
Abstract
Phosgene is a gas crucial to industrial chemical processes with widespread production (∼1 million tons/year in the USA, 8.5 million tons/year worldwide). Phosgene’s high toxicity and physical properties resulted in its use as a chemical warfare agent during the First World War with a designation of CG (‘Choky Gas’). The industrial availability of phosgene makes it a compound of concern as a weapon of mass destruction by terrorist organizations. The hydrophobicity of phosgene exacerbates its toxicity often resulting in a delayed toxidrome as the upper airways are moderately irritated; by the time symptoms appear, significant damage has occurred. As the standard of care for phosgene intoxication is supportive therapy, a pressing need for effective therapeutics and treatment regimens exists. Proposed toxicity mechanisms for phosgene based on human and animal exposures are discussed. Whereas intermediary components in the phosgene intoxication pathways are under continued discussion, generation of reactive oxygen species and oxidative stress is a common factor. As animal models are required for the study of phosgene and for FDA approval via the Animal Rule; the status of existing models and their adherence to Haber’s Rule is discussed. Finally, we review the continued search for efficacious therapeutics for phosgene intoxication; and present a rapid post-exposure response that places exogenous human heat shock protein 72, in the form of a cell-penetrating fusion protein (Fv-HSP72), into lung tissues to combat apoptosis resulting from oxidative stress. Despite significant progress, additional work is required to advance effective therapeutics for acute phosgene exposure.
History
Phosgene (carbonyl chloride, COCl2, CAS registry 75-44-5), the chemical whose name is so inextricably tied to the trenches of World War I (WWI); it is easy to forget that the molecule has been around for over 200 years. First ‘photosynthesized’ in 1812 by Cornish chemist John Davy by exposing a mixture of chlorine and carbon monoxide to sunlight, he bestowed the popular moniker upon his creation by merging the Greek words for light (phos) and born (gene) (Davy Citation1812). It proved useful to the textile industry in the 1880s when German chemist Heinrich Caro and Swiss chemist Alfred Kern treated phosgene with dimethylaniline for the first step in the synthesis of the brilliant purple dye, Crystal violet (Reinhardt and Travis Citation2000). Ironically, a toxic and colorless gas at ambient temperature (BP = 8.3 °C https://www.cdc.gov/niosh/npg/npgd0504.html), was used as a reactant in the synthesis of a rich dye. But the wide potential of phosgene as a high-volume industrial feed stock chemical was fully realized in the 20th century in the synthesis of plastics (polyurethanes, polycarbonates, and polyureas), dyes, pharmaceuticals, and agrochemicals. The majority of phosgene is currently used in the synthesis of monomers, such as toluene diisocyanate (TDI), for polyurethane foam. During this process, phosgene is typically prepared on site from the reaction of carbon monoxide and chlorine with an activated carbon catalyst rather than sunlight (Holmes et al. Citation2016). Its ready availability and high toxicity make it an agent of concern to the military and to homeland security authorities (Bast and Glass-Mattie Citation2015; Baggett and Simpkins Citation2018).
The destructive potential of phosgene was highlighted by WWI. Although the first chemical attack during the war was launched by the Germans at the battle of Ypres, Belgium on 22 April 1915; they used chlorine, a pungent yellow-green gas that allowed easy visual and olfactory detection. And yet, the surprise of the attack killed more than 1100 people and left 7000 injured (Evert Citation2015). Over the following months, the German army tested phosgene, a colorless gas. The phosgene was mixed with chlorine and used on the approaches to Warsaw in the Eastern Front. The magnitude of this type of attack was seen on 19 October 1915 when the Germans released 275 tons of a mixture of one part phosgene and four parts chlorine at Champagne on the Western Front from about 14,000 cylinders over 12 km (Ryan et al. Citation1996). By the time of the oft-cited attack over 4-5 km of the front at Ypres on 19 December 1915, the British had issued gas helmets and the phosgene attack killed only 116 and injured approximately 1000 despite the presence of 25,000 troops in the area (Ryan et al. Citation1996). Both the physical and chemical characteristics of phosgene are responsible for the observed lethality during WWI. Because of its density (ρ = 3.5 g/mL, NIOSH (National Institute of Occupational Safety and Health Citation1976) https://www.cdc.gov/niosh/nioshtic-2/00052750.html), it tended to collect and persist in low lying areas (e.g. trenches) during WWI and thus resulted in the greatest number of chemical warfare agent related fatalities (Bast and Glass-Mattie Citation2015; Summerhill et al. Citation2017). Although the Germans were the first to use it on the battlefield, it became the chemical weapon of choice for the Allies, predominantly by the French (Ryan et al. Citation1996). Phosgene is often cited as being responsible for 85% of fatalities caused by this class of weapons during WWI (Ministry of Defense Citation1987); however, authoritative sources dispute that claim (Ryan et al. Citation1996). The absolute number of WWI casualties resulting from exposure to all Chemical Warfare Agents (CWAs) is difficult to determine due to the lack of analytical equipment and the presence of multiple injuries to soldiers. Furthermore, determining which agent is responsible for the casualty is even more difficult due to the use of CWA mixtures by all armies. It is estimated that of the 518,000 casualties resulting from CWA exposure, between 17,000 and 26,000 resulted in death (Ryan et al. Citation1996).
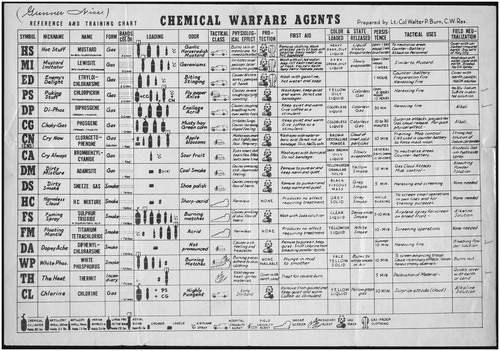
In the scientific literature today, phosgene’s odor (safety classification E) has been reported to resemble newly mowed hay or grass with a threshold for human detection between 0.5 and 1.5 ppm; however, less than 10% can detect this odor at this threshold value (Amoore and Hautala Citation1983). The NIOSH’s Immediately Dangerous to Life or Health (IDLH) level for phosgene has been set at 2 ppm (8 mg/m3) (https://www.cdc.gov/niosh/idlh/75445.html). Thus, a sizeable majority of humans may be already in immediate danger as they detect phosgene’s odor. During WWI, the odor was characterized as ‘musty hay’ or ‘green corn’ (). As scientists, we should reflect on that for a moment. Despite the repeated use of this description in the literature on phosgene, chances are the reader (as well as these authors) have not had the opportunity to regularly smell musty hay, yet it must have been an all too common experience for the legions of farm boys-turned-dough boys in the trenches of WWI. With the advent of chemical warfare, the uninitiated had to be quickly trained into a field chemist, and with that, in the post-WWI era, a chart was distributed to help soldiers determine the nature of a chemical attack (). Besides the odor, the soldier was given another description to look out for: ‘Choky-Gas’. And the CG codename for phosgene is born. An apt term for a gas which attacks the respiratory system. First aid to be rendered in the event of phosgene inhalation: ‘Keep quiet and warm. Give coffee as a stimulant.’ ().
Figure 1. Reference and Training Chart. Since most combatants in the trenches lacked a doctorate in chemistry, charts summarizing the litany of poison gasses a soldier could be exposed to were prepared. This poster was prepared by Lieutenant Colonel Walter P. Burns, a US Army chemical warfare expert. Note the phosgene entry in the 6th row from the top. This chart was issued to Lieutenant William Frederick Nice, 49th Co 5th US Marine Regiment. Image courtesy of the Veterans History Project of the American Folklife Center at the Library of Congress. William F. Nice (AFC 2001/001/1339), Veterans History Project Collection, Library of Congress http://memory.loc.gov/diglib/vhp/story/loc.natlib.afc2001001.01339/.
Modern risk to society
In 2003, the EPA identified 123 chemical plants in the nation where a terrorist attack or accident could potentially expose more than 1 million people to a cloud of toxic gas (Homeland Citation2003). This includes high-priority threat agents, such as phosgene, of which 8.5 million tons were produced worldwide in 2015 [China 36.64%, Europe 30.67%, North America 19.83% https://www.marketresearch.com/Gen-Consulting-Company-v4078/Global-Phosgene-Outlook-10248194/; Retrieved September 25, 2020]. The dangers of handling phosgene require its manufacture and consumption within the same plant (Environmental Protection Agency Citation2003), hence, all facilities producing more than 30 metric tons per year must be declared to the Organization for the Prohibition of Chemical Weapons [OPCW, Schedule 3 Annex on Chemicals. https://www.opcw.org/chemical-weapons-convention/annexes/annex-chemicals/schedule-3; Retrieved September 25, 2020]. Unfortunately, we must view such plants as ground zero in the event of a terrorist attack or industrial accident.
Although phosgene will react with water to produce hydrochloric acid and carbon monoxide (t1/2 = 0.26 s) (Environmental Protection Agency 2003), its low solubility in water significantly reduces the rate of the reaction and its rapid hydrolysis mitigates the reaction with the mucus layer manifesting its presence with moderate irritation in the upper airways (Borak and Diller Citation2001). The lack of immediate symptoms combined with the lack of alarming odor characteristics exacerbates the toxicity of phosgene as many that are exposed are not immediately aware of their peril (Bast and Glass-Mattie Citation2020). Those exposed to a sub-lethal dose can suffer chronic symptoms causing a long-term burden to health care systems and the economy. Three years after phosgene exposure in WWI, 83 British soldiers were examined and despite 53% of the men having no physical lung abnormalities, they exhibited shortness of breath (70%), cough with expectoration (54%), a tight feeling in the chest (25%), sporadic giddiness (14%) and nausea (12%) (Sandall Citation1922; Bast and Glass-Mattie Citation2015). These factors have led to extensive research into the mechanisms of toxicity, development of appropriate animal models for research, and efforts toward efficient treatments and therapeutics for phosgene exposure.
