
Abstract
Chlorine is a toxic industrial chemical produced in vast quantities globally, being used in a range of applications such as water purification, sanitation and industrial processes. Its use and transport cannot be restricted; exposure may occur following accidental or deliberate releases. The OPCW recently verified the use of chlorine gas against civilians in both Syria and Iraq. Chlorine inhalation produces damage to the lungs, which may result in the development of an acute lung injury, respiratory failure and death. Treatment remains an intractable problem. Our objective was to develop a clinically relevant pre-clinical model of a moderate to severe lung injury in the pig. This would enable future assessment of therapeutic drugs or interventions to be implemented in the pre-hospital phase after exposure. Due to the irritant nature of chlorine, a number of strategies for exposing terminally anesthetized pigs needed to be investigated. A number of challenges (inconsistent acute changes in respiratory parameters; early deaths), resulted in a moderate to severe lung injury not being achieved. However, most pigs developed a mild lung injury by 12 h. Further investigation is required to optimize the model and enable the assessment of therapeutic candidates. In this paper we describe the exposure strategies used and discuss the challenges encountered in establishing a model of chlorine-induced lung injury. A key aim is to assist researchers navigating the challenges of producing a clinically relevant model of higher dose chlorine exposure where animal welfare is protected by use of terminal anesthesia.
Introduction
Chlorine (Cl2) is an important, commonly used toxic industrial chemical produced and transported in large quantities, globally. One of its main uses is in water purification; as such its availability cannot be restricted. The easy availability and inherent toxicity make it attractive to aggressors (both state and non-state or terrorist groups) willing to disrupt infrastructure or cause mass panic and casualties. The Organization for the Prohibition of Chemical Weapons (OPCW) have confirmed the use of chlorine against civilians in a number of villages in Syria and Iraq (OPCW Citation2014) as well as its use by Iraqi insurgents conducting chemical attacks against Iraqi security forces and civilians (Morris Citation2014). Chlorine was also used against coalition troops using vehicle borne improvised explosive devices (Weitz et al. Citation2007; Jones et al. Citation2010). Whether produced for military or industrial use, chlorine gas represents a credible threat.
It is important to understand the chemical reactivity and physiochemical properties of chlorine to understand the molecular and biochemical mechanisms of the injury caused. Due to its highly reactive nature and water solubility, on inhalation, chlorine reacts with the moist mucous coated epithelial lining fluid in the airways, forming hydrochloric acid (HCl) and hypochlorous acid (HOCl). Chlorine exposure produces an excessive amount of reactive oxygen species (ROS) resulting in mitochondrial dysfunction and oxidative stress. This causes cellular and organ damage (Murphy Citation2009; Jurkuvenaite et al. Citation2015; Carlisle et al. Citation2016; Achanta and Jordt Citation2019), as well as causing damage to cell membranes and proteins (as a direct effect of the inflammatory response), leading to injury of the epithelium, endothelium, capillaries, and cell death (Martin et al. Citation2003; Leustik et al. Citation2008; Tian et al. Citation2008; O’Koren et al. Citation2013). Inflammation, damage and respiratory problems occur in both the upper and lower airway depending on concentration (Evans Citation2005; Summerhill et al. Citation2017). Clinical signs vary, depending on, for example, the concentration of the gas inhaled, the duration of exposure and the minute ventilation of the individual, with symptoms ranging from mild (e.g. mucous membrane irritation) to severe (e.g. chest tightness and difficulty breathing). Exposure to chlorine at high concentrations can result in the development of acute respiratory distress syndrome (ARDS) which, in severe cases, may result in death (Van Sickle et al. Citation2009; White and Martin Citation2010).
Human exposure to chlorine occurs through industrial accidents and spills during transport as well as following deliberate release. Following a train derailment in Graniteville, South Carolina in 2005 resulting in the release of chlorine in the town, many surviving victims developed significant pulmonary signs and severe airway inflammation. Of hospitalized individuals 53% met PaO2/FiO2 ratio criteria, a commonly used indicator of lung function, for ARDS, there being an inverse relationship between PaO2/FiO2 ratio and duration of hospital stay. Those who were hypoxic on room air on first assessment stayed in hospital 3 times longer than those who were not hypoxic. Pulse oximetry and arterial blood gas analysis provide early indications of outcome severity (Van Sickle et al. Citation2009).
Treatment of lung injury following exposure to chlorine remains an intractable problem and there are currently no scientifically validated or clinically proven pharmacological treatments available, or identified interventions to reverse or prevent the long-term effects of chlorine exposure, as also reported by Wang et al. (Citation2005). Currently, casualties who are exposed to high concentrations of chlorine are likely to require supplemental oxygen, beta-adrenoceptor agonists and supportive ventilation in an intensive care facility (Nelson and Odujebe Citation2002; Zellner and Eyer Citation2020). Such supportive measures are based on the signs and symptoms following exposure. These treatment strategies are resource intensive and are demanding on medical facilities following a mass casualty chemical-incident. Therefore, there is a need to develop post-exposure therapeutic strategies that reduce the number of casualties and severity of injury, reducing the potential burden of managing exposed individuals within both civilian and military medical facilities.
There is no clinical population in which treatments can be assessed following exposure to lung damaging chemicals such as chlorine. Animal models that replicate key aspects of exposure and lung injury seen in humans and reported in the literature are therefore essential (Gunnarsson et al. Citation1998; Wang et al. Citation2002, Citation2004). While Wang et al. (Citation2005) established a ventilated model of acute chlorine exposure, our intention was to replicate human exposure more closely by using a spontaneously breathing pig.
Many studies have investigated the pathophysiological mechanisms of action of chlorine in the lung (Summerhill et al. Citation2017; Achanta and Jordt Citation2019) and many have shown, through various therapeutic regimens, efficacy in treating acute chlorine injury in small animals (Zarogiannis et al. Citation2011; Chang et al. Citation2012; Chen et al. Citation2013; Balakrishna et al. Citation2014; Zarogiannis et al. Citation2014; Lazrak et al. Citation2015; Wigenstam et al. Citation2016). The establishment of a clinically relevant large animal model of chlorine-induced lung injury will enable potential therapies to be investigated in a species which more closely resembles man, and help translate animal efficacy studies, as well as enabling more direct extrapolation of clinical effects. As part of the development of new treatments to be approved for human use, two promising compounds R-107 (Radikal Therapeutics Citation2016; Fukuda et al. Citation2016) and TRPV4 inhibitors (Global-Biodefense Citation2017; Achanta et al. Citation2019; Achanta and Jordt Citation2020) are now in advanced development in large animal species for treatment of chlorine-induced lung injury (Summerhill et al. Citation2017).
The pig has been used widely to investigate mechanisms of lung injury caused by inhalation of a range of toxic chemicals as well as therapeutic strategies to ameliorate the injury: phosgene (Brown et al. Citation2002; Smith et al. Citation2009; Grainge and Rice Citation2010; Graham et al. Citation2018; Rendell et al. Citation2018), sulfur mustard (Fairhall et al. Citation2010; Jugg et al. Citation2013, Citation2016) and chlorine (Gunnarsson et al. Citation1998; Wang et al. Citation2002, Citation2004, Citation2005). Historically, studies exposing pigs to chlorine have used mechanically ventilated pigs with the gas delivered from the cylinder or reservoir directly to the lungs via the ventilator; the animals had neuromuscular blockade during exposure (Wang et al. Citation2002; Wang et al. Citation2004; Wang et al. Citation2005). Assessment of chlorine dose has been inferred from chlorine passed through a bubbler and pH changes or other wet chemistry techniques (Gunnarsson et al. Citation1998). Measurement of real-time chlorine concentration is preferred to post-exposure analysis as it allows for accurate dose delivery by targeting the inhaled dose (ID). This is achieved by adjusting the real-time inhaled gas concentration in response to changes in the animals’ minute volume, with the aim of reducing inter-individual variability. This approach has resulted in delivery of highly accurate inhaled doses of phosgene (Graham et al. Citation2018; Rendell et al. Citation2018) and sulfur mustard (Fairhall et al. Citation2008) in established pig models.
Using this approach, an unventilated, spontaneously breathing pig model was used to mimic human exposure more closely (pre-hospital where casualties would not be ventilated but spontaneously breathing). The induced injury was to be commensurate with a moderate to severe lung injury in man, within the 12 h timeframe. The purpose of developing this model was for the evaluation of acute (within the first few hours) therapies or strategies to mitigate chlorine induced lung injury in a relatively resource constrained, pre-hospital, military environment which might reduce the logistical burden of casualty management further down the line. This paper explores the challenges encountered during the development of an anesthetized, spontaneously breathing pig model of chlorine-induced lung injury.
