
Abstract
Lung cancer cells show inherent and acquired resistance to chemotherapy. The lack of good predictive markers/novel targets and the incomplete understanding of the mechanisms of resistance limit the success of lung cancer response to chemotherapy. In the present study, we used an isogenic pair of lung adenocarcinoma cell lines; A549 (wild-type) and A549DOX11 (doxorubicin resistant) to study the role of epigenetics and miRNA in resistance/response of non-small cell lung cancer (NSCLC) cells to doxorubicin. Our results demonstrate differential expression of epigenetic markers whereby the level of HDACs 1, 2, 3 and4, DNA methyltransferase, acetylated H2B and acetylated H3 were lower in A549DOX11 compared to A549 cells. Fourteen miRNAs were dys-regulated in A549DOX11 cells compared to A549 cells, of these 14 miRNAs, 4 (has-mir-1973, 494, 4286 and 29b-3p) have shown 2.99 – 4.44 fold increase in their expression. This was associated with reduced apoptosis and higher resistance of A549DOX11cells to doxorubicin and etoposide. Sequential treatment with the epigenetic modifiers trichostatin A or 5-aza-2'-deoxycytidine followed by doxorubicin resulted in: (i) enhanced sensitivity of both cell lines to doxorubicin especially at low concentrations, (ii) enhanced doxorubicin-induced DNA damage in both cell lines, (iii) dysregulation of some miRNAs in A549 cells. In conclusion, A549DOX11 cells resistant to DNA damaging drugs have epigenetic profile and miRNA expression different from the sensitive cells. Moreover, epigenetic modifiers may reverse the resistance of certain NSCLC cells to DNA damaging agents by enhancing induction of DNA damage. This may open the door for using epigenetic profile/miRNA expression of some cancer cells as resistance markers/targets to improve response of resistant cells to doxorubicin and for the use of combination doxorubicin/epigenetic modifiers to reduce doxorubicin toxicity.
Abbreviations
| 5AZA | = | 5-aza-2′-deoxycytidine |
| BSA | = | bovine serum albumin |
| DMSO | = | dimethyl sulfoxide |
| DNMT | = | DNA methyltransferase |
| HAT | = | histone acetyl transferase |
| HDAC | = | histone deacetylase |
| NSCLC | = | non-small cell lung cancer |
| PBS | = | phosphate-buffered saline |
| SCLC | = | small-cell lung cancer |
| TSA | = | trichostatin A |
| 5mc | = | 5-methyl cytosine |
Introduction
In spite of recent advances in patient management, lung cancer is still a leading cause of cancer death worldwide.Citation1 Histologically, lung cancer is grouped into Non-small (NSCLC) and small-cell lung cancer (SCLC). Platinum-based therapy is commonly used for NSCLC in combination with a tubulin-binding agent, a camptothecin analog, gemcitabine, or pemetrexed.Citation2 Doxorubicin and other anthracyclins have been used in several historical schemes for NSCLC. The response rate achieved for these old combinations was 20 to 23%.Citation3 Inherent and acquired resistance and the requirement of large doses, that may cause toxicity, lead to the diminished application of anthracyclins in NSCLC.Citation4
Doxorubicin has different modes of action such as topoisomerase II poisoning, DNA adduct formation, oxidative stress, histones eviction, and ceramide overproduction.Citation5 Contrary to its high activity against SCLC, it is relatively ineffective against NSCLC. Even in SCLC, doxorubicin-based chemotherapy regimens frequently fail due to inherent and acquired resistance.
Resistance to anticancer drugs is a major problem limiting the success of lung cancer therapy. In NSCLC, the drug resistance is frequently inherent, whereas SCLC acquires resistance with continued drug administration.Citation6
The mechanisms of lung cancer resistance to anticancer agents include increased drug efflux by ATP binding cassette superfamily, drug inactivation and/or sequestration by enzymes, increased DNA repair activity, apoptosis defects, and alteration of drug target.Citation2 The genetic basis of these mechanisms has been intensively studied whereas a link between these mechanisms and epigenetic profile of cancer cells is still lacking. Epigenetics and miRNA expression profile have recently emerged as important mediators of response to anticancer agents where some studies have shown significant alterations in epigenetics and miRNA expression in drug-resistant cancer cells.Citation7
Epigenetics, the study of heritable changes in gene expression that are independent of alterations in the DNA base sequence, is regulated by different mechanisms such as DNA methylation, histone modification and non-coding RNA.
