 mRNA expression levels were quantified by RT-PCR normalized to β-actin housekeeping gene. The values represent mean of fold change ± SEM (n = 6, triplicate). *p < 0.05 compared to control; #p < 0.05 compared to Pb treatment.
4. Discussion
The present study demonstrates that Pb exposure-induced oxidative stress and apoptosis in A549 human lung cells are mediated through NF-κB and AhR signaling pathways. This is supported by the following findings: (a) Pb-exposure induces up-regulation of apoptotic genes such as P53 which was associated with an increase in the percentage of apoptotic cells, (b) Pb-exposure increases the oxidative stress in lung cells by inhibiting the expression of the antioxidant gene GSTA1 and increasing the ROS generation, (c) Pb-exposure increases the activation of NF-κB and AhR pathways and their downstream targets at the mRNA and protein levels, and (d) chemical and genetic inhibition of NF-κB or AhR attenuates the Pb-induced oxidative stress and apoptosis.
Pb toxicity is associated with lung tissue damage, including induction of oxidative stress and apoptosis (Singh et al. Citation1999; Farkhondeh et al. Citation2014; Lu et al. Citation2015; Zeng et al. Citation2017). Several studies have shown that Pb-exposure increases the risk of lung cancer (Anttila et al. Citation1995; Wang et al. Citation2008; Wang et al. Citation2013). Although Pb-induced oxidative stress and apoptosis in rat lungs have been demonstrated before, the underlying molecular mechanisms remain unclear (Shabani and Rabbani Citation2000; Samarghandian et al. Citation2013). In this study, gene expression and flow cytometric analyses were used to determine the molecular and cellular apoptogenic and oxidant mechanism of Pb in human lung A549 cells.
A549 cell line model has characteristic features of Type II cells of the pulmonary epithelium, including endocytic ability, metabolic properties, and lamellar bodies that consistent with the same type cells. The in vitro A549 cell line was selected in the current work as a study model for several reasons. First, the basal and inducible expression of the transcription factors AhR and NF-κB, CYP1A, and the proinflammatory cytokines and chemokines are much higher in A549 cell line compared with common normal bronchial cell line (BEAS-2B) (Döhr et al. Citation1997; Hukkanen et al. Citation2000; Hillyer et al. Citation2018). Second, although A549 cell line is cancerous, it is a valuable model for studying the mechanism of lung infections, asthma, and allergies. In addition, they have been extensively used with success as an experimental model for investigating the toxic effect of environmental toxicants such as heavy metals. For example, in 2018, Choi et al., has examined the combined effects of heavy metals using in vitro A549 human lung cancer cells (Choi et al. Citation2018).
Initially, we have reported here that exposure of the lung cancer cells to Pb induced lung toxicities as evidenced by induction of apoptosis and oxidative stress. Apoptosis is a crucial process that is important in cancer pathology and pharmacotherapy. It is regulated by several cell cycle progression regulatory proteins, including P53. In this study, the induction of apoptosis in A549 cells after Pb-exposure was monitored by two approaches. First by the ability of Pb to increase the percentage of cells underwent apoptosis/necrosis using flow cytometry. Second, by the induction of the gene expression of apoptotic marker P53 using RT-PCR. In agreement with our results, Xu and his coworkers have identified that Pb-induced apoptosis can be mediated through an increase of P53 in mice PC 12 cells (Xu et al. Citation2006, Citation2008). Moreover, Tousson et al. (Citation2011) reported that Pb-exposure causes a significant increase in apoptotic P53 protein in rabbit liver tissues (Tousson et al. Citation2011) and that P53 knockdown attenuates apoptosis induction (Jung et al. Citation2012; Choi et al. Citation2015; Liu et al. Citation2015). These observations not only indicate that Pb is a strong trigger for apoptosis but also strongly suggest that P53 plays a key role in Pb-induced apoptosis. In this context, a previous study had reported a similar P53-mediated apoptosis in epidermal cells exposed to the heavy metal cadmium (Son et al. Citation2010).
