
Abstract
TrkB activation by brain-derived neurotrophic factor (BDNF) contributes to chemo-resistance in neuroblastoma (NB). AZD6918 is a novel potent and selective inhibitor of the Trk tyrosine kinases. In this study we evaluated the effect of AZD6918 on the sensitivity of TrkB-expressing NB cells or tumors to etoposide, a topoisomerase II inhibitor. TrkB-expressing NB cells were treated with AZD6918 and etoposide in the presence or absence of BDNF in vitro and cell survival was determined. NB xenograft tumors were treated with AZD6918 and etoposide, either alone or in combination in vivo, and the anti-tumor growth effect or mice survival advantage was evaluated. Our study showed that AZD6918 induced cell death as a single agent and attenuated BDNF/TrkB-induced protection from etoposide in vitro. Although AZD6918 alone didn't show anti-tumor growth effect or survival advantage in vivo, a combination of AZD6918 and etoposide had a statistically significant stronger anti-tumor growth effect and survival advantage compared to treatment with either agent alone. Our data indicate that as a Trk inhibitor AZD6918 increased the sensitivity of NB to etoposide. These results extend the spectrum of cytotoxic drugs whose efficacy is increased in combination with Trk inhibitors and support the combination of Trk inhibitors and cytotoxic drugs for NB treatment.
Abbreviations
| BDNF | = | brain-derived neurotrophic factor |
| NB | = | neuroblastoma |
| TET | = | tetracycline |
| Trk | = | tropomyosin-related kinase |
Introduction
Neuroblastoma (NB) is the most common extracranial solid tumor in childhood and is derived from neural crest precursor cells. It accounts for nearly 8% of pediatric malignancies, yet is responsible for approximately 15% of all pediatric cancer deaths.Citation1-3 Spontaneous regression, differentiation, and a good response to current therapeutic regimens such as surgery and chemotherapy are common in infants or those with low-risk tumors. However, children, typically those aged >18 months, fail to achieve sustained responses to intensive multimodality chemotherapy and stem cell transplantation. Although patients may initially have a response to chemotherapy, they eventually develop disease recurrence with multifocal metastatic disease and resistance to chemotherapy. Despite the aggressive treatment, the prognosis of these patients is poor and their long-term survival is <40%.Citation4 More effective treatment strategies are urgently needed for this high-risk group of patients with NB. A better understanding of the genetics and biologic behavior of poor-prognosis NB tumors should lead to new molecular targets that can be used to develop more specific, more effective, and less toxic therapies.
Brain-derived neurotrophic factor (BDNF) and its tyrosine kinase receptor TrkB (Tropomyosin-related kinase B) are often detected in NB tumors in patients with an unfavorable prognosis.Citation5 Our previous studies identified that BDNF activation of TrkB induced resistance to chemotherapy in NB cells and NB xenografts and that by targeting the critical downstream mediators of activated Trk we could reverse the chemoresistance.Citation6-9 In this study we evaluated whether specifically targeting Trk in vivo would enhance the efficacy of chemotherapy.
Tropomyosin-related kinases (Trks) are a family of receptor tyrosine kinases with 3 isoforms (TrkA, TrkB and TrkC) that are activated by neurotrophins. AZD6918 is an oral inhibitor of Trk tyrosine kinase receptors. Its ability to inhibit Trks makes it potentially relevant for the treatment of children with neuroblastoma. In the present study, we evaluated the effect of AZD6918 on cell survival in TrkB-expressing neuroblastoma in vitro, and investigated whether AZD6918 attenuated the chemoresistance of neuroblastoma to chemotherapy.
Results
Effect of AZD6918 on the survival of NB cells in vitro
To study the effect of AZD6918 on NB cell survival, TrkB-expressing TB3 cells, BE2 and KCNR cells were treated with different doses of AZD6918 (1.25–60 μM) for 24 hrs and cell viability was evaluated by MTS assay. showed that AZD6918 treatment induced cell death in a dose-dependent manner at 24 hours in NB cells.
