
Abstract
Autophagy related gene 5 (ATG5) was lost in 23% of the patients with colorectal cancer (CRC) and the role of loss of ATG5 in the pathogenesis of CRC remains unclear. Knockdown of ATG5 in cancer cells enhances the antitumor efficacy of lots of chemotherapeutic agents. However, there is still no animal model to validate these in vitro observations in vivo. In this study, we found that heterozygous deletion of ATG5 in ApcMin/+ mice increased the number and size of adenomas as compared with those in ApcMin/+ATG5+/+ mice. To investigate whether ATG5 deficiency could sensitize tumors to chemotherapies, we compared the antitumor effects of Interferon-gamma (IFN-γ) between ApcMin/+ATG5+/+ and ApcMin/+ATG5+/− mice, as IFN-γ is a potential tumor suppressor for CRC and has been used clinically as an efficient adjuvant to chemotherapy of cancer. We revealed that heterozygous deletion of ATG5 significantly enhanced the antitumor efficacy of IFN-γ. Early treatment of ApcMin/+ATG5+/− mice with IFN-γ decreased tumor incidence rate to 16.7% and reduced the number of adenomas by 95.5% and late treatment led to regression of tumor. Moreover, IFN-γ treatment did not cause any evident toxic reaction. Mechanistic analysis revealed that heterozygous deletion of ATG5 activated EGFR/ERK1/2 and Wnt/β-catenin pathways in adenomas of ApcMin/+ mice and enhanced the effects of IFN-γ-dependent inhibition of these 2 pathways. Our results demonstrate that ATG5 plays important roles in intestinal tumor growth and combination of IFN-γ and ATG5 deficiency or ATG5-targeted inhibition is a promising strategy for prevention and treatment of CRC.
Abbreviations
| ATG5 | = | autophagy related gene 5 |
| CRC | = | colorectal cancer |
| IFN-γ | = | Interferon-gamma |
| 5-FU | = | 5-fluorouracil |
| Apc | = | adenomatous polyposis coli |
| siRNAs | = | small interfering RNAs |
| EGFR | = | epidermal growth factor receptor |
| Erk | = | extracellular signal-regulated kinase |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| PCNA | = | proliferating cell nuclear antigen |
Introduction
Colorectal cancer (CRC) is the second most common cancers and is the third leading cause of cancer-related death.Citation1,2 Surgical resection in combination with chemotherapy is the current standard treatment for CRC, but it is always unsatisfactory and many patients have to receive chemotherapy to reduce tumor recurrence and prolong postoperative survival.Citation3 Therefore, it is of high importance to develop novel and effective agents for the prevention and therapy of CRC. Adenomatous polyposis coli (Apc) mutation is the initiating alteration in most colorectal cancers and mutant Apc activates the Wnt/β-catenin signaling pathway.Citation4 C57BL/6J ApcMin/+ mouse model, which contains a point mutation in the Apc gene and develops numerous intestinal adenomas, provides a valuable in vivo system to study the tumorigenesis, prevention and treatment of human CRC.Citation5
Autophagy related gene-5 (ATG5), an essential ATG protein in autophagy, contributes to the early autophagosome formation. The formation of a double membrane autophagosome initiates autophagy, which is an evolutionarily conserved catabolic pathway and degrades cytoplasmic components such as damaged organelles, macromolecules and long-lived proteins.Citation6,7 ATG5 protein is well expressed in normal colon cells, while it is lost in 23% of the patients with CRC.Citation8 Despite the low incidences, loss of ATG5 might play an important role in the pathogenesis of intestinal tumor. However, homozygous knockout of ATG5, which is essential for autophagosome formation, led to early postnatal lethal in mice.Citation9 These facts prompted us to use the Apc and ATG5 double heterozygous mouse model to investigate the role of loss of ATG5 in Apc-mediated intestinal tumors.
Recent in vitro studies have showed that knockdown of ATG5 by specific small interfering RNAs (siRNAs) enhances the antitumor efficacy of lots of chemotherapeutic agents.Citation10-12 ATG5 is a potential target in adjuvant chemotherapy for cancer. Indeed, when ATG5 gene was knocked down by specific siRNAs, colon cancer cells HCT-116 and SW480 became sensitized toward propionate-induced apoptosis through activation of caspase-7 and caspase-3.Citation13 However, there is still no animal model to validate these in vitro observations in vivo.
To investigate whether deficiency for ATG5 could sensitize intestinal tumors to chemotherapies, we compared the antitumor effects of Interferon-gamma (IFN-γ) between the ApcMin/+ATG5+/+ and ApcMin/+ATG5+/- mice, as IFN-γ is a potential tumor suppressor for CRC and has been used clinically as an efficient adjuvant to the chemotherapy of cancer.Citation14 For CRC, the potential usefulness of Interferons is less evident. Nevertheless, there are good reasons to assume that type II Interferons may provide better prospects in this respect. IFN-γ, the only member of type II Interferons, contributes to cancer surveillance and suppression and is thus an important tumor suppressor.Citation15,16 It was reported that IFN-γ displayed a marked inhibitory effect on colonic cancer cells by inducing Fas.Citation17,18 Furthermore, low serum levels of IFN-γ in cancer patients are very common and may be relate to the failure of anti-tumor immunity in colon cancer patients.Citation19 Thus, IFN-γ is considered a potential therapeutic option in colorectal cancer as this cytokine exhibits important anti-oncogenic properties. Translation in into clinical application, however, awaits proof that IFN-γ treatment is effective in relevant in vivo models of this disease.