Increasing evidence suggests that changes in the oxidation balance in tissues trigger apoptosis and play a major role in inflammatory responses both in vitro and in vivo (Rahman et al. Citation2002; Rivas-Arancibia et al. Citation2015). The ability of Pb to induce oxidative stress in lung cells in the current study is evidenced by the ability of Pb treatment to decrease the expression of GSTA1, a well-known antioxidant gene, and to increase the ROS generation. Our results were in agreement with previous studies showed that changes in the expression levels of antioxidant protein GSTA1 play a key role in the induction of lung inflammation (Rahman and Adcock Citation2006; Sohn et al. Citation2013). Furthermore, a significant reduction in the antioxidant gene, GSTA1 mRNA expression level was demonstrated in people chronically exposed to environmental heavy metals (Al Bakheet et al. Citation2013). Taken together, these reports clearly suggest that down-regulation of GSTA1 gene could be another mechanism by which immune reactions are triggered after Pb-exposure.
Transcriptional factors AhR and NF-κB play significant roles in the regulation of several physiological processes including apoptosis and oxidative stress (Nebert et al. Citation2000; Suzuki et al. Citation2009). In that, it has been demonstrated that the up-regulation of AhR and NF-κB promotes oxidative stress and apoptosis in cancer and non-cancer human lung epithelial cells (Zhang et al. Citation2015; Jaligama et al. Citation2018). Moreover, AhR knockdown protected zebrafish against cardiac toxicity (Van Tiem and Di Giulio Citation2011), whereas, NF-κB knockdown protects the lung from lipopolysaccharide-induced inflammation in rats (Li et al. Citation2016).
However, the question of whether NF-κB or AhR is controlling Pb-induced oxidative stress and apoptosis in human lung cells was not addressed before. To answer this question, we have utilized two approaches. First, we tested the effect of Pb treatment on the expression and activity of NF-κB and AhR and their downstream targets, iNOS and CYP1A1, respectively, in A549 cells. Our results demonstrated significant increases in the mRNA and protein expression of NF-κB and AhR with Pb-exposure which was also associated with a significant increase in their regulated genes iNOS and CYP1A1, respectively. Similarly, previous studies have demonstrated increase in P53, iNOS, and CYP1A1 mRNA expression levels with Pb exposure in different cell lines (Song et al. Citation2001; Korashy and El-Kadi Citation2008; Tousson et al. Citation2011; Al Bakheet et al. Citation2013). These results suggest a possible role for NF-κB and AhR in Pb-triggered immune responses (Yoshizawa et al. Citation2005; Wang et al. Citation2018; Zhao et al. Citation2006, Citation2018). This conclusion is supported by the observations that AhR and NF-κB signaling pathways mediate lung toxicities (Zhao et al. Citation2006; Yu et al. Citation2013).
The second approach was to examine whether inhibition of AhR or NF-κB either chemically or genetically would attenuate the Pb-mediated effects. In the current study, Res, a natural chemical found in red wine, has been selected as a chemical inhibitor for both pathways AhR and NF-κB, which is well documented and reported in the literature. In that, Res has been shown to protect the lungs from the apoptotic effect of benzo[a]pyrene that is an AhR agonist through direct block of the AhR receptor (Revel et al. Citation2003). This was further supported by the observations that AhR-knockout mouse did not shown any significant apoptosis after exposure to AhR agonist, TCDD, whereas Res fully protected against apoptosis induction by TCDD in wild-type mouse (Sanchez-Martin et al. Citation2011). Furthermore, a mechanistic study has reported that overexpression of AhR, increases RelA nuclear translocation causing AhR/RelA complexation which then binds to the κB element causing NF-κB pathway activation (Tsay et al. Citation2013).
Chemical inhibition of AhR and NF-κB by Res significantly attenuated the Pb-induced oxidative stress by reducing the formation of ROS and restored the expression of GSTA1, iNOS, and CYP1A1 in A549 cells exposed to Pb. In agreement with our results, it has been reported that Res prevents nanoparticles-induced inflammation and oxidative stress in A549 cells via inhibition of ROS production and oxidative stress (Hsu et al. Citation2018). In addition, since activation of AhR/CYP1A1 pathway by Pb is always associated with increased oxidative stress, we postulate here that blocking of AhR would explain the inhibitory effect of Res on ROS production.