Figure 1. Effect of AZD6918 on NB cell survival in vitro. TB3, BE2 and KCNR cells were treated with different concentrations of AZD6918 for 24 hours and MTS assay was used to detect cell survival (A), or treated for 16 hours and caspase 3/7 activity was detected (B). *P < 0.05, **P < 0.01, compared to the control in each cell line. (C):KCNR, BE2 cells were treated with AZD6918 at concentration of 2.5 μM for 2 hours, then NB cells were harvested and total protein was extracted for the evaluation of P-TrkB, P-Akt, P-mTOR and P-GSK-3. GAPDH was used as control.
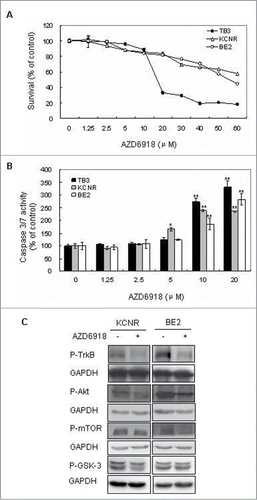
To assess whether AZD6918-induced cell death was mediated via a caspase-dependent apoptotic pathway, we performed a caspase 3/7 activity assay. Caspase-3 and caspase-7 are the common effector caspases of both intrinsic and the extrinsic apoptotic pathways. Our result showed that AZD6918 treatment increased the caspase-3/7 activity in a dose-dependent manner in TB3 cells, as well as in BE2 and KCNR cells ().
We also evaluated the inhibition of AZD6918 on P-TrkB, P-Akt, P-mTOR and P-GSK-3 in NB cells. In , 2.5 μM AZD6918 inhibited the P-TrkB, P-Akt, P-mTOR expression in KCNR and BE2 cells, there was an inhibition of P-GSK3 expression in KCNR cells after AZD6918 treatment, but no obvious inhibition of P-GSK3 was detected in BE2 cells.
Effect of AZD6918 on BDNF/TrkB-mediated rescues of cell death in vitro
Our previous study indicated that BDNF activation of TrkB protected TB3 cells from chemotherapeutic drugs-induced cell death. To determine whether AZD6918 altered BDNF/TrkB protection of TB3 cells from chemotherapeutic drugs-induced cell death, we pretreated TB3 cells with AZD6918 (2.5 μM) for 2 hours followed by etoposide (1 μg/ml or 3 μg/ml) treatment in the absence or presence of BDNF (100 ng/ml) for an additional 24 hours. The results indicated that BDNF protected TB3 cells from etoposide-induced cell death, however, prior treatment with AZD6918 blocked the BDNF/TrkB-mediated rescue of TB3 cells from etoposide-induced cell death ().
Figure 2. Effect of AZD6918 on BDNF/TrkB-mediated rescues of cell death in vitro. TB3 cells were pretreated with AZD6918(2.5 μM) for 2 hours followed by BDNF (100 ng/ml) treatment for 1 hour, and then treated with etoposide (A:1 μg/ml; B and C: 3 μg/ml) for 24 hours. To inhibit the expression of TrkB, TB3 cells were cultured in media with tetracycline (TET, 1 μg/ml) for at 3 d before experiment. MTS assay was used to assess cell survival. ** P < 0.01, etopside-treated cells via BDNF + Etoposide treated cells; ## P < 0.01, BDNF + Etoposide treated cells via BDNF + Etoposide + AZD6918 treated cells. ns: no statistical difference. (D) TB3 cells were pretreated with AZD6918 (2.5 μM) for 2 hours followed by BDNF(100 ng/ml) treatment for 1 hour, and then harvested for the evaluation of P-TrkB, P-Akt, P-mTOR, P-GSK-3 and GAPDH by Western Blotting. E: TB3 cells were transfected with activated Akt or empty vector plasmids by lipofectamine 2000 and treated with AZD6918 at 24 h after transfection. MTS assay was used to assess cell survival. ** P < 0.01, activated Akt-transfected cells via empty vector transfected cells.