In this study, we generated ApcMin/+ATG5+/− mouse model by breeding ApcMin/+ mice with ATG5+/− mice and assessed the antitumor effects of IFN-γ in ApcMin/+ATG5+/+ and ApcMin/+ATG5+/− mice. We found that heterozygous deletion of ATG5 in ApcMin/+ mice increased the number and size of intestinal adenomas as compared with those in ApcMin/+ATG5+/+ mice. Furthermore, heterozygous deletion of ATG5 enhanced the antitumor efficacies of IFN-γ. Surprisingly, we found that treatments of ApcMin/+ATG5+/− mice with IFN-γ were effective for preventing and treating intestinal adenomas and early treatment decreased tumor incidence rate to 16.7% and reduced the number of adenomas by 95.5%. Importantly, IFN-γ treatment did not cause any evident toxic reaction.
Results
Heterozygous deletion of ATG5 promotes tumor growth in ApcMin/+ mice
Though it is widely accepted that autophagy protects cells from stressful conditions, the functions of autophagy during the multiple processes of tumorigenesis is still under debate, probably genetic background dependent. To examine the role of loss of ATG5 in the context of Apc-mediated intestinal tumor progression, we crossed ApcMin/+ mice with ATG5+/− mice to generate ATG5+/−, ApcMin/+ATG5+/+ and ApcMin/+ATG5+/− littermates. In order to control the variation of genetic background, littermates were used for this study. The ATG5+/− littermates never developed intestinal tumors until sacrificed. In both ApcMin/+ATG5+/+ and ApcMin/+ATG5+/- mice, multiple adenomas in small intestine were detectable, whereas few adenomas were found in colon and rectum.
Firstly, we detected the protein levels of ATG5 in adenomas of both ApcMin/+ATG5+/+ and ApcMin/+ATG5+/- mice. As shown in , heterozygous deletion of ATG5 inhibited the expression of ATG5 (P < 0.01). Upon induction of autophagy, the microtubule-associated protein 1 light chain 3 (LC3-I) is processed to LC3-II, which is essential for the initiation of autophagosome formation.Citation20,21 Accordingly, an increase of LC3-II is a marker for autophagy.Citation22 In SDS-PAGE, LC3-I and LC3-II was detected at a molecular mass around 16 kDa and 14 kDa, respectively.Citation23 To test if ATG5 deficiency results in the inhibition of autophagy in adenomas of ApcMin/+ mice, we detected the levels of LC3-I and LC-II by immunoblotting. We revealed that heterozygous loss of ATG5 did not affect the expression of LC3. Therefore, the effects induced by ATG5 loss might be independent of autophagy.
Figure 1. Heterozygous deletion of ATG5 promotes tumor growth in ApcMin/+ mice. (A) ATG5 and LC-3 protein levels in adenomas were determined by Western blotting with β-actin as a loading control. (B) Representative images of intestinal tracts from ApcMin/+ATG5+/+ and ApcMin/+ATG5+/- mice at age 6 months. (C) Micrographs of hematoxylin and eosin stained colonic tumor sections. Histological analysis showed that dysplasia, loss of nuclear polarity and complex crypt outlines with cribriform glands in adenomas of both ApcMin/+ATG5+/+ mice (a and a‘) and ApcMin/+ATG5+/− mice (b and b’). Heterozygous deletion of ATG5 had no significant effect on the malignant progression of Apc-mediated intestinal tumor. Images a‘ and b’ (×200 magnification) are high magnification of insets in a and b (×40 magnification), respectively.
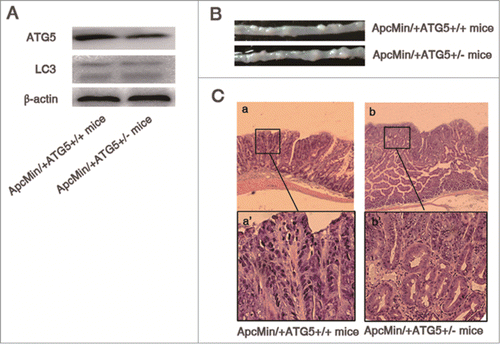
As shown in , at 4.5 months of age, heterozygous deletion of ATG5 does not significantly impact on the number and size of adenomas of ApcMin/+ mice (all P > 0.05). At the age of 6 months, compared with ApcMin/+ATG5+/+ mice, the size of intestinal tumors were increased in mice bearing Apc and ATG5 mutations (P < 0.05, and ). While approximately 23.4 tumors were found in the intestine of ApcMin/+ATG5+/+ mice, on average, 33.4 tumors were observed in the intestine of ApcMin/+ATG5+/- littermates. The difference of the number of adenomas between ApcMin/+ATG5+/+ and ApcMin/+ATG5+/− mice also reached statistical significance at the age of 6 months (P < 0.05).
Table 1. Effect of IFN-γ on the incidence, number and size of ApcMin/+ATG5+/+ and ApcMin/+ATG5+/- intestinal adenomas
Histological analysis showed that dysplasia, loss of nuclear polarity and complex crypt outlines with cribriform glands were seen in adenomas of both ApcMin/+ATG5+/+ (, a and a′) and ApcMin/+ATG5+/− mice (, b and b’). In contrast to the ApcMin/+ATG5+/+ mice, adenomas in ApcMin/+ATG5+/− mice showed no difference in histopathology according to previous reports.Citation24 These results suggest that ATG5 plays a significant role, but is insufficient to cause malignant progression of Apc-mediated intestinal tumor.