At the genetic inhibition level, NF-κB or AhR knockdown using siRNA down-regulated the basal and Pb-induced expression of AhR and NF-κB. Perhaps the most interesting findings are that AhR or NF-κB knockdown significantly blocked the Pb-induced apoptosis and oxidative stress as evidenced by reducing the percentage of apoptotic cells, formation of ROS, and restoring the apoptotic and oxidative stress genes. Our results are inline with previous studies which demonstrated that P53-mediated apoptosis and GSTA1-mediated oxidative stress are both controlled by AhR and NF-κB pathways (Mathieu et al. Citation2001; Brauze et al. Citation2014; Wohak et al. Citation2016; Zhang et al. Citation2016).
In conclusion, the current study demonstrates that Pb-exposure-induced lung toxicities are mediated through the transcription factors AhR and NF-κB. It also reveals that the transcription factor AhR and NF-κB could be used as early biomarkers for detecting Pb lung toxicities. However, there are two limitations in this study that could be addressed in future research. First, the in vitro A549 cell model utilized in the current study provides a limited and incomplete data because it does not mimic the physiological and metabolic responses of the organism. Second, the in vitro Pb concentrations used in this study are above the feasible plasma level and not directly applicable to humans. Thus the utilization of in vivo animal models is necessary to provide a whole-body assessment and an understanding of the gene expression alterations.
Acknowledgment
The authors would like to thank the College of Pharmacy and the Faculty of Graduate Studies, King Saud University, for their endless support. The authors thank the Deanship of Scientific Research and RSSU at King Saud University for providing technical support. The publication of this article was funded by the Qatar National Library, Qatar.
Disclosure statement
There are no financial or other interests with regard to this manuscript that might be constructed as a conflict of interest. All of the authors are aware of and agree to the content of the manuscript and their being listed as an author on the manuscript.
Correction Statement
This article has been republished with minor change. This change do not impact the academic content of the article.
Additional information
Funding
References
- Abdullah ML, Hafez MM, Al-Hoshani A, Al-Shabanah O. 2018. Anti-metastatic and anti-proliferative activity of eugenol against triple negative and HER2 positive breast cancer cells. BMC Complement Alternat Med. 18(1):321.
- Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. 2003. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther. 2(5):471–478.
- Agency for Toxic Substances and Disease Registry (ATSDR). 2017. The ATSDR 2017 Substance Priority List. Public Health Service; Available from: https://www.atsdr.cdc.gov/spl/index.html
- Al Bakheet SA, Attafi IM, Maayah ZH, Abd-Allah AR, Asiri YA, Korashy HM. 2013. Effect of long-term human exposure to environmental heavy metals on the expression of detoxification and DNA repair genes. Environ Pollut. 181:226–232.
- Ansari MA, Maayah ZH, Bakheet SA, El-Kadi AO, Korashy HM. 2013. The role of aryl hydrocarbon receptor signaling pathway in cardiotoxicity of acute lead intoxication in vivo and in vitro rat model. Toxicology. 306:40–49.
- Anttila A, Heikkila P, Pukkala E, Nykyri E, Kauppinen T, Hernberg S, Hemminki K. 1995. Excess lung cancer among workers exposed to lead. Scand J Work Environ Health. 21(6):460–469.
- Beamer CA, Shepherd DM. 2013. Role of the aryl hydrocarbon receptor (AhR) in lung inflammation. Semin Immunopathol. 35(6):693–704.
- Brauze D, Fijalkiewicz K, Szaumkessel M, Kiwerska K, Bednarek K, Rydzanicz M, Richter J, Grenman R, Jarmuz-Szymczak M. 2014. Diversified expression of aryl hydrocarbon receptor dependent genes in human laryngeal squamous cell carcinoma cell lines treated with β-naphthoflavone. Toxicol Lett. 231(1):99–107.
