
Abstract
An accumulating understanding of the complex pathogenesis of acute myeloid leukemia (AML) continues to lead to promising therapeutic approaches. Among the key aberrant intracellular signaling pathways involved in AML, the phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) axis is of major interest. This axis modulates a wide array of critical cellular functions, including proliferation, metabolism, and survival. Pharmacologic inhibitors of components of this pathway have been developed over the past decade, but none has an established role in the treatment of AML. This review will discuss the preclinical data and clinical results driving ongoing attempts to exploit the PI3K/AKT/mTOR pathway in patients with AML and address issues related to negative feedback loops that account for leukemic cell survival. Targeting the PI3K/AKT/mTOR pathway is of high interest for the treatment of AML, but combination therapies with other targeted agents may be needed to block negative feedback loops in leukemia cells.
Abbreviations
| AML | = | acute myeloid leukemia |
| HDAC | = | histone deacetylase |
| MNK | = | MAPK-interacting kinase |
| mTOR | = | mammalian target of rapamycin |
| PDK1 | = | phosphoinositide-dependent protein kinase-1 |
| PI3K | = | phosphatidylinositol 3-kinase |
Introduction
The pathogenesis and pathophysiology of hematological malignancies involve aberrant regulation and function of cell signaling pathways, among which the phosphatidylinositol 3-kinase /AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) axis has a central role.Citation1,2 Dysregulation of this pathway resulting from oncogene amplification, oncogene activating mutations, inactivation of tumor suppressor genes, or upstream activation of receptor tyrosine kinases (RTK), has been demonstrated in a range of human malignancies.Citation1–3 As this axis is clearly an important target for developmental therapeutics, there are a number of small molecule inhibitors of elements of this network in development, including PI3K inhibitors (pan- and isoform-specific), AKT inhibitors (pan-, allosteric-, and ATP-competitive), mTOR inhibitors (rapalogs and TORC1/2 inhibitors), and dual PI3K-mTOR inhibitors.Citation3 Of these agents, the rapalog mTOR inhibitors, temsirolimus and everolimus, are FDA-approved for the treatment of renal cell carcinoma.Citation4 Everolimus is also approved for pancreatic neuroendocrine tumors and advanced hormone receptor-positive breast cancer.Citation5 The PI3Kδ inhibitor idelalisib is FDA-approved for the treatment of certain low-grade B-cell lymphomas.Citation6 None of these agents is FDA-approved for the treatment of acute myeloid leukemia (AML), a disease in which there is very strong preclinical evidence implicating this pathway in its pathogenesis and pathophysiology.Citation7-11 In one study, more than 90% of primary AML blasts had evidence of activation of AKT, a critical substrate of PI3K, as determined by the presence of phosphorylation or a positive kinase assay. Citation12 Recher et al showed that S6K and 4E-BP1, both downstream effectors of mTOR, were constitutively phosphorylated among the majority of freshly isolated AML samples from 23 patients but were not phosphorylated in normal freshly isolated CD34 control cells.Citation13 A number of potential mechanisms for the constitutive activation of this pathway have been implicated, including activating RAS mutations, FLT3-ITD mutations, activating C-KIT mutations, and PTEN phosphorylation or downregulation.Citation14
AML is characterized by clonal expansion of early myeloid progenitor cells and some subtypes can be classified by distinct molecular and/or cytogenetic abnormalities. The annual incidence of AML in the United States is 30 to 40 cases per million individuals.Citation15 With a spectrum of failure to respond to induction therapy and relapse in the majority of adult patients who attain initial remissions, the overall prognosis is poor.Citation15 In some patients, leukemia-initiating stem cells may be responsible for relapse - their inherent resistance to standard cytotoxic agents and unique heterogeneity, resulting from clonal evolution involving quiescent subclones, pose a major problem for therapeutic strategies in AML.Citation16,17 Prognosis varies considerably according to cytogenetic and molecular abnormalities, but survival remains generally poor, emphasizing an unmet need for new effective therapies.Citation18-20 This review will discuss the scientific rationale for targeting PI3K/AKT/mTOR and related feedback pathways in AML. It will also review ongoing treatment strategies that include drugs with the ability to inhibit individual components of the pathway, combinations of these inhibitors for additive or synergist effects, and novel drugs with dual inhibitory activity. Data from clinical trials involving some of these agents for the treatment of patients with relapsed or refractory AML are also summarized.
PI3K/AKT/mTOR Pathway
PI3K (phosphatidylinositol 3′-kinase)
The important role of constitutive activation of the PI3K pathway in the pathogenesis of AML has been extensively documented.Citation12,21-24 While activating mutations in PI3K p85 regulatory subunit and p110 catalytic subunit have been described in Hodgkin lymphoma and various solid tumors, they are rarely seen in AML.Citation25,26 However, RAS mutations have been identified in 10–15% of AML and 25% of juvenile myelomonocytic leukemia cases and they can activate the PI3K/AKT/mTOR pathway with potential implications beyond promoting cell survival and proliferation, including remodeling of tumor microenvironment and modulation of tumor-induced immune suppression.Citation20,27-29 Nevertheless, there is no strong or definitive evidence of clinical benefit with single agent PI3K inhibitors in AML, but they hold promise when used in combination with inhibitors of other pathways, as described below.
Both inhibitors of class I PI3K isoforms (pan-PI3K) and isoform specific compounds are being investigated. A phase I trial is evaluating the pan-PI3K inhibitor BKM120 (buparlisib) in advanced acute leukemias based on preclinical evidence showing promising activity in acute lymphoblastic leukemia (ALL)(NCT01396499).Citation30 Isoform specific inhibitors of PI3K such as idelalisib have shown remarkable activity in lymphoid malignancies based on the importance of PI3Kδ in B-cell receptor signaling. Of note, idelalisib, a PI3Kδ-specific inhibitor, was recently FDA-approved for the treatment of relapsed chronic lymphocytic leukemia, follicular lymphoma, and small lymphocytic lymphoma. Despite frequent expression of PI3K-δ in AML cells and preclinical results showing activity of selective inhibitors, there is a lack of evidence of clinical efficacy of PI3K-δ inhibition in AML.Citation21,31 A dose-escalation trial involving idelalisib for the treatment of various hematologic malignancies, including relapsed/refractory AML, has been completed (NCT00710528) – the data has not yet been reported. The combinatorial approach of p110α specific inhibition with MEK inhibition in RAS-mutated AML seems promising as evidenced by the remarkable anti-tumor activity of BYL719 in combination with MEK162 in RAS-mutated AML cell lines and xenograft models.Citation32
Another combinatorial strategy of specific interest in AML includes the blockade of both PI3K and downstream mTOR to block the escape mechanism of mTORC2 upregulation that can result in phosphorylation of AKT on Ser473, followed by activation and anti-apoptotic responses.Citation33 The similarities between the PI3K p110 subunit and mTOR catalytic domains have allowed the development of dual PI3K/mTOR inhibitors including BEZ-235.Citation34 BEZ-235 was found to induce significant apoptosis of primary AML blasts to a greater magnitude than the selective AKT and mTOR inhibitors, MK-2206 and rapamycin, respectively.Citation34 Notably, there was particular sensitivity among the AML blasts with MLL translocations, a subset in which 52% contained an NRAS or KRAS mutation as compared to 28% in cytogenetically normal AML blasts.Citation34 These results are in agreement with reports showing that RAS mutations can activate the PI3K/AKT/mTOR pathway.Citation35 As another example of the regulatory complexity of this pathway, treatment with BEZ-235 resulted in increased phosphorylation of ERK (extracellular regulated kinase) suggesting an escape mechanism for PI3K/AKT/mTOR inhibition. In turn, the combination of BEZ-235 with the MEK inhibitor AZD6244 demonstrated synergistic pro-apoptotic effects, providing additional rationale for the clinical development of BEZ-235.Citation34
A phase I trial evaluated BEZ-235 in a cohort of 22 patients with refractory acute leukemia has been conducted.Citation36 The most frequent non-hematologic drug-related adverse events (AE) were stomatitis and GI toxicities. One patient with AML had stable disease for 4 months, and 3 responses were documented among patients with ALL (NCT01756118).Citation36 BEZ-235 is also being investigated in combination with nanoparticle formulations of chemotherapeutic agents (i.e. 5-fluorouracil) in other diseases, providing an innovative strategy that may be attractive in AML, especially if the nanoparticle platform can enhance drug delivery to the blasts or bone marrow microenvironment.Citation37 Based on extensive preclinical data, other dual inhibitors are in clinical development.Citation38-40
AKT
The protein kinase B family of serine/threonine kinases (AKT1, AKT2, and AKT3) is a key effector of the PI3K pathway. PI3K phosphorylates phosphatidylinositol 4,5-biphosphate, generating phosphatidylinositol 3,4,5-triphosphate that in turn recruits proteins containing pleckstrin homology domains, such as AKT, to the cell membrane.Citation41,42 While in the cell membrane, AKT is phosphorylated at Thr308 and Ser473, events that lead to its activation and generation of downstream biological responses, including promotion of cell proliferation and anti-apoptotic signaling via several effectors.Citation41-48
The importance of AKT in leukemogenesis was highlighted by the demonstration of constitutive activation of AKT in 70–86% of primary AML patient samples tested and the correlation of AKT activation with inferior survival.Citation9,49 The mechanisms of constitutive activation of AKT in AML seem to rely partially upon active upstream FLT3 as opposed to activating mutations in PI3K and AKT.Citation49 In fact, AKT is critical for the proliferative phenotype induced by FLT3-ITD.Citation49 AKT also contributes to chemotherapy resistance of AML blasts, while specific phosphorylation at Thr308 correlates with a high-risk cytogenetic profile.Citation50,51 Taken together, these results provide a firm rationale for the development of AKT inhibitors as therapies for AML.