Mechanisms of toxicity
The primary exposure route for phosgene is inhalation, although depending on the initial concentration, eye irritation, lacrimation, and upper airway irritation (coughing) has been reported (Glass et al. Citation2009; Grainge and Rice Citation2010). Observations at the macroscopic level since WWI and at the microscopic level since the 1970s allow us to understand phosgene’s interactions with lung tissue.
At the macroscopic level: Decades of studies in mice, rats, guinea pigs and dogs have generated myriad of Lethal Concentration (LC) values as seen in Table 23.3 of Bast and Glass-Mattie (Citation2020, p. 345). Human data has been more challenging to acquire given the sporadic nature of phosgene leakage in the workplace (see Human exposure below). Categorizing exposure to phosgene according to toxic load dosages determined by the strict linear application of concentration × time has allowed the establishment of exposure guidelines by the International Program on Chemical Safety (IPCS) (). Under the assumption of Haber’s rule, the toxidrome of phosgene exposure is linearly dependent on dose and exposure duration, hence, an acute exposure to a high concentration of CG is no more toxic than a chronic exposure to a low concentration. At concentrations >30 ppm x min (>120 mg/m3×min), exposure results in pulmonary edema and a breakdown in the blood-air barrier causing an increase in fluids (Li and Pauluhn Citation2019; Bast and Glass-Mattie Citation2020). This damage results in further accumulation of protein rich fluids or what is termed “Extra Vascular Lung Water” by some (Li and Pauluhn Citation2019). Due to a lack of physiological cues and symptoms, even at low concentrations, immediate damage from phosgene in the lower respiratory tract can remain undetected in what is termed the ‘initial’ phase of exposure (Bast and Glass-Mattie Citation2015; Public Health England Citation2016). Furthermore, clinical recognition of exposure is complicated by an asymptomatic period (Diller Citation1985). During this 2 − 24 hour (h) ‘latent’ period, patients appear symptom free (Russell et al. Citation2006; Smith et al. Citation2009). The duration of the latent phase is inversely proportional to the exposure dose (; (Public Health England Citation2016)). Workers near an undetectable gas leak exposed to 1 ppm for 2.5 h will have inhaled a high dose (>150 ppm × min; >600 mg/m3×min), resulting in neutrophil infiltration, pulmonary edema and oxidative stress in the lungs (Ghio et al. Citation1991; Russell et al. Citation2006). Based on observations from workplace accidents, inhalation of higher doses (≥300 ppm × min; ≥1200 mg/m3×min) may also result in mortality (Diller Citation1985). At higher doses (∼500 ppm × min; ∼2000 mg/m3×min), pulmonary edema becomes evident within 3 h with death following between 12 and 25 h (Li and Pauluhn Citation2019). This pattern appears to break down in the event a victim is acutely exposed (e.g. 2 min) to 300 ppm or more of phosgene (see yellow box in ), resulting in death within minutes (Borak and Diller Citation2001). There are differing views about the apparent cause of death with one source indicating rapid occlusion of the pulmonary vasculature likely due to intravascular hemorrhaging (Public Health England Citation2016) and another indicating acute overdistension of the right heart before any pulmonary edema can develop (Bast and Glass-Mattie Citation2020). Either scenario may be academic to the dead, but it should be of concern given the possibility of a terrorist attack at an industrial plant.
Table 1. Concentration dependent toxidrome of phosgene exposure in humans modified from a summary chart by Bast and Glass-Mattie (Citation2020). Below those exposure times listed as ‘Acute’ by the original authors, the rows are organized according to ascending Dosages (CxT values).
Table 3. Summary of cell-based assays used to study phosgene.
At the microscopic level: Since the mid-20th Century, the symptoms of toxicity are being dissected at the microscopic level to understand the macroscopic observations. Phosgene’s density and low aqueous solubility results in accumulation in the lower lungs where there is a more amenable hydrophobic environment (Li and Pauluhn Citation2019). Diffusion of phosgene in an aqueous solution is about 8.8 μm; 4–8 times the thickness of all three layers of the air–blood barrier in the alveoli (Nash and Pattle Citation1971). Phosgene is the molecule responsible for the damage that ensues, not hydrochloric acid resulting from hydrolysis of phosgene with water (Nash and Pattle Citation1971). According to one model detailed in Holmes et al. (Citation2016) and summarized in , upon penetrating lung tissue, it acylates nucleophilic groups (i.e. amino, hydroxyl, sulfhydryl moieties) present in proteins, lipids, and nucleic acids. At low phosgene concentrations, proteins and phosphatidylcholine constituting the alveolar surfactants are the ones acylated. At >30 ppm × min, the disruption of the surfactants and acylation of biomolecules produces a pulmonary edema as the air-blood barrier is permeabilized to blood plasma during the latent period (). Clinical manifestations at this stage include decreased efficiency in gas exchange with blood cells, labored breathing, a frothy expectorant and the accumulation of proteins in the bronchoalveolar lavage fluid (BALF) (Bast and Glass-Mattie Citation2020). Phosgene also undergoes homolytic cleavage resulting in the reactive chloro carbamoyl radical (Arroyo et al. Citation1993; Holmes et al. Citation2016). These highly reactive species can further alter the surfactants in the lungs through lipid peroxidation, a process that, if left unchecked can damage the cell membranes of lung and blood tissue. Chloro carbamoyl radicals can lead to increased oxidation of glutathione and, with the reduced antioxidant capacity in the lungs, reactive oxygen (ROS) and reactive nitrogen (RNS) species are generated. This results in a plethora of reactions that affect various systems at the molecular level: including epithelial cells needed for gas exchange (Type I and II alveoli); neuronal cells innervating the lungs; endothelial and blood cells (Holmes et al. Citation2016) ().
Figure 2. Mechanism proposed by Holmes et al (Citation2016) for phosgene injury. GSH: glutathione; TRPA1: transient receptor potential cation channel: member A1; NMDA: N-methyl-D-aspartic acid; Met: metabotropic; ET-1: endothelin 1; ETA: endothelin receptor A; ETB: endothelin receptor B; Pmv: pulmonary microvascular pressure; SOCE: store-operated calcium entry; Rxns: reactions. Used with permission, this figure was published in Toxicology Letters, 244, Holmes et al (Citation2016), p 11, Copyright Elsevier (2016).
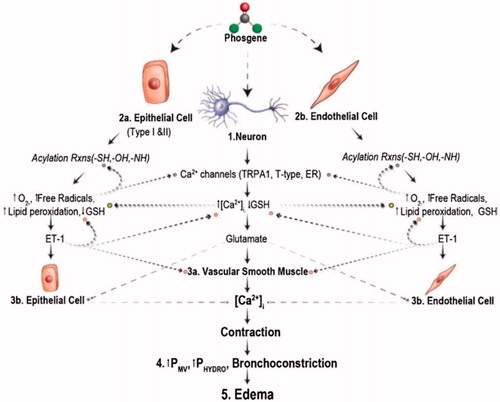
Table 2. Biological effects of phosgene.
There is some disagreement among researchers regarding whether cyclooxygenase-2 (COX-2) and inflammation plays a role in the mechanisms of vasopermeability and pulmonary edema. Studies in rabbits have demonstrated that pulmonary edema induced by phosgene intoxication is caused by increased capillary permeability and not due to any alteration in atrial pressure or other hemodynamic factors (Russell et al. Citation2006). That model relies on the phosphorylation of the JNK/SAPK and p38 MAPK pathways, which increase the expression of COX-2 and, in turn, prostaglandin E2 which contributes to inflammation by enhancing edema and immune cell infiltration. BALF analysis of phosgene intoxicated rats has shown marked increases in prostaglandin E2 (Chen et al. Citation2013) and cytokine mediators (Sciuto et al. Citation2003), as well as antioxidant enzymes (Sciuto et al. Citation2003). Phosphorylation of the same pathways also increases expression of inducible nitric oxide synthase (iNOS), leading to vasodilation, oxygen radicals and oxidative stress. Support for this model comes from the use of ethyl pyruvate, which inhibits COX-2 and iNOS expression, as wells as phosgene-induced pulmonary edema (Chen et al. Citation2013). However, others point to the fact that there are no changes in COX-2 activity post-phosgene exposure (Guo et al. Citation1990; Holmes et al. Citation2016).
Changing perspectives on the mechanistic details of phosgene intoxication impacts which biomarkers remain relevant to the scientific community as drug developers screen new therapeutic compounds. For instance, some have proposed that measuring the Extra Vascular Lung Water is more predictive of subsequent severe pulmonary edema than BALF proteins (Li and Pauluhn Citation2019). While aspects of each model are certainly correct, the clearest common denominator for both models is the formation of ROS leading to oxidative stress.
Cell-based assays for phosgene
Changing perspectives on mechanistic details also confound the ability to replace in vivo toxicity testing with either in vitro (cell-based) or in silico approaches (Rim Citation2020), despite economic and political pressure to do so for many chemicals (e.g., USA: Tox21, European Union: REACH). For example, a recognized limitation of these systems is the inability to mimic the inflammatory response in the early stages of exposure and the consequent tissue damage caused by free radicals (Arroyo et al. Citation1993; Holmes et al. Citation2016; Olivera et al. Citation2017). Within this limitation, a variety of in vitro studies have examined the effect of phosgene ().