DNA methylation can potently suppress gene activity through 2 mechanisms; by interference with binding of transcription factors through steric hindrance of methyl-CpGs or by methyl-CpG binding proteins that can bind to methylated DNA.Citation8 Methylation of genes involved in drug responses has been shown to have a potential role in determining clinical outcome following chemotherapy.Citation9
Five-Aza-2′-deoxycytidine (5AZA) is a cytidine analog and methyltransferase (DNMT) inhibitor. The formation of a covalent complex between 5AZA-DNA and DNMT1 inhibits this enzyme and results in DNA hypo methylation, gene activation and the induction of cellular differentiation, senescence, and/or apoptosis. 5AZA may also sensitize cancer cells to anticancer therapies by causing re-expression of genes involved in drug sensitivity.Citation10 However, at high concentrations 5AZA may have direct cytotoxic effects through its incorporation into DNA.Citation11
Histones, proteins that package the DNA into nucleosomes, are subject to numerous post-translational modifications that can influence their interaction with DNA and other proximal non-histone proteins, which in turn can affect transcriptional expression, DNA repair and genome stability. Histone acetylation is highly dynamic and is controlled by 2 enzymatic families, the histone acetyltransferases (HATs) and the histone deacetylases (HDACs). Acetylation is often associated with a looser chromatin conformation. Contrary to HATs, HDACs inhibit histone acetylation. It is observed that inhibitors of histone deactylases (HDAC-I) are able to reverse some of the aberrant gene repression seen in some malignancies and induce growth arrest, differentiation, and apoptosis.Citation9
Trichostatin A (TSA) is a potent inhibitor of HDAC with cytostatic and differentiating properties in mammalian cells.Citation12 Various studies have suggested that TSA may have a promising therapeutic effect on cancer cells along with radiotherapy or chemotherapy.Citation13
MicroRNAs are endogenous, small, noncoding RNA molecules that regulate multiple cellular processes by modulating gene expression through translational inhibition.Citation7 In addition to their role in tumor suppression or promotion, miRNAs also play role in regulation of vital genes that control mechanisms of chemo-resistance.
The lack of good predictive markers and novel targets and the incomplete understanding of the mechanisms of resistance to anticancer agents limit the success of chemotherapy of NSCLC. The aim of the present study is to investigate the role of epigenetics and miRNA in resistance/response of NSCLC cells to anticancer agents. The ultimate goal is to study the possibility of using these markers as predictors/targets to overcome resistance of NSCLC to cancer chemotherapy.
Results
Sensitivity of A549 and its doxorubicin-resistant variant (A549DOX11) to doxorubicin and etoposide
A549DOX11 cells were developed in our lab by exposing the parental A549 cells to repeated cycles of doxorubicin and isolating resistant clones. To confirm the development of doxorubicin resistance, both cell types (A549 and A549DOX11) were seeded in 96 well plates and treated with different concentrations of doxorubicin or etoposide (0.01 – 10 µM) and fraction of cells remaining viable relative to untreated cells was determined using sulforhodamine B assay. As shown in , surviving fraction of A549 cells is lower than that of A549DOX11 at all doxorubicin concentrations which indicates the development of doxorubicin resistance in A549DOX11 cells. The doxorubicin IC50 was 0.05 and 5.5 µM for A549 and A549DOX11 cells, respectively, indicating that the A549DOX11 cells are 110 times more resistant to doxorubicin than the A549 parental cells. Likewise, the A549DOX11 cells were more resistant to etoposide (with an IC50 of 4 µM) than the parental cells A549 (with an IC50 of 0.33 µM).
