
Abstract
Mitogen activated protein kinase phosphatase-1 (MKP-1) has emerged as an important protein mediating breast cancer oncogenesis and chemoresistance to cancer chemotherapies, especially proteasome inhibitors. In this in vitro study, we utilized the breast cancer epithelial cell lines MCF-7 and MDA-MB-231, in comparison to MCF-10A control cells, to examine the impact of MKP-1 on breast cancer cell growth and repression by proteasome inhibitors. We confirm that proteasome inhibitors MG-132 and bortezomib induce MKP-1 protein upregulation and we show that one of the ways in which bortezomib increases MKP-1 in breast cancer cells, in addition to inhibition of ubiquitin-proteasome system, is via upregulation of MKP-1 mRNA expression in p38 MAPK-mediated manner. Notably, these effects are specific to cancer cells, as bortezomib activated p38 MAPK and induced MKP-1 in MCF-7 and MDA-MB-231 breast cancer cells, but not in control cells (MCF-10A). We took a dual approach toward targeting MKP-1 to show that bortezomib-induced effects are enhanced. Firstly, treatment with the non-specific MKP-1 inhibitor triptolide reduces breast cancer cell growth and augments proteasome inhibitor-induced effects. Secondly, specific knock-down of MKP-1 with siRNA significantly repressed cell viability by reduced cyclin D1 expression, and enhanced repression of cancer cell growth by proteasome inhibitors. Taken together, these results indicate that removing the unwanted (MKP-1-inducing) effects of bortezomib significantly improves the efficacy of proteasome inhibition in breast cancer cells. Thus, future development of drugs targeting MKP-1 offer promise of combination therapies with reduced toxicity and enhanced cell death in breast cancer.
Abbreviations
| ERK | = | extracellular signal-regulated kinase |
| MAPK | = | mitogen activated protein kinase |
| MKP-1 | = | mitogen activated protein kinase phosphatase-1 |
| TTP | = | tristetraprolin |
Introduction
Inhibition of the ubiquitin-proteasome system has emerged as an anti-cancer drug target. Proteasome inhibitors targeting the 26S proteasome have shown clinical efficacy as monotherapy and in combination therapy in a broad range of malignancies. Bortezomib was the first-in-class to receive FDA approval for the treatment of relapsed multiple myelomaCitation1 or refractory mantle cell lymphoma.Citation2 But despite demonstration of the anti-proliferative effects of bortezomib in a variety of cell lines in vitro, as well as preclinical models and clinical trials, widespread therapeutic use may be hampered by development of resistance.Citation3 Several mechanisms of resistance have been proposed.Citation4 In breast cancer, one of the important mechanisms underlying resistance is the upregulation of mitogen activated protein kinase phosphatase-1 (MKP-1).Citation5-7
MKP-1 is a dual-specificity protein which inactivates mitogen activated protein kinases (MAPK) by dephosphorylating both phospho-threonine and phospho-tyrosine residues. By preferentially inactivating the c-Jun-NH2-terminal kinases (JNK) subfamily, MKP-1 is considered to diminish pro-apoptotic signaling, thereby promoting oncogenesis and/or chemoresistance.Citation7,8 MKP-1 is linked with poor prognosis in breast cancer.Citation7,9 Thus, further understanding of the effect of MKP-1 on breast cancer cell growth, the action of bortezomib in the context of breast cancer and strategies to target the unwanted MKP-1-inducing effects are warranted.
MKP-1 is a 367 amino acid protein expressed by an immediate-early gene.Citation10 It can be regulated at multiple levels; transcriptionally, post-transcriptionally and post-translationally. Stimuli which activate MAPK (such as p38 MAPK) also induce MKP-1 expression.Citation11,12 It can be rapidly translated, however, the increase in MKP-1 protein levels is usually transient, as MKP-1 protein then undergoes rapid degradation by the proteasomal machinery, controlled by post-translational modifications of regulatory motifs on MKP-1.Citation13 In models of breast cancer utilizing MDA-MB-231 cells, Shi et al.Citation6 previously demonstrated that proteasome inhibitors induce a p38 MAPK-mediated MKP-1 dependent anti-apoptotic program. Moreover, as MKP-1 is proteasomally degraded, the possibility exists that blocking the proteasome activity will allow a build-up up of MKP-1 in breast cancer cells. These studies indicate that p38 MAPK-mediated expression of MKP-1 is important in breast cancer cells, as it is in other cell typesCitation11,12,14; thus examination of the underlying mechanisms may provide further information to aid understanding of proteasome inhibitor action.
Therefore, in this study we investigated the upregulation of MKP-1 in response to proteasome inhibition in MCF-7 and MDA-MB-231 breast cancer cells, compared to a non-tumorigenic epithelial cell line MCF-10A and demonstrated that targeting MKP-1 enhances bortezomib-induced effects. Specifically, we show that when the proteasome is inhibited by MG-132 and bortezomib, MKP-1 levels increase in a time-dependent manner. We show that when the proteasome is inhibited by bortezomib, MKP-1 mRNA increases in a p38 MAPK-mediated manner. The p38 MAPK-mediated upregulation of MKP-1 induced by bortezomib appears specific to breast cancer cells, as MKP-1 was not upregulated by bortezomib treatment in MCF-10A. Taking two approaches to target MKP-1: triptolide (a non-specific pharmacological inhibitor of MKP-1 upregulation) as well as siRNA against MKP-1; we demonstrated that in the absence of MKP-1, breast cancer cell growth is significantly repressed. Finally, we show that treating cells with bortezomib when cells are unable to make MKP-1 (because it has been knocked down) results in an additional repression of cell viability. Taken together, these results indicate that removing the unwanted (MKP-1-inducing) effects of bortezomib significantly enhances repression of breast cancer cell growth in vitro.