- Chen P, Chang H, Chang J, Lin P. 2012. Aryl hydrocarbon receptor in association with RelA modulates IL-6 expression in non-smoking lung cancer. Oncogene. 31(20):2555.
- Choi EY, Shin KC, Lee J, Kwon TK, Kim S, Park JW. 2015. Treatment with a small synthetic compound, KMU-193, induces apoptosis in A549 human lung carcinoma cells through p53 up-regulation. Asian Pac J Cancer Prev. 16(14):5883–5887.
- Choi Y, Park K, Kim I, Kim SD. 2018. Combined toxic effect of airborne heavy metals on human lung cell line A549. Environ Geochem Health. 40(1):271–282.
- Csavina J, Field J, Taylor MP, Gao S, Landázuri A, Betterton EA, Sáez AE. 2012. A review on the importance of metals and metalloids in atmospheric dust and aerosol from mining operations. Sci Total Environ. 433:58–73.
- Döhr O, Sinning R, Vogel C, Münzel P, Abel J. 1997. Effect of transforming growth factor-β1 on expression of aryl hydrocarbon receptor and genes of AhGene battery: clues for independent down-regulation in A549 cells. Mol Pharmacol. 51(5):703–710.
- Farkhondeh T, Boskabady MH, Jalali S, Bayrami G. 2014. The effect of lead exposure on tracheal responsiveness to methacholine and ovalbumin, total and differential white blood cells count, and serum levels of immunoglobulin E, histamine, and cytokines in guinea pigs. Hum Exp Toxicol. 33:325–333.
- Ghosh S, May MJ, Kopp EB. 1998. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 16(1):225–260.
- Hillyer P, Shepard R, Uehling M, Krenz M, Sheikh F, Thayer KR, Huang L, Yan L, Panda D, Luongo C, et al. 2018. Differential responses by human respiratory epithelial cell lines to respiratory syncytial virus reflect distinct patterns of infection control. J Virol. 92(15):2202.
- Hsu HT, Tseng YT, Wong WJ, Liu CM, Lo YC. 2018. Resveratrol prevents nanoparticles-induced inflammation and oxidative stress via downregulation of PKC-alpha and NADPH oxidase in lung epithelial A549 cells. BMC Complement Alternat Med. 18(1):211.
- Hukkanen J, Lassila A, Paivarinta K, Valanne S, Sarpo S, Hakkola J, Pelkonen O, Raunio H. 2000. Induction and regulation of xenobiotic-metabolizing cytochrome P450s in the human A549 lung adenocarcinoma cell line. Am J Respir Cell Mol Biol. 22(3):360–366.
- Jaligama S, Patel VS, Wang P, Sallam A, Harding J, Kelley M, Mancuso SR, Dugas TR, Cormier SA. 2018. Radical containing combustion derived particulate matter enhance pulmonary Th17 inflammation via the aryl hydrocarbon receptor. Part Fibre Toxicol. 15(1):20.
- Jarup L. 2003. Hazards of heavy metal contamination. Br Med Bull. 68:167–182.
- Jemal A, Torre LA. 2018. The global burden of cancer. In: The American Cancer Society, editor. The American Cancer Society's principles of oncology: prevention to survivorship. Hoboken, NJ: Wiley:pp. 33–44.
- Jung HY, Joo HJ, Park JK, Kim YH. 2012. The blocking of c-met signaling induces apoptosis through the increase of p53 protein in lung cancer. Cancer Res Treat. 44(4):251–261.
- Kimura A, Naka T, Nakahama T, Chinen I, Masuda K, Nohara K, Fujii-Kuriyama Y, Kishimoto T. 2009. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J Exp Med. 206(9):2027–2035.
- Kobayashi S, Okamoto H, Iwamoto T, Toyama Y, Tomatsu T, Yamanaka H, Momohara S. 2008. A role for the aryl hydrocarbon receptor and the dioxin TCDD in rheumatoid arthritis. Rheumatology. 47(9):1317–1322.