Figure 1. Sensitivity of A549 and its doxorubicin-resistant variant A549DOX11 to doxorubicin and etoposide. Cells were exposed to either vehicle (DMSO), doxorubicin or etoposide (0.01 – 10 μM) for 48 h. Cells were fixed, stained with sulphorhodamine-B (SRB) and the optical density was measured spectrophotometrically. (A) sensitivity of both cell lines to doxorubicin, (B) sensitivity of both cell lines to etoposide. Each point is the mean ± SEM of 6 separate experiments. *, significantly different from the parental cells A549 (P < 0.05)
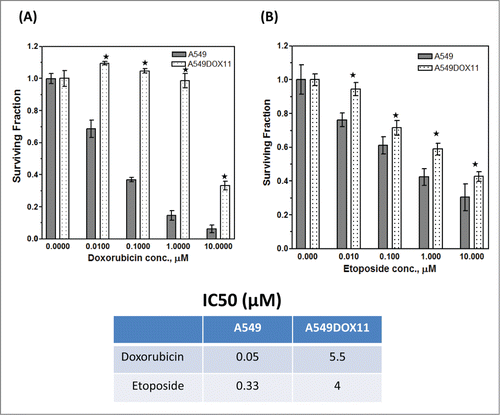
Differential expression of histone deacetylases (HDACs), acetylated histones and DNA methyl transferase (DNMT1) in A549 cells and its doxorubicin-resistant variant (A549DOX11)
To investigate the role of some epigenetic markers in the acquired resistance of A549 cells to doxorubicin, we measured the expression level of HDAC1, 2, 3, 4, 6, phosphoHDAC4, phosphoHDAC5, phosphoHDAC7 and DNMT1 in A549 and its doxorubicin resistant variant using western blot. As shown in , the expression level of HDAC1, 2, 3, 4 and the level of DNMT1 was higher in the wild-type A549 cells than the dox-resistant cells. The phosphoHDAC4 and the phosphoHDAC5 were higher in the dox-resistant cells than the A549 cells. On the other hand, no differences in the expression level of HDAC6 nor phosphoHDAC7 were detected between the 2 cell lines. Acetylated histones H2B and H3 were higher in the parental A549 cells than the dox-resistant whereas the same level of acetylated H4 was detected in both cell lines (). Global DNA methylation was higher in the parental cells (A549) than the dox-resistant ones (A549DOX11) ().
Figure 2. Expression of different HDACs, acetylated histones and global DNA methylation in A549 and A549DOX11 cells. Protein extract from A549 and A549DOX11 cells was subjected to Western blot analysis using the following antibodies: (A) HDAC1,2,3,4,6, phosphor-HDAC4, 5 and 7 and DNMT1 antibodies or (B) acetyl-histone H4 (Lys8), acetyl-H3 (lys9), and acetyl-histone H2B (Lys5) antibodies. The same blot was reprobed with Actin antibody as loading control. Left side: representative blots, Right side: quantification of the relative intensity of individual bands using the image studio™ Lite-Western blot analysis Program (LI-COR Biosciences, Lincoln, NE) normalized to the corresponding actin level. (C) global DNA methylation of both cell lines, genomic DNA was extracted and global DNA methylation was detected. ★, significantly different from the A549 cells (P < 0.05). 5mC =5-methylcytosine
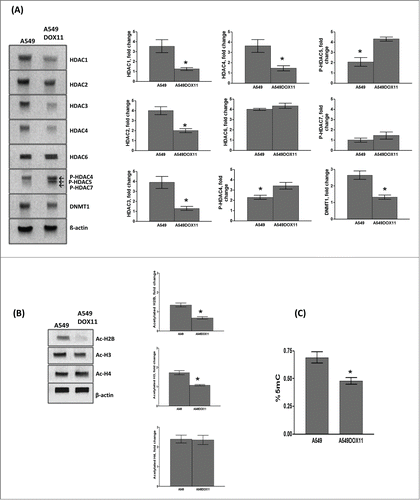
MiRNA expression profile in A549 and A549DOX11 cells
To test whether these epigenetic differences are associated with changes in the level of miRNA expression and to investigate the role of miRNA in the development of acquired chemotherapy resistance, we analyzed the miRNA expression profile of parental (A549) and its doxorubicin resistant variant (A549DOX11).