Results
Proteasome inhibitors induce upregulation of MKP-1 protein, while MKP-1 is non-specifically inhibited by triptolide
To examine the effect of proteasome inhibition on MKP-1 protein upregulation, we treated the metastatic breast cancer epithelial cell line MDA-MB-231 with MG-132 (10 μM), or the more potent derivatized dipeptide boronate bortezomib (10 nM), for up to 24 h. As demonstrated in , proteasome inhibitors robustly induced upregulation of MKP-1 over time. Both MG-132 and bortezomib induced an increase in MKP-1 protein after 1 – 2 h that was sustained for 24 h. In contrast, when cells were treated with triptolide, a tri-epoxide phenanthrene purified from Tripterygium wilfordii and a non-specific inhibitor of MKP-1,Citation12,15-17 basal MKP-1 protein expression was repressed. As demonstrated in , MKP-1 levels were reduced after 1 h treatment with triptolide (1 μM), and after 4 h of treatment MKP-1, protein was undetectable by Western blotting.
Figure 1. Proteasome inhibitors induce upregulation of MKP-1 protein, while MKP-1 is non-specifically inhibited by triptolide. MDA-MB-231 cells were treated with vehicle, 10 μM MG-132, 10 nM bortezomib, or 1 μM triptolide for up to 24 h. MKP-1 protein was measured by Western blotting (with α-tubulin used as the loading control) and results shown are representative blots of n = 3 independent experiments.

MKP-1 mRNA is augmented following bortezomib treatment
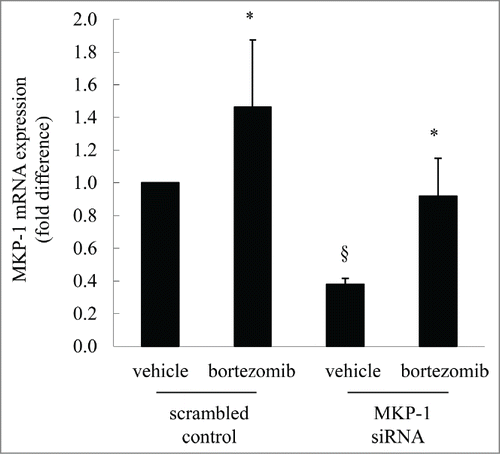
The data shown above serves to confirm that proteasome inhibitors increase protein levels of MKP-1. Although this upregulation may be due, in part, to accumulation of MKP-1 (a protein regulated by the ubiquitin-proteasome system) we were intrigued to explore whether proteasome inhibitors also induce upregulation of MKP-1 at the mRNA level. To address this, we transfected cells with MKP-1 siRNA to knock-down MKP-1, compared to scrambled control, and measured resultant MKP-1 mRNA expression. We now show, for the first time, that bortezomib increases MKP-1 mRNA expression (). Specifically, bortezomib treatment in cells transfected with the scrambled control significantly increased MKP-1 mRNA by 1.46 ± 0.41-fold (P < 0.05). MKP-1 expression in scrambled control cells can be significantly knocked-down to 0.38 ± 0.04-fold following transfection with siRNA against MKP-1 (P < 0.05). The degree of MKP-1 knock-down can be rescued by treatment with bortezomib; this is shown in , where the MKP-1 expression levels in cells transfected with MKP-1 siRNA are significantly increased by 242.2% after treatment with bortezomib (P < 0.05).
Figure 2. MKP-1 mRNA is augmented following bortezomib treatment. MDA-MB-231 cells were transfected with scrambled control or MKP-1 siRNA for 24 h in antibiotic-free media. Media was then changed to antibiotic-containing media and cells incubated for further 18 h, before treatment for 6 h with vehicle or bortezomib (10 nM). MKP-1 mRNA was quantified by real-time RT-PCR and results expressed as fold difference compared to cells transfected with scrambled control treated with vehicle (designated as 1). Statistical analysis was performed using the Student's unpaired t test, where * denotes a significant increase in MKP-1 mRNA expression after bortezomib treatment and § denotes significant knockdown of MKP-1 by siRNA (P < 0.05). Data are mean+SEM values from n = 7 independent experiments.
Bortezomib increases p38 MAPK phosphorylation and MKP-1 protein upregulation
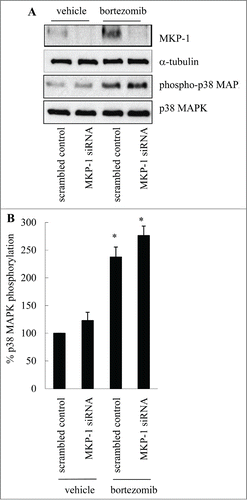
The protein results () are in accord with the mRNA results (). Western blotting demonstrates that bortezomib increases MKP-1 protein in cells transfected with the scrambled control alone (). This can be substantially knocked-down by siRNA against MKP-1. Additionally, we were interested to demonstrate the involvement of p38 MAPK as Shi et al.Citation6 had previously reported that MKP-1 upregulation in MDA-MB-231 cells was p38 MAPK-mediated. As shown in , and confirmed by densitometry in , treatment with bortezomib significantly increases p38 MAPK phosphorylation in MDA-MB-231 cells (P < 0.05). These results are in alignment with earlier studies in MDA-MB-231,Citation6 as well as in cell types apart from MDA-MB-231.Citation11,12,14
Figure 3. Bortezomib increases p38 MAPK phosphorylation and MKP-1 protein upregulation. MDA-MB-231 cells were transfected with scrambled control or MKP-1 siRNA for 24 h in antibiotic-free media. Media was then changed to antibiotic-containing media and cells incubated for further 18 h, before treatment for 6 h with vehicle or bortezomib (10 nM). Western blotting was performed using specific antibodies against MKP-1 (with α-tubulin used as the loading control), phosphorylated (Thr180/Tyr182) and total p38 MAPK. (A) Results are representative Western blots, while (B) demonstrates densitometric analysis where results are expressed as% p38 MAPK phosphorylation compared to cells transfected with scrambled control treated with vehicle (designated as 100%). Statistical analysis was performed using the Student's unpaired t test, where * denotes a significant increase in p38 MAPK phosphorylation (P < 0.05). Data are mean+SEM values from n = 3 independent experiments.