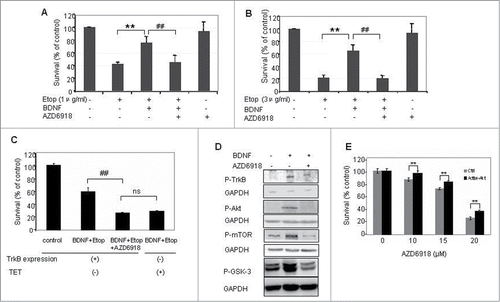
To further investigate the role of TrkB in the AZD6918 effect, we used our tetracycline (TET)-regulated TrkB expressing system of TB3 cells. In the absence of TET, TrkB is expressed in TB3 cells, while in the presence of TET, TrkB expression is inhibited.Citation8 As shown in , when TrkB was expressed in TB3 cells (TET-), the cell survival in BDNF + etoposide treated group was about 60%, pretreatment with AZD6918 reduced the cell survival to about 27%; When TrkB expression was inhibited by TET, the cell survival in BDNF+etoposide treated group was about 29%, which was similar to the AZD6918 pretreatment group in TrkB-expressing cells (27%). We also evaluated the inhibition of P-TrkB by AZD6918 (), and found that the BDNF-induced increased expression of P-TrkB was inhibited by AZD6918 in TB3 cells. These data indicate that AZD6918 attenuated BDNF/TrkB-induced resistance to etoposide via inhibition of TrkB.
In our previous study we reported that BDNF/TrkB activated PI3K/Akt signaling pathway and Akt was the key target that mediated the BDNF/TrkB protection from chemotherapy-induced cell death in TB3 cells.Citation9 In the present study, we evaluated the effect of AZD6918 on P-Akt, as well as its downstream targets P-mTOR and P-GSK-3. As shown in , pretreatment of TB3 cells with AZD6918 blocked the BDNF/TrkB-induced increase of P-Akt, P-mTOR and P-GSK-3. We further evaluated the role of Akt in the AZD6918-induced cell death. Active Akt plasmid was transfected into TB3 cells (with about 30–40% transfection rate) and then treated with different concentrations of AZD6918. There was a statistical increase of cell survival (about 10%) in TB3 cells transfected with active Akt compared to cells transfected with empty vector control ().
To study whether autophagy is involved in AZD6918 effect, we observed the changes of LC3, LAMP and P62, the autophagy indicators, in neuroblastoma cells after AZD6918 treatment and no consistent changes among the 3 neuroblastoma cell lines were detected, also we didn't detect the formation of autophagosome after AZD6918 treatment (data no shown). Together with the increases of caspase 3/7 activity after AZD6918 treatment, we considered that AZD6918 induced cell death via apoptosis.
The effect of AZD6918 as a single agent on tumor growth and mice survival in vivo
To determine whether AZD6918 affects TB3 tumor growth in vivo, mice bearing TB3 tumors were treated with AZD6918 at 70 mg/kg or 100 mg/kg dose. showed that neither dose of AZD6918 had a statistically significant anti-tumor growth effect on TB3 xenograft growth compared to xenografts in the untreated control group.
Figure 3. Effect of AZD6918 as a single agent on TB3 xenograft tumor growth and mice survival. Mice with TB3 subcutaneous tumors were treated with vehicle or AZD6918 (70 mg/kg, 100 mg/kg) twice a day, 7 d a week. At day 29 of treatment, the tumor sizes in each group were compared (A). The mice survivals in each group were plotted by Kaplan-Meier analysis (B).
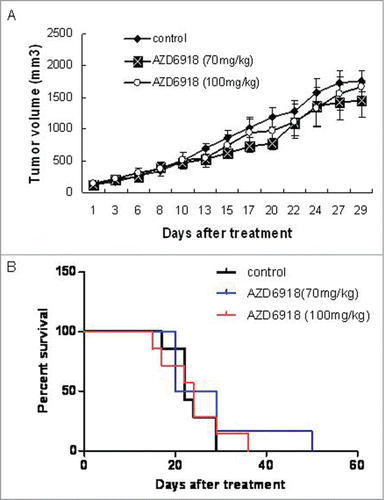
We generated Kaplan-Meier curves of the survival of mice and found that there was no statistically significant survival advantage for the AZD6918-treated mice (at either the 70 mg/kg or the 100 mg/kg dose) compared to the control group.