The purine nucleoside analog triciribine (TCN-PM) inhibits AKT phosphorylation by interfering with AKT's PH domain and preventing its membrane localization.Citation52 Based on preclinical evidence of AKT inhibition, triciribine was evaluated among 41 patients with advanced hematological malignancies, including 36 patients with AML. The treatment had an acceptable toxicity profile and correlative studies demonstrated a significant reduction in the levels of pAKT (Ser473) and pBAD (Ser112).Citation52 While no objective responses were documented, stable disease among 17 patients suggested a potential role for this inhibitor.Citation52 The phospholipid analog, perifosine, inhibits AKT by altering lipid rafts and preventing the membrane localization of AKT, resulting in apoptosis of AML cell lines and enhancement of etoposide cytotoxicity.Citation53 These results provided the impetus for ongoing clinical studies of perifosine in refractory AML and other malignancies (NCT00391560).Citation54-57
Another strategy utilized to block AKT has been the development of small molecule inhibitors such as MK-2206 and GSK690693.Citation58,59 MK-2206 is an oral non-ATP competitive allosteric inhibitor of AKT1, 2, and 3 and induces apoptosis and cell cycle arrest of AML cell lines. However, it demonstrated limited efficacy during a phase I study with only one response among 18 patients, leading to early termination of the trial.Citation60 The adverse AE profile was notable for grade 3/4 rash in 33% of patients.Citation60 Correlative studies suggested that the lack of efficacy resulted from limited inhibition of AKT and downstream targets (i.e., pFOX3A, pS6K, p4EBP1), coupled with upregulation of escape mechanisms including Bcl−2, Smad3, and STAT-3.Citation60 Nevertheless, MK-2206 might have a role in combinations with other pathway inhibitors and/or chemotherapy, as suggested by preclinical evidence in other diseases.Citation58,61-63 GSK690693 has shown promising preclinical activity in acute lymphoblastic leukemia, but a phase I trial in advanced hematological malignancies was cancelled prior to enrollment of patients, and no data have been published in AML (NCT00666081).Citation64 Another pan-AKT small molecule inhibitor, GSK2110183, is being investigated in clinical trials for patients with multiple myeloma or refractory chronic lymphocytic leukemia (NCT01532700; NCT01428492).
Novel strategies attempting to enhance the efficacy of AKT inhibitors include use of nanoparticle formulations, combinations with other agents, and the development of AKT isoform-specific inhibitors.Citation65 In addition, testing these compounds specifically in patients with increased levels of pAKT or activating AKT mutations may lead to improved efficacy outcomes than would be observed among an unselected population.
PDK1 (phosphoinositide-dependent protein kinase-1)
PDK1 is part of the large AGC (protein kinase A, protein kinase G and protein kinase C) kinase family and regulates several other downstream effector kinases, including S6K (p70 ribosomal S6 kinase), SGK (serum- and glucocorticoid-induced protein kinase), PKC and AKT.Citation66 PDK1 is constitutively active and also has a PH domain that recruits this kinase to the cell membrane upon binding to PIP3.Citation67 Upregulation of PDK1 downstream kinases is frequently observed in cancers with PTEN and PIK3CA mutations.Citation66,68 Hence, there is growing interest in the development of small molecule PDK1 inhibitors for the treatment of several malignancies including AML.Citation69-71
Overexpression of PDK1 was observed in approximately 45% of AML patient samples and correlated strongly with protein kinase C (PKC) activation.Citation72 The highest levels of PDK1 were seen among the monocytic subtype of AML in agreement with the important role of PKC pathways in monocytic differentiation.Citation73 PDK1 overexpression was detected among 42% of patients with myelomonocytic AML and it was associated with worse overall survival (OS).Citation74 Furthermore, overexpression of PDK1 promoted survival of AML blasts by a PKC-dependent mechanism and these cells were sensitive to the PDK1 inhibitor BX-795.Citation74 The clinical-translational implications of these results, however, are limited by the lack of specificity of BX-795.Citation75,76 Lack of specificity has been a major drawback of several PDK1 inhibitors.Citation70 Nevertheless, other PDK1 inhibitors have shown preclinical efficacy in AML cell lines and xenograft models and more specific and potent PDK1 inhibitors might be incorporated into clinical trials for hematological malignancies in the near future.Citation71,77
mTORC1/mTORC2
The mTOR pathway controls a number of critically important cell processes.Citation78 The mTOR kinase is present in 2 unique multiprotein complexes, mTOR complex 1 (mTORC1) and 2 (mTORC2).Citation79 In addition to mTOR kinase itself, mTORC1 consists of the following proteins/elements: RAPTOR, PRAS40, mLST8, and DEPTOR.Citation79 In general, mTORC1 promotes mRNA translation, inhibits autophagy, and acts as a signal integrator for various incoming signals. S6K1 and 4E-BP1 are known downstream mTORC1 effectors involved in the initiation of mRNA translation.Citation80 The mTORC2 complex is comprised of mTOR, RICTOR, PROTOR1, PROTOR2, mSIN1, mLST8, and DEPTOR and acts as a mediator of actin cytoskeletal organization, cell polarization, and pro-survival signals.Citation79
The rapamycin analogs (rapalogs) act by selectively targeting mTORC1 by binding to its immunophilin, FK506 binding protein 12 (FKBP12), leading to inhibition of downstream mTOR signals.Citation14 Rapalogs have been shown to have antileukemic activity in AML, in the preclinical and clinical settings, as monotherapy and in combination with chemotherapy or other inhibitors of the PI3K/AKT/mTOR axis.Citation13,81-84 In general, the rapalogs' clinical activity in AML is modest at best ().Citation83,85-87 In a phase I trial that assessed the rapalog deferolimus among 23 AML patients, none achieved a response.Citation88 A phase I/II study of everolimus in 9 AML patients also resulted in no bone marrow responses.Citation81 However, the combination of everolimus with chemotherapy in the first relapse setting resulted in 68% complete response and median OS of 19 months.Citation89 Similarly, the combination of temsirolimus with clofarabine in elderly patients with AML showed encouraging results with an overall remission rate of 21% and median disease free survival of 3.5 months.Citation90
Table 1. Clinical trials involving inhibitors of PI3K/AKT/mTOR pathway in AML