The complexity of the respiratory system and the technical challenges involved with direct exposure of cells to airborne toxicants has limited the use of in vitro assays to the elucidation of phosgene’s mechanism of action, identifying biomarkers of intoxication or for the screening of potential therapeutics (for a discussion of the latter see Rim Citation2019). Recent phosgene studies have tried to utilize a more realistic alveolar architecture and environment by growing cells on semi-permeable membranes and lowering the insert into a well so that the basolateral (lower) side contacts the culture media and the apical (upper) side has an air-liquid interface (Wijte et al. Citation2011; Olivera et al. Citation2017). When cells are grown to confluency on the insert’s membrane, such studies allow the added ability to test for cell integrity by measuring the transepithelial electrical resistance (TEER) established once a confluent cell barrier forms. Any disruption of the barrier results in a decrease of the TEER, hence providing a quantitative measure of cell viability. While other observations from such studies have the potential of being developed into cell-based quality control assays for testing the efficacy of therapeutic agents (), the use of TEER to measure cell viability is likely to remain in the realm of research given the challenges of reproducibly validating such a procedure as a lung barrier integrity assay.
Regardless of the cell type or the architecture, little correlation in toxicity between lung cell-based assays and animal models were noted with 19 toxicants (Sauer et al. Citation2013). Finally, in a comprehensive review on the use of in vitro assays for chemical warfare agents, the limitations of these assay have been noted for phosgene by no less than the National Academies of Sciences, Engineering, and Medicine (Citation2015): ‘There does not appear to be a particularly good in vitro model for lung damage associated with chemicals (for example, phosgene) that are known to increase pulmonary permeability that results in noncardiogenic pulmonary edema.’ Hence, although continued development of cell-based assays and exposure systems is necessary for accelerated development of therapeutics and to further elucidate biomarkers and the mechanism of phosgene toxicity, in vivo models are still the primary method to screen new medical countermeasures.
Animal models
While strides have been made in clarifying phosgene’s mechanism of action, capitalizing on it to develop a medical countermeasure has failed so far. A key reason for the lack of progress is its steep toxicity curve, which was once aptly characterized as approaching ‘a step function’ (see in Pauluhn Citation2006 for an excellent illustration of curve steepness). This makes it difficult to determine a consistent LC50 value in an animal model, in turn, affecting the proper determination of a toxic load equation (Concentration [C]×Time [t]) for a toxicologist to rely on when assessing the risk of exposure to a subject in a given period of time (). At the scientific level, the correct choice of a model for acute versus chronic exposure is crucial in the development of therapeutics for phosgene poisoning, given the observation that the toxicity of phosgene does not linearly follow Haber’s Rule (Hobson et al. Citation2019). At the regulatory level, the FDA’s requirement for sufficiently well-characterized animal model(s) for predicting the response in humans for new drug applications filed under the ‘Animal Rule’ has also prompted research into appropriate model species (Park and Mitchel Citation2016; Chemical Warfare Toxicology: Volume 2: Management of Poisoning Citation2018).
Figure 3. The phenomenon of toxicity for poison gas is mathematically described by multiplying the concentration (C) of the gas in a cubic meter of air by the time (t) in minutes (min) that the subject has to breathe the air before death ensues. Today, we often refer to the product of the equation as the Toxic Load Dosage (TL). (A) Haber’s Rule is a special case in the continuum of toxic load modeling where survival depends on a linear relationship between concentration and time. Mathematically speaking, the equation describing such a curve assumes the concentration has a toxic load exponent (ne) equal to 1. In the real world, that means a phosgene release from a chemical plant (represented by the refinery skyline in our schematic) would have similar survival rates for by-standers during a short-duration, high-concentration exposure 
 occurring in surrounding neighborhoods, for example, t = 400 min after the explosion. In our example, we assume the concentration released is theoretically at the LC50 (C = LC50) hence, the same number of survivors
occurring in surrounding neighborhoods, for example, t = 400 min after the explosion. In our example, we assume the concentration released is theoretically at the LC50 (C = LC50) hence, the same number of survivors  and fatalities
and fatalities  are shown in this graphic. (B) When there is an uneven distribution of survivors in the high and low concentration zones, despite an LC50 release of gas, then the concentration value in the toxic load model is modified. If the short-duration, high-concentration exposure is more toxic and has a greater number of fatalities and the long-duration, low-concentration zone has far fewer deaths, than the toxic load exponent for C is ne >1. (C) If the opposite occurs and the acute, high-concentration exposure is not as toxic compared to a chronic, low-concentration exposure that has greater lethality, than the ne <1.
are shown in this graphic. (B) When there is an uneven distribution of survivors in the high and low concentration zones, despite an LC50 release of gas, then the concentration value in the toxic load model is modified. If the short-duration, high-concentration exposure is more toxic and has a greater number of fatalities and the long-duration, low-concentration zone has far fewer deaths, than the toxic load exponent for C is ne >1. (C) If the opposite occurs and the acute, high-concentration exposure is not as toxic compared to a chronic, low-concentration exposure that has greater lethality, than the ne <1.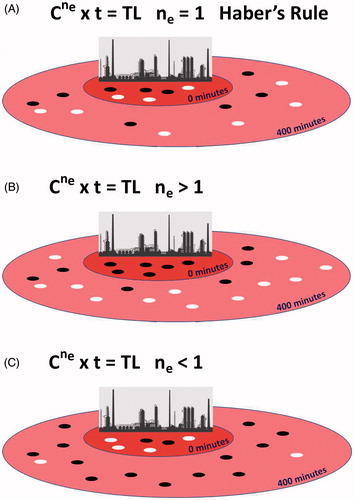
Historically, Haber’s Rule has been a guiding hypothesis when interpreting toxicity results in an animal model (Miller et al. Citation2000). Mathematically predicting the physiological effects of a toxin on an organism in proportion to the toxin’s concentration and the organism’s duration of exposure was first proposed by Warren (Warren Citation1900) regarding the toxicity of salt solutions that kill the planktonic crustacean, Daphnia magna. However, it is Fritz Haber whose name graces the iconic formula C × t, based on work he did exposing cats to phosgene (Haber Citation1924). Ironically, Haber was not an adherent to the idea that C × t applied linearly to all intoxications; he appreciated the importance of the time factor, particularly for chronic exposures, where metabolism and detoxification (toxicodynamics and toxicokinetics) would begin to influence the toxic load (Rozman and Doull Citation2000). To improve the accuracy of the dose response equation, ten Berge et al. (ten Berge et al. Citation1986) introduced the ‘toxic load exponent’ (ne) for the concentration parameter (). An exponent for the time parameter (p) improves toxic load modeling further, particularly for chronic studies; however, if p < 1 the toxicity relies more on concentration than exposure duration, and conversely if p > 1 the toxic effect may not be linear over an extended period of time. In a survey of 21 inhaled toxicants, 14 had an estimated p < 0.45 and only 3 had p > 1 (Belkebir et al. Citation2011). Those with a p > 1 did not exceed p = 1.25. While the survey did not cover phosgene, the results suggest our primary focus here on the concentration parameter with Toxic Load = ![]() × t is not unreasonable. An excellent discussion of all the power curves generated by the equation
× t is not unreasonable. An excellent discussion of all the power curves generated by the equation ![]() × tp is presented in (Miller et al. Citation2000).
× tp is presented in (Miller et al. Citation2000).
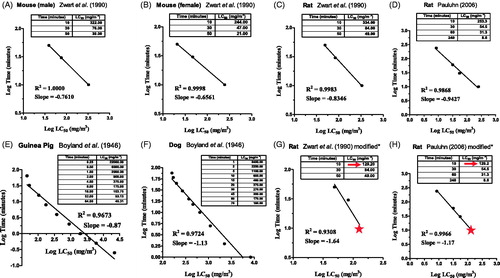
Improved survival after exposing rodents to the LC50 of a toxic inhalant is commonly used to screen drug efficacy in early preclinical development. Inaccurate LC50 values, particularly when studying acute phosgene exposures, can result in costly disruptions. This experience led the authors to develop a more robust nose-only inhalation model for rats to study the effects of short-term exposures (Hobson et al. Citation2019). Earlier systems have provided whole body and nose-only exposures, however, the LC50 data has been mixed. In 1990, (Zwart et al. Citation1990) intoxicated rats and mice with phosgene in custom made horizontal glass cylinders for whole-body exposure and reported LC50 values much higher at a 10 min exposure than for longer ones at 30 and 50 min (). A similar result was obtained in rats using a nose-only inhalation system, albeit with a ne closer to 1 (Pauluhn Citation2006) (). The ne < 1 determination from such data sets can lead to an underestimation of phosgene toxicity in an acute exposure study, as was the case when the authors of this article ran a similar analysis in rats [compare our rat survival curves to Pauluhn’s in of (Hobson et al. Citation2019)]. Variation in phosgene toxicity between small and large mammals has been noted since the 1940s [see in (Boyland et al. Citation1946)]. Whole body exposure studies in guinea pigs (), rats and mice (data not shown), provided ne values of 0.87, 0.65 and 0.73, respectively (Boyland et al. Citation1946). This was not the case for dogs. In Table 4 of Boyland et al. (Citation1946), the authors report an amalgamation of LCt50 results from several studies, including their own (). The ne > 1 seen in dogs is more reflective of the toxicity seen with phosgene in our rat studies using a more robust system.
