
Abstract
Although HER2 targeted therapies have substantially improved outcomes in HER2 overexpressing (HER2+) breast cancer, resistance to these therapies remains a clinical challenge. To better understand the mechanisms of resistance to lapatinib, a HER2 and EGFR dual kinase inhibitor, we treated HER2+ breast cancer cells with lapatinib for an extended period to generate a lapatinib-resistant (LapR) cell line model and examined cancer-promoting signaling activation in LapR cells. We found that LapR cells possess enhanced mTOR activation, which was independent of PI3K and other known mTOR activators. Lapatinib resistance could be reversed by mTOR kinase inhibition. Intriguingly, LapR cells had constitutive cytosolic cytochrome C, indicating that LapR cells suppress lapatinib-induced apoptosis downstream of cytochrome C release from mitochondria into the cytosol rather than by preventing its release into the cytosol. Consistent with this notion, LapR cells possessed increased levels of 2 of the inhibitors of apoptosis (IAPs), survivin and c-IAP-2, which are reported to block caspase activation downstream of cytosolic cytochrome C release. Further, treatment with the mTOR kinase inhibitor AZD8055 or the Hsp90 inhibitor 17-AAG reversed expression of IAPs and overcame lapatinib resistance in LapR cells. Together, these data suggest that suppression of apoptosis downstream of cytosolic cytochrome C release, possibly through increased expression of IAPs or other caspase-suppressing proteins, may promote lapatinib resistance. Further, PI3K is thought to be the main driver of lapatinib resistance, but our findings indicate that PI3K inhibitors may be ineffective in some lapatinib-resistant HER2+ breast cancers with PI3K-independent activation of mTOR kinase, which may instead benefit from mTOR or Hsp90 inhibitors.
Introduction
The HER2 receptor tyrosine kinase is amplified and/overexpressed (HER2+) in 20–25% of breast cancersCitation1 and treatment with HER2 targeted therapy is frequently successful.Citation2 However, a substantial proportion of patients develop acquired resistance to HER2 targeted therapy after an initial response, while others are intrinsically resistant.Citation3,4 Enhanced understanding of resistance to HER2 targeted therapies, such as the monoclonal antibody trastuzumab and the HER2 kinase inhibitor lapatinib, may guide development of additional therapies for patients with treatment-refractory HER2+ breast cancer.Citation5-9
Many reported lapatinib resistance mechanisms involve PI3K activation, including mutational activation of PI3K,Citation10 PTEN loss,Citation11 and activation of alternative receptor tyrosine kinases.Citation3,12 This suggests the use of PI3K inhibitors in lapatinib-resistant breast cancer. Intriguingly, PI3K-independent mTOR activation has also been suggested to promote lapatinib resistance.Citation13 HER2+ breast cancers with PI3K-independent mTOR activation are unlikely to respond to inhibitors of PI3K, the most frequent signaling node implicated in resistance to HER2 targeted therapy, and thus may require a different treatment strategy.
Here we generated an acquired laptinib resistance breast cancer cell model and found that PI3K-independent mTOR activation indeed leads to lapatinib resistance. Additionally, we found that breast cancer cells with this lapatinib resistance mechanism are resistant to PI3K inhibition. Further, we found that mTOR activation promotes expression of the inhibitors of apoptosis (IAP) family of proteins, which was effectively reversed by the mTOR kinase inhibitor AZD8055. We found that Hsp90 inhibition using 17-AAG could also reverse mTOR-dependent IAP expression and inhibit the growth of lapatinib-resistant breast cancer cells. These findings uncover a potential mechanism of mTOR/IAP-mediated resistance to HER2 targeted therapy and suggest 2 therapeutic options, AZD8055 and 17-AAG, for overcoming lapatinib resistance in HER2+ breast cancers with PI3K-independent mTOR activation.
Results
AU565 breast cancer cells with acquired lapatinib resistance rely on PI3K-independent mTOR activation
We developed acquired lapatinib resistance breast cancer cells by treating the AU565 HER2+ breast cancer cell line with lapatinib for longer than 6 months. AU565 lapatinib-resistant (LapR) cells were highly resistant to lapatinib compared to parental cells ().
Figure 1. Lapatinib-resistant cells possess enhanced mTOR activation and are dependent on mTOR but not PI3K. (A) AU565 parental and LapR cells were treated with indicated concentrations of lapatinib or vehicle in triplicate in 96-well plates. After four days of treatment, MTT assay was performed. O.D. denotes optical density and is indicative of cell number. (B) AU565 parental and LapR cells were treated with DMSO or 1μM lapatinib for 2 hours, followed by analysis of various phospho-RTKs and phosphoproteins using PathScan RTK Signaling Antibody Arrays (Cell Signaling). (C) AU565 parental and LapR cells were treated with 1μM lapatinib for indicate times followed by western blot analysis. (D) AU565 parental and LapR cells were treated with the PI3K inhibitor GDC-0941 or the mTOR kinase inhibitor AZD8055 for 4 days, followed by MTT assay. Error bars indicate s.e.m.
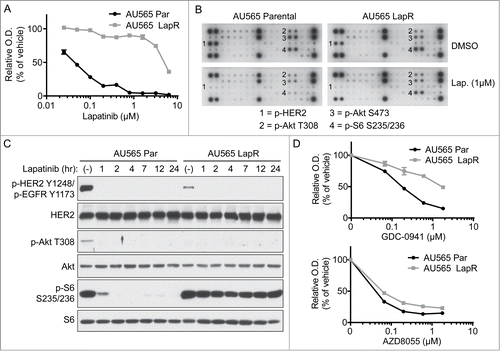
To investigate potential cancer signaling pathways promoting lapatinib resistance of the LapR cells, we analyzed AU565 parental and LapR cells for the phosphorylation of 28 RTKs and various downstream signaling molecules using Cell Signaling's PathScan RTK Signaling Antibody Arrays, which include capture antibodies recognizing total protein and detection antibodies recognizing phosphorylated proteins (). We did not detect any upregulated phospho-RTKs in AU565 LapR cells compared with parental cells. However, S6 phosphorylation remained high in LapR cells even under lapatinib treatment, whereas in parental cells S6 phosphorylation was inhibited by lapatinib.