The p38 MAPK inhibitor SB203580 inhibits bortezomib-induced MKP-1 protein upregulation
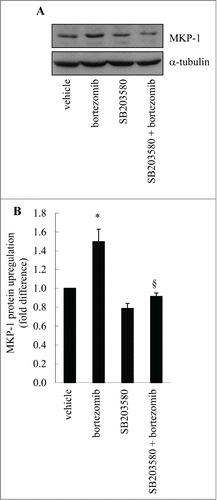
To demonstrate that p38 MAPK activation is responsible for bortezomib-induced MKP-1, we pretreated MDA-MB-231 cells with the p38 MAPK inhibitor SB203580 before treatment with bortezomib and examined MKP-1 protein upregulation by Western blotting. As shown in , bortezomib increased MKP-1 protein upregulation and this was repressed by SB203580. SB203580 alone was without effect. These data were confirmed by densitometric analysis, as shown in , where bortezomib increased MKP-1 protein upregulation by 1.5 ± 0.1 fold, and this was significantly repressed by SB203580 (: P < 0.05). These data provide further support that bortezomib induces MKP-1 in a p38 MAPK-mediated manner.
Figure 4. The p38 MAPK inhibitor SB203580 inhibits bortezomib-induced MKP-1 protein upregulation. MDA-MB-231 cells were pretreated with vehicle or SB203580 (10 μM), before treatment for 6 h with vehicle or bortezomib (10 nM). Western blotting was performed using specific antibodies against MKP-1 (with α-tubulin used as the loading control). (A) Results are representative Western blots, while (B) demonstrates densitometric analysis where results are expressed as fold difference compared to vehicle-treated cells (designated as 1). Statistical analysis was performed using the Student's unpaired t test, where * denotes a significant increase in MKP-1 upregulation after bortezomib treatment and § denotes significant repression by SB203580 (P < 0.05). Data are mean+SEM values from n = 3 independent experiments.
The non-specific MKP-1 inhibitor triptolide reduces breast cancer cell growth and augments proteasome inhibitor-induced effects
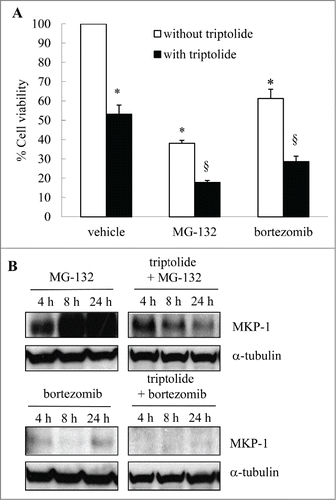
In vivo increased MKP-1 is linked with a poor prognosis in breast cancer. Increased MKP-1 has also been reported as one of the ways in which cancers become refractory to the beneficial effects of proteasome inhibition. This suggests that separating the wanted (growth-inhibitory) effects of bortezomib from the unwanted (MKP-1-inducing) effects of bortezomib may result in improved chemotherapeutics in the future. To examine this, we repressed MKP-1 levels with the non-specific pharmacological inhibitor triptolide and examined the effect of bortezomib, alongside MG-132, on cell viability in MDA-MB-231. As shown in , the first generation proteasome inhibitor, MG-132, repressed cell viability to 38.1 ± 1.5% of vehicle-treated control (P < 0.05). Bortezomib also inhibited cell viability by 38.7 ± 4.8%. Triptolide alone significantly repressed cell viability in confirmation of earlier studies.Citation18,19 When MKP-1 was non-specifically inhibited by triptolide, repression of breast cancer cell growth by MG-132 and bortezomib was significantly enhanced (P < 0.05). It is important to note that the beneficial effect of triptolide and proteasome inhibitors co-treatment appear to be additive and not synergistic. This suggests that MKP-1 is only one of the ways in which proteasome inhibitors exert chemoresistance. Triptolide also has many cellular targets, including the NF-κB pathway.Citation20,21 Western blotting shown in confirms that triptolide represses MKP-1 upregulation induced by MG-132 or bortezomib.
Figure 5. The non-specific MKP-1 inhibitor triptolide reduces breast cancer cell growth and augments proteasome inhibitor-induced effects. MDA-MB-231 cells were treated with vehicle, MG-132 (10 μM) or bortezomib (10 nM); in the absence or presence of triptolide (1 μM) for 4, 8 and 24 h. (A) Cell viability was measured by MTT assay at 24 h and results expressed as% cell viability compared to cells treated with vehicle alone (designated as 100%). Statistical analysis was performed using the Student's unpaired t test, where * denotes a significant effect on cell viability, and § denotes enhanced effects of proteasome inhibitors in the presence of triptolide (P < 0.05). Data are mean+SEM values from n = 4 independent experiments. (B) Western blotting was performed at 4, 8 and 24 h using specific antibodies against MKP-1 (with α-tubulin used as the loading control) where results are representative Western blots of n = 3 independent experiments.
Targeting MKP-1 with siRNA reduces breast cancer cell growth and enhances bortezomib-induced effects in MDA-MB-231 cells in a manner linked with reduced cyclin D1 expression
Because triptolide is a non-specific inhibitor of MKP-1, and has many cellular targets,Citation20,21 we extended our study to specifically knock-down MKP-1 with siRNA and assess cell viability (measured by MTT). MDA-MB-231 cells were transfected with either scrambled control or siRNA against MKP-1, and then treated with bortezomib or vehicle-control. As shown in , cells transfected with scrambled control grew over time to reach 406.8 ± 56.3% at 72 h, compared to 24 h (designated as 100%). When MKP-1 was knocked-down by siRNA the growth rate was significantly repressed (: P < 0.05). We found that targeting MKP-1 enhanced the repressive effects of bortezomib. Treating cells transfected with scrambled control with bortezomib decreased MDA-MB-231 cell growth; the percentage cell viability at 72 h was reduced to 165.9 ± 29.8% (: P < 0.05), when compared to scrambled control at the same time point (: P < 0.05). Notably, we also demonstrate that removing MKP-1with siRNA enhances bortezomib-induced effects in MDA-MB-231, with the cell viability at 72 h significantly reduced to 110.6 ± 15.6% (P < 0.05).