Interest in mTOR inhibition in AML has shifted to using mTOR catalytic inhibitors (also referred to as active site inhibitors) or agents that have dual PI3K/mTOR or AKT/mTOR activity and has been matched with more promising results.Citation7,91 The dual catalytic mTORC1/2 inhibitor OSI-027 has been shown to block mTORC1 and mTORC2 activities and to suppress mRNA translation of genes that mediate proliferation in AML cells, resulting in more potent antileukemic properties in vitro than rapamycin.Citation91 PP242, another dual catalytic mTORC1/2 inhibitor, led to apoptosis in primary AML samples and suppressed chemokine receptor CXCR4 expression in primary leukemic cells and in stromal cells co-cultured with leukemic cells.Citation11 In a mouse model, PP242 inhibited mTOR signaling in leukemic cells and reduced leukemia burden to a greater degree than that of rapamycin.Citation11 Promising findings were also observed with agent AZD8055, supporting its role as an mTORC1/2 inhibitor.Citation92 Among bone marrow samples from AML patients, it fully inhibited phosphorylation of 4EBP1, prevented the mTORC1-dependent PI3K/AKT feedback activation that has been observed with rapamycin, and promoted potent antileukemic effect in in vitro and in vivo preclinical models.Citation93 Decreased proliferation and induction of apoptosis among AML stem cells with high AKT/mTOR activity by another mTORC1/2 inhibitor, MLN0128, was recently reported.Citation94 MLN0128 was shown to selectively target mTORC1/2 downstream signaling in these cells with minimal effects on other pathways and without affecting non-leukemic stem cells. Compensatory activation of multiple pro-survival proteins was noted, among which was HDAC3. The use of the combination of MLN0128 and the histone deacetylase (HDAC) inhibitor vorinostat led to increased apoptosis as compared to that associated with single-agent MLN0128.Citation94
Despite the promising results seen thus far with the dual catalytic mTORC1/2 inhibitors, there is concern that over time, resistance will develop by activation of other pathways. Therefore, there is interest in concomitantly inhibiting multiple pathways within the PI3K/AKT/mTOR axis and other escape mechanisms including autophagy, RAS/RAF/MEK/ERK pathway, MAPK-interacting kinases (MNKs), HDAC pathways, and PIM kinases.Citation95-99
Combinatorial approaches to overcome escape mechanisms of resistance
As study of the PI3K/AKT/mTOR pathway has intensified, multiple regulatory escape mechanisms have been identified (). For instance, treatment with the dual mTORC1/2 inhibitors OSI-027 and AZD-2014 results in induction of autophagy in AML cell lines, as suggested by increased expression of LC3II, suppression p62/SQSTM1 proteins, and formation of autophagic structures, documented by electron microscopy.Citation99 Induction of autophagy in response to mTORC1/2 inhibition was also noted among samples from patients with AML. The addition of chloroquine, a known inhibitor of autophagy, potentiated the effects of mTORC1/2 inhibitors in reducing colony formation of primary AML cell lines. Additional experiments also suggested that induction of autophagy resulted during treatment with OSI-027 or AZD-2014 by preventing the inhibitory phosphorylation of ULK1 kinase (Ser757) that regulates autophagy.Citation100 These results have significant clinical implications in light of evidence showing the role of autophagy in chemotherapy resistance in AML models and might explain the limited efficacy of mTORC1 inhibitors in AML.Citation81,101,102 Furthermore, they provide support to develop combinations of autophagy inhibitors and mTORC1/mTORC2 blocking agents and/or chemotherapy for treatment of AML. The availability of chloroquine and more potent autophagy inhibitors (i.e., Lys05) coupled with encouraging preliminary results from trials in solid tumors will hopefully stimulate the conduct of related clinical studies in AML.Citation103-106
Figure 1. PIK3/AKT/mTOR signaling pathway in acute myeloid leukemia. Red boxes depict several inhibitors of specific pathway components undergoing preclinical or clinical studies. Dashed lines represent escape mechanisms of cell survival emerging after blockade of mTORC1 and/or mTORC2.
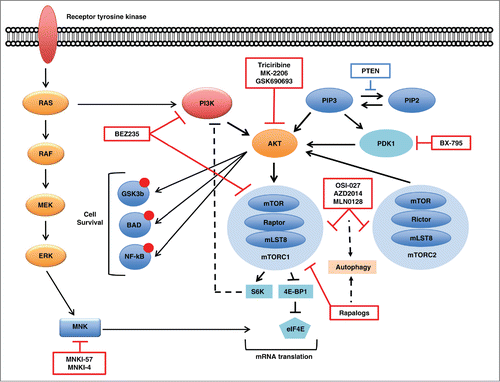
Recent results have demonstrated paradoxical AKT phosphorylation in AML cells treated with AKT or PI3K inhibitors for 24 hours compared to the shorter duration treatment of 4 hours.Citation10 These inhibitors also increased the expression of IGF-1R, PDGFR and insulin receptors and induced their autophosphorylation. These effects ultimately result in sustained activation of the PI3K/AKT/mTOR pathway by a mechanism involving upregulation of the adaptor protein insulin receptor substrate 1. This feedback loop was successfully blocked by the combination of an AKT inhibitor with the PDGFR inhibitor sunitinib, the IGF-1R/IR inhibitor linsitinib, or the FLT3 inhibitor quizartinib.Citation10 Hence, these findings suggest that turning off the PI3K/AKT/mTOR axis goes beyond combining individual inhibitors of axis nodes and may require strategies accounting for the plasticity of feedback loops that emerge after exposure to certain drugs.Citation107 Altogether, these results epitomize the complexity of PI3K/AKT/mTOR pathway regulatory network and highlight the potential importance of incorporating receptor tyrosine kinase inhibitors into genomic-based treatment plans for AML patients.
Another important escape mechanism emerging from mTORC1 inhibition consists of activation of MAPK-interacting kinases (MNK1 and MNK2) that increase eIF4E phosphorylation and trigger protein synthesis and pro-survival signals.Citation108 Based on the convergence of RAS/RAF/MAPK and PI3K/AKT/mTOR pathways in oncogenesis and the evolving role of MNKs in hematological malignancies, the effect of MNK inhibitors alone or in combination with mTOR inhibitors is being investigated.Citation97,98,109 MNK2-specific and dual MNK1/2 inhibitors (MNKI-57 and MNKI-4) displayed potent anti-proliferative activity in chronic myeloid leukemia (CML) and AML cells. Combination of rapamycin and MNK inhibitors led to a significant reduction of rapamycin-induced eIF4E phosphorylation and synergistically reduced proliferation of CML blasts (KYO-1 cells).Citation97 These findings are consistent with the emerging role of MNKs in CML blastic phase disease.Citation110
Another example of the close relationship between these pathways is provided by ERK upregulation in AML cells treated with the PI3K/mTOR inhibitor BEZ-235.Citation34 In agreement with this result, the combination of BEZ-235 with a MEK inhibitor (AZD6244) demonstrated a synergistic pro-apoptotic effect. In fact, the limited efficacy of single agent selumetinib (MEK inhibitor) in refractory AML observed in a phase II trial might be explained by the concomitant activation of PI3K/AKT/mTOR as suggested by preclinical studies.Citation95,111 Lastly, combined treatment of AML cells with PIM (AZD1897) and AKT (AZD5363) inhibitors potentiated the blockade of the mTOR axis and resulted in marked anti-leukemic effects and induction of apoptosis, providing a blueprint for clinical development of this combination.Citation96 Another PIM inhibitor (LGH447) is currently undergoing a phase I trial for refractory AML and high-risk myelodysplastic syndrome as single agent (NCT02078609), but we are not aware of any trials exploring the combination of PIM and AKT inhibitors.