We also validated the antibody array findings by western blot for p-S6 as well as one of its upstream activators, Akt, in AU565 parental and LapR cells (). We found that Akt and S6 phosphorylation were abolished by lapatinib treatment in parental cells. However, S6 phosphorylation in AU565 LapR cells was maintained after lapatinib treatment, in spite of decreased Akt phosphorylation, which was even turned off under basal conditions in LapR cells. S6 phosphorylation is mTOR-dependent and p-S6 is an indicator of mTORC1 activation.Citation14,15 The data suggest that mTOR may be activated in a HER2- and PI3K/Akt-independent manner in AU565 LapR cells. Indeed, S6 phosphorylation was due to mTOR activation in LapR cells as knocking down mTOR or the mTOR complex 1 (mTORC1) upstream activator RhebCitation15,16 reduced lapatinib-resistant S6 phosphorylation in a single-cell clone of AU565 LapR cells with signaling representative of pooled AU565 LapR cells (Supplemental ). Moreover, the mTORC1 inhibitor RAD001 blocked S6 phosphorylation in AU565 LapR cells (single-cell clone; Supplemental ), further demonstrating that S6 phosphorylation in AU565 LapR cells is mTOR-dependent. Phosphorylation of 4E-BP1, another important mTOR effector,Citation14,15 also remained high after lapatinib treatment in LapR cells compared to parental cells, but this mainly applied to the highest molecular weight form (hyperphosphorylated form) of 4E-BP1 and not less phosphorylated forms (Supplemental ). We used S6 phosphorylation as an indicator of mTOR activation in the remainder of this study as p-S6 was more dramatically enhanced in LapR cells than p-4E-BP1. Additionally, because heterogenous cell populations may manifest drug resistance from different mechanisms as previously reported by others Citation17,18, we used a single-cell clone of AU565 LapR cells with signaling representative of the pooled AU565 LapR cells in some of our remaining studies to minimize these potential complications.
Figure 2. mTOR activation in lapatinib-resistant cells is independent of PI3K and other canonical mTOR activators. (A) AU565 parental and LapR cells were treated with the PI3K inhibitor GDC-0941 or the Akt inhibitor MK-2206 for 4.5 hours, followed by protein gel blot. (B) AU565 parental and LapR (single-cell clone, denoted by *) cells were treated with lapatinib for 26 hours, followed by western blot. p-HER2 antibody also recognizes p-EGFR Y1173 as in .
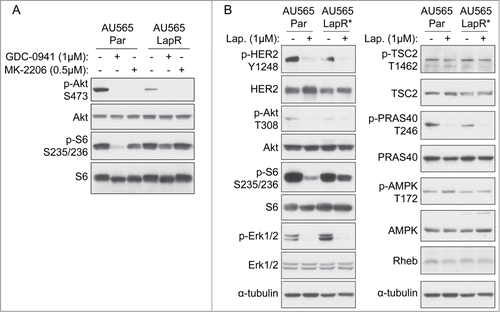
Figure 3. Inhibitors of apoptosis proteins (IAPs) are increased in lapatinib-resistant cells in an mTOR-dependent manner. (A) AU565 parental and LapR cells were treated with DMSO or 1μM lapatinib for 3 days, followed by protein gel blot. Cleaved PARP antibody recognizes PARP cleaved at aspartate 214. (B) Indicated cells were treated with lapatinib for 2 d followed by fractionation to isolate cytosol + membrane fraction using a sucrose-containing buffer; this fraction and whole-cell extract were analyzed by western blot. Hsp90 was used as a cytosolic control. (C) AU565 parental and LapR cells were treated with indicated drugs for 2 days, followed by protein gel blot. (D) Model summarizing molecular changes in lapatinib-resistant breast cancer cells. Left, lapatinib-sensitive cells suppress apoptosis through HER2-induced suppression of mitochondrial cytochrome C release, which may be through PI3K. Right, lapatinib-resistant cells have decreased HER2 and PI3K activity, which may result in constitutive cytochrome C release from mitochondria. Cytochrome C-induced apoptosis is apparently suppressed by IAPs and/or other mechanisms.
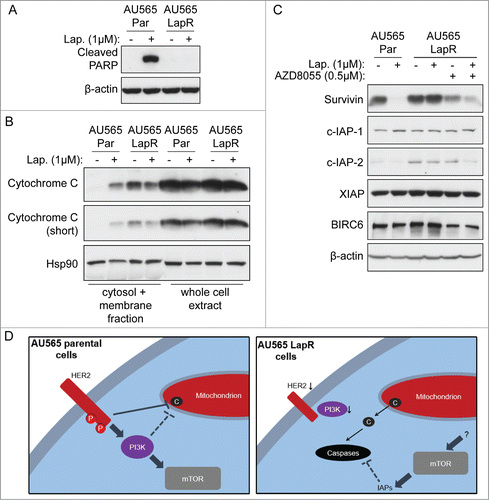
Figure 4. Pharmacological inhibition of only some IAPs is insufficient to reverse lapatinib resistance. (A) AU565 LapR cells were treated with indicated drugs for 4 days, followed by MTT assay. (Note that AU565 LapR cell growth was normalized to vehicle-treated AU565 parental cells rather than to vehicle-treated LapR cell growth. Basal proliferation of AU565 LapR cells is lower than parental cells, and this shifts the AU565 LapR drug sensitivity curve upward; thus we normalized the LapR curve to parental to show that absolute cell number after 17-AAG treatment was similar between parental and LapR cells.) (B) AU565 LapR cells were treated with 1μM birinapant for 2 days, followed by western blot.