Figure 4. Determining the correct toxic load exponent (ne) is crucial to ensure the level of risk is correctly assessed in the event of an acute exposure. Plotting exposure time versus the LC50 on a logarithmic scale allows for regression analysis to determine ne (Weinrich et al. Citation2008). Using the method of Sweeney et al. (Citation2015), both time and LC50 values are converted to Log10 before plotting (Sweeney et al. Citation2015). In these studies, concentration (C) is the more independent variable than the time (t) it takes to produce a given effect, hence, C is typically plotted on the x-axis and the negative slope is the ne value (Miller et al. Citation2000). (A–C) Rodent results reported by Zwart et al. (Citation1990) show phosgene does not adhere to Haber’s Rule; with ne <1 their data suggest short-duration, high-concentration exposures are not as toxic as long-duration, low concentration ones. (D) Pauluhn (Citation2006) conducted studies in rats using nose-only inhalation and obtained a lower LC50 at 10 min compared to Zwart et al. (Citation1990). This brought the results closer in line to Haber’s Rule (ne =1). (E) Early small animal studies using whole body exposures of rodents and guinea pigs appeared to confirm phosgene’s lower acute toxicity (Boyland et al. Citation1946). Readings taken at 0.5 and 0.25 min appear as negative log time. (F) This is not the case for dogs also immersed in the same type of whole-body chamber. With ne >1 their data suggest acute, high-concentration exposures more toxic than chronic low concentration ones. (G–H) Using an improved procedure for nose-only inhalation in rats (Hobson et al. Citation2019), a lower LC50 was obtained for 10 min exposures that align better with the realistic toxicity seen during phosgene exposure. *Modifying the tables by replacing this value with the 10 min values in the Zwart et al. (Citation1990) and Pauluhn (Citation2006) data sets suggests ne >1 and, thus, more toxic during acute, high concentration exposures than previously thought. Replaced values denoted by red star and arrow (compare with C,D).
While researchers have generally determined phosgene’s LC50 by varying the exposure concentration for a specific length of time, Plahovinsak et al (Citation2015) took a different approach by varying the exposure time male mice inhaled an 8 ppm (32.36 mg/m3) dosage from a nose-only inhalation tower. One of their key results further illustrate phosgene’s steep toxicity curve. 50% mortality occurs at 48 hours when mice are intoxicated with 8 ppm for 26.9 min, but that LC50 is reached in 24 hours when intoxication occurs for 28.3 min (for both p < 0.001); a mere difference in exposure of 1.4 minutes! They report a 48 hour LCt50 of 215.2 ppm × min (8 ppm × 26.9 min) and a 24 hour LCt50 of 226.4 ppm × min (8 ppm × 28.3 min); the latter LCt50 translating to 915.8 mg/m3×min (32.36 mg/m3×28.3 min). This result highlights differences seen from one laboratory to the next. For example, Plahovinsak et al (Citation2015) achieve 50% mortality in 24 hours when male mice are exposed to 32.36 mg/m3 of phosgene for ∼30 min, yet Zwart et al. (Citation1990) expose male mice to more than double that concentration (76 mg/m3) for 30 min () and they achieve 50% mortality in 14 days.
Our nose-only inhalation system is designed to rapidly deliver the target concentration of phosgene in a step function rather than gradually increasing to the target, thus, recreating a more likely acute, high-concentration exposure following an accident or explosion (Hobson et al. Citation2019). The exact concentration of phosgene delivered to the rats is verified via Fourier Transform Infrared spectrometry, a level of sophistication not used by any of the other researchers in . Equally important, we acclimated our rats to the confining environment of the exposure tubes for 10 min on two consecutive days prior to exposure in order to minimize any confounding effects caused by stress proteins and to better recreate the calm demeanor and early-stage behavior of victims at ground zero. For a 10 min exposure to a series of phosgene concentrations, a significantly lower LC50 in rats was calculated by Probit analysis (129.2 mg/m3) along with tighter 95% confidence limits (CL) of [109.2 − 145.7 mg/m3]. In comparison, wider limits were reported by Zwart et al. (Citation1990) [LC50: 334 mg/m3; 95% CL: 306–363 mg/m3] and Pauluhn (Citation2006) [LC50: 253.3 mg/m3; 95% CL: 194–331 mg/m3]. Exposing another set of rats (n = 9) to the calculated LC50 of 129.2 mg/m3 affirmed the toxicity of this dosage in terms of 24-h survival and lung/body weight ratios (Hobson et al. Citation2019). Replacing this value for the 10 min results reported by Zwart et al. () and Pauluhn (), provides a ne > 1 and in agreement with a model of greater toxicity during short-term high-concentration exposures. Our LCt50 value of 1292 mg/m3×min corresponds to the expected range of 1000 − 2000 mg/m3×min seen for phosgene in many experimental animals and the accepted LCt50 in humans (∼2000 mg/m3×min in ) as determined by the IPCS (Public Health England Citation2016). Such a result should not be surprising. We note that the Acute Exposure Guideline Levels (AEGL) from the Environmental Protection Agency (Citation2002) for phosgene states (pg. 16) ‘Haber’s law has been shown to be valid for phosgene within certain limits.’
Picking models that will approximate human intoxication requires more than accounting for an acute or chronic duration. One must account for physiological similarities as well. The need to power screening studies so they are statistically significant will continue to require the use of rodents; however, rodents are obligate nasal breathers and exhibit physiological differences from humans and dogs, which breath both from the nasal or oral cavity. As an example, Pauluhn (Citation2006) attributes the increased survival times in rats and, hence, greater LC50 values obtained for 10 min phosgene exposures to ‘an initial and concentration-dependent increase in the apnea time, accompanied by decreased respiratory minute volumes.’ This reflexive behavior in rats for acute exposures is believed to be caused by phosgene’s stimulation of the vagal C-fibers innervating the lower airways and controlling spontaneous breathing. Evidence of phosgene’s interaction with the vagal nerves is seen in dogs exposed to doses > 7000 mg/m3. There is a cessation of respiration with the lungs deflated; a phenomenon that does not occur when the vagus nerve is cut prior to phosgene exposure (Li and Pauluhn Citation2014). Interference from this reflex in rodent models may subside for exposures greater than 10 min. For more physiologically relevant phosgene studies, it has been reported that the dog (beagle) more closely matches the pathophysiology of humans post-exposure (Li and Pauluhn Citation2019). With these data in mind, the agreement between our ne in rats using part of Pauluhn’s Citation2006 data (ne = 1.17) and the ne determined for dogs by Boyland et al. in 1946 (ne = 1.13) () suggests that our robust nose-only inhalation system correctly predicts a greater toxicity when a subject is exposed to a short-duration, high concentration dose of phosgene than a long-duration, low concentration. The dog is an expensive model to be utilized in late stages of preclinical drug development; however, in the early stages, our acute exposure system provides an alternative option (Hobson et al. Citation2019).
Human exposure
Perhaps the most important gaps in our knowledge regarding phosgene involves human exposure. For example, determination of the exact concentration and exposure time for casualties exposed to phosgene during WWI is complicated by the nature of the exposure and the lack of analytical/diagnostic equipment in use (vide supra). Since phosgene’s use as a CWA, a number of industrial accidents have occurred with surprisingly few deaths. The largest incident in which phosgene is implicated is the Union Carbide Bhopal accident in which ∼2500 people died (Ryan et al. Citation1996). Both the cause and the main component of the leak (methyl isocyanate) makes it improbable that phosgene was involved. Further investigation into Union Carbide revealed allegations that workers were used to ‘sniff out the sources of phosgene leaks’ in plants located in Bhopal and West Virginia (Ryan et al. Citation1996). More recently, in 2010 at a DuPont facility in Belle, WV, a faulty hose that was transferring pressurized liquid phosgene ruptured exposing three workers, one of whom died (Johnson Citation2011). Finally, from the 1920s to the 1950s, there was an extensive offensive and defensive CWA research program at Porton Down in the United Kingdom; during this time, both US and UK military volunteers were exposed to various levels of CWAs including phosgene (Isenberg Citation2001).
consolidates observations of human exposure incidents described by W.F. Diller and colleagues; however, a perusal of the NIOSH’s webpage for the phosgene IDLH finds other historic human exposure data. Some observations support the IPCS’s threshold toxicity values (), such as a 30 min exposure to 17 ppm (510 ppm × min) being lethal (Diller Citation1978). Some observations are contradictory, such as a 30 min exposure to 5 ppm (150 ppm × min) being ‘probably’ lethal (Jacobs Citation1967). And some lack sufficient information: ‘brief exposure to 50 ppm may be rapidly fatal’ (Henderson and Haggard Citation1943). How long is ‘brief’?
Given the paucity of human data and the results from some animal data suggesting that phosgene may not follow Haber’s rule under all circumstances, it may be time to refine the IPCS guidelines that are currently based on a linear relationship of dose and exposure duration. Before any such action is taken, we suggest further experimentation with a single species that closely mimics the pathophysiology of humans, most likely a non-human primate. In large-scale simulations, phosgene can be released from the center of a large sealed room blanketed with state-of-the-art detectors and in which multiple test subjects have been positioned at varying distances from the center. This format allows investigation of acute and chronic intoxication scenarios to determine the overall pattern of lethality (as seen schematically in ) and, hence, derivation of robust power curves that mathematically allow us to calculate precise exposure thresholds. Experimentation on such a scale is not for testing therapeutics. The experimental strategy may be elaborate and of concern regarding the use of so many animals, yet the alternative of waiting for a large-scale release of phosgene to study the pattern of lethality in humans is not a better strategy.
Development of medical countermeasures
Despite its industrial prevalence, there is no FDA-licensed therapeutic treatment for toxic phosgene exposure (Sciuto and Hurt Citation2004; Russell et al. Citation2006; Smith et al. Citation2009; Holmes et al. Citation2016; Summerhill et al. Citation2017). While current hypothesized mechanisms of action de-emphasize the role of inflammation (Holmes et al. Citation2016), previous attempts have focused on the inflammatory processes that underlie edema, such as curbing neutrophil infiltration using cyclophosphamide, the 5-lipoxygenase inhibitor AA861, or the microtubular poison colchicine; all have been unsuccessful (Ghio et al. Citation1991). The acid-resistant protease inhibitor, ulinastatin, suppresses neutrophil accumulation, and it has shown efficacy in small animal studies, but its exact mechanism of action remains unclear (Shen et al. Citation2014). The efficacy of ibuprofen, given prophylactically 30 minutes (min) prior to phosgene exposure, for the reduction of observed lung edema in rats has been reported (Sciuto et al. Citation1996), although such a pretreatment strategy is unrealistic given the nature of ‘no-warning’ chemical exposure events, like those expected on the battlefield, a terrorist attack, or workplace accident.