Figure 6. Targeting MKP-1 with siRNA reduces breast cancer cell growth and enhances bortezomib-induced effects in MDA-MB-231 cells in a manner linked with cyclin D1 expression. (A) MDA-MB-231 cells were transfected with scrambled control or MKP-1 siRNA for 24 h in antibiotic-free media. Media was then changed to antibiotic-containing media containing vehicle or bortezomib (10 nM) and cell viability measured by MTT assay at 24, 48 and 72 h. Results are expressed as% cell viability compared to cells transfected with scrambled control and treated with vehicle at 24 h (designated as 100%). Statistical analysis was performed using 2-way ANOVA then Bonferroni's post-test, where § denotes a significant effect of MKP-1 knockdown on cell viability, and * denotes enhanced bortezomib-induced effects in cells transfected with siRNA against MKP-1 (P < 0.05). Data are mean ± SEM values from n = 4 independent experiments. (B, C) MDA-MB-231 cells were transfected with scrambled control or MKP-1 siRNA for 24 h in antibiotic-free media. Media was then changed to antibiotic-containing media and cells incubated for further 18 h, before treatment for 6 h with vehicle or bortezomib (10 nM). (B) Cyclin D1 mRNA and (C) TTP were quantified by real-time RT-PCR and results expressed as fold difference compared to cells transfected with scrambled control treated with vehicle (designated as 1). Statistical analysis was performed using the Student's unpaired t test, where * denotes a significant decrease in cyclin D1 mRNA expression after bortezomib treatment and § denotes significant effect of MKP-1 siRNA (P < 0.05). Data are mean+SEM values from n = 7 independent experiments.
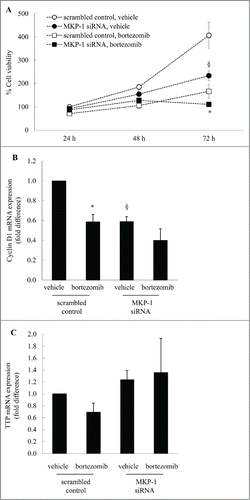
The effect of treatments on cell proliferation was linked to expression of a key cell cycle component – cyclin D1. Cyclin D1 drives G1-to-S-phase progression and as shown in , cyclin D1 mRNA expression in scrambled control cells was significantly knocked-down to 0.59 ± 0.05-fold following transfection with siRNA against MKP-1 (P < 0.05). Bortezomib also significantly reduces cyclin D1 expression (to 0.59 ± 0.08: P < 0.05) and this can be reduced further when MKP-1 is knocked-down by siRNA (to 0.40 ± 0.12), although the difference is not significant. These studies show that knocking down MKP-1 correspondingly decreases the mRNA expression of a key cell cycle component – cyclin D1. How MKP-1 regulates cyclin D1 may be due to the close association between MKP-1 and the mRNA destabilizing molecule – tristetraprolin (TTP). TTP is a RNA binding protein that binds to the 3′-untranslated region of many target mRNAs to confer instability, including cyclin D1.Citation22 Intriguingly, we found that knocking down MKP-1 results in a corresponding upregulation of TTP expression. We propose that TTP acts to repress cyclin D1 expression and cell growth in a manner linked to MKP-1. However, although we have focused on cyclin D1 in this study, it is important to note that cyclin D1 expression is expressed during G1 phase of the cell cycle and a decrease in cell viability can be due to accumulation of cells in another phase of the cell cycle. These studies reveal, in part, the downstream effectors responsible for repression of breast cancer cell growth by MKP-1 down-regulation.
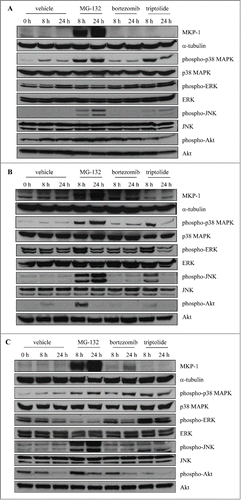
Bortezomib induces MKP-1 in a p38 MAPK-dependent manner in MCF-7 and MDA-MB-231 breast cancer cells, but not control cells (MCF-10A)
Taken together, these results indicate that by removing unwanted (MKP-1-inducing) effects in MDA-MB-231 cells, bortezomib-induced repression of breast cancer cell growth can be enhanced. In order to broaden our study on the role of MKP-1 in cancer cell proliferation, we were intrigued to see if this effect was specific to MDA-MB-231 cells, or whether this bortezomib induced-increase in MKP-1 was a generalizable effect shown to occur in other breast cancer cell lines. To address this, we performed additional experimentation in MCF-7 breast cancer cells. Moreover, in order to demonstrate the significance of these effects on breast cancer cells compared to "normal" cells, control experiments were run with MCF-10A; a non-tumorigenic epithelial cell line. We then performed parallel studies to examine the effect of bortezomib on all 3 cell lines (see ). Interestingly, bortezomib induced MKP-1 in a p38 MAPK-dependent manner in MCF-7 () and MDA-MB-231 () breast cancer cells, but not control cells (MCF-10A) (). Additionally, we assessed the effect of the non-specific proteasome inhibitor, MG-132, as well as the non-specific MKP-1 inhibitor triptolide. MG-132 was equally effective at activating p38 MAPK phosphorylation and upregulating MKP-1 in all 3 cell lines. There was no MKP-1 detected in cells treated with triptolide and notably, we observed a corresponding increase in p38 MAPK phosphorylation that was consistent with loss of MAPK-deactivating phosphatase. These results demonstrate that bortezomib induces MKP-1 in a p38 MAPK-dependent manner in breast cancer cell lines, when compared to control cells. For completeness, we have included Western blots for phospho- and total ERK, phospho- and total JNK, phospho- and total Akt for each cell line.
Figure 7. Bortezomib induces MKP-1 in a p38 MAPK-dependent manner in MCF-7 and MDA-MB-231 breast cancer cells, but not control cells (MCF-10A). (A) MCF-10A, (B) MCF-7 and (C) MDA-MB-231 were treated with vehicle, 10 μM MG-132, 10 nM bortezomib, or 1 μM triptolide for the indicated time points. Western blotting was performed using specific antibodies against MKP-1 (with α-tubulin used as the loading control), phosphorylated (Thr180/Tyr182) and total p38 MAPK, phosphorylated (Thr202/Tyr204) and total ERK, phosphorylated (Thr183/Tyr185) and total JNK, phosphorylated (Ser473) and total Akt and results shown are representative.