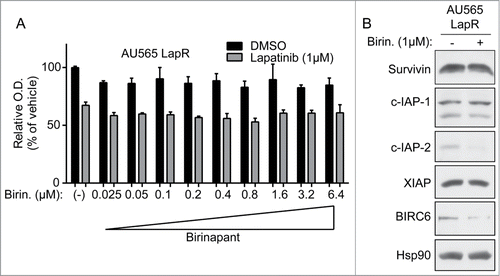
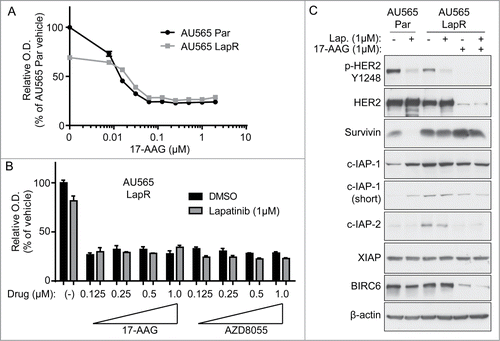
Figure 5. Hsp90 blockade inhibits the growth of lapatinib-resistant cells. (A) AU565 parental and LapR cells were treated with indicated concentrations of 17-AAG for 3 days, followed by MTT assay. All reading were normalized to vehicle-treated AU565 parental cells. (B) AU565 LapR cells were treated with indicated drugs for 3 days, followed by MTT assay. (C) Cells were treated with indicated drugs for 2 days, followed by protein gel blot analysis. Error bars on all graphs indicate s.e.m.
Because AU565 LapR cells appeared to possess PI3K/Akt-independent mTOR activation (), we tested whether LapR cells might be sensitive to mTOR inhibitors but resistant to PI3K inhibitors. We treated parental and LapR cells with the mTOR kinase inhibitor AZD8055, which blocks the catalytic activity of mTOR,Citation19 or the PI3K inhibitor GDC-0941.Citation20 AU565 LapR cells were resistant to the PI3K inhibitor GDC-0941 compared to parental cells, indicating that LapR cells have decreased dependence on PI3K (, top). On the other hand, AU565 LapR cells were sensitive to AZD8055 to a degree similar to AU565 parental cells, indicating that both cell lines are mTOR-dependent (, bottom). These data reinforce the notion that AU565 LapR cells possess PI3K-independent mTOR activation that is required for their proliferation. We also found that catalytic inhibitors of mTOR which block both mTORC1 and mTORC2, such as AZD8055 and BEZ235, inhibited proliferation of AU565 LapR cells more effectively than rapalogs which block mTORC1 only, suggesting that both mTOR complexes may be important for the proliferation of AU565 LapR cells (data not shown). Therefore, we used the dual mTOR kinase inhibitor AZD8055 in subsequent experiments.
PI3K-independent mTOR activation in AU565 LapR cells is independent of canonical mTOR activators
To further explore the mechanism of PI3K/Akt-independent mTOR activation and S6 phosphorylation in AU565 LapR cells, we treated LapR cells (and parental cells as a control) with the PI3K inhibitor GDC-0941 or the Akt inhibitor MK-2206Citation21 and analyzed S6 phosphorylation (). GDC-0941 blocked S6 phosphorylation in AU565 parental cells but not in AU565 LapR cells, reaffirming that S6 phosphorylation in AU565 LapR cells is indeed PI3K-independent. Interestingly, Akt inhibition did not significantly inhibit S6 phosphorylation in parental cells suggesting that mTOR activation in parental cells is dependent on PI3K but not via Akt. Not surprisingly, Akt inhibition did not significantly inhibit the high level of S6 phosphorylation in AU565 LapR cells, which is independent of PI3K/Akt.
Multiple known mTOR upstream activators can lead to mTOR activation.Citation15 For example, Erk, Akt, IKKβ activate and AMPK and GSK-3β inhibit the TSC1/2 mTOR-suppressive complex, while Rheb and PRAS40 directly activate or inhibit mTORC1, respectively.Citation15 We next tested whether these known mTOR upstream regulators were increased or decreased in LapR cells and confer mTOR activation and increased S6 phosphorylation compared with parental cells. We detected Erk, TSC2, PRAS40, AMPK, Rheb (), GSK-3 and IKK (data not shown) and found that none of these signaling nodes were significantly different at total or phospho- protein levels in LapR cells compared to parental cells ().
To identify potential activating mutations in mTOR pathway genes and other mutational events that confer resistance in AU565 LapR cells, we performed whole-exome sequencing of AU565 parental and LapR cells. However, AU565 LapR cells did not gain apparent cancer-associated mutations or copy-number variations compared to parental cells (data not shown). Specifically, we carefully analyzed genes known to be involved in the mTOR pathwayCitation15 and genes from the Cancer Gene CensusCitation22 by looking for known point mutations present in LapR but not parental cells, and by comparing read depth as an indicator of copy number changes. We found that LapR cells did not possess mutations or copy number changes in mTOR pathway genes (or known oncogenes) compared to parental cells (data not shown). Together, these data indicate that a potentially novel, and as yet unclear, mechanism of mTOR activation may be involved in lapatinib resistance in LapR cells.
PI3K-independent mTOR activation upregulates the inhibitors of apoptosis (IAP) family proteins, which can be reversed by the mTOR inhibitor AZD8055
Because known mTOR upstream activators did not suggest any obvious therapeutic opportunities in AU565 LapR cells, we sought to identify mTOR downstream events, such as suppression of cell death,Citation23 that may promote lapatinib resistance in AU565 LapR cells, in order to uncover potential therapeutic vulnerabilities. We hypothesized that AU565 LapR cells may have acquired lapatinib resistance by suppressing apoptosis. Indeed, we found that lapatinib induced PARP cleavage, which indicates activation of caspase-dependent apoptosis, in parental cells but not in LapR cells (). Thus LapR cells apparently had a suppressed lapatinib treatment-induced caspase activity that can lead to apoptosis resistance.