Summary
The aberrant activity of the PI3K/AKT/mTOR axis, modulating critical cellular functions such as proliferation, metabolism, and survival, has been well established as a major component of AML pathogenesis. Deregulation of this pathway may be the result of oncogene amplification and activating mutations, inactivation of tumor suppressor genes, or activation of RTKs. A number of classes of pharmacologic inhibitors of various nodes of this pathway in AML have been evaluated in the preclinical setting, resulting in promising signals of activity. Despite these results and the number of years spent accumulating evidence of this pathway's role in AML pathogenesis, there is still no evidence of meaningful clinical effectiveness for pharmacologic inhibition of this pathway as a treatment for AML. Nonetheless, the prospect of inhibiting this pathway in combination with other novel agents, based on previously discussed data to support this approach, remains highly encouraging and might be able to overcome some of the biological challenges of AML pathogenesis.
The complexity of this pathway's multiple regulatory mechanisms and feedback loops is evident. Aside from escape mechanisms inherent to inhibition of this axis, such as cross-talk from compensatory oncogenic pathways (e.g., RAS or MYC), feedback leading to rebound activation of PI3K after mTOR inhibition, and upregulation of upstream receptor tyrosine kinases, AML stem cells may be become resistant to very targeted interventions by recruiting other signaling molecules, modifying gene expression through epigenetic mechanisms, or altering components of the stromal microenvironment. Therefore, despite the remarkable advances secondary to the knowledge acquisition from AML genetic profiling and novel targeted therapies blocking PI3K/AKT/mTOR pathway, combinatorial approaches capable of blocking multiple escape mechanisms of resistance may be required for achieving clinically meaningful benefits. A phase III study demonstrating improved OS with the combination of BRAF and MEK inhibition over BRAF inhibition alone in patients with advanced BRAF-mutated melanoma provides proof of principle for mechanistically rational combinations of targeted therapies.Citation112
What makes the combinatorial approach a challenging endeavor for designing clinical trials for most malignancies, however, is the increasing number of potential drug combinations in the setting of limited resources. In the case of targeting PI3K/AKT/mTOR in AML, one must consider the variety of classes of inhibitors available for each node of the pathway, including pan-PI3K inhibitors, isoform-specific PI3K inhibitors (and possibly agents that selectively target just 2 or 3 out of the 4 class I isoforms of PI3K), rapalogs, catalytic dual mTORC1/2 inhibitors, pan-PI3K/mTOR inhibitors, and AKT inhibitors. When considering targeting PI3K, for example, it may seem attractive to target a specific PI3K isoform, in an attempt to improve efficacy and decrease off-target toxicity. However, out of concern for functional redundancy of all 4 class I PI3K isoforms, pan-PI3K inhibition may be more efficacious albeit possibly more toxic, especially considering the vital role of PI3K in normal cells. The success of idelalisib, a PI3Kδ isoform inhibitor, among certain B-cell neoplasms may be explained by the differential expression of PI3Kδ among haematopoietic cells as compared to cells of other origin. In regard to mTOR inhibition, the further pursuit of the use of rapalogs in AML should likely be abandoned in favor of catalytic dual mTORC1/2 inhibitors (for reasons already discussed), but the question of whether to pursue rapalog use in combination with targets of other pathways (e.g.,, autophagy or PIM inhibitors) remains to be answered. Further adding to the complexity of designing appropriate trials is deciding among the many compensatory oncogenic pathways (e.g., RAS-RAF-MEK-ERK) to concurrently target, the interindividual genetic heterogeneity of the patients in the trial, and the intraindividual genetic heterogeneity of AML cells that can result from subclonal evolution.
One could consider performing multiple phase I trials in parallel, to identify optimal dosing for combinations of PI3K/AKT/mTOR inhibitors with one another and/or with agents that target other compensatory pathways, prioritizing the selection of agents and combinations based upon biologic rationale and preexisting preclinical evidence, followed by a phase II “pick the winner” protocol involving multiple combinations. The incorporation of correlative studies, performed at baseline and after initiation of treatment, such as pS6, p4EBP1, other established pharmacodynamic markers, tumor microenvironment markers, and high-throughput whole genome sequencing (i.e., next generation sequencing) will be imperative to help identify biomarkers for predicting response to therapy and toxicity. These challenges highlight the need for novel trial designs and collaboration among multiple institutions, cooperative groups, and biopharmaceutical companies.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
The research of Dr. Platanias is supported by grants CA77816, CA155566, CA161196, and CA121192 from the National Institutes of Health and by grant I01CX000916 from the Department of Veterans Affairs.
References
- Gentzler RD, Altman JK, Platanias LC. An overview of the mTOR pathway as a target in cancer therapy. Expert Opin Ther Targets 2012; 16:481–9; PMID:22494490; http://dx.doi.org/10.1517/14728222.2012.677439
- Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008; 27:5497–510; PMID:18794884; http://dx.doi.org/10.1038/onc.2008.245
- Porta C, Paglino C, Mosca A. Targeting PI3K/AKT/mTOR Signaling in Cancer. Front Oncol 2014; 4:64; PMID:24782981; http://dx.doi.org/10.3389/fonc.2014.00064
- Kwitkowski VE, Prowell TM, Ibrahim A, Farrell AT, Justice R, Mitchell SS, Sridhara R, Pazdur R. FDA approval summary: temsirolimus as treatment for advanced renal cell carcinoma. Oncologist 2010; 15:428–35; PMID:20332142; http://dx.doi.org/10.1634/theoncologist.2009-0178
- Lebwohl D, Anak O, Sahmoud T, Klimovsky J, Elmroth I, Haas T, Posluszny J, Saletan S, Berg W. Development of everolimus, a novel oral mTOR inhibitor, across a spectrum of diseases. Ann N Y Acad Sci 2013; 1291:14–32; PMID:23659703; http://dx.doi.org/10.1111/nyas.12122
- Markham A. Idelalisib: first global approval. Drugs 2014; 74:1701–7; PMID:25187123; http://dx.doi.org/10.1007/s40265-014-0285-6