Heterozygous deletion of ATG5 enhances the antitumor efficacy of IFN-γ
Recent in vitro studies have shown that knockdown of ATG5 by specific siRNAs enhances the antitumor efficacy of lots of chemotherapeutic agents.Citation10-13 Herein, we validated these in vitro observations using animal models. To investigate whether deficiency for ATG5 could sensitize tumor to chemotherapies, we compared the antitumor effects of IFN-γ between the ApcMin/+ATG5+/+ and ApcMin/+ATG5+/− mice. For early treatment, administration of IFN-γ began at age 1.5 months and continued until 4.5 months of age. Late treatment did not begin until age 3 months, when adenomas already developed, and continued until 6 months of age. There were very few adenomas in control mice at age 1.5 months (when early treatment began). As shown in , early treatment of ApcMin/+ATG5+/+ mice with IFN-γ decreased the number and size of adenomas, compared with treatment with saline (all P < 0.05). Following early treatment with IFN-γ, the tumor incidence rate for ApcMin/+ATG5+/+ mice was 75% and the number of adenomas was decreased by 43.3%, compared with treatment with vehicle. Interestingly, early treatment of ApcMin/+ATG5+/− mice with IFN-γ reduced tumor incidence rate to 16.7% and decreased the number of adenomas by 95.5%. Moreover, early treatment of ApcMin/+ATG5+/− mice with IFN-γ resulted in more powerful suppressive effects on the size of intestinal adenomas and led to disappearance of macroscopic tumor nodules (). Further histological analysis of intestinal adenomas demonstrated that following early treatment with vehicle, ApcMin/+ATG5+/− mice formed adenomas with severe dysplasia (, a and a‘). However, following early treatment with IFN-γ, most of the polypoid area in intestinal mucosa of ApcMin/+ATG5+/− mice showed hyperplastic morphology without obvious dysplasia (, b and b’).
Figure 2. IFN-γ treatments effectively prevent intestinal adenomas in ApcMin/+ATG5+/− mice. (A) Representative images of intestinal tracts from ATG5 deficient ApcMin/+ mice after early treatment with IFN-γ. (B) Micrographs of hematoxylin and eosin stained colonic tumor sections. Histological analysis of intestinal adenomas in ApcMin/+ATG5+/− mice receiving vehicle revealed well-formed adenomas with severe dysplasia (a and a‘). Following early treatment with IFN-γ, adenomas of ApcMin/+ATG5+/− mice mostly exhibited hyperplastic morphology without obvious dysplasia in the polypoid area of mucosa of intestine (b and b’). Images a‘ and b’ (×200 magnification) are high magnification of insets in a and b (×40 magnification), respectively.
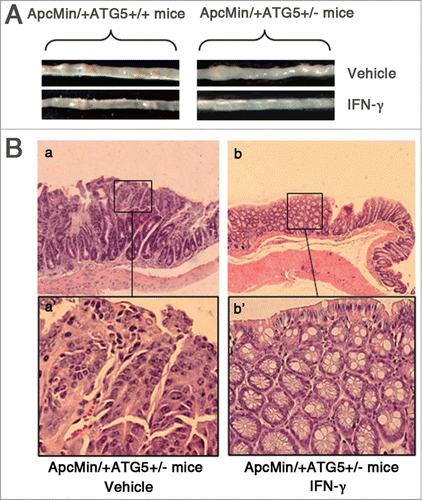
Moreover, late treatment of ApcMin/+ATG5+/+ mice with IFN-γ significantly decreased the number of adenomas by 15.4%, compared with treatment with vehicle (P < 0.05) (). In contrast, late treatment of ApcMin/+ATG5+/− mice with IFN-γ significantly decreased the number of adenomas by 69.8% and also decreased the size of adenomas (all P < 0.001). These results indicate that heterozygous deletion of ATG5 enhances the efficacy of IFN-γ on the suppression of adenomas in ApcMin/+ mice. Thus, IFN-γ is a potent preventive and therapeutic agent for intestinal adenomas in ATG5-deficient ApcMin/+ mice.
IFN-γ treatments do not cause any significant toxic reaction in ApcMin/+ATG5+/+ and ApcMin/+ATG5+/− mice
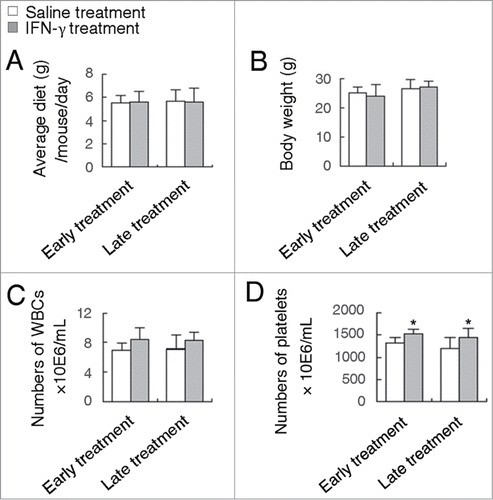
Among the chemotherapeutic agents used in the adjuvant treatment of CRC in the past 30 years, 5-fluorouracil (5-FU) and 5-FU-based chemotherapy have been the mainstream.Citation25 However, 5-FU resistance and its side effects during the course of treatment are very common, thus new drugs for CRC prevention and treatment need to be explored.Citation26,27 It is well documented that 5-FU exhibits a high toxicity such as severe gastrointestinal toxicity and myelosuppression.Citation28 It was reported that 5-FU-treated ApcMin/+ mice produced a potent antitumor effect, but resulted in a significant decrease in body weight and food consumption.Citation29 In contrast to 5-FU, treatment of ApcMin/+ mice with IFN-γ did not cause any apparent decrease in body weight and food consumption (all P > 0.05) (). In addition, except for gastrointestinal toxicity, 5-FU suppressed bone marrow hematopoiesis including a significant decrease in the number of white blood cells (WBCs) in the peripheral blood.Citation29 However, surprisingly, administration of IFN-γ did not induce the decrease of the number of WBCs in the peripheral blood of ApcMin/+ mice (). Moreover, administration of IFN-γ resulted in an improvement of bone marrow hematopoiesis as demonstrated by the increase of PLT numbers (). Similar results were observed for treatment of ApcMin/+ATG5+/− mice with IFN-γ (Data not shown). These results suggest that IFN-γ is very potential for the treatment of CRC.