The microarray analysis revealed significant changes in miRNA expression profile in A549DOX11 cells compared to A549 cells. The results showed 14 miRNA genes (12 up-regulated and 2 down-regulated) that were differentially expressed in A549DOX11 cells compared with parental A549 cells ( and ). Fold change greater or equal to 1.5 have been considered as significant change.Citation14 To validate the results of miRNA microarray analysis, 4 miRNAs that showed highest level of differential expression (has-mir-1973, 494, 4286 and 29b-3p) were selected for RT-PCR analysis. The results of RT-PCR confirmed the miRNA array results whereby the expression level of the selected miRNA was higher in the A549DOX11 cells than the A549 cells ().
Figure 3. miRNA expression profile in A549DOX11 cells compared to the parental A549 cells. Total RNA was extracted from both cell lines and MicroRNA expression analysis was performed with Agilent® Unrestricted Human miRNA V16.0 8 × 60 K Microarray as described under the materials and methods section. (A) miRNA microarray analysis (B) miRNA microarray results validated by RT-PCR.
Table 1. Fold change of miRNA expression in A549 and A549DOX11 cells with and without epigenetic modifiers
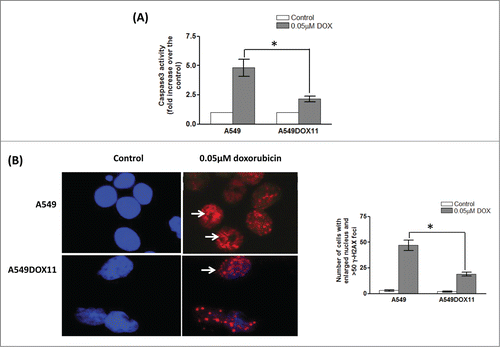
Apoptosis induction in A549 and A549DOX11 after treatment with doxorubicin
To investigate how these epigenetic differences and miRNA differential expression translates into different responses to genotoxic stress, we tested induction of apoptosis in both cell lines after treatment with 0.05 µM doxorubicin for 48 h. Caspase3 activity and formation of uncountable number of γ–H2AX foci were used as markers for apoptosis. As shown in , doxorubicin increased the caspase 3 activity 5 folds in the A549 cells and only 2 folds in A549DOX 11 over the control cells. Similarly, the number of cells with uncountable number of γ–H2AX foci was higher in A549 cells than A549DOX11 after treatment with 0.05 µM doxorubicin for 48 h ().
Figure 4. Caspase 3 activity and γ–H2AX induction as apoptosis markers in A549 and A549DOX11 cells after treatment with doxorubicin. Exponentially growing cells were treated with 0.05 µM doxorubicin for 48h and cells were collected by tripsinization. (A) Activity of caspase-3 was measured colorimetrically. (B) γ–H2AX foci formation was detected by anti-phospho-H2AX (Ser139) antibody (Upstate, Lake Placid, NY USA) and Alexa Fluor-594 goat anti-mouse. Left is a representative photo for cells with γ–H2AX foci and right is quantification of number of cells with enlarged nucleus and more than 50 γ–H2AX foci. *, significantly different from the dox-resistant A549DOX11 cells (P < 0.05).