These data indicated that as a single agent, AZD6918 at the indicated doses didn't have a significant anti-tumor growth effect or alter the survival of mice bearing NB xenografts.
The combination effects of AZD6918 and etoposide on tumor growth in vivo
Since we didn't observe anti-tumor growth effect or mice survival advantage when tumor-bearing nude mice were treated with AZD6918 alone, we evaluated whether AZD6918 would affect the sensitivity of TB3 tumors to etoposide. Mice bearing TB3 tumors were treated with AZD6918 (70 mg/kg) and etoposide (10 mg/kg, or 20 mg/kg) alone or in combination. showed the combination effect of AZD6918 with low dose of etoposide (10 mg/kg) on tumor growth. Neither AZD6918 nor etoposide (10 mg/kg) had any statistical anti-tumor growth effect compared to control group. However there was a 40% inhibition of tumor growth at day 29 of treatment in etoposide (10 mg/kg) + AZD6918 combination group compared to control group. This difference was statistically significant (P = 0.023).
Figure 4. The combination effects of AZD6918 and etoposide on tumor growth in vivo. Mice with TB3 tumors were treated with vehicle, AZD6918 (70 mg/kg), etoposide (10 mg/kg in A, 20 mg/kg in B) or a combination of AZD6918 and etoposide. The tumor sizes were compared among these groups. *P < 0.05, **P < 0.01.
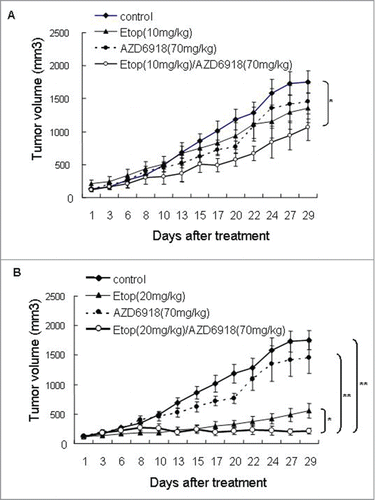
showed the combination effect of AZD6918 with high dose of etoposide (20 mg/kg) on tumor growth. At day 29 of treatment, there was an 88% inhibition of tumor growth in the group receiving the combination of etoposide (20 mg/kg) and AZD6918. This was significantly greater than the 68% inhibition of tumor growth in the group treated with etoposide alone (P = 0.037) and the 17% inhibition of tumor growth in the group treated with AZD6918 alone (P = 0.0009). These data indicated that AZD6918 sensitized TB3 tumors to etoposide treatment.
The combination effect of AZD6918 and etoposide on mice survival in vivo
To study the combination effect of AZD6918 and etoposide on mice survival in vivo, we generated Kaplan-Meier curves of the mice survival. showed the effect of the combination of AZD6918 and low dose of etoposide (10 mg/kg) on mice survival. The median survival days were 22 for mice in the control group, 32.5 for mice in the etoposide-treated group, 24.5 for mice in the AZD6918-treated group and 34 for mice in the etoposide and AZD6918 combination group. Compared to control group, both etoposide (10 mg/kg)-treated group (P = 0.032) and etoposide (10 mg/kg) and AZD6918 combination group (P = 0.021) had significant survival advantage.
Figure 5. The combination effects of AZD6918 and etoposide on mice survival in vivo. Mice with TB3 tumors were treated with vehicle, AZD6918 (70 mg/kg), etoposide (10 mg/kg in A, 20 mg/kg in B) or a combination of AZD6918 and etoposide. The mice survivals in each group were plotted by Kaplan-Meier analysis. Median survival days in each group were calculated. *P < 0.05, **P < 0.01.