Cytochrome C release from mitochondria into the cytosol is a key initiating event in apoptosis as it leads to caspase activation.Citation24 It is regulated by the Bcl-2 family of proteins, which act on upstream cellular signals (such as the PI3K and MAPK pathways, which activate pro-survival Bcl-2 family proteins to suppress cytochrome C releaseCitation25 or inhibit activation of pro-death Bcl-2 family proteinsCitation26,27) to combinatorially determine whether to allow cytochrome C release into the cytosol and thus induce the intrinsic apoptosis pathway.Citation28 Because lapatinib induced PARP cleavage in parental but not LapR cells, we postulated that cytochrome C was released into the cytosol after lapatinib treatment in parental but not LapR cells.
To analyze cytochrome C release into the cytosol, we removed the mitochondrial fraction from AU565 parental and LapR cells by sucrose fractionation and analyzed the presence of cytochrome C in the cytosolic fraction. After 2 d of lapatinib treatment, AU565 parental cells released cytochrome C into the cytosol (). We expected that AU565 LapR cells would have no cytochrome C release into the cytosol under basal or lapatinib-treated as AU565 LapR cells showed no significant PARP cleavage. Unexpectedly, however, we found that cytochrome C was constitutively present in the cytosol of AU565 LapR cells, even under basal conditions without lapatinib treatment (). This may be due to the reduced basal levels of HER2 and Akt activation in AU565 LapR cells (), which could theoretically allow cytochrome C release into the cytosol through loss of Akt's inhibitory interactions with the pro-apoptotic Bcl-2 family member Bad.Citation27
The constitutive cytochrome C release into the cytosol of AU565 LapR cells suggest that AU565 LapR cells were capable of blocking apoptosis after cytochrome C release. The inhibitors of apoptosis (IAP) family proteinsCitation29,30 are capable of preventing apoptosis after cytochrome C release from mitochondria by directly or indirectly inhibiting caspase activation.Citation31 Therefore, we identified from RNA-Seq dataCitation32 the 5 most highly expressed IAPs in HER2-associated breast cancers, i.e., survivin, c-IAP-1, c-IAP-2, XIAP, and BIRC6. We compared these IAPs' expression between AU565 LapR cells and parental cells and found that although basal (without lapatinib treatment) survivin expression levels were similar, lapatinib dramatically decreased survivin expression in parental but not LapR cells (). Basal c-IAP-2 expression was significantly increased in AU565 LapR cells, which was not inhibited by lapatinib treatment.
Other than the dramatic difference in survivin and c-IAP-2, c-IAP-1, XIAP, and BIRC6 were not significantly changed (). Further, treatment with the mTOR kinase inhibitor AZD8055 in combination with lapatinib decreased both survivin and c-IAP-2 expression in LapR cells but did not dramatically affect other IAPs, suggesting that survivin and c-IAP-2 increase in LapR cells was mTOR-dependent. As previously mentioned, AZD8055 alone or combined with lapatinib significantly inhibited the growth of AU565 LapR cells (), and these data suggest that this inhibition may result from, at least partly, decreased expression of some IAPs, including survivin and c-IAP-2.
The above data indicate that, in parental cells, HER2 and PI3K are highly active and HER2 suppresses apoptosis by blocking mitochondrial cytochrome C release (, left). Lapatinib treatment inhibits HER2 and PI3K and allows release of cytochrome C into the mitochondria. In contrast, AU565 LapR cells have decreased basal HER2 and PI3K activation (, right), which permits the constitutive release of cytochrome C into the cytosol in these cells. However, mTOR activation is maintained in LapR cells through a HER2- and PI3K/Akt-independent unknown mechanism. This mTOR signaling contributes to the increased expression of IAPs (e.g., surviving and c-IAP-2), which block caspase activation induced by constitutive cytosolic cytochrome C. It is conceivable that other mechanisms (other than IAPs) may also play a role in suppression of caspase activation and apoptosis in LapR cells.
Pharmacological inhibition of some IAPs is insufficient to reverse lapatinib resistance
Since we detected increased IAPs that could potentially block caspase activation and apoptosis in LapR cells, we next tested whether pharmacological inhibition of IAP expression may allow caspase activation and induce apoptosis in LapR cells. Birinapant is a potent inhibitor of the IAPs c-IAP-1, c-IAP-2, and XIAPCitation33–35 under preclinical and clinical study. Although birinapant has not been reported to inhibit survivin, we reasoned that inhibition of a spectrum of IAPs, including c-IAP-2 that is increased in LapR cells, might still induce apoptosis by increasing caspase activity in LapR cells. Therefore, we treated LapR cells with birinapant, alone or combined with lapatinib, followed by MTT assay (). Unfortunately, birinapant did not substantially inhibit the growth of LapR cells, alone or in combination with lapatinib, even though birinapant effectively inhibited c-IAP-2 expression, which is consistent with previous reports.Citation33-35 However, in contrast to previous findings, birinapant did not inhibit c-IAP-1 expression in AU565 LapR cells. Birinapant did not inhibit XIAP expression in LapR cells, which is expected as birinapant was known not to inhibit XIAP expression but to inhibit XIAP activity.Citation33-35 Expectedly, birinapant did not inhibit survivin, whereas it unexpectedly decreased BIRC6 expression in AU565 LapR cells. Together, these data indicate that inhibition of c-IAP-2 and some other IAPs is insufficient to reverse lapatinib resistance in AU565 LapR cells. It may be that inhibition of survivin (or other novel caspase-inhibiting factors increased in LapR cells) is required to induce apoptosis in LapR cells.