The use of a nebulized β2 adrenergic receptor agonist, salbutamol, has shown a deleterious effect on arterial oxygenation (Grainge et al. Citation2009). Neither treatment with nebulized nor intravenous steroids improves survival (Smith et al. Citation2009; Grainge and Rice Citation2010). And draining the lungs with the diuretic furosemide also did not increase survival (Grainge et al. Citation2010).
In an ambitious screening program using whole body exposure of outbred CD-1 male mice to 32-40.5 mg/m3 (8-10 ppm) phosgene for 20 min, a wide array of compounds was administered at a series of doses post-exposure. These included: antioxidants; an ETA receptor blocker; NMDA and muscarinic receptor antagonists; a radioprotectant; an ACE inhibitor; an antihistamine; an AMPK activator; and inhibitors for PDE4, PDE5, 5-lipoxygenase, a couple for GABA transaminase and three TRP channel inhibitors (Holmes et al. Citation2016). Conclusions of this wide screen indicated that neuromodulation and interruption of vascular tone/permeability pathways (PDEs, angiotensin, and endothelin) are promising leads. However, limiting ROS formation, as seen with some of the antioxidants tested, may be key in preventing mortality following phosgene exposure (Holmes et al. Citation2016).
Targeting oxidative stress and apoptotic pathways
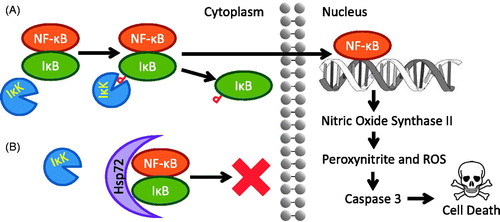
In separate studies, inducible nitric oxide synthase (NOS2, iNOS) inhibitors have shown efficacy inhibiting phosgene induced acute lung injury with inbred C57BL mice (Filipczak et al. Citation2015); meanwhile, angiopoietin-1 transfection and induction has shown similar results in rats (He et al. Citation2014). Answers as to why such strategies work in phosgene exposure models can be found not in pulmonary research, but in neurology. Using an epileptic seizure model in rats, researchers have shown that sustained seizures activate nuclear factor-kappa B (NF-κB), a transcription factor with multiple roles, including the upregulation of NOS2, which can lead to the biosynthesis of peroxynitrite from nitric oxide when in an environment rich with ROS such as the superoxide radical (O2•−) (Chang et al. Citation2014). As noted above, phosgene depletes the lung of glutathione, the main defense against reactive oxygen species (Bast and Glass-Mattie Citation2020). Peroxynitrite and ROS generation in the lungs in turn activate caspase cascades that ultimately lead to apoptosis. However, angiopoietin-1 suppresses the NF-κB transcription pathway in phosgene induced acute lung injury (He et al. Citation2014). The suppression of NOS2 and NF-κB can involve multiple intermediaries along a metabolic pathway, and yet there is a more direct suppression mechanism now under investigation in our laboratories, Heat Shock Protein 72 (HSP72) (Parseghian et al. Citation2016).
HSP72 is a pleiotropic agent with multiple anti-stress, anti-apoptotic roles and has been investigated as a cytoprotectant in a wide variety of tissues, including in the pulmonary system (Parseghian et al. Citation2016). HSP72 is known to inhibit three apoptotic pathways that can occur with traumatic injury to any tissue: (1) During ATP-dependent apoptosis, mitochondria release cytochrome C, triggering the construction of a molecular machine known as an apoptosome. Apoptosomes cleave procaspase-9 proteins into their active caspase-9 form, which go about triggering a cascade of cellular destruction leading to cell death. HSP72 binds the apoptosome, preventing conversion of procaspase-9 into its active caspase-9 form (Apoptosome) (Beere et al. Citation2000). (2) During ATP-independent apoptosis, mitochondria release apoptosis inducing factor (AIF). Sufficient AIF in the cell cytoplasm can also trigger cell death. Unlike the apoptosome, this apoptotic process triggered by AIF does not require ATP, therefore it occurs even under low energy conditions. HSP72 binds AIF, inhibiting this ATP- and Caspase-independent apoptotic pathway. (Cande et al. Citation2002). (3) Most importantly, in the context of this discussion, HSP72 has also been shown to directly bind NF-κB and prevent its release from sequestration with IκB in the cytoplasm (Zheng et al. Citation2008). If released from the NF-κB: IκB complex, NF-κB can translocate to the nucleus and upregulate NOS2, leading eventually to apoptosis caused by oxidative stress (). HSP72 induction is one of the body’s responses to ROS synthesis caused by oxidative stress (Madamanchi et al. Citation2001); however, in certain cases, an ROS can actually impair HSP72 expression through RNA interference with microRNA intermediaries (Adachi et al. Citation2009; Spiró et al. Citation2012). NF-κB also plays a role in the induction of inflammatory pathways. In fact, while the use of angiopoietin-1 helps attenuate inflammation in phosgene injured lungs (He et al. Citation2014), another of the body’s natural responses is the induction of HSP72 via IL-6 (Levada et al. Citation2018).
Figure 5. (A) Transcription factor NF-κB is activated and triggers a cell death cascade upon phosphorylation of IκB by its kinase, IκK, under conditions of neural cell stress. (B) Immunoprecipitation data indicates Hsp72 blocks IκK’s access to IκB, preventing NF-κB’s translocation to the nucleus (Zheng et al. Citation2008). Image from Parseghian et al. (Citation2016) courtesy of the Annals of the New York Academy of Sciences/John Wiley & Sons.
Based on acute lung injury studies, the induction process for HSP72 requires 12 h after the initial insult to the lungs, an unacceptably long period of time in the event of phosgene exposure, although it remains at a level above baseline for up to 72 h (Villar et al. Citation1993). We have been developing a strategy that rapidly delivers human HSP72 into lung tissue using the scFv fragment of a proprietary cell-penetrating antibody, mAb 3E10, as an intracellular transport system for protein therapeutics (Weisbart et al. Citation2000; Hansen et al. Citation2007; Weisbart et al. Citation2015). The 3E10 monoclonal is a well-characterized antibody with a unique cell penetration pathway (Hansen et al. Citation2007) that has been found to be safe in an FDA approved Phase I clinical trial (Spertini et al. Citation1999). 3E10 binds DNA allowing its penetration through a specific nucleoside salvage channel found in most cells, known as the equilibrative nucleoside transporter 2 (ENT2) (Lu et al. Citation2004). This 100 kD fusion of a humanized 3E10 to human HSP72, formerly known as Fv-Hsp70 (Zhan et al. Citation2010), and now known as Fv-HSP72, targets extracellular DNA, to deliver HSP72 to damaged lung cells. Nucleoside targets are abundant, stable and quite accessible during tissue damage where there is cell necrosis (Chen et al. Citation1990; Parseghian and Luhrs Citation2006; Weisbart et al. Citation2015). Selectivity of this agent in vivo is based on the simple concept that tissues undergoing significant cell injury possess a high concentration of extracellular DNA. Salvaging of the DNA by surrounding cells, through the ENT2 channel, provides 3E10 the opportunity to enter those energy-deficient cells still hanging on to life in a diffusion-driven process that does not depend on ATP-fueled endocytosis.
Rapid post-exposure response
Any successful therapeutic to treat phosgene exposure requires the following equally important technical characteristics: (1) efficient storage, distribution and drug administration, (2) efficacy when delivered post-exposure, (3) efficacy in the broadest population possible, and (4) rapid delivery and localization to the lungs. Let us briefly ponder each of these concepts in the context of the HSP72 approach just discussed.
Efficient storage, distribution and drug administration
Recently, independent laboratory confirmation that delivery of HSP72 to the lungs is a viable option comes from a group that virally transduced mesenchymal stem cells (MSC) to overexpress HSP70 before intra-tracheally delivering 1 × 106 cells/50 μL into the lungs of phosgene intoxicated Sprague Dawley rats (Jin et al. Citation2020). The results of the study showed that exogenous HSP70 production in these MSCs can significantly reduce damage associated with phosgene, including (1) the lung wet/dry ratio; (2) protein and cell content in the BALF; and (3) the levels of inflammatory cytokine TNFα. The same strategy also increased the anti-inflammatory cytokine IL-10 in the serum and BALF. These results were statistically significant even compared to those rats receiving MSC without any HSP70 transduction (Jin et al. Citation2020). While they verify the HSP72 approach, it is hard to imagine the utility of stockpiling millions of vials of MSCs in liquid nitrogen tanks throughout the country for distribution to hospitals that can deliver the product intra-tracheally to victims after an exposure event. The authors of the study point out that 90% of MSCs can undergo apoptosis after transplantation, coupled with poor targeting of the cells to the areas of greatest injury, and only a small number of MSCs are going to reach the damaged cells. Development of the Fv-HSP72 fusion protein allows reformulation into existing modes of delivery, such as inhalers, that can be deployed close to phosgene vulnerable sites. In that case, one must consider the option of delivering a therapeutic dose intranasally (IN) versus intratracheally (IT) and weigh the amount of drug needed for either administration. In mice, IN delivery of volumes less than 35 μL have been reported to be distributed primarily to the upper respiratory tract, whereas a 50 μL volume was predominantly deposited in the lower respiratory tract, based on studies tracking radiolabeled colloids (Southam et al. Citation2002; Turner et al. Citation2011). Our laboratory is exploring dry powder formulations that can be placed into inhalers, stored at room temperature and distributed to paramedics, front line troops, police/fire units for rapid deployment.