Materials and methods
Animals
Large white juvenile female pigs (48–60 kg, n = 20, 10–12 weeks old) were obtained from a commercial source approved as a supplier by Dstl’s Animal Welfare Ethical Review body. Animals were housed in pairs where possible and allowed access to food and water ad libitum as previously described (Brown et al. Citation2002). All experiments were carried out in accordance with the UK Animals (Scientific Procedures) Act, 1986.
Anesthesia and surgery
Experimental procedures have previously been described in detail (Graham et al. Citation2018; Rendell et al. Citation2018). Briefly, following pre-medication with midazolam (Hypnovel® Roche, Welwyn Garden City, UK) (0.5 mg·kg−1 intramuscular) animals were anesthetized via a face mask using up to 5% isoflurane (Isocare®, Animalcare LTD, York, UK) and up to 16 L·min−1 oxygen and medical air delivered using a non-rebreathing system. Once anesthetized the animal was intubated with a cuffed endotracheal tube (ET) size 7–9 mm (Portex, Smiths Medical International Ltd, UK). After intubation anesthesia was maintained with isoflurane in oxygen enriched air (fraction of inspired oxygen (FiO2) 40–50%) via a Penlon AV-S ventilator (Penlon Ltd, Abingdon, UK) set to volume control mode. Ventilator settings were 5–8 mL·kg−1 tidal volume, 10–25 breaths/min. Adjustments were made as required to keep end-tidal and arterial carbon dioxide levels within normal physiological limits (4.5 − 5.5 KPa). The pig was placed in the supine position.
The left internal jugular vein and common carotid artery were cannulated via open technique using an 8 F Portex Dog catheter (Smiths Medical, Ashford, UK) and intravenous anesthesia and continuous systemic blood pressure monitoring were commenced. Intravenous anesthesia was maintained throughout the study using a mixture of propofol 2% (Fresenius Propoven 2%, Fresenius Kabi, Cheshire, UK) remifentanil hydrochloride (Ultiva, Aspen Pharma Trading Ireland Limited, Ireland) and midazolam. Infusion rates of the individual components were started at 5 mg·kg−1·h−1 (propofol), 0.2 µg·kg−1·min−1 (not exceeding 0.4 µg·kg−1·min−1; remifentanil), 0.2 mg·kg−1·h−1 (midazolam) and were adjusted as required to maintain adequate ventilation whilst maintaining surgical anesthesia.
The right internal jugular vein was cannulated with an 8 F Desivalve introducer sheath (Vygon, Swindon, UK) and the right common carotid artery cannulated with a 7 F Desivalve introducer sheath using the Seldinger technique. A balloon tipped flow-directed cannula (744MF75 Swan Ganz, Edwards Life Sciences Ltd, Newbury, UK) was placed in each of the right vessels to enable measurement of continuous real time arterial and venous oxygen saturation via Vigilance II (Vigilance Volumetrics CEDV, Edwards Lifesciences, US). As soon as intravenous access was available an infusion of Vetivex 11 (Hartmann’s solution) (2.0 ml·kg−1·h−1) was delivered to replace insensible fluid losses. Finally, a Bonanno catheter was introduced into the bladder via an open cystotomy.
Following surgery, animals were weaned off the ventilator and allowed to breathe spontaneously.
Study protocol
On completion of the surgical procedures, baseline physiological and biochemical measurements were taken 1 h pre-exposure. The animals were randomly assigned to the following groups:
Air-exposed control animals (n = 5)
Chlorine-exposed animals (n = 15)
Chlorine exposure
Animals were transferred to the exposure facility and connected to the exposure port in the exposure apparatus () via the ET tube. For chlorine exposures, pure (>99%) chlorine gas was metered into the exposure tube from a lecture bottle (CK Special Gases, UK) using a fine-control metering valve (Bristol Fluid Systems Technologies Ltd, UK). Dilution (room) air was drawn through the exposure tube at a continuous flow rate of 85 ± 4 L·min−1; this high carrier flow was sufficient to ensure that the animal’s peak inspiratory and expiratory flow rates did not affect the rate or direction of flow during the exposure (Fairhall et al. Citation2008). The concentration of chlorine gas within the exposure tube was measured continuously, upstream of the connection port for animal exposure, thus there can be no effect of breath humidity on chlorine measurements made using an ultraviolet spectrometer (X-Stream continuous gas analyzer; Emerson Process Management, UK). The spectrometer was zeroed and calibrated prior to each challenge using an 899 ppm chlorine in nitrogen span gas. A mass flow controller (MFC) (1 L·min−1; approximately 1.2% of total flow down exposure tube) was used to control the sample flow to the X-Stream analyzer, upstream of the animal, however a critical orifice was used to control the gas flow down the exposure tube, downstream from the animal.
Figure 1. Schematic of the chlorine gas exposure system. MFC: mass flow controller.
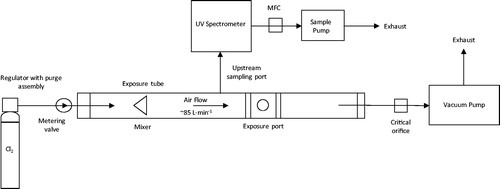
Each animal was allowed to acclimatize to the exposure apparatus (10 min) before being exposed to either air or chlorine gas diluted into room air. The animal’s respiratory parameters (including, but not limited to minute volume, tidal volume and respiration rate) were measured throughout using a Fleisch pneumotach in combination with data acquisition software (eDacq; Electro-medical Measurement Systems, UK). The expected exposure profile was plotted in advance. The actual exposure profile was plotted in real-time and compared against the expected profile. Where significant deviation was detected, or the animal was struggling to maintain respiration, then chlorine concentration was adjusted accordingly. The inhaled dose was calculated from the sum of the product of minute volume and corresponding chlorine concentration every minute divided by the body weight. Three exposure strategies were attempted; targeting an inhaled dose, a tolerable dose and a peak chlorine concentration. Following the exposure period, the animal was disconnected from the exposure apparatus. Animals experiencing prolonged periods of apnea during, or immediately after, exposure were immediately disconnected from the exposure apparatus and manually ventilated using a bag valve mask manual resuscitator (without pressure relief valve; Intersurgical Ltd, UK) connected to the ET tube for a maximum of 30 min.
Physiological measurements and sampling
Continuous observation and monitoring of the animals’ physiology was performed throughout the study (Dräger Infinity Delta XL, Hemel Hempstead, UK). Physiological measurements were recorded at the start of exposure and then every 30 min until the end of the experimental period (12 h post exposure or death). Arterial and venous blood samples were taken at hourly intervals and immediately analyzed (pH, blood gases, electrolytes, co-oximetry, calculated parameters e.g. shunt fraction and base excess) using a GEM Premier 4000 blood-gas and co-oximetry analyzer (Werfen UK Ltd, UK). Temperature adjusted values are reported. Haematological analysis was performed on EDTA blood samples using an Advia 2120i hematology analyzer (Siemens, UK).
Post mortem and histopathology
At the end of the 12 h monitoring period, or when the animal became moribund (defined as periods of asystole and a venous oxygenation <15%), the animal was humanely killed by an overdose of sodium pentabarbitone (200 mg·mL−1; IV) (‘Euthatal’ Rhone Merieux Ltd., UK). A full post mortem examination was carried out. The trachea was clamped distal to the end of the ET tube to prevent fluid loss from the trachea. The thorax was opened and the lungs and heart removed en bloc.
Bronchoalveolar lavage (BAL) of the right medial lobe was performed as previously described (Grainge et al. Citation2009). BAL fluid was analyzed for total white blood cell (WBC) count using a Siemens Advia 2120i hematology analyzer and for differential cell counts (Shandon Cytospin, 1800 rpm, 10 min). Slides were stained with Thermo Scientific Shandon Kwik-Diff stain and 100 cells counted. Protein content of BAL fluid supernatant was determined using the Coomassie blue method (Bradford Citation1976).
Following lavage, the lungs were weighed for lung wet weight to body weight (LWW:BW) ratio determinations (the weight of the remaining lavage saline being taken into account) and lung wet weight to dry weight (LWW:DW) ratios. Three sections taken from each lung lobe (left and right apical, left medial, left and right caudal) were fixed by immersion in neutral buffered formalin and processed for histopathological examination using standard processing techniques. The right medial lobe was not processed due to being lavaged.
Hematoxylin and eosin (H&E) stained sections of respiratory tract from each animal were examined by a veterinary pathologist blinded to the study design. Four levels of interest were identified, from conducting to respiratory airways; trachea, bronchi, bronchioles, and the alveoli and interstitium. A spectrum of lesions was observed, and each region was assigned a score, as outlined below.