Sensitivity of A549 and its doxorubicin-resistant variant (A549DOX11) to the chromatin modifying agents TSA and 5AZA
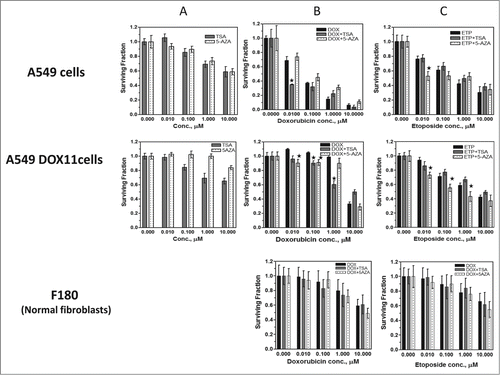
To select the optimum concentration of TSA and 5AZA that can be used for molecular studies, the effect of different concentrations of these 2 agents on the 2 cell lines was tested using the SRB assay. shows that both agents have weak concentration-dependent reduction of the survival of the 2 cell lines. The effect on the survival of A549 was more pronounced than A549DOX11 cells. 0.1 µM TSA and 1 µM 5AZA were selected for further investigations because these concentrations show low effect on survival of both cell lines and also in accordance with previous investigations.Citation14,15
Figure 5. Sensitivity of A549 cells, its doxorubicin-resistant variant A549DOX11 and normal fibroblasts F180 cells to anticancer drugs ±TSA or 5AZA. Cells were exposed to either vehicle (DMSO) or drug(s) for 48 h. (A) sensitivity of the two cell lines to TSA or 5AZA. (B) sensitivity of the three cell lines to doxorubicin ±TSA or 5AZA. (C) sensitivity of the three cell lines to Etoposide ±TSA or 5AZA. Each point is the mean ±SEM of six separate experiments. *, significantly different from cells treated with doxorubicin or etoposide alone (P < 0.05)
Effect of chromatin structural modifications (by TSA and 5AZA) on sensitivity of A549, A549DOX11 and normal fibroblast cells to doxorubicin and etoposide
Results of our initial experiments showed that induction of resistance to doxorubicin in A549 cells resulted in epigenetic and miRNA profile changes (). To confirm the interrelationship between epigenetic status, miRNA expression and response to anticancer agents we tried the opposite way, that is inducing epigenetic modifications by chemicals and studying the effects of these modifications on the miRNA expression and on the response of A549 and its doxorubicin resistant variant to DNA damaging anticancer drugs. To this end, the 2 cell lines were treated with either 0.1 µM TSA or 1 µM 5AZA for 24 h followed by doxorubicin or etoposide (0.01 – 10 µM) for 48 h and the fraction of cells remaining survival relative to control cells was measured by SRB assay. Pretreatment of the parental A549 cells with 5AZA did not alter their response to all doxorubicin concentrations whereas TSA enhanced the sensitivity of A549 cells to 0.01 µM doxorubicin where the survival using combination of 0.01 µM doxorubicin + 0.1 µM TSA was 0.35 compared to 0.7 after doxorubicin alone with a sensitizing factor of 2 (). On the other hand, sensitivity of the doxorubicin resistant A549DOX11 cells to doxorubicin has been enhanced by pretreatment with either TSA or 5AZA. The survival of A549DOX11 cells was reduced from 1.15 (after 0.01 µM doxorubicin alone) to 0.9 (after 1 µM 5AZA or 0.1 µM TSA + 0.01 µM doxorubicin) with a sensitizing factor of 1.33. A similar sensitizing factor of 1.3 was also detected after combining TSA or 5AZA with 0.1 µM doxorubicin. At higher doxorubicin concentrations (1 and 10 µM), 5AZA did not change the response of the resistant cells to doxorubicin whereas TSA enhanced the sensitivity of the cells by a factor of 1.5 at 1 µM doxorubicin (, middle panel).
Etoposide is another DNA damaging anticancer drug. Here, we investigated the effect of pretreatment with TSA or 5AZA on the sensitivity of both cell lines to etoposide. TSA did not alter the response of both cell lines to etoposide whereas 5AZA have increased the sensitivity of both cell lines to etoposide whereby it increased the sensitivity of A549 cells only at low etoposide concentrations (0.01 and 0.1 µM) and increased the sensitivity of A549DOX11 cells at all etoposide concentrations (0.01 – 10 µM)().
As a normal control, we investigated the effects of TSA and 5AZA on the sensitivity of the normal fibroblast cells F180 to doxorubicin and etoposide. Opposite to cancer cells, neither of the 2 agents (TSA and 5AZA) enhanced the toxicity of the 2 drugs (doxorubicin and etoposide) on the normal fibroblasts ( lower panel).