Inhibition of Hsp90 reverses IAP expression and inhibits the growth of AU565 LapR cells
Because survivin is a known client of the Hsp90 molecular chaperone,Citation36 we considered using Hsp90 inhibitors to reverse lapatinib resistance. Hsp90 stabilizes certain proteins ("clients") during protein translation,Citation37 and small-molecule inhibitors that block Hsp90-induced stabilization of these proteins leads to degradation of Hsp90 clients.Citation38 We hypothesized that the clinically applicable Hsp90 inhibitor 17-AAGCitation39,40 may block survivin expression in AU565 LapR cells (and potentially other IAPs as well) and thus allow cytosolic cytochrome C to fully activate caspases and induce apoptosis. Inhibition of IAPs may also inhibit the cell cycle as several IAPs have known cell cycle-promoting functions.Citation41,42
We tested whether 17-AAG could inhibit the growth of AU565 LapR cells by MTT assay (). Seventeen-AAG inhibited AU565 parental and LapR cells at a similar level; 17-AAG may inhibit parental cell growth via HER2 inhibition.Citation38 Further, 17-AAG alone or combined with lapatinib inhibited AU565 LapR proliferation to a degree similar to the mTOR kinase inhibitor AZD8055 (). Interestingly, 17-AAG inhibited the same IAPs as birinapant in LapR cells, c-IAP-2 and BIRC6 (). However, 17-AAG did not inhibit expression of survivin, notwithstanding previous reports indicating that survivin is an Hsp90 client.Citation36 These data, along with our findings in , suggest that the stability of IAPs and their susceptibility to pharmacological inhibition may be cell context-dependent. Additionally, 17-AAG may inhibit other non-IAP targets that inhibit apoptosis and promote proliferation in LapR cells, as 17-AAG blocks the IAPs that were also blocked by birinapant yet has much better efficacy (). Indeed, 17-AAG has been reported to inhibit a variety of proteins.Citation43–46 For example, HER2 expression in LapR cells was also inhibited by 17-AAG, consistent with previous findings that HER2 is an Hsp90 client.Citation38,47 17-AAG alone inhibited IAP and HER2 expression () as well as proliferation of AU565 LapR cells () similar to the lapatinib plus 17-AAG combination.
Our findings suggest that mTOR kinase inhibition or Hsp90 inhibitors may be effective in inhibition of lapatinib-resistant breast cancers, including those LapR cells with PI3K-independent mTOR activation and subsequent mTOR-mediated upregulation of IAPs or other (potentially novel) caspase inhibitors that inhibit apoptosis and confer lapatinib resistance.
Discussion
In this study, we report PI3K-independent mTOR activation promotes lapatinib resistance in AU565 LapR cells. Another group previously reported PI3K-independent mTOR activation in another acquired lapatinib-resistant breast cancer cell line (SKBR3 LapR).Citation48 Cell signaling in SKBR3 LapR and AU565 LapR cells appears to be strikingly similar, including decreased basal HER2 and Akt phosphorylation along with enhanced S6 phosphorylation in both LapR cell lines. Interestingly, the AU565 and SKBR3 cell lines were derived from the same patient (primary versus metastatic sample, respectively), suggesting that the PI3K-independent mTOR activation clone may represent a subclone that pre-existed in the breast cancer patient from which these cell lines were derived. It would be of interest to determine whether the IAP signaling and Hsp90 sensitivity is also similar between the 2 cell lines. Importantly, our findings together provide further evidence that PI3K inhibition is unlikely to reverse lapatinib resistance in certain lapatinib-resistant breast cancers. To block PI3K-independent mTOR activation, we used the mTOR kinase inhibitor AZD8055 as it is more effective than rapalogs in inhibiting the growth of AU565 LapR cells (data not shown). However, mTOR kinase inhibitors demonstrated high toxicitiesCitation49,50 and may be too toxic to use at doses sufficient to effectively block the target of interest. Therefore, it is important to identify additional strategies to effectively target lapatinib-resistant breast cancer cells.
It is remarkable that Hsp90 inhibitors effectively suppressed AU565 LapR proliferation., i.e., can overcome lapatinib resistance. Hsp90 inhibitors have shown efficacy in HER2+ breast cancer, including those resistant to HER2 targeted therapy.Citation39,40 Numerous other Hsp90 inhibitors with potentially improved properties are under clinical developmentCitation51 and may be promising alternatives to 17-AAG, which must be delivered intravenously and has somewhat suboptimal pharmacokinetics.Citation52 Although the mechanism by which 17-AAG inhibits LapR cancer cell growth in our study is not entirely clear, inhibition of IAPs may play a role. It would be of interest to test whether clinical post-17-AAG-treatment specimens have decreased IAP expression compared to matched pre-treatment samples.
Determining the upstream mTOR activator in AU565 LapR cells also has the potential to reveal novel signaling mechanisms in this important cellular pathway. We performed exome sequencing of AU565 parental and LapR cells to look for point mutations and copy-number variations in LapR but not present in parental cells that might account for mTOR activation. However, there were no point mutations in known mTOR pathway members in LapR cells or copy number variations that could account for mTOR activation. Thus there may be novel mTOR pathway activators activated genetically, epigenetically, or at the protein level in AU565 LapR cells to be further investigated in future studies.
Our finding that birinapant inhibits c-IAP-2 and BIRC6, but not c-IAP-1, in our model merits a closer look at birinapant's mechanism of action. To our knowledge, this is the first report that birinapant inhibits BIRC6 expression. Its activity against specific IAPs may be cell context-dependent and the spectrum of IAPs inhibited in a certain cell type may correlate with drug response. Thus, the effect of birinapant should be carefully evaluated for each specific cell and model system.
In summary, lapatinib resistance can be promoted by suppression of caspase activation downstream of cytochrome C release from mitochondria by IAPs or other novel caspase inhibitors. Further, we found that PI3K-independent mTOR activation leads to upregulation of IAPs that may promote lapatinib resistance; thus PI3K inhibitors are not effective in these cells. Finally, we provide evidence that mTOR kinase inhibitors or Hsp90 inhibitors are clearly superior to PI3K inhibitors in reversing lapatinib resistance in the lapatinib-resistant breast cancers having PI3K-independent mTOR activation.
Materials and Methods
Cell lines and cell culture
AU565 parental cells were from ATCC. AU565 LapR cells were generated by treating AU565 parental cells with 2.1μM lapatinib for longer than 6 months. Cells were grown in DMEM/F12 in 9% FBS. Lapatinib was withdrawn generally for at least one week prior to each experiment. Cells were tested for mycoplasma periodically and found to by mycoplasma-negative in each case.