Efficacy postexposure
A prophylactic is unrealistic if responding to an accident or act of terrorism. Case in point, there are small molecules that can be provided to induce HSP72, such as geranylgeranyl acetone (GGA), which requires lag times varying from 8 to 24 h in in vivo models, depending on the organ being targeted (Zhang et al. Citation2009). Based on the exposure models discussed above, the victim may be fully symptomatic or dead within that time frame. The Fv-HSP72 strategy is not prophylactic and has no lag time. Immediate activity has already been proven in vivo with cerebral and myocardial infarction models in which delivery of the Fv-HSP72 postreperfusion has reduced the infarct volume by 68% in the brain (Zhan et al. Citation2010) and reduced cardiomyocyte death by 43% in the heart (Tanimoto et al. Citation2017), respectively. Early proof-of-concept data has been obtained in rats using the acute phosgene exposure model developed by our team (Hobson et al. Citation2019) followed by intravenous (IV) delivery of Fv-HSP72 30 min post-exposure (unpublished results). We are now in the process of developing a formulation that delivers the therapeutic agent directly into the lungs for testing in our acute exposure model.
Efficacy in the broadest population
Induction of many HSP70 protein family members is attenuated with aging (Fargnoli et al. Citation1990; Pardue et al. Citation1992; Heydari et al. Citation1993), dampening the effectiveness of any small molecule strategy in older individuals. The Fv-HSP72 approach rapidly rescues lung tissue by placing therapeutic doses of exogenous human HSP72 into cells at risk regardless of the age of the patient.
Rapid delivery and localization to the lungs
A small molecule inducer of HSP72 can lack specificity and may suffer from a double-edged problem. Without targeting specific cells, product potency can be diminished unless excess material is delivered to the lungs. Conversely, excessive material delivered to the lungs can result in dangerous side effects. For a small molecule that induces gene expression, the need to evaluate unintended induction of other genes is critical. Thus, finding an optimal therapeutic window can often be challenging. On the other hand, with Fv-HSP72, we are targeting a cytoprotectant to the alveolar and bronchial cells that interface with the air; we are not delivering our biologic to the systemic circulation or going beyond the cells lining lungs and airways. Fv-HSP72 is designed to congregate in regions of traumatic injury (Hansen et al. Citation2006), which it should be able to do in the lungs when it is delivered by dry powder or by aerosol. A further advantage is the use of an scFv fragment and not the entire 3E10 antibody. If an intact antibody were to be delivered to the lungs, one must consider the work conducted by the Sakagami lab which determined a relatively low rate of IgG transport across the pulmonary epithelial barrier (80 ng/h in rats) using the FcRn system (Sakagami et al. Citation2006). Such an absorption rate, if allometrically scaled by body weight to a 70 kg human would equate to ∼3 μg/h, far below what may be delivered to a patient minutes after exposure. Furthermore, there are an estimated 25 million alveolar macrophages and their Fc receptors which can act as an IgG sink in the lungs (Sakagami and Gumbleton Citation2006). Since Fv-HSP72 does not possess the Fc portion of the 3E10 antibody, it should largely remain in the lungs.
Future needs
Over 200 years after it was first synthesized, one of the most toxic, yet critically important industrial reagents, has no standard efficacy testing model for potential therapeutic countermeasures that would allow for a clear FDA approval pathway. Questions have arisen even about the applicability of Haber’s Rule with or without modifications to determine accurately phosgene’s toxicity at various exposure concentrations and times. Establishing a consensus will be critical to allowing more testing of potential countermeasures under the Animal Rule. To that end, our laboratory has developed a robust model for acute, short-term, high-concentration inhalation of phosgene that is likely to occur at ground zero of an industrial accident or terrorist attack. Furthermore, working with our collaborators, we believe the rapid intracellular delivery of exogenous Fv-HSP72 can protect cells from stress-based injury and this simple solution avoids the lag times needed for endogenous HSP72 induction post-phosgene exposure. Although comprehensive toxicity studies of Fv-HSP72 in all potential vulnerable populations (i. e. pediatric, geriatric, pregnant), have not been completed, it is not anticipated that the addition of exogenous human HSP72 is going to have a deleterious effect. A recent maximum tolerated dose study of 3 Fv-HSP72 variants did not find any signs of gross toxicity in either males or female Sprague Dawley rats (unpublished results).
Finally, one may ask what is the existing standard of care any new therapy that is going to seek approval have to compete against? Ironically, despite the potential role of oxidative stress and inflammation in phosgene-induced acute lung injury, clinical protocols currently advise providing victims with supplemental oxygen, which appears to be the only current treatment that results in improved survival, improved arterial oxygenation and reduced lung edema (Russell et al. Citation2006; Grainge et al. Citation2010). That and “Keep quiet and warm. Give coffee as a stimulant.”
Acknowledgements
The authors wish to thank Dr. Robert P. Casillas and Dr. Glenn T. Reynolds for excellent and sage advice during a technical reading of the manuscript.
Disclosure statement
MHP, RAR, and STH are compensated by Rubicon Biotechnology. MHP and RAR are co-owners of Rubicon.
Additional information
Funding
References
- Adachi M, Liu Y, Fujii K, Calderwood SK, Nakai A, Imai K, Shinomura Y. 2009. Oxidative stress impairs the heat stress response and delays unfolded protein recovery. PLoS One. 4(11):e7719
- Aggarwal S, Jilling T, Doran S, Ahmad I, Eagen JE, Gu S, Gillespie M, Albert CJ, Ford D, Oh JY, et al. 2019. Phosgene inhalation causes hemolysis and acute lung injury. Toxicol Lett. 312:204–213.
- Amoore JE, Hautala E. 1983. Odor as an aid to chemical safety: odor thresholds compared with threshold limit values and volatilities for 214 industrial chemicals in air and water dilution. J Appl Toxicol. 3(6):272–290.
- Arroyo CM, Feliciano F, Kolb DL, Keeler JR, Millette SR, Stotts RR. 1993. Autoionization reaction of phosgene (OCCl2) studied by electron paramagnetic resonance/spin trapping techniques. J Biochem Toxicol. 8(2):107–110.
- Baggett RK, Simpkins BK. 2018. Chapter 6, Critical Infrastructure Threats and Hazards. Homeland Security and Critical Infrastructure Protection. 2nd ed. Santa Barbara, Denver: Praeger, ABC-CLIO, LLC; p. 146–170.
- Bast CB, Glass-Mattie DF. 2015. Chapter 25, Phosgene. In: Gupta RC, editor. Handbook of toxicology of chemical warfare agents. 2nd ed. New York, San Francisco, London: Academic Press, Inc.; p. 327–335.
- Bast CB, Glass-Mattie DF. 2020. Chapter 23, Phosgene. In: Gupta RC, editor. Handbook of toxicology of chemical warfare agents. 3rd ed. New York, San Francisco, London: Academic Press; p. 341–351.
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. 2000. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2(8):469–475.
- Belkebir E, Rousselle C, Duboudin C, Bodin L, Bonvallot N. 2011. Haber's rule duration adjustments should not be used systematically for risk assessment in public health decision-making. Toxicol Lett. 204(2-3):148–155.
- Borak J, Diller WF. 2001. Phosgene exposure: mechanisms of injury and treatment strategies. J Occup Environ Med. 43(2):110–119.
- Boyland E, McDonald FF, Rumens MJ. 1946. The variation in the toxicity of phosgene for small animals with the duration of exposure. Br J Pharmacol Chemother. 1:81–89.
- Cande C, Cohen I, Daugas E, Ravagnan L, Larochette N, Zamzami N, Kroemer G. 2002. Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie. 84(2-3):215–222.
- Chang CC, Chen SD, Lin TK, Chang WN, Liou CW, Chang AY, Chan SH, Chuang YC. 2014. Heat shock protein 70 protects against seizure-induced neuronal cell death in the hippocampus following experimental status epilepticus via inhibition of nuclear factor-κB activation-induced nitric oxide synthase II expression. Neurobiol Dis. 62:241–249.
- Chemical Warfare Toxicology: Volume 2: Management of Poisoning 2018. Worek F, Jenner J, Thiermann H, editors. London: Royal Society of Chemistry.
- Chen F-M, Wisner JR, Jr., Omachi H, Renner IG, Taylor CR, Epstein AL. 1990. Localization of monoclonal antibody TNT-1 in experimental kidney infarction of the mouse. Faseb J. 4(12):3033–3039.
- Chen HL, Bai H, Xi MM, Liu R, Qin XJ, Liang X, Zhang W, Zhang XD, Li WL, Hai CX. 2013. Ethyl pyruvate protects rats from phosgene-induced pulmonary edema by inhibiting cyclooxygenase2 and inducible nitric oxide synthase expression. J Appl Toxicol. 33(1):71–77.
- Cowan FM, Smith WJ, Moran TS, Paris MM, Williams AB, Sciuto AM. 2005. Sulfur Mustard-and Phosgene-Increased IL-8 in Human Small Airway Cell Cultures: Implications for Medical Countermeasures Against Inhalation Toxicity. Technical Report USAMRICD-TR-04-04. US Army Medical Research Institute of Chemical Defense. Aberdeen Proving Ground, MD.
- Currie WD, Hatch GE, Frosolono MF. 1987. Pulmonary alterations in rats due to acute phosgene inhalation. Fundam Appl Toxicol. 8(1):107–114.
- Currie WD, Pratt PC, Frosolono MF. 1985. Response of pulmonary energy metabolism to phosgene. Toxicol Ind Health. 1(2):17–27.