Trachea and bronchi
At least one section of trachea and a section of each bronchus were examined from each animal. These were assigned grades of epithelial injury (). Artifacts introduced during sampling and processing included loss of epithelium, precluding examination of the entire mucosal surface of the sections (detachment without evidence of inflammation or hemorrhage), thus scoring was achieved using intact sections only. This artifact, accompanied by the segmental sampling (i.e. if an injury falls outwith the sections of trachea/bronchi sampled), may result in a lower score for animals if lesions are missed.
Table 1. Grades of epithelial injury scoring within trachea and bronchi.
Bronchioles
Sections of bronchioles present in at least 15 sections of H&E stained lung tissue were examined from each animal. Lymphoid cells and tissue were not quantified in this scoring system ().
Table 2. Grades of injury scoring within bronchioles.
Parenchyma, alveoli and interstitium
Scoring of the lung tissue was adapted from previously published systems for ARDS in animal models (Matute-Bello et al. Citation2011; Wang et al. Citation2005) to reflect the expected changes in chlorine induced lung injury (Wang et al. Citation2005) and incorporating a more objective assessment (Matute-Bello et al. Citation2011) to the scoring process. Scoring was also undertaken using the Matute-Bello et al. scheme as published (Matute-Bello et al. Citation2011). At least 15 sections of H&E stained lung tissue were examined from each animal. The entirety of each section was scanned at low power to assess congestion, edema, hemorrhage and consolidation. Interstitial/alveolar edema as well as congestion, hemorrhage and consolidation were graded as defined in .
Table 3. Grades of injury scoring within parenchyma, alveoli and interstitium.
Thirty (2 from each of the 15 lung sections), random, high power (x400) fields were examined to assess neutrophil infiltration. Fields were excluded if they included large airways, large vessels, alveolar collapse, or pulmonary parenchyma did not fill the field. All neutrophils within the field, including alveolar spaces, interstitium and small vessels, were counted in each high power field. The average across the 30 fields was calculated for each animal. Neutrophil infiltration was scored based on average counts of 0, 1–5, or greater than 5 neutrophils per high power field.
Alveolar collapse was not included in the scoring, as this may have had multiple causes, including blockage of small airways by debris, effects of anesthesia, agonal and post mortem changes. Alveolar macrophages were not quantified in this scoring system. The histopathological scoring undertaken in this study was performed on tissues fixed by immersion. This resulted in the inability to assess alveolar wall thickness in all sections as described by Matute-Bello et al. (Matute-Bello et al. Citation2011).
Statistical analysis
Statistical analysis was carried out using the statistical analysis package NCSS (Version 11) on data collected from animals challenged with three doses of chlorine (low, medium, or high inhaled doses; ) and an air-exposed control group. This dose-effect time profile for key measurement variables was contrasted up to 12 h post-exposure. A sub-group analysis was undertaken using the identical dataset to contrast air-exposed controls and chlorine-exposed survivors. Chlorine non-survivors were not statistically compared to air-exposed controls or chlorine-exposed survivors, since these animals all died within 2 h of exposure. All data are expressed as mean ± SD.
Time-to-death data across all groups were contrasted using a Kaplan-Meier survival analysis followed by a Log Rank test of median survival time with an alpha level of rejection of p < 0.05. Absolute mortality (comparing each chlorine dose group with air controls) recorded at 12 h post challenge was determined using a one-sided Fisher Exact test at an alpha level of rejection of p < 0.05.
For physiological data, in animals surviving to 12 h post exposure, a percentage change from baseline followed by Area Under Curve (AUC) analysis was determined to contrast air-exposed animals with chlorine exposed animals at 12 h. In the dose-effect study AUC group means were contrasted following one way ANOVA using a Tukey-Kramer procedure at an alpha level of rejection of p < 0.05 and polynomial contrasts generated to investigate any linear trend. In the sub-group study comparing chlorine survivors with air controls, AUC group means were contrasted using a two sample t test.
For terminal parameters and histopathological scoring One Way ANOVA (Kruskal-Wallis or Tukey’s multiple comparisons test) was used to determine differences between air controls and chlorine-exposed survivors for terminal parameters, with p < 0.05 considered significant.
Results
Chlorine exposure strategies
Dosimetry
Previous studies in unventilated animals have targeted the ID to reduce the variability of dosing due to differences in the animals respiration under anesthesia (Graham et al. Citation2018; Rendell et al. Citation2018). Exposure to chlorine gas caused changes in the respiratory response in spontaneously breathing individual animals. Some animals were observed to increase minute volume while others decreased minute volume, or displayed periods of apnea when challenged with chlorine. It was therefore not always possible to continue with the planned length of exposure in some animals due to periods of profound apnea. For these reasons, the concentration x time (Ct) profile was not related to dose in the same way as it would be if a ventilated exposure was used to standardize the respiratory parameters of the animals and it is therefore not a good measure of dosimetry in our studies. The ID approach provided a more direct and accurate measure of how much chlorine the lungs were exposed to, in each individual animal.
Strategy 1 - target inhaled dose 2.4 mg·kg−1
Wang et al. (Citation2004) describe severe injury in pigs exposed to 4.8 mg·kg−1 (1200 mg·m−3 for 20 min) where one animal died two hours post exposure with signs of respiratory failure (Wang et al. Citation2004) therefore pigs were exposed to a target ID of chlorine, starting at 2.4 mg·kg−1.
A dose of 2.4 mg·kg−1 was targetted by increasing the chlorine concentration rapidly and targeting a 20 min exposure, whilst titrating the concentration to the animal’s minute volume. The exposures were challenging and resulted in a range of respiratory responses from apnea to tachypnea in 5 animals. Therefore some exposures were aborted early with a consequent failure to attain the target ID (resulting in an ID of 1.81 ± 0.96 mg·kg−1 (Strategy 1, ) for the 5 animals). Of 5 animals exposed to chlorine in this way, 1 survived to 12 h with no intervention, 4 required manual resuscitation immediately post exposure in an attempt to get the animal past the acute crisis, 1 of which died at 1 h, the others surviving to 12 h, with only mild lung injury. A representative exposure profile for an animal exposed using this approach is shown (). It can be seen how the chlorine concentration was changed in line with changes in the animal’s respiratory parameters. This animal stopped breathing and died 1 h post exposure, despite manual resuscitation for approximately 30 min.
Figure 2. (A) A representative example of an exposure profile of an animal exposed to chlorine, where an inhaled dose was targeted by titrating the chlorine concentration [Cl2] to the animal’s minute volume (Strategy 1). (B) A representative example of an exposure profile of an animal exposed to chlorine, where the exposure time was extended and chlorine concentration slowly increased (Strategy 2). (C) A representative example of an exposure profile of an animal exposed to chlorine, where the exposure time was short and chlorine concentration increased quickly (Strategy 3).
![Figure 2. (A) A representative example of an exposure profile of an animal exposed to chlorine, where an inhaled dose was targeted by titrating the chlorine concentration [Cl2] to the animal’s minute volume (Strategy 1). (B) A representative example of an exposure profile of an animal exposed to chlorine, where the exposure time was extended and chlorine concentration slowly increased (Strategy 2). (C) A representative example of an exposure profile of an animal exposed to chlorine, where the exposure time was short and chlorine concentration increased quickly (Strategy 3).](/cms/asset/2b401c89-519c-46f8-a255-b09832c9468e/itxm_a_1906808_f0002_b.jpg)
Table 4. Dosimetry metrics for Strategies 1, 2 and 3.
Table 5. Dosimetry metrics for low, medium and high inhaled dose groups.
Strategy 2 – targeting a tolerable exposure
In an attempt to achieve a more tolerable exposure, the chlorine concentration was increased more slowly to reduce large changes in respiratory parameters and the exposure duration increased, effectively targeting dose to effect rather than targeting an ID (). However, the animals experienced similar changes to Strategy 1 in their respiratory parameters. Of 5 animals exposed to chlorine in this way, 2 survived to 12 h with no intervention and mild lung injury, 3 required manual resuscitation immediately post exposure, 2 of which died at 1 and 1.5 h post exposure. Using these exposure conditions the ID achieved was 0.48 ± 0.30 mg·kg−1 (Strategy 2, ).