Figure 3. IFN-γ treatments do not cause any significant toxic reaction. Effects of early or late treatment of ApcMin/+ATG5+/+ mice with IFN-γ on body weight (A), average diet (B) and the number of WBCs (C) and platelets (D). *P < 0.05.
Heterozygous deletion of ATG5 promotes cell proliferation, activates EGFR/Erk1/2 and Wnt/β-catenin pathways and enhances the effects of IFN-γ-dependent suppression of these 2 pathways
Firstly, we investigated the proliferation and apoptosis-related protein levels in intestinal adenomas of ApcMin/+ATG5+/+ and ApcMin/+ATG5+/− mice. We found that heterozygous loss of ATG5 significantly increased the protein level of proliferating cell nuclear antigen (PCNA), a cell proliferation marker (). However, western blotting revealed that heterozygous loss of ATG5 had no significant effect on the levels of apoptosis-related protein including bax, bcl-2 and PARP (). Therefore, heterozygous loss of ATG5 promotes tumor cell proliferation, but has no significant effect on apoptosis.
Figure 4. Heterozygous deletion of ATG5 promotes cell proliferation, activates Wnt/β-catenin and EGFR/ERK1/2 pathways and enhances the effects of IFN-γ-dependent suppression of these 2 signaling pathways. Western blotting assay showed the protein levels of PCNA (A), apoptosis-related protein including bax, bcl-2 and PARP (B), Wnt signaling-related protein including nuclear β-catenin, cyclin D1 and Survivin (C) and EGFR/Erk1/2 signaling-related protein including EGFR, phospho-EGFR and phospho-Erk1/2 (D). The protein β-actin served as a loading control and for the study involving phospho-Erk1/2, total Erk1/2 served as a control. The data presented is representative of 3 experiments. Significant differences are not shown.
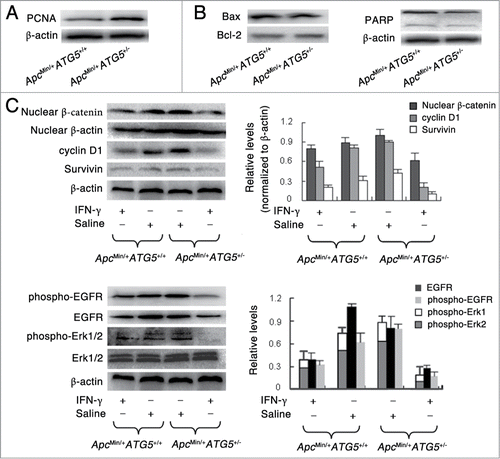
Apc is part of the β-catenin destruction complex and thus loss of Apc protein leads to an accumulation of free β-catenin and nuclear translocation.Citation30 In the nucleus, β-catenin acts as a transcription factor and induces the up-regulation of genes like the proto-oncogene cyclin D1, c-myc and Survivin, leading to cell proliferation. Deregulation of the β-catenin signaling pathway promotes the tumorigenesis in ApcMin/+ mice. To explore the potential association between cell proliferation and Wnt/β–catenin signaling, we studied the protein levels of Wnt/β-catenin signaling molecules in adenomas of ApcMin/+ATG5+/+ and ApcMin/+ATG5+/− mice. Considering most of the adenomas were located in the small intestine, protein levels in adenomas of small intestine were investigated. As shown in , adenomas of ApcMin/+ATG5+/− mice exhibited increased nuclear levels of β-catenin as compared with those of ApcMin/+ATG5+/+ mice, suggesting that heterozygous deletion of ATG5 increased the nuclear translocation of β-catenin and thus might activate Wnt/β-catenin signaling. Moreover, heterozygous deletion of ATG5 also increased the downstream protein levels of cyclin D1 and Survivin. Besides, early treatment with IFN-γ suppressed the expression levels of nuclear β-catenin, cyclin D1 and Survivin by 11.1%, 37.5% and 33.3% in adenomas of ApcMin/+ATG5+/+ mice, respectively. In contrast, early treatment with IFN-γ suppressed the expressions of nuclear β-catenin, cyclin D1 and Survivin by 40%, 77.8% and 75% in adenomas of ApcMin/+ATG5+/− mice, respectively. Hence, heterozygous deletion of ATG5 enhanced the inhibitory effect of IFN-γ on the protein levels of Wnt/β-catenin signaling molecules.