- Davy J. 1812. On a gaseous compound of carbonic oxide and chlorine. Philosophical Transactions of the Royal Society of London. 102:144–151.
- Diller WR. 1978. Medical phosgene problems and their possible solution. J Occup Med. 20(3):189–193.
- Diller WF. 1985. Pathogenesis of phosgene poisoning. Toxicol Ind Health. 1(2):7–15.
- Diller WF, Bruch J, Dehnen W. 1985. Pulmonary changes in the rat following low phosgene exposure. Arch Toxicol. 57(3):184–190.
- Environmental Protection Agency 2002. Acute Exposure Guideline Levels for Selected Airborne Chemicals Volume 2. Washington, D.C: The National Academies Press. https://www.epa.gov/sites/production/files/2014-09/documents/tsd7.pdf
- Environmental Protection Agency. 2003. Phosgene: USEPA HPV Challenge Program Test Plan Submission. Washington DC. EPA/201-14578A.
- Evert S. 2015. A brief history of chemical war. Distillations. Science History Institute. https://www.sciencehistory.org/distillations/a-brief-history-of-chemical-war.
- Fargnoli J, Kunisada T, Fornace AJ, Jr., Schneider EL, Holbrook NJ. 1990. Decreased expression of heat shock protein 70 mRNA and protein after heat treatment in cells of aged rats. Proc Natl Acad Sci USA. 87(2):846–850.
- Filipczak PT, Senft AP, Seagrave J, Weber W, Kuehl PJ, Fredenburgh LE, McDonald JD, Baron RM. 2015. NOS-2 inhibition in phosgene-induced acute lung injury. Toxicol Sci. 146(1):89–100.
- Frosolono MF, Pawlowski R. 1977. Effect of phosgene on rat lungs after single high-level exposure. Arch Environ Health. 32(6):271–277.
- Ghio AJ, Kennedy TP, Hatch GE, Tepper JS. 1991. Reduction of neutrophil influx diminishes lung injury and mortality following phosgene inhalation. J Appl Physiol (1985)). 71(2):657–665.
- Glass D, McClanahan M, Koller L, Adeshina F. 2009. Provisional advisory levels (PALs) for phosgene (CG). Inhal Toxicol. 21 Suppl 3:73–94.
- Grainge C, Brown R, Jugg BJ, Smith AJ, Mann TM, Jenner J, Rice P, Parkhouse DA. 2009. Early treatment with nebulised salbutamol worsens physiological measures and does not improve survival following phosgene induced acute lung injury. J R Army Med Corps. 155(2):105–109.
- Grainge C, Jugg BJ, Smith AJ, Brown RF, Jenner J, Parkhouse DA, Rice P. 2010. Delayed low-dose supplemental oxygen improves survival following phosgene-induced acute lung injury. Inhal Toxicol. 22(7):552–560.
- Grainge C, Rice P. 2010. Management of phosgene-induced acute lung injury. Clin Toxicol (Phila). 48(6):497–508.
- Grainge C, Smith AJ, Jugg BJ, Fairhall SJ, Mann T, Perrott R, Jenner J, Millar T, Rice P. 2010. Furosemide in the treatment of phosgene induced acute lung injury. J R Army Med Corps. 156(4):245–250.
- Guo YL, Kennedy TP, Michael JR, Sciuto AM, Ghio AJ, Adkinson NF, Jr., Gurtner GH. 1990. Mechanism of phosgene-induced lung toxicity: role of arachidonate mediators. J Appl Physiol (1985)). 69(5):1615–1622.
- Gurtner GH. 1996. Development and testing of treatments for battlefield phosgene poisoning. Final report, 1 August 1994 – 31 January 1996 (No. AD-A-314742/8/XAB). New York Medical Coll., Valhalla, NY (United States) (https://apps.dtic.mil/dtic/tr/fulltext/u2/a314742.pdf).
- Haber F. 1924. Zur geschichte des gaskrieges (On the history of gas warfare). In Fünf Vorträge aus den Jahren 1920-1923 (Five Lectures from the Years 1920-1923). Springer, Berlin. pp p. 76–92.
- Hansen JE, Sohn W, Kim C, Chang SS, Huang NC, Santos DG, Chan G, Weisbart RH, Nishimura RN. 2006. Antibody-mediated Hsp70 protein therapy. Brain Res. 1088(1):187–196.
- Hansen JE, Tse CM, Chan G, Heinze ER, Nishimura RN, Weisbart RH. 2007. Intranuclear protein transduction through a nucleoside salvage pathway. J Biol Chem. 282(29):20790–20793.
- He DK, Shao YR, Zhang L, Shen J, Zhong ZY, Wang J, Xu G. 2014. Adenovirus-delivered angiopoietin-1 suppresses NF-κB and p38 MAPK and attenuates inflammatory responses in phosgene-induced acute lung injury. Inhal Toxicol. 26(3):185–192.
- Henderson Y, Haggard HW. 1943. Noxious gases. 2nd ed. New York, NY: Reinhold Publishing Corporation. p. 137–138.
- Heydari AR, Wu B, Takahashi R, Strong R, Richardson A. 1993. Expression of heat shock protein 70 is altered by age and diet at the level of transcription. Mol Cell Biol. 13(5):2909–2918.
- Hobson ST, Casillas RP, Richieri RA, Nishimura RN, Weisbart RH, Tuttle R, Reynolds GT, Parseghian MH. 2019. Development of an acute, short-term exposure model for phosgene. Toxicol Mech Methods. 29(8):604–615.
- Holmes WW, Keyser BM, Paradiso DC, Ray R, Andres DK, Benton BJ, Rothwell CC, Hoard-Fruchey HM, Dillman JF, Sciuto AM, et al. 2016. Conceptual approaches for treatment of phosgene inhalation-induced lung injury. Toxicol Lett. 244:8–20.
- Homeland S. 2003. Voluntary initiatives are under way at chemical facilities, but the extent of security preparedness is unknown. GAO-03-24R ed. Washington DC: United States Government Accounting Office.
- Isenberg D. 2001. Guinea pigs. Bull at Sci. 57(3):72–73.
- Jacobs MB. 1967. The analytical toxicology of industrial inorganic poisons. New York, NY: Interscience Publishers. p. 648–649.
- Jin C, Zhou F, Zhang L, Shen J. 2020. Overexpression of heat shock protein 70 enhanced mesenchymal stem cell treatment efficacy in phosgene-induced acute lung injury. J Biochem Mol Toxicol. 34(8):e22515. doi:10.1002/jbt.22515.
- Johnson J. 2011. Investigating a fatal phosgene leak. Chem Eng News Archive. 89(41):34–35.
- Levada K, Guldiken N, Zhang X, Vella G, Mo FR, James LP, Haybaeck J, Kessler SM, Kiemer AK, Ott T, et al. 2018. Hsp72 protects against liver injury via attenuation of hepatocellular death, oxidative stress, and JNK signaling. J Hepatol. 68(5):996–1005.
- Li W, Pauluhn J. 2014. Chapter 19, Mechanisms involved in the inhalation toxicity of phosgene. In: Salem H, Katz SA, editors. Inhalation toxicology. 3rd ed. Boca Raton, Florida: CRC Press; p. 459–483.
- Li W, Pauluhn J. 2019. Phosgene-induced lung edema: Comparison of clinical criteria for increased extravascular lung water content with postmortem lung gravimetry and lavage-protein in rats and dogs. Toxicol Lett. 305:32–39.
- Lu H, Chen C, Klaassen C. 2004. Tissue distribution of concentrative and equilibrative nucleoside transporters in male and female rats and mice. Drug Metab Dispos. 32(12):1455–1461.
- Madamanchi NR, Li S, Patterson C, Runge MS. 2001. Reactive oxygen species regulate heat-shock protein 70 via the JAK/STAT pathway. Arterioscler Thromb Vasc Biol. 21(3):321–326.
- Madden MC, Friedrnan M, Keyes LL, Koren HS, Burleson GR. 1991. Effects of phosgene exposure on lung arachidonic acid metabolism. Inhal Toxicol. 3(1):73–90.
- Miller FJ, Schlosser PM, Janszen DB. 2000. Haber's rule: a special case in a family of curves relating concentration and duration of exposure to a fixed level of response for a given endpoint. Toxicology. 149(1):21–34.
- Ministry of Defense 1987. Medical manual of defence against chemical agents. London: Ministry of Defence D/Med (F&S) (2)/10/1/1.
- Nash T, Pattle RE. 1971. The absorption of phosgene by aqueous solutions and its relation to toxicity. Ann Occup Hyg. 14(3):227–233.
- National Academies of Sciences, Engineering, and Medicine 2015. Application of Modern Toxicology Approaches for Predicting Acute Toxicity for Chemical Defense. Washington, DC: The National Academies Press. https://doi.org/10.17226/21775.
- National Institute of Occupational Safety and Health 1976. Criteria for a Recommended Standard: Occupational Exposure to Phosgene. NIOSH-76-137.
- Olivera DS, Hoard-Fruchey H, Sciuto AM. 2017. Evaluation of an in vitro screening model to assess phosgene inhalation injury. Toxicol Mech Methods. 27(1):45–51.
- Pardue S, Groshan K, Raese JD, Morrison-Bogorad M. 1992. Hsp70 mRNA induction is reduced in neurons of aged rat hippocampus after thermal stress. Neurobiol Aging. 13(6):661–672.
- Park GD, Mitchel JT. 2016. Working with the U.S. Food and Drug Administration to obtain approval of products under the Animal Rule. Ann NY Acad Sci. 1374(1):10–16.
- Parseghian MH, Hobson ST, Richieri RA. 2016. Targeted heat shock protein 72 for pulmonary cytoprotection. Ann NY Acad Sci. 1374(1):78–85.