- Altman JK, Platanias LC. Acute myeloid leukemia: potential for new therapeutic approaches targeting mRNA translation pathways. Int J Hematol Oncol 2013; 2:10.2217/ijh.13.23; PMID:24319589; http://dx.doi.org/10.2217/ijh.13.23
- Martelli AM, Evangelisti C, Chiarini F, Grimaldi C, Manzoli L, McCubrey JA. Targeting the PI3K/AKT/mTOR signaling network in acute myelogenous leukemia. Expert Opin Investig Drugs 2009; 18:1333–49; PMID:19678801; http://dx.doi.org/10.1517/14728220903136775
- Min YH, Eom JI, Cheong JW, Maeng HO, Kim JY, Jeung HK, Lee ST, Lee MH, Hahn JS, Ko YW. Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: its significance as a prognostic variable. Leukemia 2003; 17:995–7; PMID:12750723; http://dx.doi.org/10.1038/sj.leu.2402874
- Bertacchini J, Guida M, Accordi B, Mediani L, Martelli AM, Barozzi P, Petricoin E, 3rd, Liotta L, Milani G, Giordan M, et al. Feedbacks and adaptive capabilities of the PI3K/AKT/mTOR axis in acute myeloid leukemia revealed by pathway selective inhibition and phosphoproteome analysis. Leukemia 2014; 28:2197–205; PMID:24699302; http://dx.doi.org/10.1038/leu.2014.123
- Zeng Z, Shi YX, Tsao T, Qiu Y, Kornblau SM, Baggerly KA, Liu W, Jessen K, Liu Y, Kantarjian H, et al. Targeting of mTORC1/2 by the mTOR kinase inhibitor PP242 induces apoptosis in AML cells under conditions mimicking the bone marrow microenvironment. Blood 2012; 120:2679–89; PMID:22826565; http://dx.doi.org/10.1182/blood-2011-11-393934
- Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood 2003; 102:972–80; PMID:12702506; http://dx.doi.org/10.1182/blood-2002-11-3429
- Recher C, Beyne-Rauzy O, Demur C, Chicanne G, Dos Santos C, Mas VM, Benzaquen D, Laurent G, Huguet F, Payrastre B. Antileukemic activity of rapamycin in acute myeloid leukemia. Blood 2005; 105:2527–34; PMID:15550488; http://dx.doi.org/10.1182/blood-2004-06-2494
- Martelli AM, Tazzari PL, Evangelisti C, Chiarini F, Blalock WL, Billi AM, Manzoli L, McCubrey JA, Cocco L. Targeting the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin module for acute myelogenous leukemia therapy: from bench to bedside. Curr Med Chem 2007; 14:2009–23; PMID:17691943; http://dx.doi.org/10.2174/092986707781368423
- Dores GM, Devesa SS, Curtis RE, Linet MS, Morton LM. Acute leukemia incidence and patient survival among children and adults in the United States, 2001–2007. Blood 2012; 119:34–43; PMID:22086414; http://dx.doi.org/10.1182/blood-2011-04-347872
- Valent P. Targeting of leukemia-initiating cells to develop curative drug therapies: straightforward but nontrivial concept. Curr Cancer Drug Targets 2011; 11:56–71; PMID:21062243; http://dx.doi.org/10.2174/156800911793743655
- Zeijlemaker W, Gratama JW, Schuurhuis GJ. Tumor heterogeneity makes AML a “moving target” for detection of residual disease. Cytometry B Clin Cytom 2014; 86:3–14; PMID:24151248; http://dx.doi.org/10.1002/cyto.b.21134
- Shah A, Andersson TM, Rachet B, Bjorkholm M, Lambert PC. Survival and cure of acute myeloid leukaemia in England, 1971–2006: a population-based study. Br J Haematol 2013; 162:509–16; PMID:23786647; http://dx.doi.org/10.1111/bjh.12425
- Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, Wheatley K, Harrison CJ, Burnett AK. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010; 116:354–65; PMID:20385793; http://dx.doi.org/10.1182/blood-2009-11-254441
- Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012; 366:1079–89; PMID:22417203; http://dx.doi.org/10.1056/NEJMoa1112304
- Sujobert P, Bardet V, Cornillet-Lefebvre P, Hayflick JS, Prie N, Verdier F, Vanhaesebroeck B, Muller O, Pesce F, Ifrah N, et al. Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood 2005; 106:1063–6; PMID:15840695; http://dx.doi.org/10.1182/blood-2004-08-3225
- Billottet C, Grandage VL, Gale RE, Quattropani A, Rommel C, Vanhaesebroeck B, Khwaja A. A selective inhibitor of the p110delta isoform of PI 3-kinase inhibits AML cell proliferation and survival and increases the cytotoxic effects of VP16. Oncogene 2006; 25:6648–59; PMID:16702948; http://dx.doi.org/10.1038/sj.onc.1209670
- Altman JK, Sassano A, Platanias LC. Targeting mTOR for the treatment of AML. New agents and new directions. Oncotarget 2011; 2:510–7; PMID:21680954
- Sanja P, Ivo U, Boris L, Damir N, Josip B, Renata Z, Suncica R, Koraljka GK, Sanja D, Drago B. Prognostic significance of constitutive PI3K/Akt and MAPK phosphorylation in acute myeloid leukemia. Leuk Lymphoma 2014; published on line December 29 2014, http://dx.doi.org/10.3109/10428194.2014.990012
- Jucker M, Sudel K, Horn S, Sickel M, Wegner W, Fiedler W, Feldman RA. Expression of a mutated form of the p85alpha regulatory subunit of phosphatidylinositol 3-kinase in a Hodgkin's lymphoma-derived cell line (CO). Leukemia 2002; 16:894–901; PMID:11986952; http://dx.doi.org/10.1038/sj.leu.2402484
- Muller CI, Miller CW, Hofmann WK, Gross ME, Walsh CS, Kawamata N, Luong QT, Koeffler HP. Rare mutations of the PIK3CA gene in malignancies of the hematopoietic system as well as endometrium, ovary, prostate and osteosarcomas, and discovery of a PIK3CA pseudogene. Leuk Res 2007; 31:27–32; PMID:16764926; http://dx.doi.org/10.1016/j.leukres.2006.04.011
- Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood 2012; 120:3397–406; PMID:22898602; http://dx.doi.org/10.1182/blood-2012-05-378596
- Johnson DB, Smalley KS, Sosman JA. Molecular pathways: targeting NRAS in melanoma and acute myelogenous leukemia. Clin Cancer Res 2014; 20:4186–92; PMID:24895460; http://dx.doi.org/10.1158/1078-0432.CCR-13-3270
- Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 2011; 11:761–74; PMID:21993244; http://dx.doi.org/10.1038/nrc3106
- Lonetti A, Antunes IL, Chiarini F, Orsini E, Buontempo F, Ricci F, Tazzari PL, Pagliaro P, Melchionda F, Pession A, et al. Activity of the pan-class I phosphoinositide 3-kinase inhibitor NVP-BKM120 in T-cell acute lymphoblastic leukemia. Leukemia 2014; 28:1196–206; PMID:24310736; http://dx.doi.org/10.1038/leu.2013.369
- Nguyen LX, Sesay A, Mitchell BS. Effect of CAL-101, a PI3Kdelta inhibitor, on ribosomal rna synthesis and cell proliferation in acute myeloid leukemia cells. Blood Cancer J 2014; 4:e228; PMID:25014775; http://dx.doi.org/10.1038/bcj.2014.49
- Gritsman K, Yuzugullu H, Von T, Yan H, Clayton L, Fritsch C, Maira SM, Hollingworth G, Choi C, Khandan T, et al. Hematopoiesis and RAS-driven myeloid leukemia differentially require PI3K isoform p110alpha. J Clin Invest 2014; 124:1794–809; PMID:24569456; http://dx.doi.org/10.1172/JCI69927