Epidermal Growth Factor Receptor (EGFR) signaling plays an important role in maintaining the cell proliferation of epidermis by activating the extracellular signal-regulated kinase (ERK) pathway.Citation31 To explore a potential mechanism for the observation, we studied the effects of heterozygous deletion of ATG5 on EGFR/ERK1/2 pathway. As shown in , molecular analyses of the adenomas suggested that compared with ApcMin/+ATG5+/+ mice, heterozygous deletion of ATG5 in ApcMin/+ mice decreased the level of EGFR and increased the phosphorylation of EGFR. Furthermore, the level of phosphorylated-Erk1/2 was significantly increased (all P < 0.001). Therefore, heterozygous deletion of ATG5 increased the level of phosphorylated-EGFR, and thus stimulated the downstream ERK1/2 activation and cell proliferation. Besides, following early treatment of IFN-γ, the levels of EGFR, phosphorylated-EGFR and phosphorylated-ERK1/2 were decreased by 61.5%, 42% and 42.1% in adenomas of ApcMin/+ATG5+/+ mice, respectively. In contrast, levels of EGFR, phosphorylated-EGFR and phosphorylated-ERK1/2 were decreased by 63.3%, 75% and 77.4% in adenomas of ApcMin/+ATG5+/− mice after early treatment of IFN-γ, respectively. Hence, heterozygous deletion of ATG5 enhanced the effects of IFN-γ on the suppression of EGFR-ERK1/2 signaling pathway.
Discussion
ATG5 gene encodes ATG5 protein in human and is necessary for autophagy due to its role in autophagosome elongation.Citation20 ATG5 is activated by ATG7 and forms a complex with ATG12 and ATG16L1. Nishiumi S et al.Citation32 have demonstrated that conditional deletion of Atg7 in the intestinal epithelium did not alter the number of intestinal polyps in ApcMin/+ mice. Moreover, Nishida Y et al.Citation33 discovered the Atg5/Atg7-independent alternative autophagy. In our studies, we found that heterozygous deletion of ATG5 in ApcMin/+ mice down-regulated the protein level of ATG5 in adenomas, but had no effect on levels of LC3-I and LC3-II. We also found that heterozygous deletion of ATG5 promoted intestinal tumor growth and sensitized the tumor to chemotherapy. Therefore, the effects induced by heterozygous loss of ATG5 might be independent of autophagy, or alternatively, that loss of ATG5 in another tissue could be responsible for the phenotype observed.
Several in vitro studies have showed that knockdown of ATG5 by specific siRNAs could enhance the antitumor efficacy of chemotherapeutic agents. It has been reported that ablation of ATG5 in gastric cancer cells using siRNA augmented quercetin-induced cell death.Citation12 Similarly, after genetically knockdown of ATG5, the colon cancer cells became sensitized toward propionate-induced apoptosis.Citation13 Moreover, silencing ATG5 enhanced resveratrol-induced caspase activation in breast and colon cancer cells.Citation11 In myeloid leukemia cells, cell death induced by DNR was greatly enhanced by using siRNA targeting ATG5.Citation10 Consistent with these results from in vitro studies, our results demonstrated that heterozygous deletion of ATG5 could enhance the antitumor efficacy of IFN-γ in vivo. Therefore, ATG5 might play an important role in promoting cell adaptation and survival against antitumor therapy. Wnt/β-catenin signaling is vital to the development and progression of CRC. Targeting Wnt/β-catenin signaling and blocking cytoplasmic accumulation of β-catenin and translocation to the nucleus has been shown to decrease CRC incidence and development.Citation34,35 We revealed that ATG5 deficiency combined with IFN-γ treatment significantly inhibited Wnt/β-catenin and EGFR/ERK1/2 signaling pathways. Thus suppression of the Wnt/β-catenin and EGFR/ERK1/2 signaling pathways is one of the underlying mechanisms by which heterozygous depletion of ATG5 sensitizes adenoma to IFN-γ.
Furthermore, treatments of ApcMin/+ATG5+/− mice with IFN-γ were very effective for preventing intestinal adenomas. Following early treatment of ApcMin/+ATG5+/− mice with IFN-γ, few tumors were observed by gross examination of the intestinal mucosa and microscopic examination demonstrated that residual tumors had a “flattened” and regressed appearance. If a drug is to be considered for prevention and treatment of cancer, the risk of toxicity is an important consideration. In particular, cancer preventative agents should have low systemic toxicity and minimal adverse effects on healthy tissue.Citation36 To our surprise, IFN-γ did not cause any evident side effects such as the reduction of body weight, food consumption and the number of blood cells. Thus, IFN-γ is a very promising agent for the prevention and treatment of CRC in combination of ATG5 deficiency or ATG5-targeted inhibition.
In summary, our findings indicated that heterozygous deletion of ATG5 promoted intestinal tumor growth in ApcMin/+ mice, which might be independent of autophagy. Loss of ATG5 in another tissue could be responsible for the phenotype observed. Knockdown of ATG5 may be advantageous in a combination therapy setting to sensitize CRC to chemotherapeutic agents. Importantly, IFN-γ exerted powerful suppressive effects on adenomas of ApcMin/+ATG5+/− mice and did not cause any evident side effects. Our findings may provide novel strategies such as combination of IFN-γ and ATG5 deficiency or ATG5-targeted inhibition for the prevention and treatment of CRC.