MTT assay
Cells were plated at 3,000 to 5,000 cells per well (or were uncounted and plated empirically) in 96-well plates in triplicate in 50–100μL of medium. The next day 2x drug was added to give the final 1x concentrations indicated. Three–5 d after drug addition, MTT assay was performed by adding 20–50μL per well of filtered 5mg/mL MTT (Sigma) for 1–3 hours at 37º C. Medium was removed from wells and 100μL of DMSO per well was added. After formazan crystals were sufficiently dissolved by DMSO (overnight incubation or 15–30 minutes on a shaker), plates were read at OD570 minus OD650. Readings were normalized to the vehicle-treated samples (=100%).
RTK array
Cells were treated as indicated in figures and legends and harvested per manufacturer's instruction using the PathScan RTK Signaling Array Kit (Cell Signaling). Lysates were incubated on arrays with capture antibodies and detection antibody cocktail was added. After washes, arrays were incubated with enhanced chemiluminescence (ECL) reagents (Pierce) to visualize phosphoprotein levels.
Western blot analysis
Cells were treated as indicated followed by removal of medium and addition of cold PBS on ice. Cells were harvested in cold PBS using a scraper and pelleted at 3220 x g for 2 minutes. Supernatant was removed and cells were lysed in lysis buffer (0.1% SDS, 1% Triton X-100, 150mM NaCl, 50mM Tris pH 8.0 with protease inhibitor cocktail from Sigma and PhosSTOP from Roche). After at least 15 minutes of lysis, lysates were centrifuged at maximum speed for 10–15 minutes at 4º C. Supernatant was saved and protein content was quantified using the BCA assay (Pierce). Protein was diluted to 1μg/μL using lysis buffer and 1x loading buffer (diluted to 1x from 6x loading buffer containing 40% glycerol, 10% β-mercaptoethanol, 0.25M Tris pH 6.8, 8% SDS). This was then boiled for 10 minutes and 30μg of protein was run by SDS-PAGE and transferred to nitrocellulose or PVDF membranes using semi-dry transfer (Bio-Rad). Blocking with 5% PBS-T or TBS-T milk lasted for 30 minutes, followed by 2 5-minute washes in PBS-T or TBS-T. Primary antibody diluted in 5% BSA in PBS-T or TBS-T (generally at 1:1000) was added to membrane overnight at 4º C. Membranes were washed 3 times in PBS-T or TBS-T and secondary antibody (GE) was added at 1:3000 in blocking buffer. After three more washes membranes were exposed to ECL reagents (Pierce) followed by film exposure.
Antibodies used were from Cell Signaling except for Rheb (Santa Cruz), α-tubulin (Sigma), and β-actin (Sigma).
siRNA transfection
Sigma MISSION siRNAs (Sigma) were transfected using PepMute siRNA Transfection Reagent (SignaGen) according to manufacturer's instructions. Cells were harvested 3 d later for analysis of knockdown efficiency. In some cases a drug was also added a few hours before harvest as indicated.
Cell fractionation
Cytosolic/membrane fraction was separated from mitochondria and other organelles by lysing cells in a hypotonic sucrose-containing buffer (250 mM sucrose, 20 mM HEPES (pH 7.4), 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and protease inhibitor cocktail from Sigma) and passing cells through 18 to 27 gauge needles 10 to 20 times to shear plasma membrane. Samples were left on ice for at least 20 minutes. Nuclei were removed by centrifuging at 720 x g for 5 minutes. Supernatant (containing mitochondria and cytosol/membrane) was then centrifuged at 10,000 x g for 5–10 minutes to pellet and remove mitochondria. The cytosolic and membrane fraction in the supernatant was removed, concentrated using Microcon 10 kDa molecular weight cutoff filters (Sigma), quantified by BCA assay (Pierce), and run by protein gel blot (30μg per well). Whole cell extract was also run as a control.
Small-molecule inhibitors
Small-molecule inhibitors were from Selleck (17-AAG, RAD001, temsirolimus, AZD8055, and birinapant).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1002693_Supplementary_Materials.zip
Download Zip (405.5 KB)Acknowledgments
We thank Dr. Siyuan Zhang for helpful discussion, Ping Li for aid developing LapR cells, and Dr. Cheryl Walker and Dr. Jiangwei Zhang for suggestions regarding mTOR pathway antibodies.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was partially supported by NIH/NCI F31CA165819 (S.W.B.), NIH grants P30-CA 16672 (MDACC), PO1-CA099031 project 4 (D.Y.), RO1-CA112567–07 (D.Y.), Susan G. Komen Foundation KG091020 (D.Y.). D. Yu is the Hubert L. & Olive Stringer Distinguished Chair in Basic Science at MD Anderson Cancer Center.
References
- Slamon D, Godolphin W, Jones L, Holt J, Wong S, Keith D, Levin W, Stuart S, Udove J, Ullrich A, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989; 244:707-12; PMID:2470152; http://dx.doi.org/10.1126/science.2470152.
- Esteva FJ, Yu D, Hung M-C, Hortobagyi GN. Molecular predictors of response to trastuzumab and lapatinib in breast cancer. Nat Rev Clin Oncol 2010; 7:98-107; PMID:20027191; http://dx.doi.org/10.1038/nrclinonc.2009.216.
- Garrett JT, Arteaga CL. Resistance to HER2-directed antibodies and tyrosine kinase inhibitors: Mechanisms and clinical implications. Cancer Biol Ther 2011; 11:793-800; PMID:21307659; http://dx.doi.org/10.4161/cbt.11.9.15045.
- Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol 2012; 9:16-32; http://dx.doi.org/10.1038/nrclinonc.2011.177.
- Wang Q, Li S-H, Wang H, Xiao Y, Sahin O, Brady SW, Li P, Ge H, Jaffee EM, Muller WJ, et al. Concomitant targeting of tumor cells and induction of T-cell response synergizes to effectively inhibit trastuzumab-resistant breast cancer. Cancer Res 2012; 72:4417-28; PMID:22773664; http://dx.doi.org/10.1158/0008-5472.CAN-12-1339-T.