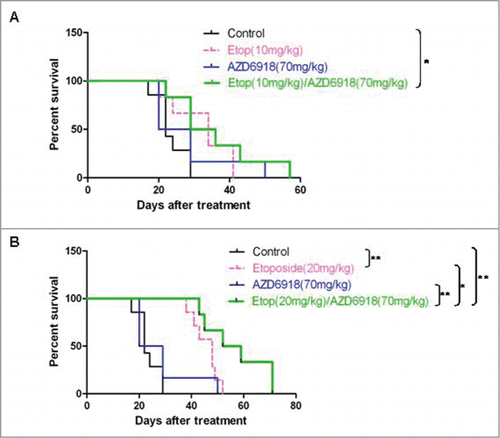
showed the effect of the combination of AZD6918 and high dose of etoposide (20 mg/kg) on mice survival. The median survival days were 22 for mice in the control group, 48 for mice in the etoposide-treated group, 24.5 for mice in the AZD6918-treated group and 55.5 for mice in the etoposide and AZD6918 combination group. Compared to control group, both etoposide (20 mg/kg)-treated group (P = 0.002) and etoposide (20 mg/kg) and AZD6918 combination group (P = 0.0005) had significant survival advantage. Furthermore, the etoposide (20 mg/kg) and AZD6918 combination group had survival advantage compared to etoposide (20 mg/kg)-treated group (P = 0.046), or to AZD6918-treated group (P = 0.0053). These data indicated that a combination of AZD6918 and etoposide increased the survival of mice compared to each individual treatment.
Discussion
In this study, we show that Trk inhibitor AZD6918 inhibited TB3 cell growth in a caspase-dependent manner, and blocked BDNF/TrkB-induced chemoresistance in TB3 cells in vitro. AZD6918 increased the sensitivity of TB3 xenograft tumors to etoposide treatment in vivo.
Clinical observation has indicated that TrkB and BDNF are expressed in a subset of aggressive or unfavorable NB.Citation5,10 To understand how activation of the BDNF/TrkB pathway affected the biology of NB tumors, we and other investigators determined that BDNF activation of TrkB did not significantly alter NB cell growth but enhanced cell survival under stress conditions such as serum starvation.Citation11,12 Subsequently, we reported that BDNF activation of TrkB induced NB cell resistance to the cytotoxic drugs.Citation7-9 Given the role of TrkB in the biological and clinical behavior of NB, it was reasonable to pursue inhibition of the TrkB receptor as an important adjunct to therapy. Lestaurtinib (CEP-701) is a Trk tyrosine kinase inhibitor that blocks activated Trk and has shown efficacy alone or in combination with conventional chemotherapy in NB cells.Citation13 Other Trk inhibitors, AZ623 and AZ64, were found to inhibit activation of TrkB in vitro and had a synergistic effect on tumor growth with topotecan in vivo.Citation14,15 In our current study, we found that while AZD6918 may not be sufficient to inhibit tumor growth or increase mice survival as a single agent at the tested doses (70 mg/kg, 100 mg/kg), it did increase the sensitivity of TB3 xenograft tumors to etoposide, a topoisomerase II inhibitor that is currently used in the treatment of NB. This result is consistent with the studies of other Trk inhibitors Lestaurtinib or AZ623 and AZ64.Citation13-15
While CEP-701 has shown activity against a range of in vivo models including prostate, pancreatic and thyroid cancer xenografts,Citation16-18 CEP-701 inhibits several additional tyrosine kinase targets such as PDGFR, VEGFR, RET and PKC,Citation18,19 and remains active in trials in clinical settings in which Trk kinases are not believed to pathogenic, such as acute myeloid leukemia and myeloproliferative disease.Citation20,21 Therefore, development of potent and selective Trk kinase inhibitors remains critical to evaluate the role of Trk signaling pathway inhibition in treatment of patients with NB. Our previous studies have shown that in the absence of tetracycline, TrkB was expressed in the TB3 cells in vitro and TB3 xenograft tumors in vivo.Citation7,8 TB3 cells don't secrete BDNF, so in our previous studies and in this study we added BDNF to the cells to activate TrkB in vitro.Citation7,8 From our previous in vivo study, TrkB and its downstream targets such as Akt are activated in situCitation8 most probably because the levels of BDNF in the blood and tissues is sufficiently high enough to activate TrkB.Citation22 We didn't observe any obvious side effect of AZD6918 at either the 70 mg/kg or 100 mg/kg doses except a slight decrease in body weight. While we did not observe any enhanced activity when we increased the dose to 100 mg/kg, there were solubility issues at higher concentrations. Compared to AZD6918, the AZ623 Trk inhibitor appears to have better solubility.Citation14
Previously, Trk inhibitors have been shown to increase the sensitivity of NB cells to a topoisomerase I inhibitor (topotecan) aloneCitation14 or in combination with an alkylating agent (irinotecan and temozolimide).Citation15 In this study we show that inhibition of the Trk activity can also increase the cytotoxic activity of etoposide, a topoisomerase II inhibitor. This study extends the spectrum of cytotoxic agents whose in vivo activity in pre-clinical models can be increased by inhibition of Trk activity.Citation14 Moreover, we found a survival advantage for mice in the group treated with a combination of AZD6918 and etoposide compared to the groups treated with either agent alone. Our results and the previous studies Citation14,15 provide compelling evidence that the combination of a Trk inhibitor with current cytotoxic therapies will improve treatment efficacy.