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307:1098–101; PMID:15718470; http://dx.doi.org/10.1126/science.1106148
- Sandhofer N, Metzeler KH, Rothenberg M, Herold T, Tiedt S, Groiss V, Carlet M, Walter G, Hinrichsen T, Wachter O, et al. Dual PI3K/mTOR inhibition shows antileukemic activity in MLL-rearranged acute myeloid leukemia. Leukemia 2014; Oct 17; [Epub ahead of print]; PMID:25322685, http://dx.doi.org/10.1038/leu.2014.305
- Castellano E, Downward J. Role of RAS in the regulation of PI 3-kinase. Curr Top Microbiol Immunol 2010; 346:143–69; PMID:20563706
- Wunderle L, Badura S, Lang F, Wolf A, Schleyer E, Serve H, Goekbuget N, Pfeifer H, Bug G, Ottmann OG. Safety and Efficacy Of BEZ235, a Dual PI3-Kinase /mTOR Inhibitor, In Adult Patients With Relapsed Or Refractory Acute Leukemia: Results Of a Phase I Study. Blood 2013; 122: abstract 2675
- Chen J, Shao R, Li L, Xu ZP, Gu W. Effective inhibition of colon cancer cell growth with MgAl-layered double hydroxide (LDH) loaded 5-FU and PI3K/mTOR dual inhibitor BEZ-235 through apoptotic pathways. Int J Nanomedicine 2014; 9:3403–11; PMID:25075187
- Badura S, Tesanovic T, Pfeifer H, Wystub S, Nijmeijer BA, Liebermann M, Falkenburg JH, Ruthardt M, Ottmann OG. Differential effects of selective inhibitors targeting the PI3K/AKT/mTOR pathway in acute lymphoblastic leukemia. PLoS One 2013; 8:e80070; PMID:24244612; http://dx.doi.org/10.1371/journal.pone.0080070
- Jabbour E, Ottmann OG, Deininger M, Hochhaus A. Targeting the phosphoinositide 3-kinase pathway in hematologic malignancies. Haematologica 2014; 99:7–18; PMID:24425689; http://dx.doi.org/10.3324/haematol.2013.087171
- Kampa-Schittenhelm KM, Heinrich MC, Akmut F, Rasp KH, Illing B, Dohner H, Dohner K, Schittenhelm MM. Cell cycle-dependent activity of the novel dual PI3K-MTORC1/2 inhibitor NVP-BGT226 in acute leukemia. Mol Cancer 2013; 12:46; PMID:23705826; http://dx.doi.org/10.1186/1476-4598-12-46
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 1997; 7:261–9; PMID:9094314; http://dx.doi.org/10.1016/S0960-9822(06)00122-9
- Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J 2000; 346:561–76; PMID:10698680; http://dx.doi.org/10.1042/0264-6021:3460561
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997; 91:231–41; PMID:9346240; http://dx.doi.org/10.1016/S0092-8674(00)80405-5
- Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999; 401:86–90; PMID:10485711; http://dx.doi.org/10.1038/43474
- Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 1998; 12:3499–511; PMID:9832503; http://dx.doi.org/10.1101/gad.12.22.3499
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 2002; 4:648–57; PMID:12172553; http://dx.doi.org/10.1038/ncb839
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999; 96:857–68; PMID:10102273; http://dx.doi.org/10.1016/S0092-8674(00)80595-4
- Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci 2013; 126:1713–19; PMID:23641065; http://dx.doi.org/10.1242/jcs.125773
- Brandts CH, Sargin B, Rode M, Biermann C, Lindtner B, Schwable J, Buerger H, Muller-Tidow C, Choudhary C, McMahon M, et al. Constitutive activation of Akt by Flt3 internal tandem duplications is necessary for increased survival, proliferation, and myeloid transformation. Cancer Res 2005; 65:9643–50; PMID:16266983; http://dx.doi.org/10.1158/0008-5472.CAN-05-0422
- Grandage VL, Gale RE, Linch DC, Khwaja A. PI3-kinase/Akt is constitutively active in primary acute myeloid leukaemia cells and regulates survival and chemoresistance via NF-kappaB, Mapkinase and p53 pathways. Leukemia 2005; 19:586–94; PMID:15703783
- Gallay N, Dos Santos C, Cuzin L, Bousquet M, Simmonet Gouy V, Chaussade C, Attal M, Payrastre B, Demur C, Recher C. The level of AKT phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia 2009; 23:1029–38; PMID:19158829; http://dx.doi.org/10.1038/leu.2008.395
- Sampath D, Malik A, Plunkett W, Nowak B, Williams B, Burton M, Verstovsek S, Faderl S, Garcia-Manero G, List AF, et al. Phase I clinical, pharmacokinetic, and pharmacodynamic study of the Akt-inhibitor triciribine phosphate monohydrate in patients with advanced hematologic malignancies. Leuk Res 2013; 37:1461–67; PMID:23993427; http://dx.doi.org/10.1016/j.leukres.2013.07.034
- Papa V, Tazzari PL, Chiarini F, Cappellini A, Ricci F, Billi AM, Evangelisti C, Ottaviani E, Martinelli G, Testoni N, et al. Proapoptotic activity and chemosensitizing effect of the novel Akt inhibitor perifosine in acute myelogenous leukemia cells. Leukemia 2008; 22:147–60; PMID:17928881; http://dx.doi.org/10.1038/sj.leu.2404980
- Krawczyk J, Keane N, Swords R, O'Dwyer M, Freeman CL, Giles FJ. Perifosine–a new option in treatment of acute myeloid leukemia? Expert Opin Investig Drugs 2013; 22:1315–27; PMID:23931614; http://dx.doi.org/10.1517/13543784.2013.826648
- Richardson PG, Nagler A, Ben-Yehuda D, Badros A, Hari P, Hajek R, Spicka I, Kaya H, Le Blanc R, Yoon S-S, et al. Randomized Placebo-Controlled Phase III Study Of Perifosine Combined With Bortezomib and Dexamethasone In Relapsed, Refractory Multiple Myeloma Patients Previously Treated With Bortezomib. Blood 2013; 122: abstract 3189; http://dx.doi.org/10.1182/blood-2013-01-481325
- Figg WD, Monga M, Headlee D, Shah A, Chau CH, Peer C, Messman R, Elsayed YA, Murgo AJ, Melillo G, et al. A phase I and pharmacokinetic study of oral perifosine with different loading schedules in patients with refractory neoplasms. Cancer Chemother Pharmacol 2014; 74:955–67; PMID:25183650; http://dx.doi.org/10.1007/s00280-014-2569-7
- Gojo I, Perl A, Luger S, Baer MR, Norsworthy KJ, Bauer KS, Tidwell M, Fleckinger S, Carroll M, Sausville EA. Phase I study of UCN-01 and perifosine in patients with relapsed and refractory acute leukemias and high-risk myelodysplastic syndrome. Invest New Drugs 2013; 31:1217–27; PMID:23443507; http://dx.doi.org/10.1007/s10637-013-9937-8
- Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 2010; 9:1956–67; PMID:20571069; http://dx.doi.org/10.1158/1535-7163.MCT-09-1012
- Rhodes N, Heerding DA, Duckett DR, Eberwein DJ, Knick VB, Lansing TJ, McConnell RT, Gilmer TM, Zhang SY, Robell K, et al. Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res 2008; 68:2366–74; PMID:18381444; http://dx.doi.org/10.1158/0008-5472.CAN-07-5783
- Konopleva MY, Walter RB, Faderl SH, Jabbour EJ, Zeng Z, Borthakur G, Huang X, Kadia TM, Ruvolo PP, Feliu JB, et al. Preclinical and early clinical evaluation of the oral AKT inhibitor, MK-2206, for the treatment of acute myelogenous leukemia. Clin Cancer Res 2014; 20:2226–35; PMID:24583795; http://dx.doi.org/10.1158/1078-0432.CCR-13-1978