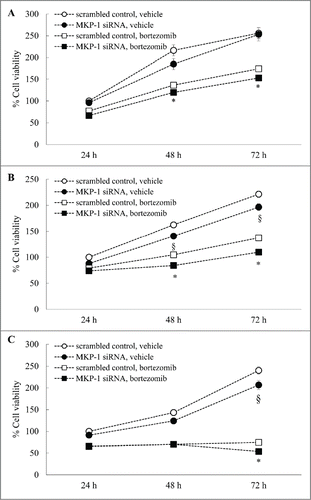
Reducing MKP-1 by siRNA has an impact on the effects of proteasome inhibition on MCF-7 and MDA-MB-231 breast cancer cell growth, but not control cells (MCF-10A)
We then compared the effect of specifically knocking-down MKP-1 with siRNA on the viability of all 3 cell lines measured by MTT, in the absence and presence of bortezomib (). In the non-tumorigenic epithelial cell line MCF-10A () there was no significant effect of knocking-down MKP-1 on the rate of growth of these control cells. In contrast, the rate of growth was significantly repressed in the breast cancer cell lines MCF-7 after siRNA against MKP-1 (: P < 0.05) and MDA-MB-231 (: P < 0.05). These data are in accord with in vivo studies linking MKP-1 with a breast cancer oncogenesis and are supported by previous studies linking MKP-1 to the promotion of the pro-proliferative phenotype in MCF-7 and MDA-MB-231 breast cancer cells.Citation7,9 We then examined the impact of bortezomib on cell viability and whether targeting MKP-1 enhances the repressive effects of bortezomib. Importantly, like MDA-MB-231 cells (), proteasome inhibition by bortezomib has a significant impact on the viability of MCF-7 cells (). Notably, the rate of growth of MCF-7 breast cancer cells can be further reduced by removing MKP-1with siRNA. As shown in , the percentage cell viability in MCF-7 treated with MKP-1 siRNA at 72 h was significantly reduced to 109.7 ± 5.7%, when compared to scrambled control at the same time point (137.4 ± 5.9%: P < 0.05). Knocking-down MKP-1with siRNA enhanced repression of MDA-MB-231 cell growth by bortezomib (: P < 0.05), in accordance with our earlier data (). There was also an effect of siRNA knockdown in control cells but this may be MKP-1-independent given previous results ().
Figure 8. Reducing MKP-1 by siRNA has an impact on the effects of proteasome inhibition on MCF-7 and MDA-MB-231 breast cancer cell growth, but not control cells (MCF-10A). (A) MCF-10A, (B) MCF-7 and (C) MDA-MB-231 cells were transfected with scrambled control or MKP-1 siRNA for 24 h in antibiotic-free media. Media was then changed to antibiotic-containing media containing vehicle or bortezomib (10 nM) and cell viability measured by MTT assay at 24, 48 and 72 h. Results are expressed as% cell viability compared to cells transfected with scrambled control and treated with vehicle at 24 h (designated as 100%). Statistical analysis was performed using 2-way ANOVA then Bonferroni's post-test, where § denotes a significant effect of MKP-1 knockdown on cell viability, and * denotes enhanced bortezomib-induced effects in cells transfected with siRNA against MKP-1 (P < 0.05).
Discussion
Success with bortezomib in the treatment of multiple myeloma has encouraged the exploration of small molecules targeting the ubiquitin-proteasome system as promising chemotherapeutics for solid tumors, including breast cancer. However, clinical trials utilizing bortezomib, either as monotherapyCitation23,24 or in combination therapy,Citation25 have demonstrated that further refinement of the treatment regimens are necessary to generate superior anti-tumor responses with proteasome inhibitors.
Proteasomes are multicatalytic 26S proteases consisting of a 20S central core catalytic subunit bordered by 2 19S components which hydrolyze peptide bonds of a multitude of proteins tagged by ubiquitination for degradation. By controlling the level, activity and location of various cellular proteins, proteasome inhibitors, alone or in combination with other chemotherapeutics, enhance death of cancer cells by apoptosis. Several generations of proteasome inhibitors have been developed: they range from MG-132, a cell permeable tripeptide aldehyde used widely to explore the proteasomal degradative machinery in vitro, to bortezomib (Velcade®) the first-in-class proteasome inhibitor approved by the FDA in 2003 as cancer therapy.
Recent evidence has suggested that clinical outcomes following treatment with proteasomal inhibitors may be limited by emergence of proteasome inhibitor-chemoresistance. As the multi-catalytic proteasome is capable of regulating the proteolysis of a large number of cellular proteins, it is not surprising that a concomitant effect of proteasome inhibition is to increase level of proteins that interfere with cell growth and apoptotic signaling. One important protein shown to interfere with chemotherapy-mediated signaling is MKP-1.Citation5–7 MKP-1 is the prototypical member of the MKP family that dephosphorylates members of the MAPK superfamily (p38 MAPK, JNK, extracellular signal-regulated kinase (ERK), in a cell-type specific manner. MKP-1 interferes with chemotherapy-mediated cell death through deactivation of the MAPK superfamily members responsible for repressing growth and driving apoptosis in cancer, the latter predominately considered to be JNK-driven.Citation26
Taken together these earlier studies implicate an important role for MKP-1 in breast cancer chemoresistance. To address this further we have performed a study that firstly examines the upregulation of MKP-1 by proteasome inhibitions and secondly, demonstrates the impact of bortezomib-induced MKP-1 upregulation on cell growth.