Strategy 3 – targeting short high intensity exposure
In an attempt to increase injury severity, and since chlorine concentration is key to generating injury within the distal lung with short high intensity exposures producing more significant effects (Hoyle et al. Citation2010), a final strategy assessed targeting a peak chlorine concentration quickly over a shorter exposure duration (). Exposure to chlorine was again not well tolerated (prolonged periods of apnea during exposure) and the chlorine dose was again limited by the animal’s response to exposure. Of 5 animals exposed to chlorine in this way, 2 survived to 12 h with no intervention, 3 required manual resuscitation immediately post exposure, all of which died at between 1 and 2 h post exposure. Using these exposure conditions the ID achieved was 0.28 ± 0.15 mg·kg−1 (Strategy 3, ).
Despite attempting to standardize the exposure methodology using 3 different strategies, it was apparent that, in all cases, exposure was not well tolerated and the animal’s response limited the exposure dose. In all strategies used, some animals required manual resuscitation, but this did not ensure survival beyond 2 h post exposure. The doses achieved overlapped between strategies ().
In order to analyze the physiological response to chlorine exposure, the data were grouped based on dose achieved, regardless of exposure strategy. This resulted in three groups of 5 chlorine-exposed animals receiving a low, medium or high ID of chlorine, based on actual dose delivered rather than target inhaled dose, where the range of doses achieved for each group did not overlap (). These exposures ranged in duration from 4 to 26.5 min (15.06 ± 6.86 min). Air exposed control animals (n = 5) were exposed for 24 ± 4.18 min.
Survival
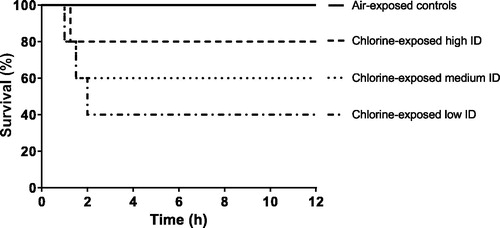
All five air-exposed control animals survived to the end of the 12 h monitoring period (). Four of 5 chlorine-exposed high dose animals, 3 of 5 chlorine-exposed medium dose animals and 2 of 5 chlorine-exposed low dose animals survived to 12 h. All animals which succumbed died between 1 and 2 h post exposure, despite manual resuscitation efforts. There was no significant difference in survival between any of the groups.
Figure 3. Kaplan-Meier survival graph of air-exposed control animals and chlorine-exposed animals (n = 5/group). ID: inhaled dose.
Oxygenation
Air-exposed control animals maintained partial pressure of oxygen in arterial blood (PaO2) at stable levels over the course of the study. All animals exposed to chlorine showed an immediate, sharp fall in PaO2 following exposure which did not return to normal levels in animals surviving to 12 h (). Deaths in all chlorine exposure groups occurred between 0.5 and 2 h post exposure (). There was a significant linear trend of effect, (drop in PaO2; p < 0.05) with increasing doses of chlorine. There was no significant difference in the drop in PaO2 between chlorine exposure groups.
Figure 4. Changes in temperature adjusted PaO2 (A) and PaCO2 (B) in animals grouped as air-exposed controls, chlorine-exposed low ID, chlorine-exposed medium ID and chlorine-exposed high ID. * Significant linear trend (p < 0.05) between increasing doses of chlorine and the air controls. ‘d’ represents the death of an animal. Change in temperature adjusted arterial blood PaO2 (C) and PaCO2 (D) in animals grouped as air-exposed controls, chlorine survivors and chlorine non-survivors. Dashed line (- - -) displays a significant difference across 12 h between the chlorine-exposed survivors and the air-exposed controls. ** Percentage change from baseline AUC survivors vs air controls (p = 0.0049). Mean ± SD.
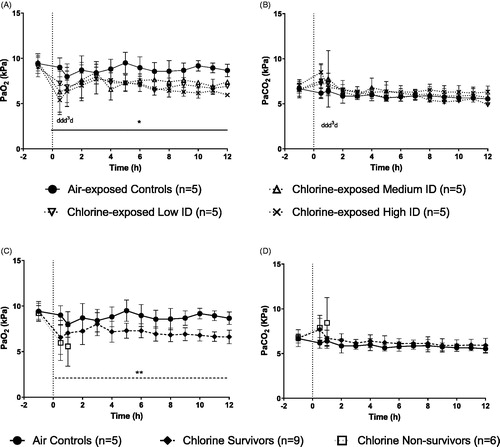
Air-exposed controls maintained partial pressure of arterial blood carbon dioxide (PaCO2) at stable levels over the course of the study. Exposure to chlorine resulted in a small transient but non-significant increase in PaCO2 levels which returned to baseline levels within two hours of exposure ().
Since no dose-related differences were observed in any measured physiological response (e.g. arterial blood gas analysis, respiratory rate, hematology, systemic blood pressure) between the chlorine-exposed animals, the data were pooled to investigate differences between air-exposed controls (n = 5), chlorine-exposed survivors (n = 9) and chlorine-exposed non-survivors (n = 6; ). The chlorine-exposed survivors and non-survivors had overlapping doses (). No statistical analysis has been performed on data from non-survivors, as these were not the focus of the study and the number of animals was small due to early deaths.
Table 6. Dosimetry metrics for survivors vs non-survivors.
As seen previously (), animals exposed to chlorine showed an immediate fall in PaO2 following exposure (). For the animals that survived to 12 h, PaO2 improved over the following 3 h, and then a steady fall in PaO2 was observed through to 12 h. Chlorine-exposed-survivors showed a significant decrease in PaO2 when compared to air-exposed controls over the 12 h post exposure (p = 0.0049).
There was no significant difference in PaCO2 between the chlorine-exposed survivors and the air-exposed controls ().
Acid base analysis
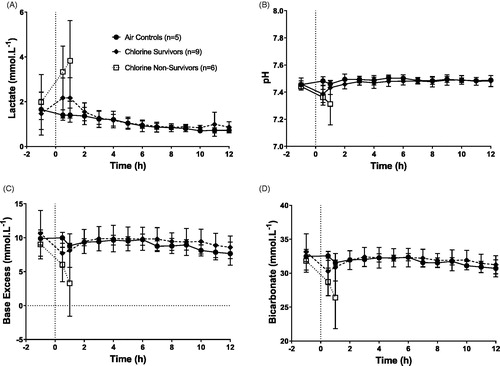
Air-exposed control animals show a small decline in lactate over the 12 h period, but maintained stable pH, base excess and bicarbonate levels over the 12 h monitoring period (). Acid base levels fluctuated in all chlorine-exposed animals immediately post exposure, but returned to levels comparable with air exposed controls in all animals that survived to 12 h. There was no significant difference in these acid base levels between the chlorine-exposed survivors and the air-exposed controls.
Figure 5. Changes in temperature adjusted arterial blood lactate (A), pH (B), base excess (C) and bicarbonate (D). Mean ± SD.
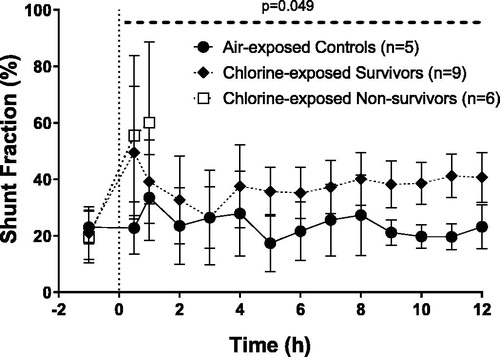
Shunt fraction
Air-exposed controls maintain stable shunt fraction over the course of the 12 h study (). Exposure to chlorine increased shunt fraction in animals which survived post exposure, reaching levels of >35% from 4 h and remaining elevated throughout the study. Chlorine-exposed survivors had a significantly higher shunt fraction when compared to air-exposed controls over the 12 h post exposure (p = 0.049).
Figure 6. Changes in percentage shunt fraction. Dashed line (- - -) displays a significant difference across 12 h between the chlorine-exposed survivors and the air-exposed controls. Percentage change from baseline AUC survivors vs air controls (p = 0.049). Mean ± SD.
While changes in heart and respiratory rate and rhythm were observed during exposure to chlorine, these rapidly resolved at the end of exposure either with or without resuscitation. No other changes in any cardiovascular (e.g. heart rate, mean arterial blood pressure) parameters between air-exposed controls and chlorine exposed surviving animals were observed following exposure.
Measures of pulmonary edema
For all terminal measurements data were pooled into chlorine-exposed survivors (12 h) and chlorine-exposed non-survivors (∼2 h). There was no observed change in LWW:BW, LWW:DW ratios (both measures of extravascular lung water and degree of alveolar permeability) and terminal BAL fluid protein concentration of all chlorine-exposed animals compared to air-exposed controls (data not shown).