- Meng J, Dai B, Fang B, Bekele BN, Bornmann WG, Sun D, Peng Z, Herbst RS, Papadimitrakopoulou V, Minna JD, et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PLoS One 2010; 5:e14124; PMID:21124782; http://dx.doi.org/10.1371/journal.pone.0014124
- Floc'h N, Kinkade CW, Kobayashi T, Aytes A, Lefebvre C, Mitrofanova A, Cardiff RD, Califano A, Shen MM, Abate-Shen C. Dual targeting of the Akt/mTOR signaling pathway inhibits castration-resistant prostate cancer in a genetically engineered mouse model. Cancer Res 2012; 72:4483–93; PMID:22815528; http://dx.doi.org/10.1158/0008-5472.CAN-12-0283
- Sun D, Sawada A, Nakashima M, Kobayashi T, Ogawa O, Matsui Y. MK2206 potentiates cisplatin-induced cytotoxicity and apoptosis through an interaction of inactivated Akt signaling pathway. Urol Oncol 2014; S1078-1439(14)00363–9 published on line December 9 2014, http://dx.doi.org/10.1016/j.urolonc.2014.10.018
- Levy DS, Kahana JA, Kumar R. AKT inhibitor, GSK690693, induces growth inhibition and apoptosis in acute lymphoblastic leukemia cell lines. Blood 2009; 113:1723–29; PMID:19064730; http://dx.doi.org/10.1182/blood-2008-02-137737
- Lucero-Acuna A, Jeffery JJ, Abril ER, Nagle RB, Guzman R, Pagel MD, Meuillet EJ. Nanoparticle delivery of an AKT/PDK1 inhibitor improves the therapeutic effect in pancreatic cancer. Int J Nanomedicine 2014; 9:5653–65; PMID:25516710
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 2010; 11:9–22; PMID:20027184; http://dx.doi.org/10.1038/nrm2822
- Flynn P, Wongdagger M, Zavar M, Dean NM, Stokoe D. Inhibition of PDK-1 activity causes a reduction in cell proliferation and survival. Curr Biol 2000; 10:1439–42; PMID:11102805; http://dx.doi.org/10.1016/S0960-9822(00)00801-0
- Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P, Kim SY, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 2009; 16:21–32; PMID:19573809; http://dx.doi.org/10.1016/j.ccr.2009.04.012
- Feldman RI, Wu JM, Polokoff MA, Kochanny MJ, Dinter H, Zhu D, Biroc SL, Alicke B, Bryant J, Yuan S, et al. Novel small molecule inhibitors of 3-phosphoinositide-dependent kinase-1. J Biol Chem 2005; 280:19867–74; PMID:15772071; http://dx.doi.org/10.1074/jbc.M501367200
- Peifer C, Alessi DR. Small-molecule inhibitors of PDK1. ChemMedChem 2008; 3:1810–38; PMID:18972468; http://dx.doi.org/10.1002/cmdc.200800195
- Najafov A, Sommer EM, Axten JM, Deyoung MP, Alessi DR. Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1. Biochem J 2011; 433:357–69; PMID:21087210; http://dx.doi.org/10.1042/BJ20101732
- Pearn L, Fisher J, Burnett AK, Darley RL. The role of PKC and PDK1 in monocyte lineage specification by Ras. Blood 2007; 109:4461–69; PMID:17255356; http://dx.doi.org/10.1182/blood-2006-09-047217
- Aihara H, Asaoka Y, Yoshida K, Nishizuka Y. Sustained activation of protein kinase C is essential to HL-60 cell differentiation to macrophage. Proc Natl Acad Sci U S A 1991; 88:11062–66; PMID:1763021; http://dx.doi.org/10.1073/pnas.88.24.11062
- Zabkiewicz J, Pearn L, Hills RK, Morgan RG, Tonks A, Burnett AK, Darley RL. The PDK1 master kinase is over-expressed in acute myeloid leukemia and promotes PKC-mediated survival of leukemic blasts. Haematologica 2014; 99:858–64; PMID:24334295; http://dx.doi.org/10.3324/haematol.2013.096487
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J 2007; 408:297–315; PMID:17850214; http://dx.doi.org/10.1042/BJ20070797
- Clark K, Plater L, Peggie M, Cohen P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J Biol Chem 2009; 284:14136–46; PMID:19307177; http://dx.doi.org/10.1074/jbc.M109.000414
- Medina JR, Becker CJ, Blackledge CW, Duquenne C, Feng Y, Grant SW, Heerding D, Li WH, Miller WH, Romeril SP, et al. Structure-based design of potent and selective 3-phosphoinositide-dependent kinase-1 (PDK1) inhibitors. J Med Chem 2011; 54:1871–95; PMID:21341675; http://dx.doi.org/10.1021/jm101527u
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274–93; PMID:22500797; http://dx.doi.org/10.1016/j.cell.2012.03.017
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011; 12:21–35; PMID:21157483; http://dx.doi.org/10.1038/nrm3025
- Hara K, Yonezawa K, Kozlowski MT, Sugimoto T, Andrabi K, Weng QP, Kasuga M, Nishimoto I, Avruch J. Regulation of eIF-4E BP1 phosphorylation by mTOR. J Biol Chem 1997; 272:26457–63; PMID:9334222; http://dx.doi.org/10.1074/jbc.272.42.26457
- Yee KW, Zeng Z, Konopleva M, Verstovsek S, Ravandi F, Ferrajoli A, Thomas D, Wierda W, Apostolidou E, Albitar M, et al. Phase I/II study of the mammalian target of rapamycin inhibitor everolimus (RAD001) in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res 2006; 12:5165–73; PMID:16951235; http://dx.doi.org/10.1158/1078-0432.CCR-06-0764
- Bohm A, Aichberger KJ, Mayerhofer M, Herrmann H, Florian S, Krauth MT, Derdak S, Samorapoompichit P, Sonneck K, Vales A, et al. Targeting of mTOR is associated with decreased growth and decreased VEGF expression in acute myeloid leukaemia cells. Eur J Clin Invest 2009; 39:395–405; PMID:19320940; http://dx.doi.org/10.1111/j.1365-2362.2009.02101.x
- Perl AE, Kasner MT, Tsai DE, Vogl DT, Loren AW, Schuster SJ, Porter DL, Stadtmauer EA, Goldstein SC, Frey NV, et al. A phase I study of the mammalian target of rapamycin inhibitor sirolimus and MEC chemotherapy in relapsed and refractory acute myelogenous leukemia. Clin Cancer Res 2009; 15:6732–9; PMID:19843663; http://dx.doi.org/10.1158/1078-0432.CCR-09-0842
- Janus A, Linke A, Cebula B, Robak T, Smolewski P. Rapamycin, the mTOR kinase inhibitor, sensitizes acute myeloid leukemia cells, HL-60 cells, to the cytotoxic effect of arabinozide cytarabine. Anticancer Drugs 2009; 20:693–701; PMID:19584709; http://dx.doi.org/10.1097/CAD.0b013e32832e89b4
- Callera F, Lopes CO, Rosa ES, Mulin CC. Lack of antileukemic activity of rapamycin in elderly patients with acute myeloid leukemia evolving from a myelodysplastic syndrome. Leuk Res 2008; 32:1633–4; PMID:18405970; http://dx.doi.org/10.1016/j.leukres.2008.02.004
- Boehm A, Mayerhofer M, Herndlhofer S, Knoebl P, Sillaber C, Sperr WR, Jaeger U, Valent P. Evaluation of in vivo antineoplastic effects of rapamycin in patients with chemotherapy-refractory AML. Eur J Intern Med 2009; 20:775–8; PMID:19892307; http://dx.doi.org/10.1016/j.ejim.2009.09.007
- Liesveld JL, O'Dwyer K, Walker A, Becker MW, Ifthikharuddin JJ, Mulford D, Chen R, Bechelli J, Rosell K, Minhajuddin M, et al. A phase I study of decitabine and rapamycin in relapsed/refractory AML. Leuk Res 2013; 37:1622–7; PMID:24138944; http://dx.doi.org/10.1016/j.leukres.2013.09.002