- Zhang S, Huang W-C, Li P, Guo H, Poh S-B, Brady SW, Xiong Y, Tseng L-M, Li S-H, Ding Z, et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med 2011; 17:461-9; PMID:21399647; http://dx.doi.org/10.1038/nm.2309.
- Nagata Y, Lan K-H, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004; 6:117-27; PMID:15324695; http://dx.doi.org/10.1016/j.ccr.2004.06.022.
- Sahin O, Wang Q, Brady SW, Ellis K, Wang H, Chang C-C, Zhang Q, Priya P, Zhu R, Wong ST, et al. Biomarker-guided sequential targeted therapies to overcome therapy resistance in rapidly evolving highly aggressive mammary tumors. Cell Res 2014; 24:542-59; PMID:24675532; http://dx.doi.org/10.1038/cr.2014.37.
- Lu C-H, Wyszomierski SL, Tseng L-M, Sun M-H, Lan K-H, Neal CL, Mills GB, Hortobagyi GN, Esteva FJ, Yu D. Preclinical testing of clinically applicable strategies for overcoming trastuzumab resistance caused by PTEN deficiency. Clin Cancer Res 2007; 13:5883-8; PMID:17908983; http://dx.doi.org/10.1158/1078-0432.CCR-06-2837.
- Wang L, Zhang Q, Zhang J, Sun S, Guo H, Jia Z, Wang B, Shao Z, Wang Z, Hu X. PI3K pathway activation results in low efficacy of both trastuzumab and lapatinib. BMC Cancer 2011; 11:248; PMID:21676217; http://dx.doi.org/10.1186/1471-2407-11-248.
- Eichhorn PJA, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, Beijersbergen RL, Valero V, Seoane J, Bernards R, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res 2008; 68:9221-30; PMID:19010894; http://dx.doi.org/10.1158/0008-5472.CAN-08-1740.
- Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, Halsey W, Sathe GM, Martin A-M, Gilmer TM. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res 2009; 69:6871-8; PMID:19671800; http://dx.doi.org/10.1158/0008-5472.CAN-08-4490.
- Jegg A-M, Ward TM, Iorns E, Hoe N, Zhou J, Liu X, Singh S, Landgraf R, Pegram MD. PI3K independent activation of mTORC1 as a target in lapatinib-resistant ERBB2+ breast cancer cells. Breast Cancer Res Treat 2012; 136:683-92; PMID:23089982; http://dx.doi.org/10.1007/s10549-012-2252-9.
- Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR Controls Cell Cycle Progression through Its Cell Growth Effectors S6K1 and 4E-BP1/Eukaryotic Translation Initiation Factor 4E. Mol Cell Biol 2003; 24:200-16; http://dx.doi.org/10.1128/MCB.24.1.200-216.2004.
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-93; PMID:22500797; http://dx.doi.org/10.1016/j.cell.2012.03.017.
- Inoki K, Li Y, Xu T, Guan K-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 2003; 17:1829-34; PMID:12869586; http://dx.doi.org/10.1101/gad.1110003.
- Qi J, McTigue MA, Rogers A, Lifshits E, Christensen JG, Jänne PA, Engelman JA. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to MET inhibitors. Cancer Res 2011; 71:1081-91; PMID:21266357; http://dx.doi.org/10.1158/0008-5472.CAN-10-1623.
- Burrell RA, Swanton C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol Oncol 2014; 8:1095-1111; PMID:25087573; http://dx.doi.org/10.1016/j.molonc.2014.06.005.
- Maira S-M, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chène P, De Pover A, Schoemaker K, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 2008; 7:1851-63; PMID:18606717; http://dx.doi.org/10.1158/1535-7163.MCT-08-0017.
- Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer . J Med Chem 2008; 51:5522-32; PMID:18754654; http://dx.doi.org/10.1021/jm800295d.
- Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan B-S, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 2010; 9:1956-67; PMID:20571069; http://dx.doi.org/10.1158/1535-7163.MCT-09-1012.
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer 2004; 4:177-83; PMID:14993899; http://dx.doi.org/10.1038/nrc1299.
- Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med 2004; 10:594-601; PMID:15156201; http://dx.doi.org/10.1038/nm1052.
- Tait SWG, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 2010; 11:621-32; PMID:20683470; http://dx.doi.org/10.1038/nrm2952.
- Wang J-M, Chao J-R, Chen W, Kuo M-L, Yen JJ-Y, Yang-Yen H-F. The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol Cell Biol 1999; 19:6195-206; PMID:10454566.
- Takezawa K, Okamoto I, Nishio K, Jänne PA, Nakagawa K. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin Cancer Res 2011; 17:2140-8; PMID:21415216; http://dx.doi.org/10.1158/1078-0432.CCR-10-2798.
- Datta S. Akt Phosphorylation of BAD Couples Survival Signals to the Cell-Intrinsic Death Machinery. Cell 1997; 91:231-41; PMID:9346240; http://dx.doi.org/10.1016/S0092-8674(00)80405-5.
- Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 2014; 15:49-63; PMID:24355989; http://dx.doi.org/10.1038/nrm3722.
- Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T, Reed JC. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res 1998; 58:5315-20; PMID:9850056.
- Hunter AM, LaCasseEC, Korneluk RG. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 2007; 12:1543-68; PMID:17573556; http://dx.doi.org/10.1007/s10495-007-0087-3.
- Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J 1998; 17:2215-23; PMID:9545235; http://dx.doi.org/10.1093/emboj/17.8.2215.
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490:61-70; PMID:23000897; http://dx.doi.org/10.1038/nature11412.
- Benetatos CA, Mitsuuchi Y, Burns JM, Neiman EM, Condon SM, Yu G, Seipel ME, Kapoor GS, Laporte MG, Rippin SR, et al. Birinapant (TL32711), a bivalent SMAC mimetic, targets TRAF2-associated cIAPs, abrogates TNF-induced NF-κB activation, and is active in patient-derived xenograft models. Mol Cancer Ther 2014; 13:867-79; PMID:24563541; http://dx.doi.org/10.1158/1535-7163.MCT-13-0798.