- Korashy HM, Attafi IM, Ansari MA, Assiri MA, Belali OM, Ahmad SF, Al-Alallah IA, Anazi FE, Alhaider AA. 2016a. Molecular mechanisms of cardiotoxicity of gefitinib in vivo and in vitro rat cardiomyocyte: role of apoptosis and oxidative stress. Toxicol Lett. 252:50–61.
- Korashy HM, Belali OM, Ansar MA, Alharbi NO. 2016b. FoxO3a is essential for the antiproliferative and apoptogenic effects of sunitinib in MDA-MB231 cell line. Anticancer Res. 36:6097–6108.
- Korashy HM, El-Kadi AO. 2005. Regulatory mechanisms modulating the expression of cytochrome P450 1A1 gene by heavy metals. Toxicol Sci. 88(1):39–51.
- Korashy HM, El-Kadi AO. 2008. The role of redox-sensitive transcription factors NF-kappaB and AP-1 in the modulation of the Cyp1a1 gene by mercury, lead, and copper. Free Radic Biol Med. 44(5):795–806.
- Korashy HM, Maayah ZH, Al Anazi FE, Alsaad AM, Alanazi IO, Belali OM, Al-Atawi FO, Alshamsan A. 2017. Sunitinib inhibits breast cancer cell proliferation by inducing apoptosis, cell-cycle arrest and DNA repair while inhibiting NF-κB signaling pathways. Anticancer Res. 37(9):4899–4909.
- Kumar S, Singh S, Mehta D, Garg RR, Garg ML, Singh N, Mangal PC, Trehan PN. 1991. Effect of automobile exhaust on the distribution of trace elements and its modulation following Fe, Cu, and Zn supplementation. Biol Trace Elem Res. 31(1):51–62.
- Li N, Song Y, Zhao W, Han T, Lin S, Ramirez O, Liang L. 2016. Small interfering RNA targeting NF-κB attenuates lipopolysaccharide-induced acute lung injury in rats. BMC Physiol. 16(1):7.
- Liu CJ, Zhang XL, Luo DY, Zhu WF, Wan HF, Yang JP, Yang XJ, Wan FS. 2015. Exogenous p53 upregulated modulator of apoptosis (PUMA) decreases growth of lung cancer A549 cells. *Asian Pac J Cancer Prev. 16(2):741–746.
- Liu CM, Sun YZ, Sun JM, Ma JQ, Cheng C. 2012. Protective role of quercetin against lead-induced inflammatory response in rat kidney through the ROS-mediated MAPKs and NF-kappaB pathway. Biochim Biophys Acta. 1820(10):1693–1703.
- Lu CF, Yuan XY, Li LZ, Zhou W, Zhao J, Wang YM, Peng SQ. 2015. Combined exposure to nano-silica and lead induced potentiation of oxidative stress and DNA damage in human lung epithelial cells. Ecotoxicol Environ Saf. 122:537–544.
- Lundstrom NG, Nordberg G, Englyst V, Gerhardsson L, Hagmar L, Jin T, Rylander L, Wall S. 1997. Cumulative lead exposure in relation to mortality and lung cancer morbidity in a cohort of primary smelter workers. Scand J Work Environ Health. 23:24–30.
- Mathieu M-C, Lapierre I, Brault K, Raymond M. 2001. Aromatic hydrocarbon receptor (AhR)·AhR nuclear translocator-and p53-mediated induction of the murine multidrug resistance mdr1 gene by 3-methylcholanthrene and benzo (a) pyrene in hepatoma cells. J Biol Chem. 276(7):4819–4827.
- Mu L, Liu L, Niu R, Zhao B, Shi J, Li Y, Swanson M, Scheider W, Su J, Chang S-C, et al. 2013. Indoor air pollution and risk of lung cancer among Chinese female non-smokers. Cancer Causes Control. 24(3):439–450.
- Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. 2000. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 59(1):65–85.
- Rahman I, Adcock I. 2006. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. 28(1):219–242.
- Rahman I, Gilmour PS, Jimenez LA, MacNee W. 2002. Oxidative stress and TNF-alpha induce histone acetylation and NF-kappaB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem. 234/235(1):239–248.