Materials and Methods
Cell culture
Human NB TB3, BE2 and KCNR cells were used in this study. TB3 cells have a transfected tetracycline (TET)-regulated rat TrkB. In the absence of TET, TrkB is expressed in the TB3 cells. NB cells were cultured in RPMI-1640 containing 10% fetal bovine serum (FBS), 2 mM of glutamine, and antibiotics as described previously.Citation7
Cell treatments
NB cells were seeded into 96-well plates in triplicate, incubated overnight and then treated with drugs. To study the effect of AZD6918 on cell survival, NB cells were treated with AZD6918 at concentrations ranging from 1.25 μM to 60 μM for 24 hrs. To study the blockage effect of AZD6918 on BDNF/TrkB protection from etoposide-induced cell death, TB3 cells were first treated with AZD6918 for 2 hrs followed by treatment with BDNF and etoposide for 24 hrs.
Cell survival analysis
The MTS assay (3-[4,5-dimethylthiazol-2-yl]-5-[3-carboxymethoxyphenyl]-2-[4-sulfophenyl]-2H-tetrazolium, inner salt assay) was performed according to the manufacturer's instructions (Promega Corporation). The percentage of cell survival (survival rate) was calculated by dividing the absorbance value of the treated samples by the absorbance value of the untreated control within each group. All experiments were repeated 2 to 3 times.
Assay of Caspase3/7 activity in vitro
TB3, BE2 and KCNR cells were seeded into 96-well plates in triplicate, incubated overnight and then treated with AZD6918 at concentrations ranging from 1.25 μM to 20 μM for 16 hrs, the combined activity of caspase-3/7 was evaluated using the Caspase-Glo 3/7 Assay Kit (Promega, WI) according to the manufacturer's instruction.
Western blotting
To evaluation the effect of AZD6918 on P-TrkB, P-Akt, P-mTOR and P-GSK-3, NB cells were treated with AZD6918 (2.5 μM) for 2 h (BE2, KCNR cells) or followed by BDNF (100 ng/ml) treatment for 1 h (TB3 cells), and then protein lysate was extracted. 20 μg protein in each condition was loaded onto SDS-PAGE gels, transferred to nitrocellulose and probed with the anti-P-TrkB antibody (1:500 dilution, Cell Signaling Tech.), anti-P-Akt, anti-P-mTOR and anti-P-GSK-3 antibody (1:250 dilution, 1:500 dilution and 1:500 dilution, Cell Signaling Tech.) or anti-GAPDH antibody(1:10000 dilution, Kangchen bio-tech). Signals were detected using enhanced chemiluminescence reagents (Amersham Life Science, Arlington Heights, IL, USA).
Transfection
Activated Akt plasmid and empty vector were transfected into TB3 cells by Lipofectamine 2000 purchased from Invitrogen. TB3 cells were treated with different concentrations of AZD6918 24 h after transfection.