Materials and Methods
Animals and genotyping
C57BL/6J mice were purchased from Vital River Laboratory. ApcMin/+ of C57BL/6 background were obtained from the Jackson Laboratory. Heterozygous ATG5-deficient mice (RBRC; 02231) were obtained from RIKEN BioResource Center. ApcMin/+ males were bred with ATG5+/− females (>10 generations on C57BL/6 background) to generate ApcMin/+ATG5+/+ and ApcMin/+ATG5+/− littermates. All the animals were bred and maintained in a pathogen-free animal facility at School of Pharmaceutical Sciences in Shandong University. Genotyping was conducted by PCR following a protocol provided by Jackson Laboratory. Southern blotting was done as described previously.Citation37 All efforts were made to minimize suffering. Experimental protocols and care of the animals were reviewed and approved by the Institutional Animal Care and Use Committee at School of Pharmaceutical Sciences in Shandong University. The permit number was SYXK (LU) 20100418.
Drug treatment protocol
Treatments started either at age 1.5 months or, in the other group, after a delay to age 3 months, when adenomas were already well established. IFN-γ (Prospec; cyt-358) (80 × 104 U/kg) was administered by abdomen injection using a 3-month schedule with 2 treatment months and one intermittent rest month.Citation38,39 A volume of 0.2 ml of saline was injected per mouse from the control group. Mice were sacrificed at age 4.5 month after early treatment and at age 6 month after late treatment.
Food intake and body weight
The mice were monitored daily for any signs of illness. During the feeding period, body weight and non-consumed food were weighed every 3 days. The remaining food was collected and weighed. The daily food intake was calculated by subtracting the remaining food from the amount of diet provided 3 days ago.
Blood cell analysis
Animals were sacrificed by cardiac puncture after anesthesia with methoxyflurane, and blood was collected. A complete blood count was performed immediately by using automatic hematology analyzer (Beckman; Counter LH750).
Tumor Counts
The small and large intestines were removed immediately from mice after sacrifice and flushed free of debris with PBS. Tumors were counted and measured using a dissecting microscope with a micrometer by a single observer who was blinded to the treatments received by the animals. Intestinal tissues and tumors were stored at −70°C for the biochemical analysis. The remaining intestinal tissues and tumors were fixed in 10% formalin.
Histopathological analysis
Intestinal segments were fixed in 10% buffered formalin and embedded in paraffin. About 5 μm paraffin sections were used for hematoxylin and eosin staining. Stained sections were viewed and evaluated by a senior pathologist without knowledge of the genotypes. Adenomas were characterized by the increase in size, the reduction in goblet cell number and the loss of mucosal architecture.
Preparation of tissue extracts
Mice were sacrificed at age 4.5 month after early treatment. Adenomas appearing small intestinal tissues of mice were acquired. Adenomas of size >2 mm were pooled within each animal for the preparation of total protein extracts. This experiment was performed in at least triplicates for 3 times. Adenomas were rinsed with PBS, lysed in ice-cold RIPA buffer at 4°C for 30 min, and centrifuged at 13,000 rpm for 15 minutes at 4°C. The supernatants were retained as a total protein extract. The nuclear proteins were extracted as described previously.Citation40 The tumors were dispersed mechanically and placed into in ice-cold lysis buffer (10 mM Tris-HCl, 1.5 mM MgClCitation2, 10 mM KCl, 0.1 mM EDTA, 1 mM Dithiothreito (DTT), 2% NP-40, 50 mM sodium fluoride, 5 mM sodium orthovanadate and protease inhibitor cocktail). After centrifugation at 2,500 rpm for 5 minutes at 4°C, the supernatants were retained as a cytoplasmic fraction. The pellets were washed with ice-cold lysis buffer and mixed with hypertonic nuclear extract buffer (20 mM Tris-HCl, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 25% glycerol, 50 mM sodium fluoride and protease inhibitor cocktail) on ice for 10 min. After centrifugation at 140,000 rpm for 10 min at 4°C, the supernatants were retained as a nuclear fraction. All these total and nuclear proteins were stored at −80°C.
Western blotting
Tumors were cut into small pieces, grinded in 300 μl RIPA lysis buffer [20 mmol/L Tris-HCl, pH 7.5; 150 mmol/L NaCl; 1 mmol/L Na2EDTA; 1 mmol/L EGTA; 1% Triton; 2.5 mmol/L sodium pyrophosphate; 1 mmol/L β-glycerophosphate; 1 mmol/L Na3VO4; 1 mg/mL leupeptin; 1 mmol/L phenylmethylsulfonylfluoride (PMSF); and 1 mmol/L PMSF] on dry ice and lysed on ice for 30 min. Cell lysates were centrifuged at 15,000 rpm for 15 min at 4°C, and the supernatants were stored at −80°C. Protein quantification was performed with the BCA protein assay (Pierce; 23225). Equal amounts of protein (30 μg) from each sample were denatured in SDS sample buffer and separated on SDS-PAGE gels (Invitrogen). Proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (IPVH00010; Millipore, USA) in buffer containing 25 mM Tris, 192 mM glycine, and 20% methanol. Immunoblotting was done using the following antibodies: β-catenin Phospho (pY1068) (1727-1), EGFR (1902-1), EGFR Phospho (1727-1) (all from Epitomics), Phospho-p44/42 MAPK (p-Erk1/2) (9101), p44/42 MAPK (Erk1/2) (9102), β-catenin (9562), cyclin D1 (2922), poly(ADP-ribose polymerase) (PARP) (9532), Bcl-2 (2872), Bax (2772), Survivin (2808) (all from Cell Signaling), PCNA (sc-2586) (from Santa Cruz), β-actin (ab52614), ATG5 (#3167-1) (both from Abcam), LC-3 (L8918)(from Sigma). The PVDF membranes were washed and then incubated with horseradish peroxidase-conjugated secondary antibody for 1 hour at room temperature. Signals were developed with the enhanced chemiluminescence detection system (Millipore, USA) and quantified by densitometry using ChemiDoc XRS+ image analyzer (Bio-Rad, USA). Protein bands were quantified with ImageJ software and results were adjusted with β-actin as loading control.Citation41 Triplicate experiments with triplicate samples were performed.