MKP-1 has been implicated as an important protagonist of chemoresistance, especially to proteasome inhibitors.Citation5-7 We provide the first demonstration that, in addition to the MKP-1 protein build-up due to proteasome inhibition, bortezomib also increases MKP-1 mRNA expression. This increase in gene expression is associated with increased p38 MAPK phosphorylation and MKP-1 protein upregulation in MCF-7 and MDA-MB-231. In contrast, bortezomib does not activate p38 MAPK phosphorylation, nor increase MKP-1 protein upregulation in non-cancer control cells. This p38 MAPK-dependence is in accord with an earlier study in MDA-MB-231 cells,Citation6 where inhibition of p38 MAPK suppressed induction of MKP-1. Moreover, p38 MAPK-mediated MKP-1 gene and protein expression have been demonstrated in other cell types; we have recently shown that MKP-1 expression in human airway smooth muscle cells is p38 MAPK-mediatedCitation11,12 and Hu et al. demonstrated similar findings in alveolar macrophages.Citation14
We have examined the upregulation and function of MKP-1in 2 breast cancer cell lines, MCF-7 and MDA-MB-231, and contrasted their response to that of a non-tumorigenic epithelial cell line, MCF-10A. In vivo increased MKP-1 is linked with a poor prognosis and development of oncogenesis.Citation7-9 The important role played by MKP-1 in the promotion of the pro-proliferative phenotype in breast cancer is supported by in vitro data in MCF-7 and MDA-MB-231 cells.Citation7,9 This is the first study to directly compare their MKP-1-dependent responses to a control cell line (MCF-10A). By taking a dual approach toward inhibition of MKP-1, we show that breast cancer cell growth is significantly attenuated in the absence of MKP-1. Firstly, we inhibit MKP-1 non-specifically with triptolide and show significant reduction in cell viability when MKP-1 is repressed. Secondly, we knock-down MKP-1 with specific siRNA and show similar repression of cancer cell growth. Notably, we show that targeting MKP-1 with siRNA reduces breast cancer cell growth in a manner linked with reduced cyclin D1 expression and increased TTP. Dual approaches to block MKP-1 are necessary as triptolide is not a specific inhibitor of MKP-1. It can also inhibit NF-κB transactivation, in addition to repressing other targets, including general transcription and the proteasome itself.Citation20,21,27 Hence, it is important to note that effects on cell viability observed with triptolide in , may be due to repression of a combination of targets (in addition to MKP-1) and thus may appear to have an exaggerated impact on cell viability. These studies underscore the importance of our approach to specifically targeting MKP-1 with siRNA.
Our study builds upon the findings originally reported by research group headed by R.Z. Orlowski and confirms their observations of MKP-1 induction by proteasome being responsible for breast cancer chemoresistance.Citation5-7 Our study adds to this literature through the novel observation of p38 MAPK-induced MKP-1 mRNA expression in MDA-MB-231 breast cancer cells. This new information adds to the known mechanisms of MKP-1 induction by proteasome inhibitors, viz, being post-translationally regulated, MKP-1 can accumulate when the proteasome is inhibited. These data support observations made by Shi et al.Citation6 where proteasome inhibitors were demonstrated to exert a p38 MAPK-mediated MKP-1 dependent anti-apoptotic program. Notably, these effects are specific to cancer cells, as bortezomib activated p38 MAPK and induced MKP-1 in MCF-7 and MDA-MB-231 breast cancer cells, but not in control cells (MCF-10A). Moreover, the earlier studies demonstrated the link for MKP-1 in breast cancer carcinogenesis through MKP-1 overexpression studies. Our study complements these earlier publications by showing the promise of inhibition of MKP-1 as a potential breast cancer treatment in the future. We specifically knock down MKP-1 with siRNA and repressed cell viability. Notably, as breast cancer cell lines express greater amounts of MKP-1 than “normal cells,” the impact of MKP-1 siRNA is greater in MDA-MB-231 and MCF-7, than in MCF-10A. Additionally, as bortezomib appears not to increase p38 MAPK in MCF-10A and correspondingly does not increase MKP-1 in control epithelial cells, this observation suggests some degree of selective toxicity may be achieved in the future with MKP-1 inhibitors.
Taken together, our results confirm that MKP-1 plays an important role in breast cancer cell oncogenesis and proteasome inhibitor-chemoresistance. We reveal a novel way in which bortezomib increase MKP-1 gene expression and take a dual approach toward inhibiting MKP-1 to demonstrate enhanced repression of breast cancer cell growth by proteasome inhibitors in vitro. Thus, combination therapies with MKP-1 inhibitors may offer promise of enhanced cancer cell death following breast cancer treatment with proteasome inhibitors.
Materials and Methods
Cell culture
MCF-10A cell lines were the generous gift of Drs. Robert Pauley and Herbert Soule, Karmanos Cancer Institute (Detroit, MI)Citation28,29 to JLM. MCF-10A cells were cultured according to previously established conditionsCitation29,30 in Dulbecco's Modified Eagle's Medium (DMEM)/Ham's F-12 mixture (1:1) supplemented with 5% horse serum, 10 μg/ml bovine insulin, 10 ng/ml epidermal growth factor, 0.5 μg/ml hydrocortisone and 100 ng/ml cholera enterotoxin. MCF-7 cells were purchased from American Type Culture Collection (Manassas, VA) and maintained in DMEM containing 10% heat-inactivated fetal bovine serum, 5 mM L-glutamine, 20 mM HEPES, 100 U/mL penicillin, 100 μg/mL streptomycin and 0.25 μg/mL amphotericin B. MDA-MB-231 cells, also from the American Type Culture Collection, were cultured in RPMI supplemented with 10% heat-inactivated fetal bovine serum, 5 mM L-glutamine, 10 μg/mL insulin, 100 U/mL pencillin, 100 μg/mL streptomycin and 0.25 μg/mL amphotericin B. All cell lines were maintained at 37°C in an atmosphere of 5% CO2 in air. A minimum of 3 independent experiments were performed.
Unless otherwise specified, all chemicals used in this study were purchased from Sigma-Aldrich (St. Louis, MO).