In vivo animal model
TB3 cells were cultured in RPMI-1640 and 10% FBS media, harvested, washed with Hank balanced salt solution (HBSS), and re-suspended in HBSS and Matrigel (Trevigen Inc., Gaithersburg, Md). A total of 100 μl of cell suspension containing 4 × 106 TB3 cells were implanted into the subcutaneous tissue of the right flank of female nude mice ages 4–5 weeks (Taconic, Germantown, NY). Treatment was initiated when tumors reached approximately 200 mm3. Etoposide or AZD6918 was given individually or in combination. Etoposide was given 3 times a week at doses of 10 mg/kg and 20 mg/kg by intraperitoneal injection. AZD6918 was given twice a day by oral gavage at doses of 70 mg/kg and 100 mg/kg. The dimensions of the resulting tumors were determined at least 3 times a week using a digital caliper and the tumor volume (mm3) was calculated as (LxW2)/4, in which L indicates length in mm and W indicates width in mm. All mice were euthanized by asphyxiation with regulated carbon dioxide.
To determine mice survival, we counted the days from the date treatment was administered to the time when tumors reached a length of 20 mm, or to the end of the experiment in treatment groups. The Kaplan–Meier method was used to determine the probability of survival as a function of time.
All xenograft studies were approved by the Animal Care and Use Committee of the National Cancer Institute, and all mouse treatments, including their housing, were in accordance with the institutional guidelines (PB-023).
Statistical analysis
Comparisons between 2 groups were performed using the Student t test; comparisons among ≥3 groups were performed using 1-way analysis of variance with Bonferroni correction. The results were shown as the means ± the standard error.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank AstraZeneca Biopharmaceutical Company for providing AZD6918.
Funding
This work was supported by NCI Intramural Research Program at National Cancer Institute in National Institute of Health, National Natural Science Foundation of China (No. 81272538, No. 81472359), Society Development Foundation of Liaoning Province (No. 2012225016), 2013 Liaoning Climbing Scholar Foundation.
References
- Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, De Bernardi B, Evans AE, Favrot M, Hedborg F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 1993; 11:1466-77; PMID:8336186
- Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol 1999; 17:2264-79; PMID:10561284
- Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 2003; 3:203-16; PMID:12612655; http://dx.doi.org/10.1038/nrc1014
- Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet 2007; 369:2106-20; PMID:17586306; http://dx.doi.org/10.1016/S0140-6736(07)60983-0
- Nakagawara A, Arima-Nakagawara M, Scavarda NJ, Azar CG, Cantor AB, Brodeur GM. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N Engl J Med 1993; 328:847-54; PMID:8441429; http://dx.doi.org/10.1056/NEJM199303253281205
- Scala S, Wosikowski K, Giannakakou P, Valle P, Biedler JL, Spengler BA, Lucarelli E, Bates SE, Thiele CJ. Brain-derived neurotrophic factor protects neuroblastoma cells from vinblastine toxicity. Cancer Res 1996; 56:3737-42; PMID:8706017
- Jaboin J, Kim CJ, Kaplan DR, Thiele CJ. Brain-derived neurotrophic factor activation of TrkB protects neuroblastoma cells from chemotherapy-induced apoptosis via phosphatidylinositol 3'-kinase pathway. Cancer Res 2002; 62:6756-63; PMID:12438277
- Li Z, Oh DY, Nakamura K, Thiele CJ. Perifosine-induced inhibition of Akt attenuates brain-derived neurotrophic factor/TrkB-induced chemoresistance in neuroblastoma in vivo. Cancer 2011; 117:5412-22; PMID:21590687; http://dx.doi.org/; http://dx.doi.org/10.1002/cncr.26133