Effect of chromatin structural modifications (by TSA and 5AZA) on chromatin acetylation, global DNA methylation and on DNA damage induction in A549 and A549DOX11 cells after doxorubicin
To establish a link between the cellular effects of TSA and its presumed mechanism of action, the degree of acetylation of histones H2B, H3 and H4 in both cell lines after treatment with 0.1 or 0.2 µM TSA was analyzed using Western blotting (). Quantification of band intensities showed that background acetylated H2B and H3 was higher in parental A549 cells than doxorubicin resistant A549DOX11 cells. Treatment of A549 cells with 0.1 µM TSA increased the degree of histones H2B, and H4 acetylation. Under the same treatment, A549DOX11 cells showed hyperacetylation of H2B and H3 but not H4 histones. To explore the different genomic methylation patterns of A549 and A549DOX11 cells at different concentrations of 5AZA, genomic DNA was isolated from 5AZA-treated A549, A549DOX11 and untreated cells. The global cytosine DNA methylation pattern was then assessed. Control A549DOX11 cells showed reduced total cytosine DNA methylation compared with parental A549 cells. Both cell lines showed decreased cytosine methylation with increased 5AZA concentration ().
Figure 6. Effect of TSA, 5AZA or their combination on chromatin acetylation, global DNA methylation and DNA damage induction in A549 and A549DOX11 cells after doxorubicin. Cells were exposed to either vehicle (DMSO) or drug(s) for 48 h, collected and: (A) tested for the expression of acetylated H2B, H3 and H4. The same blot was reprobed with Actin antibody as loading control. Statistical significance of differences in histone acetylation by different treatments was evaluated by one-way ANOVA. ★, significantly different from the control (P < 0.05). Left side: representative blots, Right side: quantification of the relative intensity of individual acetylated histone bands normalized to actin level. (B) genomic DNA was extracted from pelleted cells and global DNA methylation was detected. Statistical significance of global DNA methylation was evaluated by one-way ANOVA. ★, significantly different from the control (P < 0.05). (C) DNA fragments were separated by constant field gel electrophoresis (0.6 V/cm, 40 h). Left side: photos of UV-trans-illuminated gels, Right side: quantification of band intensities. Statistical significance of differences in DNA damage induction by different treatments was evaluated by one-way ANOVA. ★, significantly different from the control (P < 0.05); ★, significantly different from doxorubicin (P < 0.05).
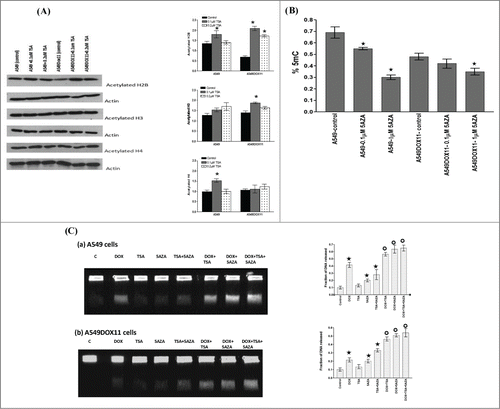
Doxorubicin exercises its cytotoxic effects through induction of DNA damage by targeting cellular DNA topoisomerases and generation of free radicles. We hypothesized that induction of chromatin hyperacetylation by TSA or hypomethylation by 5AZA may make the chromatin more open and consequently enhances accessibility of DNA by doxorubicin and hence increases the extent of doxorubicin-induced DNA damage. shows that TSA alone did not induce DNA damage in both cell lines whereas doxorubicin alone and 5AZA alone increased the degree of DNA damage in both cell lines compared to the untreated cells. It is noteworthy that doxorubicin induced higher degree of DNA damage in the parental A549 cells compared to the doxorubicin resistant A549DOX11 cells. Pretreatment of both cell lines with either TSA or 5AZA or combination thereof, increased the degree of DNA damage compared to doxorubicin alone.