- Parseghian MH, Luhrs KA. 2006. Beyond the walls of the nucleus: the role of histones in cellular signaling and innate immunity. Biochem Cell Biol. 84(4):589–604.
- Pauluhn J. 2006. Acute nose-only exposure of rats to phosgene. Part I: concentration x time dependence of LC50s, nonlethal-threshold concentrations, and analysis of breathing patterns. Inhal Toxicol. 18(6):423–435.
- Plahovinsak JL, Perry MR, Knostman KA, Segal R, Babin MC. 2015. Characterization of a nose-only inhaled phosgene acute lung injury mouse model. Inhal Toxicol. 27(14):832–840.
- Public Health England 2016. Phosgene: Toxicological overview. 2014790.
- Reinhardt C, Travis AS. 2000. Heinrich Caro and the creation of the modern chemical industry. Dordrecht, the Netherlands: Kluwer Academic.
- Rim KT. 2019. In vitro models for chemical toxicity: review of their applications and prospects. Toxicol Environ Health Sci. 11(2):94–103.
- Rim KT. 2020. In silico prediction of toxicity and its applications for chemicals at work. Toxicol Environ Health Sci. 12(3):191–202.
- Rozman KK, Doull J. 2000. Dose and time as variables of toxicity. Toxicology. 144(1-3):169–178.
- Russell D, Blain PG, Rice P. 2006. Clinical management of casualties exposed to lung damaging agents: a critical review. Emerg Med J. 23(6):421–424.
- Ryan TA, Seddon EA, Seddon KR, Ryan C. 1996. Chapter 1, History of phosgene. Phosgene and related carbonyl halides. 24th ed. Amsterdam: Elsevier; p. 3–72.
- Sakagami M, Gumbleton M, 2006. Endogenous and exogenous IgG transfer in the airways: a prospective evaluation. Vol. 1In: Dalby RN, Byron PR, Peart J. editors. Richmond, Virginia: Virginia Commonwealth University; p. 57–64.
- Sakagami M, Omidi Y, Campbell L, Kandalaft LE, Morris CJ, Barar J, Gumbleton M. 2006. Expression and transport functionality of FcRn within rat alveolar epithelium: a study in primary cell culture and in the isolated perfused lung. Pharm Res. 23(2):270–279.
- Sandall TE. 1922. The later effects of gas poisoning. Lancet. 200(5173):857–859.
- Sauer UG, Vogel S, Hess A, Kolle SN, Ma-Hock L, van Ravenzwaay B, Landsiedel R. 2013. In vivo-in vitro comparison of acute respiratory tract toxicity using human 3D airway epithelial models and human A549 and murine 3T3 monolayer cell systems. Toxicol in Vitro. 27(1):174–190.
- Sciuto AM. 1998. Assessment of early acute lung injury in rodents exposed to phosgene. Arch Toxicol. 72(5):283–288.
- Sciuto AM, Cascio MB, Moran TS, Forster JS. 2003. The fate of antioxidant enzymes in bronchoalveolar lavage fluid over 7 days in mice with acute lung injury. Inhal Toxicol. 15(7):675–685.
- Sciuto AM, Clapp DL, Hess ZA, Moran TS. 2003. The temporal profile of cytokines in the bronchoalveolar lavage fluid in mice exposed to the industrial gas phosgene. Inhal Toxicol. 15(7):687–700.
- Sciuto AM, Hurt HH. 2004. Therapeutic treatments of phosgene-induced lung injury. Inhal Toxicol. 16(8):565–580.
- Sciuto AM, Phillips CS, Orzolek LD, Hege AI, Moran TS, Dillman JF, III. 2005. Genomic analysis of murine pulmonary tissue following carbonyl chloride inhalation. Chem Res Toxicol. 18(11):1654–1660.
- Sciuto AM, Stotts RR, Hurt HH. 1996. Efficacy of ibuprofen and pentoxifylline in the treatment of phosgene-induced acute lung injury. J Appl Toxicol. 16(5):381–384.
- Sciuto AM, Strickland PT, Kennedy TP, Guo YL, Gurtner GH. 1996. Intratracheal administration of DBcAMP attenuates edema formation in phosgene-induced acute lung injury. J Appl Physiol (1985)). 80(1):149–157.
- Shen J, Gan Z, Zhao J, Zhang L, Xu G. 2014. Ulinastatin reduces pathogenesis of phosgene-induced acute lung injury in rats. Toxicol Ind Health. 30(9):785–793.
- Smith A, Brown R, Jugg B, Platt J, Mann T, Masey C, Jenner J, Rice P. 2009. The effect of steroid treatment with inhaled budesonide or intravenous methylprednisolone on phosgene-induced acute lung injury in a porcine model. Mil Med. 174(12):1287–1294.
- Southam DS, Dolovich M, O'Byrne PM, Inman MD. 2002. Distribution of intranasal instillations in mice: effects of volume, time, body position, and anesthesia. Am J Physiol Lung Cell Mol Physiol. 282(4):L833–L839.
- Spertini F, Leimgruber A, Morel B, Khazaeli MB, Yamamoto K, Dayer JM, Weisbart RH, Lee ML. 1999. Idiotypic vaccination with a murine anti-dsDNA antibody: phase I study in patients with nonactive systemic lupus erythematosus with nephritis. J Rheumatol. 26(12):2602–2608.
- Spiró Z, Arslan MA, Somogyvári M, Nguyen MT, Smolders A, Dancsó B, Németh N, Elek Z, Braeckman BP, Csermely P, et al. 2012. RNA interference links oxidative stress to the inhibition of heat stress adaptation. Antioxid Redox Signal. 17(6):890–901.
- Summerhill EM, Hoyle GW, Jordt SE, Jugg BJ, Martin JG, Matalon S, Patterson SE, Prezant DJ, Sciuto AM, Svendsen ER, ATS Terrorism and Inhalational Disasters Section of the Environmental, Occupational, and Population Health Assembly, et al. 2017. An official American thoracic society workshop report: chemical inhalational disasters. Biology of lung injury, development of novel therapeutics, and medical preparedness. Ann Am Thorac Soc. 14(6):1060–1072.
- Sweeney LM, Sommerville DR, Channel SR, Sharits BC, Gargas NM, Gut CP. Jr. 2015. Evaluating the validity and applicable domain of the toxic load model: impact of concentration vs. time profile on inhalation lethality of hydrogen cyanide. Regul Toxicol Pharmacol. 71(3):571–584.
- Tanimoto T, Parseghian MH, Nakahara T, Kawai H, Narula N, Kim D, Nishimura R, Weisbart RH, Chan G, Richieri RA, et al. 2017. Cardioprotective effects of HSP72 administration on ischemia-reperfusion injury. J Am Coll Cardiol. 70(12):1479–1492.
- ten Berge WF, Zwart A, Appelman LM. 1986. Concentration-time mortality response relationship of irritant and systemically acting vapours and gases. J Hazard Mater. 13(3):301–309.
- Turner PV, Brabb T, Pekow C, Vasbinder MA. 2011. Administration of substances to laboratory animals: routes of administration and factors to consider. J Am Assoc Lab Anim Sci. 50(5):600–613.
- Villar J, Edelson JD, Post M, Mullen JB, Slutsky AS. 1993. Induction of heat stress proteins is associated with decreased mortality in an animal model of acute lung injury. Am Rev Respir Dis. 147(1):177–181.
- Warren E. 1900. On the reaction of Daphnia magna (Straus) to certain changes in its environment. Q J Microsc Sci. 43:199–224.
- Weinrich SL, Lawrence AE, Burr JK, Grotte JH, Laviolet LL. 2008. A comparative analysis of toxicity models. Alexandria, Virginia. IDA Paper P-4327.
- Weisbart RH, Baldwin R, Huh B, Zack DJ, Nishimura R. 2000. Novel protein transfection of primary rat cortical neurons using an antibody that penetrates living cells. J Immunol. 164(11):6020–6026.
- Weisbart RH, Chan G, Jordaan G, Noble PW, Liu Y, Glazer PM, Nishimura RN, Hansen JE. 2015. DNA-dependent targeting of cell nuclei by a lupus autoantibody. Sci Rep. 5:12022
- Wijte D, Alblas MJ, Noort D, Langenberg JP, van Helden HP. 2011. Toxic effects following phosgene exposure of human epithelial lung cells in vitro using a CULTEX® system. Toxicol in Vitro. 25(8):2080–2087.
- Zhan X, Ander BP, Liao IH, Hansen JE, Kim C, Clements D, Weisbart RH, Nishimura RN, Sharp FR. 2010. Recombinant Fv-Hsp70 protein mediates neuroprotection after focal cerebral ischemia in rats. Stroke. 41(3):538–543.
- Zhang K, Zhao T, Huang X, Liu ZH, Xiong L, Li MM, Wu LY, Zhao YQ, Zhu LL, Fan M. 2009. Preinduction of HSP70 promotes hypoxic tolerance and facilitates acclimatization to acute hypobaric hypoxia in mouse brain. Cell Stress Chaperones. 14(4):407–415.
- Zhang XD, Hai CX, Cai FL, Liang X, Liu R, Chen HL, Qin XJ, Feng AJ. 2008. Time course for expression of VEGF and its receptor and regulator levels of contraction and relaxation in increased vascular permeability of lung induced by phosgene. Inhal Toxicol. 20(9):805–812.
- Zheng Z, Kim JY, Ma H, Lee JE, Yenari MA. 2008. Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J Cereb Blood Flow Metab. 28(1):53–63.
- Zwart A, Arts JHE, Klokman-Houweling JM, Schoen ED. 1990. Determination of Concentration-Time-Mortality Relationships to Replace LC50 Values. Inhal Toxicol. 2(2):105–117.