- Li Z, Jaboin J, Dennis PA, Thiele CJ. Genetic and pharmacologic identification of Akt as a mediator of brain-derived neurotrophic factor/TrkB rescue of neuroblastoma cells from chemotherapy-induced cell death. Cancer Res 2005; 65:2070-5; PMID:15781614; http://dx.doi.org/10.1158/0008-5472.CAN-04-3606
- Schramm A, Schulte JH, Astrahantseff K, Apostolov O, Limpt Vv, Sieverts H, Kuhfittig-Kulle S, Pfeiffer P, Versteeg R, Eggert A. Biological effects of TrkA and TrkB receptor signaling in neuroblastoma. Cancer Lett 2005; 228:143-53; PMID:15921851; http://dx.doi.org/10.1016/j.canlet.2005.02.051
- Nakagawara A, Azar CG, Scavarda NJ, Brodeur GM. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol Cell Biol 1994; 14:759-67; PMID:8264643
- Matsumoto K, Wada RK, Yamashiro JM, Kaplan DR, Thiele CJ. Expression of brain-derived neurotrophic factor and p145TrkB affects survival, differentiation, and invasiveness of human neuroblastoma cells. Cancer Res 1995; 55:1798-806; PMID:7712490
- Norris RE, Minturn JE, Brodeur GM, Maris JM, Adamson PC. Preclinical evaluation of lestaurtinib (CEP-701) in combination with retinoids for neuroblastoma. Cancer Chemother Pharmacol 2011; 68:1469-75; PMID:21484309; http://dx.doi.org/10.1007/s00280-011-1623-y
- Zage PE, Graham TC, Zeng L, Fang W, Pien C, Thress K, Omer C, Brown JL, Zweidler-McKay PA. The selective Trk inhibitor AZ623 inhibits brain-derived neurotrophic factor-mediated neuroblastoma cell proliferation and signaling and is synergistic with topotecan. Cancer 2011; 117:1321-91; PMID:20960503; http://dx.doi.org/10.1002/cncr.25674
- Iyer R, Varela CR, Minturn JE, Ho R, Simpson AM, Light JE, Evans AE, Zhao H, Thress K, Brown JL, et al. AZ64 inhibits TrkB and enhances the efficacy of chemotherapy and local radiation in neuroblastoma xenografts. Cancer Chemother Pharmacol 2012; 70:477-86; PMID:22623209; http://dx.doi.org/10.1007/s00280-012-1879-x
- Miknyoczki SJ, Chang H, Klein-Szanto A, Dionne CA, Ruggeri BA. The Trk tyrosine kinase inhibitor CEP-701 (KT-5555) exhibits significant antitumor efficacy in preclinical xenograft models of human pancreatic ductal adenocarcinoma. Clin Cancer Res 1999; 5:2205-12; PMID:10473107
- Weeraratna AT, Dalrymple SL, Lamb JC, Denmeade SR, Miknyoczki S, Dionne CA, Isaacs JT. Pan-trk inhibition decreases metastasis and enhances host survival in experimental models as a result of its selective induction of apoptosis of prostate cancer cells. Clin Cancer Res 2001; 7:2237-45; PMID:11489797
- Strock CJ, Park JI, Rosen M, Dionne C, Ruggeri B, Jones-Bolin S, Denmeade SR, Ball DW, Nelkin BD. CEP-701 and CEP-751 inhibit constitutively activated RET tyrosine kinase activity and block medullary thyroid carcinoma cell growth. Cancer Res 2003; 63:5559-63; PMID:14500395
- Shabbir M, Stuart R. Lestaurtinib, a multitargeted tyrosine kinase inhibitor: from bench to bedside. Expert Opin Investig Drugs 2010; 19:427-36; PMID:20141349; http://dx.doi.org/10.1517/13543781003598862
- Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, Murphy KM, Dauses T, Allebach J, Small D. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood 2004; 103:3669-76; PMID:14726387; http://dx.doi.org/10.1182/blood-2003-11-3775
- Hexner EO, Serdikoff C, Jan M, Swider CR, Robinson C, Yang S, Angeles T, Emerson SG, Carroll M, Ruggeri B, et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood 2008; 111:5663-71; PMID:17984313; http://dx.doi.org/10.1182/blood-2007-04-083402
- Grade S, Weng YC, Snapyan M, Kriz J, Malva JO, Saghatelyan A. Brain-derived neurotrophic factor promotes vasculature-associated migration of neuronal precursors toward the ischemic striatum. PLoS One 2013; 8:e55039; PMID:23383048; http://dx.doi.org/10.1371/journal.pone.0055039