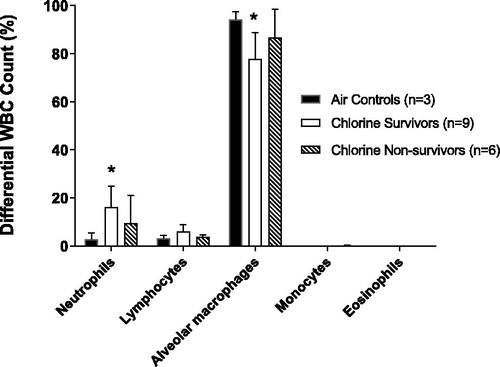
To examine neutrophilic inflammation, a known consequence of chlorine inhalation, differential WBC count analysis of terminal BAL fluid was conducted. The air-exposed control animals demonstrated a normal cellular distribution with the majority of cells being the resident alveolar macrophage, in agreement with previously published results (Graham et al. Citation2018; Jugg et al. Citation2016). The percentage () and absolute number (data not shown) of neutrophils recruited into the lungs of the chlorine-exposed survivors increased significantly when compared with air-exposed controls (p = 0.02). The percentage () and absolute number (data not shown) of alveolar macrophages in the chlorine-exposed survivors significantly decreased when compared to air-exposed controls (p = 0.04). Consistent with the BAL fluid analysis, lung tissue neutrophils were increased in chlorine-exposed survivors compared with air-exposed controls (data not shown).
Figure 7. Differential WBC count analysis in terminal lavage fluid. The air-exposed control group for BAL fluid WBC counts were reduced to 3 animals due to lung abnormalities or difficulty collecting a full sample of BAL fluid at post mortem. * indicates significant difference for neutrophils (p = 0.02) and alveolar macrophages (p = 0.04) between the chlorine-exposed survivors when compared to air-exposed controls (Kruskal-Wallis One-Way ANOVA). Mean ± SD.
Pathology
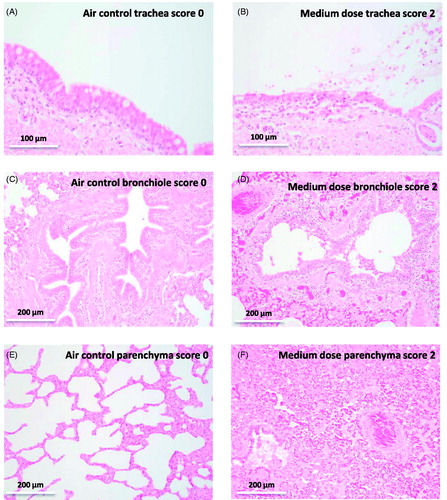
The trachea, bronchi and bronchioles were normally lined by pseudostratified, columnar, ciliated, epithelium (). Injury to the respiratory epithelium, in the animals exposed to chlorine, ranged from loss of cilia, epithelial cell degeneration and minimal loss of cells (mild injury, score 1), to loss of cells without exposure of the basement membrane (erosions; moderate injury, score 2, ), through to areas with loss or detachment of all epithelial layers and exposure or damage to the basement membrane (ulcerations; severe injury, score 3).
Figure 8. Hematoxylin and eosin stained sections (A) Air control, trachea, no injury (score 0), pseudostratified, ciliated, columnar epithelium with goblet cells. (B) Medium dose, trachea, moderate injury (score 2), erosion and degeneration of epithelium, edema, mixed inflammatory cell infiltration, and cellular debris, mucus, and erythrocytes in the lumen. (C) Air control, bronchiole, no injury (score 0). (D) Medium dose, bronchiole, moderate injury (score 2), erosion and degeneration of epithelium, mixed inflammatory cell infiltration, and sloughed epithelial cells, cellular debris, mucus and erythrocytes in the lumen. The surrounding parenchyma exhibits congestion, edema, fibrin exudation and polymorphonuclear cell infiltration. (E) Air control, lung parenchyma, no injury (score 0), (F) Medium dose, lung parenchyma, moderate injury (score 2), diffuse filling of alveolar spaces by eosinophilic fluid (edema) and fibrin strands, with congestion of alveolar capillaries and larger vessels, and numerous neutrophils within alveolar spaces and the interstitium.
In air controls, the lung parenchyma exhibited multifocal collapse and over-inflation, an artifact of the experimental methods, with less than 1 neutrophil per high power field (). In animals exposed to chlorine, changes in the parenchyma ranged from congestion, less than 50% interstitial or alveolar edema, less than 5 neutrophils per high power (x400) field, and presence of hemorrhage (mild changes, score 1), to congestion, greater than 50% interstitial or alveolar edema, more than 5 neutrophils per high power field, and presence of extravasated erythrocytes (moderate changes, score 2, ). Cell death was occasionally observed in exposed animals, and alveolar macrophages were not quantified in this scoring system.
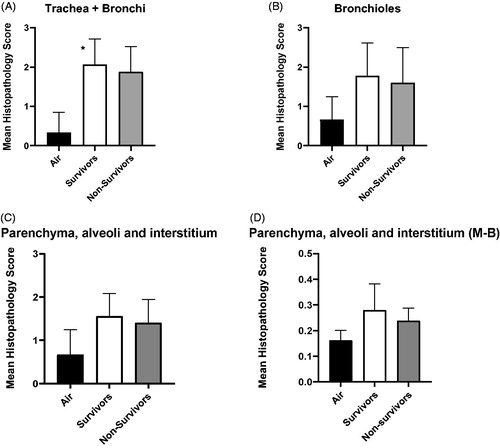
Amongst the chlorine exposed animals, mean histopathology scores between the low, medium and high ID groups were not significantly different. Mean histopathology scores were significantly higher in chlorine-exposed survivors (p < 0.0001) compared to air-exposed controls in the conducting airways (trachea and bronchi, ). Scores decreased along the length of the respiratory tract, of animals exposed to chlorine ().
Figure 9. Mean histopathology score comparing air-exposed controls with animals surviving to 12 h along the length of the respiratory tract (A – trachea and bronchi; B – Bronchioles; C – Parenchyma, alveoli and interstitium; D - Parenchyma, alveoli and interstitium using scoring system of Matute-Bello et al., Citation2011 (M-B)). Air-exposed control = 3, chlorine-exposed survivors = 9, chlorine-exposed non-survivors = 5. * indicates significant difference between air-exposed controls and chlorine-exposed survivors (p < 0.0001, Tukey’s multiple comparison, one way ANOVA). Mean ± SD.
Discussion
Our research goal was to establish a model of chlorine-induced lung injury in the spontaneously breathing pig in order to test therapies and supportive medical interventions to ameliorate the injury during the acute, pre-hospital phase after exposure. A key outcome from the study is the assessment of changes in clinical parameters with direct read-across to the management of human casualties (e.g. oxygenation status), as well as a range of other parameters defining injury within the lung (e.g. inflammatory cell analysis, blood-air barrier integrity and pathology). This study has demonstrated that we have established a mild chlorine-induced lung injury in the spontaneously breathing pig with significant changes in the following physiological and pathological parameters: PaO2, shunt fraction, histopathology and differential WBC counts. However, the experimental set up described in this paper did not produce the moderate to severe injury that we set out to achieve, therefore further investigation is required to optimize the model and enable the assessment of therapeutic candidates.
To establish a large animal model of moderate to severe chlorine-induced lung injury, a fully characterized, consistent lung injury is required. Using our previous expertise in exposing animals to toxic chemicals (Brown et al. Citation2002; Graham et al. Citation2018; Rendell et al. Citation2018; Fairhall et al. Citation2010; Jugg et al. Citation2013; Jugg et al. Citation2016) and taking account of the severe effects seen in pigs exposed to chlorine where one animal died two hours post exposure with signs of respiratory failure (4.8 mg·kg−1; (Wang et al. Citation2004)), we used a starting ID of 2.4 mg·kg−1 chlorine. However, this did not result in a reproducible model and two further chlorine exposure strategies were evaluated. None of the three strategies examined resulted in a consistent exposure profile and no apparent relationship between the exposure strategy and outcome was identified (survival, physiological and pathological assessment of injury consistent across groups). In all cases animals did not tolerate the exposure protocols, as demonstrated by immediate changes in breathing pattern during the exposure. Since there was no apparent relationship between the exposure strategy used and the experimental outcomes, and the range of doses achieved was highly variable, animals were re-grouped by total ID achieved. This resulted in three groups of 5 chlorine-exposed animals receiving a low, medium or high ID of chlorine. Some significant decrements in physiology and pathology through the respiratory tract were observed compared to air control animals, however these changes were similar between the three chlorine exposed groups. Since there were no significant differences in any parameter between the three chlorine exposed groups of animals, these data were pooled to investigate differences between air-exposed animals, chlorine-exposed survivors and chlorine-exposed non-survivors.