- Ren Z, Wang L, Cui J, Huoc Z, Xue J, Cui H, Mao Q, Yang R. 2013. Resveratrol inhibits NF-kB signaling through suppression of p65 and IkappaB kinase activities. Die Pharmazie. 68(8):689–694.
- Revel A, Raanani H, Younglai E, Xu J, Rogers I, Han R, Savouret JF, Casper RF. 2003. Resveratrol, a natural aryl hydrocarbon receptor antagonist, protects lung from DNA damage and apoptosis caused by benzo[a]pyrene. J Appl Toxicol. 23(4):255–261.
- Rivas-Arancibia S, Zimbron LF, Rodriguez-Martinez E, Maldonado PD, Borgonio Perez G, Sepulveda-Parada M. 2015. Oxidative stress-dependent changes in immune responses and cell death in the substantia nigra after ozone exposure in rat. Front Aging Neurosci. 7:65.
- Samarghandian S, Borji A, Afshari R, Delkhosh MB, Gholami A. 2013. The effect of lead acetate on oxidative stress and antioxidant status in rat bronchoalveolar lavage fluid and lung tissue. Toxicol Mech Methods. 23(6):432–436.
- Sanchez-Martin FJ, Fernandez-Salguero PM, Merino JM. 2011. Aryl hydrocarbon receptor-dependent induction of apoptosis by 2,3,7,8-tetrachlorodibenzo-p-dioxin in cerebellar granule cells from mouse. J Neurochem. 118:153–162.
- Shabani A, Rabbani A. 2000. Lead nitrate induced apoptosis in alveolar macrophages from rat lung. Toxicology. 149(2–3):109–114.
- Singh J, Pritchard DE, Carlisle DL, McLean JA, Montaser A, Orenstein JM, Patierno SR. 1999. Internalization of carcinogenic lead chromate particles by cultured normal human lung epithelial cells: formation of intracellular lead-inclusion bodies and induction of apoptosis. Toxicol Appl Pharmacol. 161(3):240–248.
- Sohn SW, Jung JW, Lee SY, Kang HR, Park HW, Min KU, Cho SH. 2013. Expression pattern of GSTP1 and GSTA1 in the pathogenesis of asthma. Exp Lung Res. 39(4–5):173–181.
- Son YO, Lee JC, Hitron JA, Pan J, Zhang Z, Shi X. 2010. Cadmium induces intracellular Ca2+- and H2O2-dependent apoptosis through JNK- and p53-mediated pathways in skin epidermal cell line. Toxicol Sci. 113(1):127–137.
- Song JS, Sim SY, Hong DP, Dal Rhee S, Song CW, Han SS, Yang SD. 2001. Lead treatment in vitro at early developmental stage of bone marrow-derived macrophages enhances NO production through IL-1beta and IL-6 but not TNF-alpha. Toxicology. 162(1):61–68.
- Suzuki T, Yamashita C, Zemans RL, Briones N, Van Linden A, Downey GP. 2009. Leukocyte elastase induces lung epithelial apoptosis via a PAR-1-, NF-kappaB-, and p53-dependent pathway. Am J Respir Cell Mol Biol. 41(6):742–755.
- Tousson E, Rafat BM, Hessien M, El Barbary AA, Sami A. 2011. P53 and Bcl2 apoptosis proteins in meso-2,3-dimercaptosuccinic acid treated lead-intoxicated rabbits. Toxicol Ind Health. 27(3):271–278.
- Tsay JJ, Tchou-Wong KM, Greenberg AK, Pass H, Rom WN. 2013. Aryl hydrocarbon receptor and lung cancer. Anticancer Res. 33(4):1247–1256.
- Van Tiem LA, Di Giulio RT. 2011. AHR2 knockdown prevents PAH-mediated cardiac toxicity and XRE- and ARE-associated gene induction in zebrafish (Danio rerio). Toxicol Appl Pharmacol. 254(3):280–287.