- Rizzieri DA, Feldman E, Dipersio JF, Gabrail N, Stock W, Strair R, Rivera VM, Albitar M, Bedrosian CL, Giles FJ. A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res 2008; 14:2756–62; PMID:18451242; http://dx.doi.org/10.1158/1078-0432.CCR-07-1372
- Park S, Chapuis N, Saint Marcoux F, Recher C, Prebet T, Chevallier P, Cahn JY, Leguay T, Bories P, Witz F, et al. A phase Ib GOELAMS study of the mTOR inhibitor RAD001 in association with chemotherapy for AML patients in first relapse. Leukemia 2013; 27:1479–86; PMID:23321953; http://dx.doi.org/10.1038/leu.2013.17
- Amadori S, Stasi R, Martelli AM, Venditti A, Meloni G, Pane F, Martinelli G, Lunghi M, Pagano L, Cilloni D, et al. Temsirolimus, an mTOR inhibitor, in combination with lower-dose clofarabine as salvage therapy for older patients with acute myeloid leukaemia: results of a phase II GIMEMA study (AML-1107). Br J Haematol 2012; 156:205–12; PMID:22082314; http://dx.doi.org/10.1111/j.1365-2141.2011.08940.x
- Altman JK, Sassano A, Kaur S, Glaser H, Kroczynska B, Redig AJ, Russo S, Barr S, Platanias LC. Dual mTORC2/mTORC1 targeting results in potent suppressive effects on acute myeloid leukemia (AML) progenitors. Clin Cancer Res 2011; 17:4378–88; PMID:21415215; http://dx.doi.org/10.1158/1078-0432.CCR-10-2285
- Willems L, Chapuis N, Puissant A, Maciel TT, Green AS, Jacque N, Vignon C, Park S, Guichard S, Herault O, et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia 2012; 26:1195–202; PMID:22143671; http://dx.doi.org/10.1038/leu.2011.339
- Tamburini J, Chapuis N, Bardet V, Park S, Sujobert P, Willems L, Ifrah N, Dreyfus F, Mayeux P, Lacombe C, et al. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: rationale for therapeutic inhibition of both pathways. Blood 2008; 111:379–82; PMID:17878402; http://dx.doi.org/10.1182/blood-2007-03-080796
- Zeng Z, Wang R-Y, Shi Y, Qiu Y, Mak DH, Coombes K, Yoo SY, Jessen K, Liu Y, Rommel C, et al. MLN0128, a second-generation mTOR kinase inhibitor, disrupts survival signaling and triggers apoptosis in AML. Blood 2014; 124: abstract 3613; PMID:25261197; http://dx.doi.org/10.1182/blood-2014-01-551457
- Jain N, Curran E, Iyengar NM, Diaz-Flores E, Kunnavakkam R, Popplewell L, Kirschbaum MH, Karrison T, Erba HP, Green M, et al. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: a University of Chicago phase II consortium trial. Clin Cancer Res 2014; 20:490–8; PMID:24178622; http://dx.doi.org/10.1158/1078-0432.CCR-13-1311
- Meja K, Stengel C, Sellar R, Huszar D, Davies BR, Gale RE, Linch DC, Khwaja A. PIM and AKT kinase inhibitors show synergistic cytotoxicity in acute myeloid leukaemia that is associated with convergence on mTOR and MCL1 pathways. Br J Haematol 2014; 167:69–79; PMID:24975213; http://dx.doi.org/10.1111/bjh.13013
- Teo T, Yu M, Yang Y, Gillam T, Lam F, Sykes MJ, Wang S. Pharmacologic co-inhibition of Mnks and mTORC1 synergistically suppresses proliferation and perturbs cell cycle progression in blast crisis chronic myeloid leukemia cells. Cancer Lett 2014; 357:612–23; PMID:25527453; http://dx.doi.org/10.1016/j.canlet.2014.12.029
- Joshi S, Platanias LC. Mnk kinase pathway: cellular functions and biological outcomes. World J Biol Chem 2014; 5:321–33; PMID:25225600; http://dx.doi.org/10.4331/wjbc.v5.i3.321
- Altman JK, Szilard A, Goussetis DJ, Sassano A, Colamonici M, Gounaris E, Frankfurt O, Giles FJ, Eklund EA, Beauchamp EM, et al. Autophagy is a survival mechanism of acute myelogenous leukemia precursors during dual mTORC2/mTORC1 targeting. Clin Cancer Res 2014; 20:2400–9; PMID:24610825; http://dx.doi.org/10.1158/1078-0432.CCR-13-3218
- Wong PM, Puente C, Ganley IG, Jiang X. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 2013; 9:124–37; PMID:23295650; http://dx.doi.org/10.4161/auto.23323
- Han W, Sun J, Feng L, Wang K, Li D, Pan Q, Chen Y, Jin W, Wang X, Pan H, et al. Autophagy inhibition enhances daunorubicin-induced apoptosis in K562 cells. PLoS One 2011; 6:e28491; PMID:22164300; http://dx.doi.org/10.1371/journal.pone.0028491
- Bosnjak M, Ristic B, Arsikin K, Mircic A, Suzin-Zivkovic V, Perovic V, Bogdanovic A, Paunovic V, Markovic I, Bumbasirevic V, et al. Inhibition of mTOR-dependent autophagy sensitizes leukemic cells to cytarabine-induced apoptotic death. PLoS One 2014; 9:e94374; PMID:24714637; http://dx.doi.org/10.1371/journal.pone.0094374
- Amaravadi RK, Winkler JD. Lys05: a new lysosomal autophagy inhibitor. Autophagy 2012; 8:1383–84; PMID:22878685; http://dx.doi.org/10.4161/auto.20958
- Kaneko M, Nozawa H, Hiyoshi M, Tada N, Murono K, Nirei T, Emoto S, Kishikawa J, Iida Y, Sunami E, et al. Temsirolimus and chloroquine cooperatively exhibit a potent antitumor effect against colorectal cancer cells. J Cancer Res Clin Oncol 2014; 140:769–81; PMID:24619662; http://dx.doi.org/10.1007/s00432-014-1628-0
- Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, Mita AC, Curiel TJ, Espitia CM, Nawrocki ST, et al. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014; 10:1403–14; PMID:24991835; http://dx.doi.org/10.4161/auto.29231
- Sehgal AR, Konig H, Johnson DE, Tang D, Amaravadi RK, Boyiadzis M, Lotze MT. You eat what you are: autophagy inhibition as a therapeutic strategy in leukemia. Leukemia. 2015 Mar; 29(3):517-525; http://dx.doi.org/10.1038/leu.2014.349
- Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012; 149:307–21; PMID:22500798; http://dx.doi.org/10.1016/j.cell.2012.02.053
- Gingras AC, Kennedy SG, O'Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev 1998; 12:502–13; PMID:9472019; http://dx.doi.org/10.1101/gad.12.4.502
- Diab S, Teo T, Kumarasiri M, Li P, Yu M, Lam F, Basnet SK, Sykes MJ, Albrecht H, Milne R, et al. Discovery of 5-(2-(phenylamino)pyrimidin-4-yl)thiazol-2(3H)-one derivatives as potent Mnk2 inhibitors: synthesis, SAR analysis and biological evaluation. ChemMedChem 2014; 9:962–72; PMID:24677692; http://dx.doi.org/10.1002/cmdc.201300552
- Lim S, Saw TY, Zhang M, Janes MR, Nacro K, Hill J, Lim AQ, Chang CT, Fruman DA, Rizzieri DA, et al. Targeting of the MNK-eIF4E axis in blast crisis chronic myeloid leukemia inhibits leukemia stem cell function. Proc Natl Acad Sci U S A 2013; 110:E2298–307; PMID:23737503; http://dx.doi.org/10.1073/pnas.1301838110
- Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM, Sellers WR, Lengauer C, Stegmeier F. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res 2009; 69:4286–93; PMID:19401449; http://dx.doi.org/10.1158/0008-5472.CAN-08-4765
- Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015; 372:30–9; PMID:25399551; http://dx.doi.org/10.1056/NEJMoa1412690