A number of difficulties were experienced during chlorine exposures, for example, respiratory depression and apnea. Some exposures had to be aborted early, and the intended dose was not achieved. The immediate but variable changes to respiratory parameters seen during the exposure may be caused by activation of nerve afferents in the airways which may elicit reflex responses in breathing pattern, including apnea, which complicates exposure. Exposures of airways to some chemicals and gases can generate a cascade of events and mediators to trigger sensory neuronal signals, which are transmitted to the afferent parasympathetic autonomic nervous system leading to constriction of airway smooth muscle (Bacsi et al. Citation2016). Activation of chemosensors (e.g. ion channels of the transient receptor potential (TRP) family) in the upper respiratory tract may occur following inhalation of toxic chemicals such as chlorine and trigger defined signaling pathways (Bessac and Jordt Citation2008; Büch et al. Citation2013). In addition, chemosensory TRP channels are also expressed in non-neuronal cells (such as the lung epithelium), where they are activated by toxic chemicals such as chlorine and promote a complex signaling network (Büch et al. Citation2013). Activation may result in initiation of pain, respiratory depression, secretions, and other protective responses such as bronchoconstriction (Bessac and Jordt Citation2010). Okponyia et al. (Citation2018) reported severe bradypnea in rats during, and following, whole body high dose chlorine exposure, where respiratory rate rapidly decreased. The authors attribute the changes seen in respiratory parameters, in part, to a direct response of the activation of TRP channels within the respiratory tract resulting in altered breathing pattern (Okponyia et al. Citation2018). Based on the evidence presented, the immediate changes in breathing pattern seen during exposure in our studies could be the result of a combination of activation of nerve fibers, such as pulmonary C-fibers, in the airways causing reflex changes in respiratory parameters as well as activation of specific TRPA1 ion channels. However, we acknowledge this is speculation as we have no definitive proof of either mechanism and such investigations were beyond the scope of the experiments reported.
Six animals died within 2 h of chlorine exposure. These animals are not the focus of this study, since we were aiming to develop a model of lung injury 12 h post exposure. All other animals survived to 12 h. While limited manual resuscitation, with the bag-valve resuscitator, was permissible under the experimental protocol, this did not ensure survival beyond the acute crisis. All non-surviving animals required manual resuscitation immediately post exposure while only 4 of 9 surviving animals required resuscitation. The lack of difference in the observed pathological response seen between survivors and non-survivors suggests that the timescales for assessment of morphological changes observed by light microscopy are too short in this study and that assessment of physiological and biological changes is more appropriate during the acute phase of the response. The changes in arterial blood gas data are consistent with hypoventilation. The severity of the respiratory failure seen was such that manual ventilation was unable to compensate for, nor improve arterial oxygenation to a level required to maintain adequate tissue oxygen delivery and sustain life.
In animals that survived to 12 h post exposure, there was a significant decrease in arterial blood PaO2 (to approx. 6.5 kPa) and SaO2 (to approx. 77%; data not shown), while the PaCO2 and acid base levels remained unchanged, suggesting the decrement in oxygenation did not overwhelm the animals which went on to develop a mild injury at 12 h. Hypoxemic respiratory failure is characterized by a PaO2 of <8 kPa (60 mmHg) with normal or low arterial PaCO2 (Roussos and Koutsoukou Citation2003). The sustained decrease in arterial blood oxygenation seen in the chlorine-exposed animals was accompanied by a significantly increased shunt fraction, suggesting a mismatch between ventilation and perfusion most likely caused by damage to the lungs rather than cardiovascular compromise. Even with low arterial oxygen there was no evidence of shock in the surviving animals (base excess and lactate within normal limits). These results are in agreement with other studies assessing chlorine induced acute lung injury in both small and large animals. Wang et al. Citation2004; Citation2005 demonstrated a rapid decrease in PaO2 to approximately 6 kPa out to 5 h following chlorine exposure of anesthetized, ventilated pigs. Similar changes were observed in anesthetized, ventilated sheep over a range of doses of chlorine; animals demonstrated rapid and sustained decreases in PaO2 and PaO2/FiO2 ratio, normal PaCO2 levels and ventilation/perfusion mismatch (Fukuda et al. Citation2016; Batchinsky et al. Citation2006). Okponyia et al. (Citation2018) demonstrated an immediate and prolonged drop in peripheral oxygen saturation (SpO2) over 6 h in rats exposed to a high dose of chlorine. The PaO2/FiO2 ratio dropped to levels consistent with severe to extreme respiratory failure. In all studies, hypoxemia appears to be the hallmark of chlorine inhalation injury.
Chlorine exposure resulted in a neutrophilic infiltration into lung tissue. Increased numbers of neutrophils were seen in the terminal lavage fluid, however, no change in terminal lavage fluid protein or other measure of extravascular lung water or degree of alveolar permeability was seen, consistent with the mild injury observed within our acute timeframe. Neutrophils migrating into airway epithelium may go on to release oxidants and proteolytic enzymes further damaging the airways. Neutrophil numbers were significantly elevated to 5 days post exposure in mice exposed to chlorine (Tuck et al. Citation2008). Jonasson et al. (Citation2013), reported on the adverse inflammatory and respiratory responses over an acute time course of injury in mice exposed to a range of doses of chlorine, observing an infiltration of inflammatory cells into the lungs. Damage to the airways was marked with neutrophil and macrophage recruitment to the airways accompanied by decreased respiratory function. Histological assessment demonstrated that the injury to the lungs followed the respiratory tract, affecting the trachea and bronchi the most, whilst having less severe effects in the deep lung parenchyma. These results are in agreement with Jonasson et al. (Citation2013), where injury was restricted to the larger airways, with little to no damage in the peripheral airways.
There are a number of limitations associated with this study, which include the use of anesthesia, species differences between the pig and human and early deaths reducing sample size. The use of anesthesia, while essential for performing studies in animals, is known to have a detrimental effect on physiological parameters such as oxygenation and respiration either through direct effects (respiratory depression) or indirect effects (lung collapse due to prolonged recumbancy) (Graham et al. Citation2018; Hockel et al. Citation2012). In addition, there is evidence that anesthetic agents such as isoflurane and propofol, used in the current study, have anti-oxidant (Braz and Karahalil Citation2015) and anti-inflammatory properties, which may reduce the severity of lung injury (Kundumani-Sridharan et al. Citation2019; Ruan et al. Citation2020). The use of anesthesia is therefore a balance between Refinement of the experimental procedures and the influence of the agents on the experimental outcome (Russell and Burch Citation1959). We have extensive experience in using long-term anesthesia in terminal studies and the detrimental physiological effects of intravenous anesthesia are kept as low as possible, as demonstrated by the air control animals which maintained stable physiology throughout the study with levels consistent with previous (non-ventilated) pig studies (Graham et al. Citation2018; Rendell et al. Citation2018). In addition, there was evidence of lung pathology following chlorine exposure when compared to air controls. One problem consistent with all animal models of chemical induced lung injury is the more highly developed olfactory system compared to humans (Brown et al. Citation2006). This means that the surface area in the upper respiratory tract is much greater and therefore capable of greater absorption of inhaled chemicals. Bypassing the upper respiratory tract is easily achieved with the use of an ET tube or tracheostomy; such exposures may more accurately reflect human inhalational injury (Jugg Citation2016). Large animals have an additional advantage over rodents as their larger blood volume allows more frequent, longitudinal, blood sampling enabling more data to be generated from a single animal. The pig was the species of choice as it has been used by a number of groups worldwide to study chlorine induced lung injury (Wang et al. Citation2002; Wang et al. Citation2004; Wang et al. Citation2005; Gunnarsson et al. Citation1998) and our group has successfully developed spontaneously breathing pig models for inhaled chemicals (Graham et al. Citation2018; Rendell et al. Citation2018; Fairhall et al. Citation2010).
The current study had small numbers of animals, the number decreasing due to early deaths; this increases the variance between groups and thus reduces the power of any statistical analysis. We only examined physiology up to 12 h after chlorine exposure; the intention is to extend this out to 24 h to enable the injury to develop further. We were unable to perform consistent exposures of pigs to chlorine, despite a number of approaches. This study highlights the challenges of developing a pig model of chlorine-induced lung injury in a spontaneously breathing animal.