- Vogel CF, Khan EM, Leung PS, Gershwin ME, Chang WW, Wu D, Haarmann-Stemmann T, Hoffmann A, Denison MS. 2014. Cross-talk between aryl hydrocarbon receptor and the inflammatory response a role for nuclear factor-κB. J Biol Chem. 289(3):1866–1875.
- Vogel CF, Matsumura F. 2009. A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-κB family. Biochem Pharmacol. 77(4):734–745.
- Wang CY, Lin YW, Yang JL. 2008. Activation of protein kinase Calpha signaling prevents cytotoxicity and mutagenicity following lead acetate in CL3 human lung cancer cells. Toxicology. 250(1):55–61.
- Wang SW, Xu Y, Weng YY, Fan XY, Bai YF, Zheng XY, Lou LJ, Zhang F. 2018. Astilbin ameliorates cisplatin-induced nephrotoxicity through reducing oxidative stress and inflammation. Food Chem Toxicol. 114:227–236.
- Wang YT, Tzeng DW, Wang CY, Hong JY, Yang JL. 2013. APE1/Ref-1 prevents oxidative inactivation of ERK for G1-to-S progression following lead acetate exposure. Toxicology. 305:120–129.
- Wohak LE, Krais AM, Kucab JE, Stertmann J, Øvrebø S, Seidel A, Phillips DH, Arlt VM. 2016. Carcinogenic polycyclic aromatic hydrocarbons induce CYP1A1 in human cells via a p53-dependent mechanism. Arch Toxicol. 90(2):291–304.
- Wong O, Harris F. 2000. Cancer mortality study of employees at lead battery plants and lead smelters, 1947–1995. Am J Ind Med. 38(3):255–270.
- Xu J, Ji L-D, Xu L-H. 2006. Lead-induced apoptosis in PC 12 cells: involvement of p53, Bcl-2 family and caspase-3. Toxicol Lett. 166(2):160–167.
- Xu J, Lian L-J, Wu C, Wang X-F, Fu W-Y, Xu L-H. 2008. Lead induces oxidative stress, DNA damage and alteration of p53, Bax and Bcl-2 expressions in mice. Food Chem Toxicol. 46(5):1488–1494.
- Yoshizawa K, Marsh T, Foley JF, Cai B, Peddada S, Walker NJ, Nyska A. 2005. Mechanisms of exocrine pancreatic toxicity induced by oral treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin in female Harlan Sprague–Dawley rats. Toxicol Sci. 85(1):594–606.
- Yu X, Pan Y, Ma H, Li W. 2013. Simvastatin inhibits proliferation and induces apoptosis in human lung cancer cells. Oncol Res. 20(8):351–357.
- Zeng X, Xu X, Boezen HM, Vonk JM, Wu W, Huo X. 2017. Decreased lung function with mediation of blood parameters linked to e-waste lead and cadmium exposure in preschool children. Environ Pollut. 230:838–848.
- Zhang D, Ma D, Yao Z, Fu C, Shi Y, Wang Q, Tang Q. 2016. ERK1/2/p53 and NF-κB dependent-PUMA activation involves in doxorubicin-induced cardiomyocyte apoptosis. Eur Rev Med Pharmacol Sci. 20(11):2435–2442.
- Zhang S, Patel A, Chu C, Jiang W, Wang L, Welty SE, Moorthy B, Shivanna B. 2015. Aryl hydrocarbon receptor is necessary to protect fetal human pulmonary microvascular endothelial cells against hyperoxic injury: mechanistic roles of antioxidant enzymes and RelB. Toxicol Appl Pharmacol. 286(2):92–101.
- Zhao H, Barger MW, Ma JK, Castranova V, Ma JY. 2006. Cooperation of the inducible nitric oxide synthase and cytochrome P450 1A1 in mediating lung inflammation and mutagenicity induced by diesel exhaust particles. Environ Health Perspect. 114(8):1253–1258.
- Zhao H, Wang Y, Shao Y, Liu J, Wang S, Xing M. 2018. Oxidative stress-induced skeletal muscle injury involves in NF-kappaB/p53-activated immunosuppression and apoptosis response in copper (II) or/and arsenite-exposed chicken. Chemosphere. 210:76–84.