- Krepler C, Chunduru SK, Halloran MB, He X, Xiao M, Vultur A, Villanueva J, Mitsuuchi Y, Neiman EM, Benetatos C, et al. The novel SMAC mimetic birinapant exhibits potent activity against human melanoma cells. Clin Cancer Res 2013; 19:1784-94; PMID:23403634; http://dx.doi.org/10.1158/1078-0432.CCR-12-2518.
- Allensworth JL, Sauer SJ, Lyerly HK, Morse MA, Devi GR. Smac mimetic Birinapant induces apoptosis and enhances TRAIL potency in inflammatory breast cancer cells in an IAP-dependent and TNF-α-independent mechanism. Breast Cancer Res Treat 2013; 137:359-71; PMID:23225169; http://dx.doi.org/10.1007/s10549-012-2352-6.
- Fortugno P, Beltrami E, Plescia J, Fontana J, Pradhan D, Marchisio PC, Sessa WC, Altieri DC. Regulation of survivin function by Hsp90. Proc Natl Acad Sci U S A 2003; 100:13791-6; PMID:14614132; http://dx.doi.org/10.1073/pnas.2434345100.
- Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 2010; 10:537-49; PMID:20651736; http://dx.doi.org/10.1038/nrc2887.
- Solit DB, Zheng FF, Drobnjak M, Munster PN, Higgins B, Verbel D, Heller G, Tong W, Cordon-Cardo C, Agus DB, et al. Seventeen-allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin Cancer Res 2002; 8:986-93; PMID:12006510.
- Modi S, Stopeck AT, Gordon MS, Mendelson D, Solit DB, Bagatell R, Ma W, Wheler J, Rosen N, Norton L, et al. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: a phase I dose-escalation study. J Clin Oncol 2007; 25:5410-7; PMID:18048823; http://dx.doi.org/10.1200/JCO.2007.11.7960.
- Modi S, Stopeck A, Linden H, Solit D, Chandarlapaty S, Rosen N, D'Andrea G, Dickler M, Moynahan ME, Sugarman S, et al. HSP90 inhibition is effective in breast cancer: a phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin Cancer Res 2011; 17:5132-9; PMID:21558407; http://dx.doi.org/10.1158/1078-0432.CCR-11-0072.
- Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998; 396:580-4; PMID:9859993; http://dx.doi.org/10.1038/25141.
- Dubrez-Daloz L, Dupoux A, Cartier J. More than just inhibitors of apoptosis proteins. Cell Cycle 2008; 7:1036-1046; PMID:18414036; http://dx.doi.org/10.4161/cc.7.8.5783.
- Nimmanapalli R, O'Bryan E, Kuhn D, Yamaguchi H, Wang H-G, Bhalla KN. Regulation of 17-AAG-induced apoptosis: role of Bcl-2, Bcl-XL, and Bax downstream of 17-AAG-mediated down-regulation of Akt, Raf-1, and Src kinases. Blood 2003; 102:269-75; PMID:12623837; http://dx.doi.org/10.1182/blood-2002-12-3718.
- Fumo G, Akin C, Metcalfe DD, Neckers L. . 17-Allylamino-17-demethoxygeldanamycin (17-AAG) is effective in down-regulating mutated, constitutively activated KIT protein in human mast cells. Blood 2004; 103:1078-84; PMID:14551138; http://dx.doi.org/10.1182/blood-2003-07-2477.
- Georgakis G V, Li Y, Younes A. The heat shock protein 90 inhibitor 17-AAG induces cell cycle arrest and apoptosis in mantle cell lymphoma cell lines by depleting cyclin D1, Akt, Bid and activating caspase 9. Br J Haematol 2006; 135:68-71; PMID:16925576; http://dx.doi.org/10.1111/j.1365-2141.2006.06247.x.
- Radujkovic A, Schad M, Topaly J, Veldwijk MR, Laufs S, Schultheis BS, Jauch A, Melo J V, Fruehauf S, Zeller WJ. Synergistic activity of imatinib and 17-AAG in imatinib-resistant CML cells overexpressing BCR-ABL-Inhibition of P-glycoprotein function by 17-AAG. Leukemia 2005; 19:1198-206; PMID:15902298; http://dx.doi.org/10.1038/sj.leu.2403764.
- Smith-Jones PM, Solit DB, Akhurst T, Afroze F, Rosen N, Larson SM. Imaging the pharmacodynamics of HER2 degradation in response to Hsp90 inhibitors. Nat Biotechnol 2004; 22:701-6; PMID:15133471; http://dx.doi.org/10.1038/nbt968.
- Johnston S, Pippen J, Pivot X, Lichinitser M, Sadeghi S, Dieras V, Gomez HL, Romieu G, Manikhas A, Kennedy MJ, et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. J Clin Oncol 2009; 27:5538-46; PMID:19786658; http://dx.doi.org/10.1200/JCO.2009.23.3734.
- Asahina H, Nokihara H, Yamamoto N, Yamada Y, Tamura Y, Honda K, Seki Y, Tanabe Y, Shimada H, Shi X, et al. Safety and tolerability of AZD8055 in Japanese patients with advanced solid tumors; a dose-finding phase I study. Invest New Drugs 2013; 31:677-84; PMID:22843211; http://dx.doi.org/10.1007/s10637-012-9860-4.
- Pallet N, Legendre C. Adverse events associated with mTOR inhibitors. Expert Opin Drug Saf 2013; 12:177-86; PMID:23252795; http://dx.doi.org/10.1517/14740338.2013.752814.
- Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 2012; 18:64-76; PMID:22215907; http://dx.doi.org/10.1158/1078-0432.CCR-11-1000.
- Banerji U, O'Donnell A, Scurr M, Pacey S, Stapleton S, Asad Y, Simmons L, Maloney A, Raynaud F, Campbell M, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol 2005; 23:4152-61; PMID:15961763; http://dx.doi.org/10.1200/JCO.2005.00.612.