Conclusions
Assessing the effects of chlorine exposure in the pig has been challenging, due in large part to the acute changes in respiratory parameters during exposure, namely respiratory depression and apnea, which were not consistent between animals. We have suggested that these respiratory changes are reflex responses following activation of sensory nerves such as pulmonary C fibers. Some animals recovered quickly from the respiratory disturbances following a brief period of manual ventilator support but some animals failed to recover spontaneous breathing and died between 1 and two hours post-exposure; the mechanism underlying this discrepancy remains unclear. The changes in respiratory parameters resulted in some exposures having to be terminated early and consequently these animals did not receive the intended ID of chlorine. While we have established a model of mild lung injury within the spontaneously breathing pig, it is currently not possible to expose spontaneously breathing animals to a consistent, reproducible dose of chlorine to enable efficacy studies to be performed. The future challenge will be in identifying methods for providing a more consistent exposure to chlorine (e.g. ventilated exposure), and allowing additional time for the severity of the injury to develop further.
Disclaimer
All experiments were carried out in accordance with the UK Animals (Scientific Procedures) Act, 1986.
| Abbreviations | ||
| OPCW | = | Organization for the Prohibition of Chemical Weapons |
| Cl2 | = | chlorine |
| HCl | = | hydrochloric acid |
| HOCl | = | hypochlorous acid |
| ROS | = | reactive oxygen species |
| ARDS | = | acute respiratory distress syndrome |
| ID | = | inhaled dose |
| ET | = | endotracheal tube |
| FiO2 | = | fraction of inspired oxygen |
| MFC | = | mass flow controller |
| Qs | = | Qt |
| shunt fraction | = | ratio of shunted blood to total cardiac output |
| BAL | = | bronchoalveolar lavage |
| LWW:BW | = | lung wet weight to body weight ratio |
| LWW:DW | = | lung wet weight to dry weight ratio |
| H&E | = | hematoxylin and eosin |
| AUC | = | area under the curve |
| ANOVA | = | analysis of variance |
| SD | = | standard deviation |
| PaO2 | = | partial pressure of oxygen (arterial) |
| PaCO2 | = | partial pressure of carbon dioxide (arterial) |
| WBC | = | white blood cell |
| TRP | = | transient receptor potential |
Acknowledgements
We thank the following for their contribution to this work: B Farnfield for anaesthesia and support; Animal Services Staff for animal husbandry, surgery and technical laboratory support; C Underwood-Fowler, C Whitmore and S Gore for clinical pathology; R Gwyther for statistical analysis.
Disclosure statement
The authors report no conflict of interest.
Additional information
Funding
References
- Achanta S, Liu B, Gentile M, Caceres A, Behm D, Burgert M, Costell M, Cheifetz I, Gunn M, Jordt S. 2019. TRPV4 inhibitor improves pulmonary function and oxygen saturation in a pig translational model of chlorine gas-induced acute lung injury. Proceedings of the American Thoracic Society 2019 International Conference, Dallas.
- Achanta S, Jordt S. 2019. Toxic effects of chlorine gas and potential treatments: a literature review. Toxicol Mech Methods. 1–13. DOI:10.1080/15376516.2019.1669244
- Achanta S, Jordt S. 2020. Transient receptor potential channels in pulmonary chemical injuries and as countermeasure targets. Ann N Y Acad Sci. 1480(1):73–103.
- Bacsi A, Pan L, Ba X, Boldogh I. 2016. Pathophysiology of bronchoconstriction: role of oxidatively damaged DNA repair. Curr Opin Allergy Clin Immunol. 16(1):59–67.
- Balakrishna S, Song W, Achanta S, Doran S, Liu B, Kaelberer M, Yu Z, Sui A, Cheung M, Leishman E, et al. 2014. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 307(2):L158–L172.
- Batchinsky A, Martini D, Dick E, Fudge J, Hardin D, Cancio L. 2006. Acute respiratory distress syndrome secondary to inhalation of chlorine gas in sheep. J Trauma. 60:944–957.
- Bessac B, Jordt S. 2008. Breathtaking TRP channels: TRPA1 and TRPV1 in airway chemosensation and reflex control. Physiology (Bethesda). 23:360–370.
- Bessac B, Jordt S. 2010. Sensory detection and responses to toxic gases: mechanisms, health effects, and countermeasures. Proc Am Thorac Soc. 7(4):269–277.
- Bradford M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 72(1–2):248–254.
- Braz M, Karahalil B. 2015. Genotoxicity of anesthetics evaluated in vivo (animals). BioMed Res Int. 2015:1–8.
- Brown R, Jenner J, Jugg B. 2006. The use of large animals in inhalation toxicology. In: Salem H & Katz S, editors. Inhalation toxicology. 2nd ed. Boca Raton (FL): CRC Press, Taylor & Francis Group.
- Brown R, Jugg B, Harban F, Ashley Z, Kenward C, Platt J, Hill A, Rice P, Watkins P. 2002. Pathophysiological responses following phosgene exposure in the anaesthetized pig. J Appl Toxicol. 22(4):263–269.
- Büch T, Schäfer E, Steinritz D, Dietrich A, Gudermann T. 2013. Chemosensory TRP channels in the respiratory tract: role in toxic lung injury and potential as "sweet spots" for targeted therapies. Rev Physiol Biochem Pharmacol. 165:31–65.
- Carlisle M, Lam A, Svendsen E, Aggarwal S, Matalon S. 2016. Chlorine-induced cardiopulmonary injury. Ann N Y Acad Sci. 1374(1):159–167.
- Chang W, Chen J, Schlueter C, Rando R, Pathak Y, Hoyle G. 2012. Inhibition of chlorine-induced lung injury by the type 4 phosphodiesterase inhibitor rolipram. Toxicol Appl Pharmacol. 263(2):251–258.
- Chen J, Mo Y, Schlueter C, Hoyle G. 2013. Inhibition of chlorine-induced pulmonary inflammation and edema by mometasone and budesonide. Toxicol Appl Pharmacol. 272(2):408–413.
- Evans R. 2005. Chlorine: state of the art. Lung. 183(3):151–167.
- Fairhall S, Brown R, Jugg B, Smith A, Mann T, Jenner J, Sciuto A. 2008. Preliminary studies of sulphur mustard-induced lung injury in the terminally anesthetized pig: exposure system and methodology. Toxicol Mech Methods. 18(4):355–362.
- Fairhall S, Jugg B, Read RW, Stubbs S, Rutter S, Smith A, Mann T, Jenner J, Sciuto A. 2010. Exposure–response effects of inhaled sulfur mustard in a large porcine model: a 6-h study. Inhal Toxicol. 22(14):1135–1143.
- Fukuda S, Ihara K, Lopez E, Cox R, Southan G, Salzman A, Salsbury J, Prough D, Enkhbaatar P. 2016. R-107 attenuates severity of acute respiratory distress syndrome induced by chlorine gas inhalation in ovine model. Am J Respir Crit Care Med, 201:A6299.
- Global-Biodefense. 2017. Chlorine gas medical countermeasures: 61M contract awarded to GlaxoSmithKline; [accessed 2020 May 28]. https://globalbiodefense.com/2017/09/26/chlorine-gas-medical-countermeasure-61m-contract-awarded-glaxosmithkline/.
- Graham S, Fairhall S, Rutter S, Auton P, Rendell R, Smith A, Perrott R, Nicholson-Roberts T, Jugg B. 2018. Continuous positive airway pressure: an early intervention to prevent phosgene-induced acute lung injury. Toxicol Lett. 293:120–126.
- Grainge C, Rice P. 2010. Management of phosgene-induced acute lung injury. Clin Tox. 48(6):497–508.
- Grainge C, Brown R, Jugg B, Smith A, Mann T, Jenner J, Rice P, Parkhouse D. 2009. Early treatment with nebulised salbutamol worsens physiological measures and does not improve survival following phosgene induced acute lung injury. JR Army Med Corps. 155(2):105–109.
- Gunnarsson M, Walther S, Seidal T, Bloom G, Lennquist S. 1998. Exposure to chlorine gas: effects on pulmonary function and morphology in anaesthetised and mechanically ventilated pigs. J Appl Toxicol. 18(4):249–255.
- Hockel K, Trabold R, Schöller K, Török E, Plesnila N. 2012. Impact of anesthesia on pathophysiology and mortality following subarachnoid hemorrhage in rats. Exp Transl Stroke Med. 4(1):5–13.
- Hoyle G, Chang W, Chen J, Schlueter C, Rando R. 2010. Deviations from Haber's Law for multiple measures of acute lung injury in chlorine-exposed mice. Toxicol Sci. 118(2):696–703.
- Jonasson S, Koch B, Bucht A. 2013. Inhalation of chlorine causes long-standing lung inflammation and airway hyperresponsiveness in a murine model of chemical-induced lung injury. Toxicology. 303:34–42.
- Jones R, Wills B, Kang C. 2010. Chlorine gas: an evolving hazardous material threat and unconventional weapon. West J Emerg Med. 11(2):151–156.