MKP-1 siRNA
Cells were transfected by reverse transfection according to the manufacturer's protocols (Invitrogen, NY, USA). Specifically, for each well of 6-well plates, 400 ng of ON-Target plus Control Non-targeting siRNA or ON-Target plus SMART pool Human DUSP1 (mixture of 4 siRNA from Dharmacon: Thermo Fisher Scientific, Waltham, MA) was diluted in 500 μL of Opti-MEM I Reduced Serum Media (Invitrogen) as scrambled sequence RNA (scrambled controls) and siRNA against MKP-1 (MKP-1 siRNA) treatments, respectively. This was followed by the addition of 5 μL of Lipofectamine RNAiMAX Reagent (Invitrogen) into each well and incubation at room temperature for 20 min. Cells were prepared in antibiotic-antimycotic-free feeding media at a concentration of 100,000 cells/mL and 2.5 mL added to each well. Plates were incubated for 24 hours then media changed to antibiotic-containing feeding media and cells used for either RT-PCR, Western blotting or cell viability experiments.
Real-time RT-PCR
Total RNA was extracted using the RNeasy Mini Kit (Qiagen Australia, Doncaster, VIC, Australia) and reverse transcription performed by using the RevertAid First strand cDNA Synthesis Kit (Fermentas Life Sciences, Hanover, MD) as per the manufacturer's protocol.
MKP-1 mRNA levels were measured using real-time RT-PCR on an ABI Prism 7500 (Applied Biosystems, Foster City, CA) with the MKP-1 (DUSP1: Hs00610256_g1), cyclin D1 (Hs00277039_m1) TaqMan® Gene Expression Assays and the eukaryotic 18S rRNA endogenous control probe (Applied Biosystems) subjected to the following cycle parameters: 50°C for 2 min, 1 cycle; 95°C for 10 min, 1 cycle; 95°C for 15 s, 60°C for 1 min, 40 cycles.
Western blotting
Western blotting was performed as per previous publicationsCitation11,12 using rabbit polyclonal antibodies against MKP-1 (C-19: Santa Cruz Biotechnology, Santa Cruz, CA) compared to α-tubulin as the loading control (mouse monoclonal IgG1, clone DM 1A: Santa Cruz). Antibodies to detect phosphorylated (Thr180/Tyr182) and total p38 MAPK, phosphorylated (Thr202/Tyr204) and total ERK, phosphorylated (Thr183/Tyr185) and total JNK, phosphorylated (Ser473) and total Akt were from Cell Signaling Technology, Danvers, MA. Primary antibodies were detected with goat anti-rabbit or goat anti-mouse HRP–conjugated secondary antibodies (Cell Signaling Technology) and visualized by enhanced chemiluminescence (PerkinElmer, Wellesley, MA).
Cell viability
Cellular respiration, an indicator of cell viability, was determined by the mitochondrial-dependent reduction of 3-(3,4-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to formazan.Citation31 The extent of the reduction of MTT to formazan within cells was quantified by measuring the optical density at 550 nm.
Statistical analysis
Statistical analysis was performed using either the Student's unpaired t-test or 2-way ANOVA followed by Bonferroni's post-test. P values < 0.05 were sufficient to reject the null hypothesis for all analyses.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 2005; 352:2487-98; PMID:15958804; http://dx.doi.org/10.1056/NEJMoa043445
- Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol 2006; 24:4867-74; PMID:17001068; http://dx.doi.org/10.1200/JCO.2006.07.9665
- Cvek B. Proteasome inhibitors. Prog Mol Biol Transl Sci 2012; 109:161-226; PMID:22727422; http://dx.doi.org/10.1016/B978-0-12-397863-9.00005-5
- McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat 2008; 11:164-79; PMID:18818117; http://dx.doi.org/10.1016/j.drup.2008.08.002
- Small GW, Shi YY, Edmund NA, Somasundaram S, Moore DT, Orlowski RZ. Evidence that mitogen-activated protein kinase phosphatase-1 induction by proteasome inhibitors plays an antiapoptotic role. Mol Pharmacol 2004; 66:1478-90; PMID:15448190; http://dx.doi.org/10.1124/mol.104.003400
- Shi YY, Small GW, Orlowski RZ. Proteasome inhibitors induce a p38 mitogen-activated protein kinase (MAPK)-dependent anti-apoptotic program involving MAPK phosphatase-1 and Akt in models of breast cancer. Breast Cancer Res Treat 2006; 100:33-47; PMID:16807678; http://dx.doi.org/10.1007/s10549-006-9232-x
- Small GW, Shi YY, Higgins LS, Orlowski RZ. Mitogen-activated protein kinase phosphatase-1 is a mediator of breast cancer chemoresistance. Cancer Res 2007; 67:4459-66; PMID:17483361; http://dx.doi.org/10.1158/0008-5472.CAN-06-2644
- Haagenson KK, Wu GS. The role of MAP kinases and MAP kinase phosphatase-1 in resistance to breast cancer treatment. Cancer Metastasis Rev 2010; 29:143-9; PMID:20111893; http://dx.doi.org/10.1007/s10555-010-9208-5
- Rojo F, Gonzalez-Navarrete I, Bragado R, Dalmases A, Menendez S, Cortes-Sempere M, Suárez C, Oliva C, Servitja S, Rodriguez-Fanjul V, et al. Mitogen-activated protein kinase phosphatase-1 in human breast cancer independently predicts prognosis and is repressed by doxorubicin. Clin Cancer Res 2009; 15:3530-9; PMID:19417026; http://dx.doi.org/10.1158/1078-0432.CCR-08-2070
- Sun H, Charles CH, Lau LF, Tonks NK. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell 1993; 75:487-93; PMID:8221888; http://dx.doi.org/10.1016/0092-8674(93)90383-2