Effects of 5AZA and TSA on miRNA expression of A549 and A549DOX11 cells
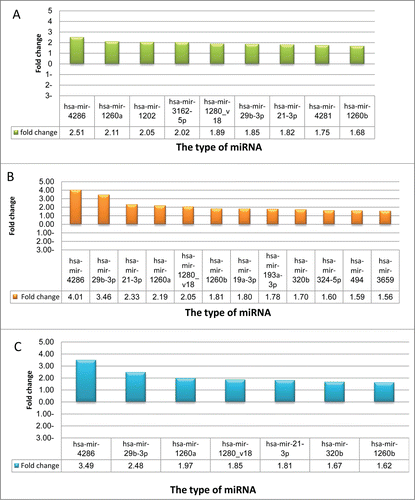
To get deeper insight into the interrelationship of chromatin structure modification, miRNA expression and response of cancer cells to chemotherapy, the miRNA expression profile in both cell lines after treatment with the chromatin modifiers TSA, 5AZA or their combination, was analyzed. In A549 cells, by comparing the miRNA expression profile of untreated and 5AZA- treated cells, 9 miRNAs were found to be up regulated in 5AZA-treated cells (, ). Expression of 12 miRNAs was found to be upregulated after treatment of A549 cells with TSA compared with untreated cells (, ) and 7 miRNAs were up regulated after 5AZA/TSA combination treatment ( and ). However, none of the miRNAs analyzed showed differential expression greater than 1.5-fold change after treatment of A549DOX11 cells with 5AZA and/or TSA. Interestingly, 6 miRNAs (Has-mir-4286, 29b-3p, 1260a, 21–3p, 1280_v18 and 1260b) that were upregulated in A549DOX11 cells due to development of resistance were also found to be upregulated in A549 cells after treatment with either 5AZA or TSA ().
Figure 7. miRNAs differentially expressed in A549 and A549DOX11 cells after treatment with TSA, 5AZA and their combination. Cells were treated with either vehicle (DMSO) TSA (0.1 μM), 5AZA (1 μM) or their combination for 48 h. Total RNA was extracted. MicroRNA expression analysis was performed with Agilent® Unrestricted Human miRNA V16.0 8×60 K Microarray (Agilent Technologies, AMADID 31181) as described under the materials and methods section. Only miRNAs showing differential expression are presented in this figure. (A) miRNA expression profile in 5AZA-treated A549 cells compared to the profile of untreated A549 cells. (B) miRNA expression profile in TSA-treated A549 cells compared to the profile of untreated A549 cells (C) miRNA expression profile in TSA+5AZA-treated A549 cells compared to the profile of untreated A549 cells.
Discussion
Lung cancer cells, especially the non-small cell type, show both inherent and acquired resistance to cancer chemotherapy. This can be attributed to both genetic and epigenetic causes. Genetic basis of resistance have been intensively investigated whereas only few studies have addressed the role of epigenetics in lung cancer resistance. Epigenetics may affect the response of cancer cells to anticancer agents through 2 main mechanisms; silencing/activation of genes involved in response to chemotherapy and altering chromatin condensation state and hence modifying the accessibility of anticancer drugs to cellular DNA. Histone acetylation and DNA methylation are the most common epigenetic alterations in cancer cells and since epigenetic modifications, opposite to the genetic ones, do not mutate the DNA sequence of genes, strategies to reverse epigenetic abnormalities are considered useful in reversing drug resistance.
The aim of the present study is to investigate the role of some epigenetic markers in mediating the response of lung cancer cells to chemotherapy. We used an isogenic pair of cancer cell lines, A549 (NSCLC) and its doxorubicin-resistant variant (A549DOX11). Because NSCLC cells are known to be intrinsically resistant to chemotherapy, this model is attractive as it allows the study of both intrinsic and extrinsic resistance.
Differential expression of epigenetic markers and miRNA reported in the present study in the 2 cell lines was associated with: (i) higher resistance of A549DOX11cells to doxorubicin and etoposide, (ii) lower level of some apoptotic markers in A549DOX11 compared to A549 cells which indicates reduced ability of A549DOX11 cells to execute apoptosis after genotoxic stress induced by doxorubicin.