- Jugg B. 2016. Toxicology and treatment of phosgene induced lung injury. In: Worek F, Jenner J, editors. Issues in toxicology: chemical warfare toxicology. Croydon (UK): The Royal Society of Chemistry.
- Jugg B, Fairhall S, Smith A, Rutter S, Mann T, Perrott R, Jenner J, Salguero J, Shute J, Sciuto A. 2013. N-acetyl-L-cysteine protects against inhaled sulfur mustard poisoning in the large swine. Clin Tox. 51(4):216–224.
- Jugg B, Hoard-Fruchey H, Rothwell C, Dillman J, David J, Jenner J, Sciuto A. 2016. Acute gene expression profile of lung tissue following sulfur mustard inhalation exposure in large anesthetized swine. Chem Res Toxicol. 29(10):1602–1610.
- Jurkuvenaite A, Benavides G, Komarova S, Doran S, Johnson M, Aggarwal S, Zhang J, Darley-Usmar V, Matalon S. 2015. Upregulation of autophagy decreases chlorine-induced mitochondrial injury and lung inflammation. Free Radic Biol Med. 85:83–94.
- Kundumani-Sridharan V, Subramani J, Raghavan S, Maiti G, Owens C, Walker T, Wasnick J, Idell S, DAS K. 2019. Short-duration hyperoxia causes genotoxicity in mouse lungs: protection by volatile anesthetic isoflurane. Am J Physiol Lung Cell Mol Physiol. 316(5):L903–L917.
- Lazrak A, Creighton J, Yu Z, Komarova S, Doran S, Aggarwal S, Emala CS, Stober V, Trempus C, Garantziotis S, et al. 2015. Hyaluronan mediates airway hyperresponsiveness in oxidative lung injury. Am J Physiol Lung Cell Mol Physiol. 308(9):L891–L903.
- Leustik M, Doran S, Bracher A, Williams S, Squadrito G, Schoeb T, Postlethwait E, Matalon S. 2008. Mitigation of chlorine-induced lung injury by low-molecular-weight antioxidants. Am J Physiol Lung Cell Mol Physiol. 295(5):L733–L743.
- Martin J, Campbell H, Iijima H, Gautrin D, Malo J-L, Eidelman D, Hamid Q, Maghni K. 2003. Chlorine-induced injury to the airways in mice. Am J Respir Crit Care Med. 168(5):568–574.
- Matute-Bello G, Downey G, Moore B, Groshong S, Matthay M, Slutsky A, Kuebler W. 2011. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 44(5):725–738.
- Morris L. 2014. Islamic state militants allegedly used chlorine gas against Iraqi security forces. Washington Post News report; [accessed 2020 May 28]. https://www.washingtonpost.com/world/middle_east/islamic-state-militants-allegedly-used-chlorine-gas-against-iraqi-security-forces/2014/10/23/c865c943-1c93-4ac0-a7ed-033218f15cbb_story.html
- Murphy M. 2009. How mitochondria produce reactive oxygen species. Biochem J. 417(1):1–13.
- Nelson L, Odujebe O. 2002. Simple asphyxiants and pulmonary irritants. In Toxicologic emergencies. 7th ed. New York (NY): McGraw-Hill: Goldfrank’s.
- O’Koren EG, Hogan BLM, Gunn MD. 2013. Loss of basal cells precedes bronchiolitis obliterans-like pathological changes in a murine model of chlorine gas inhalation. Am J Respir Cell Mol Biol. 49(5):788–797.
- Okponyia O, Mcgraw M, Dysart M, Garlick R, Rioux J, Murphy A, Roe G, White C, Veress L. 2018. Oxygen administration improves survival but worsens cardiopulmonary functions in chlorine-exposed rats. Am J Respir Cell Mol Biol. 58(1):107–116.
- OPCW. 2014. OPCW fact finding mission: “Compelling confirmation” that chlorine gas used as weapon in Syria. Press release; [accessed 2020 May 28]. https://www.opcw.org/news/article/opcw-fact-finding-mission-compelling-confirmation-that-chlorine-gas-used-as-weapon-in-syria/.
- Radikal Therapeutics. 2016. Radikal Therapeutics awarded funding by Health and Human Services (HHS) for development of inhaled chlorine antidote R-107. [Accessed 2020 May 28]. http://radikalrx.com/radikal-therapeutics-awarded-funding-health-human-services-hhs-development-inhaled-chlorine-antidote-r-107/.
- Rendell R, Fairhall S, Graham S, Rutter S, Auton P, Smith A, Perrott R, Jugg B. 2018. Assessment of N-acetylcysteine as a therapy for phosgene-induced acute lung injury. Toxicol Lett. 290:145–152.
- Roussos C, Koutsoukou A. 2003. Respiratory failure. Eur Respir J. 22(Supplement 47):3s–14s.
- Ruan H, Li W, Wang J, Chen G, Xia B, Wang Z, Zhang M. 2020. Propofol alleviates ventilator-induced lung injury through regulating the Nrf2/NLRP3 signaling pathway. Exp Mol Pathol. 114:1–8.
- Russell W, Burch R. 1959. The principles of humane experimental technique. London: Methuen.
- Smith A, Brown R, Jugg B, Platt J, Mann T, Masey C, Jenner J, Rice P. 2009. The effect of steroid treatment with inhaled budesonide or intravenous methylprednisolone on phosgene-induced acute lung injury in a porcine model. Mil Med. 174(12):1287–1294.
- Summerhill E, Hoyle G, Jordt S, Jugg B, Martin J, Matalon S, Patterson S, Prezant D, Sciuto A, Svendsen E, et al. 2017. An official American Thoracic Society Workshop report: chemical inhalational disasters. Biology of lung injury, development of novel therapeutics, and medical preparedness. Ann Am Thorac Soc. 14(6):1060–1072.
- Tian X, Tao H, Brisolara J, Chen J, Rando R, Hoyle G. 2008. Acute lung injury induced by chlorine inhalation in C57BL/6 and FVB/N mice. Inhal Toxicol. 20(9):783–793.
- Tuck S, Ramos-Barbón D, Campbell H, Mcgovern T, Karmouty-Quintana H, Martin J. 2008. Time course of airway remodelling after an acute chlorine gas exposure in mice. Respir Res. 9(1):61–76.
- VAN Sickle D, Wenck M, Belflower A, Drociuk D, Ferdinands J, Holguin F, Svendsen E, Bretous L, Jankelevich S, Gibson J, et al. 2009. Acute health effects after exposure to chlorine gas released after a train derailment. Am J Emerg Med. 27(1):1–7.
- Wang J, Winskog C, Edston E, Walther S. 2005. Inhaled and intravenous corticosteroids both attenuate chlorine gas-induced lung injury in pigs. Acta Anaesthesiol Scand. 49(2):183–190.
- Wang J, Zhang L, Walther S. 2002. Inhaled budesonide in experimental chlorine gas lung injury: influence of time interval between injury and treatment. Intensive Care Med. 28(3):352–357.
- Wang J, Zhang L, Walther S. 2004. Administration of aerosolized terbutaline and budesonide reduces chlorine gas–induced acute lung injury. J Trauma Acute Care Surg. 56:850–862.
- Weitz R, Al-Marashi I, Hilal K. 2007. Chlorine as a terrorist weapon in Iraq. In: WMD Insights - Issues and viewpoints in the international media. DTRA Publication. 15:9–14.
- White C, Martin J. 2010. Chlorine gas inhalation: human clinical evidence of toxicity and experience in animal models. Proc Am Thorac Soc. 7(4):257–263.
- Wigenstam E, Elfsmark L, Koch B, Bucht A, Jonasson S. 2016. Acute respiratory changes and pulmonary inflammation involving a pathway of TGF-beta1 induction in a rat model of chlorine-induced lung injury. Toxicol Appl Pharmacol. 309:44–54.
- Zarogiannis S, Jurkuvenaite A, Fernandez S, Doran S, Yadav A, Squadrito G, Postlethwait E, Bowen L, Matalon S. 2011. Ascorbate and deferoxamine administration after chlorine exposure decrease mortality and lung injury in mice. Am J Respir Cell Mol Biol. 45(2):386–392.
- Zarogiannis S, Wagener B, Basappa S, Doran S, Rodriguez C, Jurkuvenaite A, Pittet J, Matalon S. 2014. Postexposure aerosolized heparin reduces lung injury in chlorine-exposed mice. Am J Physiol Lung Cell Mol Physiol. 307(5):L347–L354.
- Zellner T, Eyer F. 2020. Choking agents and chlorine gas - History, pathophysiology, clinical effects and treatment. Toxicol Lett. 320:73–79.