Statistical analysis
Statistical analysis was performed by Student's t test for pair samples or 2-way using the SPSS/Win13.0 software (SPSS, Inc., Chicago, Illinois). Results are presented as mean ± standard deviation (SD). A value of P < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This research was made possible by the Grant 91229113 from Natural Science Foundation of China for Research Resources.
References
- Jacoby RF, Seibert K, Cole CE, Kelloff G, Lubet RA. Cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the Min mouse model of adenomatous polyposis. Cancer Res 2000; 60:5040-4; PMID:11016626
- Chau I, Cunningham D. Adjuvant therapy in colon cancer–what, when and how? Ann Oncol 2006; 17:1347-59; PMID:16524974; http://dx.doi.org/10.1093/annonc/mdl029
- Huerta S, Goulet EJ, Livingston EH. Colon cancer and apoptosis. Am J Surg 2006; 191:517-26; PMID:16531147; http://dx.doi.org/10.1016/j.amjsurg.2005.11.009
- Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer 2001; 1:55-67; PMID:11900252; http://dx.doi.org/10.1038/35094067
- Ishikawa TO, Tamai Y, Li Q, Oshima M, Taketo MM. Requirement for tumor suppressor Apc in the morphogenesis of anterior and ventral mouse embryo. Dev Biol 2003; 253:230-46; PMID:12645927; http://dx.doi.org/10.1016/S0012-1606(02)00020-9
- Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol 2005; 6:505-10; PMID:15928714; http://dx.doi.org/10.1038/nrm1666
- Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 2004; 16:663-9; PMID:15530778; http://dx.doi.org/10.1016/j.ceb.2004.09.011
- An CH, Kim MS, Yoo NJ, Park SW, Lee SH. Mutational and expressional analyses of ATG5, an autophagy-related gene, in gastrointestinal cancers. Pathol Res Pract 2011; 207:433-7; PMID:21664058; http://dx.doi.org/10.1016/j.prp.2011.05.002
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032-6; PMID:15525940; http://dx.doi.org/10.1038/nature03029
- Han W, Sun J, Feng L, Wang K, Li D, Pan Q, Chen Y, Jin W, Wang X, Pan H, et al. Autophagy inhibition enhances daunorubicin-induced apoptosis in K562 cells. PLoS One 2011; 6:e28491; PMID:22164300; http://dx.doi.org/10.1371/journal.pone.0028491
- Prabhu V, Srivastava P, Yadav N, Amadori M, Schneider A, Seshadri A, Pitarresi J, Scott R, Zhang H, Koochekpour S, et al. Resveratrol depletes mitochondrial DNA and inhibition of autophagy enhances resveratrol-induced caspase activation. Mitochondrion 2013; 13:493-9; PMID:23088850; http://dx.doi.org/10.1016/j.mito.2012.10.010
- Wang K, Liu R, Li J, Mao J, Lei Y, Wu J, Zeng J, Zhang T, Wu H, Chen L, et al. Quercetin induces protective autophagy in gastric cancer cells: involvement of Akt-mTOR- and hypoxia-induced factor 1α-mediated signaling. Autophagy 2011; 7:966-78; PMID:21610320; http://dx.doi.org/10.4161/auto.7.9.15863
- Tang Y, Chen Y, Jiang H, Nie D. Short-chain fatty acids induced autophagy serves as an adaptive strategy for retarding mitochondria-mediated apoptotic cell death. Cell Death Differ 2011; 18:602-18; PMID:20930850; http://dx.doi.org/10.1038/cdd.2010.117
- Borden EC, Lindner D, Dreicer R, Hussein M, Peereboom D. Second-generation interferons for cancer: clinical targets. Semin Cancer Biol 2000; 10:125-44; PMID:10936063; http://dx.doi.org/10.1006/scbi.2000.0315
- Billiau A, Heremans H, Vermeire K, Matthys P. Immunomodulatory properties of interferon-gamma. Ann N Y Acad Sci 1998; 856:22-32; PMID:9917861; http://dx.doi.org/10.1111/j.1749-6632.1998.tb08309.x
- Sawai N, Kita M, Kodama T, Tanahashi T, Yamaoka Y, Tagawa Y, Iwakura Y, Imanishi J. Role of gamma interferon in Helicobacter pylori-induced gastric inflammatory responses in a mouse model. Infect Immun 1999; 67:279-85; PMID:9864227
- Kudinov Y, Wiseman CL, Kharazi AI. Phorbol myristate acetate and Bryostatin 1 rescue IFN-gamma inducibility of MHC class II molecules in LS1034 colorectal carcinoma cell line. Cancer Cell Int 2003; 3:4; PMID:12787470
- von Reyher U, Sträter J, Kittstein W, Gschwendt M, Krammer PH, Möller P. Colon carcinoma cells use different mechanisms to escape CD95-mediated apoptosis. Cancer Res 1998; 58:526-34; PMID:9458101
- Favrot MC, Philip T, Coze C, Lenoir G. Immunodeficiency and cancer. Pediatrie 1989; 44:11-8.
- Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S, Klionsky DJ. In search of an “autophagomometer”. Autophagy 2009; 5:585-9; PMID:19411822; http://dx.doi.org/10.4161/auto.5.5.8823
- Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 2004; 36:2503-18; PMID:15325588; http://dx.doi.org/10.1016/j.biocel.2004.05.009
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/10.1016/j.cell.2010.01.028
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID:17611390; http://dx.doi.org/10.4161/auto.4600
- Takaku K, Wrana JL, Robertson EJ, Taketo MM. No effects of Smad2 (madh2) null mutation on malignant progression of intestinal polyps in Apc (delta716) knockout mice. Cancer Res 2002; 62:4558-61; PMID:12183405
- Nordman IC, Iyer S, Joshua AM, Clarke SJ. Advance in the adjuvant treatment of colorectal cancer. ANZ J Surg 2006; 76:373-80; PMID:16768699; http://dx.doi.org/10.1111/j.1445-2197.2006.03726.x
- Wils J, O'Dwyer P, Labianca R. Adjuvant treatment of colorectal cancer at the turn of the century: European and US perspectives. Ann Oncol 2001; 12:13-22; PMID:11249040; http://dx.doi.org/10.1023/A:1008-357725209
- Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, Jandik P, Iveson T, Carmichael J, Alakl M, et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet 2000; 355:1041-7; PMID:10744089; http://dx.doi.org/10.1016/S0140-6736(00)02034-1
- Macdonald JS. Toxicity of 5-fluorouracil. Oncology (Williston Park) 1999; 13:33-4; PMID:10442356
- Cao Z, Zhang Z, Huang Z, Wang R, Yang A, Liao L, DU J. Antitumor and immunomodulatory effects of low-dose 5-FU on hepatoma 22 tumor-bearing mice. Oncol Lett 2014; 7:1260-4; PMID:24660037
- Ormanns S, Neumann J, Horst D, Kirchner T, Jung A. WNT signaling and distant metastasis in colon cancer through transcriptional activity of nuclear β-Catenin depend on active PI3K signaling. Oncotarget 2014; 5:2999-3011; PMID:24930890
- Ge X, Shi Z, Yu N, Jiao Y, Jin L, Zhang J. The role of EGFR/ERK/ELK-1 MAP kinase pathway in the underlying damage to diabetic rat skin. Indian J Dermatol 2013; 58:101-6; PMID:23716797; http://dx.doi.org/10.4103/0019-5154.108035
- Nishiumi S, Fujishima Y, Inoue J, Masuda A, Azuma T, Yoshida M. Autophagy in the intestinal epithelium is not involved in the pathogenesis of intestinal tumors. Biochem Biophys Res Commun 2012; 421:768-72; PMID:22546555; http://dx.doi.org/10.1016/j.bbrc.2012.04.081
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461:654-8; PMID:19794493; http://dx.doi.org/10.1038/nature08455
- Burn J, Mathers J, Bishop DT. Genetics, inheritance and strategies for prevention in populations at high risk of colorectal cancer (CRC). Recent Results Cancer Res 2013; 191:157-83; PMID:22893205; http://dx.doi.org/10.1007/978-3-642-30331-9_9
- Esufali S, Bapat B. Cross-talk between Rac1 GTPase and dysregulated Wnt signaling pathway leads to cellular redistribution of beta-catenin and TCF/LEF-mediated transcriptional activation. Oncogene 2004; 23:8260-71; PMID:15377999; http://dx.doi.org/10.1038/sj.onc.1208007
- Li WW, Li VW, Hutnik M, Chiou AS. Tumor angiogenesis as a target for dietary cancer prevention. J Oncol 2012; 2012:879623; PMID:21977033; http://dx.doi.org/10.1155/2012/879623
- Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-γ genes. Science 1993; 259:1739-42; PMID:8456300; http://dx.doi.org/10.1126/science.8456300
- Miyahara K, Nishimura H, Hamaguchi K, Ookura N, Hamai J, Ushijima H, Yakushiji M, Kato T. Direct antitumor effect of recombinant human IFN-gamma evaluated in ovarian pseudomyxoma peritoneally transplanted into nude mice. Gan To Kagaku Ryoho 1987; 14:411-5; PMID:3101605
- Rehg JE. Effect of interferon-gamma in experimental Cryptosporidium parvum infection. J Infect Dis 1996; 174:229-32; PMID:8656002; http://dx.doi.org/10.1093/infdis/174.1.229
- Kang YJ, Park HJ, Chung HJ, Min HY, Park EJ, Lee MA, Shin Y, Lee SK. Wnt/β-catenin signaling mediates the antitumor activity of magnolol in colorectal cancer cells. Mol Pharma col 2012; 82:168-77; PMID:22550094; http://dx.doi.org/10.1124/mol.112.078535
- Park EJ, Shin JW, Seo YS, Kim DW, Hong SY, Park WI, Kang BM. Gonadotropin-releasing hormone-agonist induces apoptosis of human granulosa-luteal cells via caspase-8, -9 and -3, and poly-(ADP-ribose)-polymerase cleavage. Biosci Trends 2011; 5:120-8; PMID:21788697; http://dx.doi.org/10.5582/bst.2011.v5.3.120