- Che W, Manetsch M, Quante T, Rahman MM, Patel BS, Ge Q, Ammit AJ. Sphingosine 1-phosphate induces MKP-1 expression via p38 MAPK- and CREB-mediated pathways in airway smooth muscle cells. Biochim Biophys Acta 2012; 1823:1658-65; PMID:22743041; http://dx.doi.org/10.1016/j.bbamcr.2012.06.011
- Manetsch M, Che W, Seidel P, Chen Y, Ammit AJ. MKP-1: a negative feedback effector that represses MAPK-mediated pro-inflammatory signaling pathways and cytokine secretion in human airway smooth muscle cells. Cell Signal 2012; 24:907-13; PMID:22200679; http://dx.doi.org/10.1016/j.cellsig.2011.12.013
- Doddareddy MR, Rawling T, Ammit AJ. Targeting mitogen-activated protein kinase phosphatase-1 (MKP-1): structure-based design of MKP-1 inhibitors and upregulators. Curr Med Chem 2012; 19:163-73; PMID:22320295; http://dx.doi.org/10.2174/092986712803414196
- Hu JH, Chen T, Zhuang ZH, Kong L, Yu MC, Liu Y, Zang JW, Ge BX. Feedback control of MKP-1 expression by p38. Cell Signal 2007; 19:393-400; PMID:16978838; http://dx.doi.org/10.1016/j.cellsig.2006.07.010
- Chen P, Li J, Barnes J, Kokkonen GC, Lee JC, Liu Y. Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. J Immunol 2002; 169:6408-16; PMID:12444149; http://dx.doi.org/10.4049/jimmunol.169.11.6408
- Zhao Q, Shepherd EG, Manson ME, Nelin LD, Sorokin A, Liu Y. The role of mitogen-activated protein kinase phosphatase-1 in the response of alveolar macrophages to lipopolysaccharide: attenuation of proinflammatory cytokine biosynthesis via feedback control of p38. J Biol Chem 2005; 280:8101-8; PMID:15590669; http://dx.doi.org/10.1074/jbc.M411760200
- Tai C-J, Wu AT, Chiou J-F, Jan H-J, Wei H-J, Hsu C-H, Lin CT, Chiu WT, Wu CW, Lee HM, et al. The investigation of Mitogen-Activated Protein kinase Phosphatase-1 as a potential pharmacological target in non-small cell lung carcinomas, assisted by non-invasive molecular imaging. BMC Cancer 2010; 10:1-14; PMID:20226009; http://dx.doi.org/10.1186/1471-2407-10-95
- Kang DW, Lee JY, Oh DH, Park SY, Woo TM, Kim MK, Park MH, Jang YH, Min do S. Triptolide-induced suppression of phospholipase D expression inhibits proliferation of MDA-MB-231 breast cancer cells. Exp Mol Med 2009; 41:678-85; PMID:19478552; http://dx.doi.org/10.3858/emm.2009.41.9.074
- Liu J, Jiang Z, Xiao J, Zhang Y, Lin S, Duan W, Yao J, Liu C, Huang X, Wang T, et al. Effects of triptolide from Tripterygium wilfordii on ERalpha and p53 expression in two human breast cancer cell lines. Phytomedicine 2009; 16:1006-13; PMID:19524422; http://dx.doi.org/10.1016/j.phymed.2009.03.021
- Lu L, Kanwar J, Schmitt S, Cui QC, Zhang C, Zhao C, Dou QP. Inhibition of tumor cellular proteasome activity by triptolide extracted from the Chinese medicinal plant 'thunder god vine'. Anticancer Res 2011; 31:1-10; PMID:21273574
- Bensaude O. Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity? Transcription 2011; 2:103-8; PMID:21922053; http://dx.doi.org/10.4161/trns.2.3.16172
- Marderosian M, Sharma A, Funk AP, Vartanian R, Masri J, Jo OD, Gera JF. Tristetraprolin regulates Cyclin D1 and c-Myc mRNA stability in response to rapamycin in an Akt-dependent manner via p38 MAPK signaling. Oncogene 2006; 25:6277-90; PMID:16702957; http://dx.doi.org/10.1038/sj.onc.1209645
- Yang CH, Gonzalez-Angulo AM, Reuben JM, Booser DJ, Pusztai L, Krishnamurthy S, Esseltine D, Stec J, Broglio KR, Islam R, et al. Bortezomib (VELCADE) in metastatic breast cancer: pharmacodynamics, biological effects, and prediction of clinical benefits. Ann Oncol 2006; 17:813-7; PMID:16403809; http://dx.doi.org/10.1093/annonc/mdj131
- Engel RH, Brown JA, Von Roenn JH, O'Regan RM, Bergan R, Badve S, Rademaker A, Gradishar WJ. A phase II study of single agent bortezomib in patients with metastatic breast cancer: a single institution experience. Cancer Invest 2007; 25:733-7; PMID:17952740; http://dx.doi.org/10.1080/07357900701506573
- Cresta S, Sessa C, Catapano CV, Gallerani E, Passalacqua D, Rinaldi A, Bertoni F, Viganò L, Maur M, Capri G, et al. Phase I study of bortezomib with weekly paclitaxel in patients with advanced solid tumours. Eur J Cancer 2008; 44:1829-34; PMID:18640031; http://dx.doi.org/10.1016/j.ejca.2008.05.022
- Wu W, Pew T, Zou M, Pang D, Conzen SD. Glucocorticoid receptor-induced MAPK phosphatase-1 (MPK-1) expression inhibits paclitaxel-associated MAPK activation and contributes to breast cancer cell survival. J Biol Chem 2005; 280:4117-24; PMID:15590693; http://dx.doi.org/10.1074/jbc.M411200200
- Liu Q. Triptolide and its expanding multiple pharmacological functions. Int Immunopharmacol 2011; 11:377-83; PMID:21255694; http://dx.doi.org/10.1016/j.intimp.2011.01.012
- Soule HD, Maloney TM, Wolman SR, Peterson WD Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res 1990;50(18):6075-86; PMID:1975513
- Basolo F, Elliott J, Tait L, Chen XQ, Maloney T, Russo IH, Pauley R, Momiki S, Caamano J, Klein-Szanto AJ, et al. Transformation of human breast epithelial cells by c-Ha-ras oncogene. Mol Carcinog 1991; 4(1):25-35; PMID:2009132
- Caixeiro NJ, Martin JL, Scott CD. Silencing the mannose 6-phosphate/IGF-II receptor differentially affects tumorigenic properties of normal breast epithelial cells. Int J Cancer 2013; 133(11):2542-50; PMID:23686499
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983; 65:55-63; PMID:6606682; http://dx.doi.org/10.1016/0022-1759(83)90303-4