Because the model used in the present study is an isogenic pair of cell lines, we may conclude that these epigenetic markers are involved in the acquired resistance of A549 cells to doxorubicin and their expression has been modified through the repeated exposure of A549 cells to doxorubicin during the development of the resistant variant. This agrees with previous demonstration that HDACs are involved in DNA damage response machinery following drugs like doxorubicin.Citation16 Similarly, Calcango et al reported that exposure of breast, ovarian and colon cancer cells to low concentrations of doxorubicin for 10 days resulted in development of resistance that was associated with down-regulation of HDAC1.Citation17
MicroRNAs have been reported previously to induce chemo resistance of cancer cells by different mechanisms such as modulation of intracellular drug concentrations, modulation of drug target, enhancement of drug metabolism, inhibition of apoptosis, regulation of angiogenesis and generation of tumor stem cells.Citation18-23 Among the 14 miRNAs dysregulated in the present study in A549DOX11 cells, 4 (has-miR-1973, 494, 4286 and 29b-3p) have shown the highest level of upregulation (2.99 – 4.44 folds) compared with the A549 parental cells.
In line with our finding, a recent investigation have demonstrated that overexpression of miR-494 is associated with resistance of a panel of NSCLC cells to TRAIL-induced apoptosis.Citation24 Searching in different databases, we did not identify previous reports relating the other 13 miRNAs reported in and with resistance of NSCLC cells to doxorubicin or other chemotherapeutic agents. This finding adds new miRNAs and opens the avenue for new targets/predictors to improve the response of lung cancer cells to chemotherapy. We speculate that these miRNAs, especially those 4 showing highest level of differential expression, control the expression of proteins involved in apoptosis such as bcl2, bclx, Bax, Bak, akt and others either directly or indirectly by modifying chromatin structure nearby the promoter regions of these genes. However, further studies are needed to validate this finding and to determine the exact molecular targets of these miRNAs (some confirmed targets are presented in ). Collectively, this result indicates a relationship between epigenetic status of certain cancer cells (such as HDACS, acetylated histones, DNA methylation and miRNA expression), their ability to execute apoptosis and their sensitivity to DNA damaging agents like doxorubicin and etoposide.
Table 2. Some confirmed miRNA-target interaction for miRNAs reported in the present study (taken from Hsu et al 2014)Citation32
Initial set of experiments showed that acquired resistance to doxorubicin is associated with changes in miRNA expression and epigenetic profile of A549 cells. It was unclear whether these 2 events (miRNA expression and epigenetic profile) are linked together or they are 2 independent events. To understand this complex relationship between epigenetic profile, miRNA expression and response to doxorubicin, we changed the epigenetic profile of both cell lines using epigenetic modifiers and investigated its effects on the expression of miRNA and on the response to doxorubicin. Pretreatment of both cell lines with TSA increased their sensitivity to low but not high concentrations of doxorubicin. This effect was more pronounced in A549DOX11than A549 cells. TSA is a potent HDAC inhibitor and its combination with doxorubicin may enhance its effects by different mechanisms such as relaxation of cellular chromatin and enhancing accessibility of the DNA to doxorubicin and/or induction of apoptosis and cell cycle arrest.Citation25 The variable effects of TSA at different concentrations of doxorubicin may be explained by the fact that low doxorubicin concentrations has no effects on histones and hence TSA enhances doxorubicin effects in both cell lines but at higher concentrations, doxorubicin itself may alter the epigenetic state of the cells by causing histone eviction and consequently TSA will not add more effects on cellular response to doxorubicin. The effects of doxorubicin on histone has been recently published by Pang et al who showed that anthracyclines promote histone eviction which is associated with attenuated DNA repair.Citation15 TSA effects on doxorubicin cytotoxicity were more pronounced on A549DOX11 than A549 cells. This could be due to epigenetic changes developed in A549DOX11 cells during the establishment of doxorubicin resistance which can be reversed upon treatment with TSA. This explanation is supported by the finding that background level of acetylated H2B is higher in A549 than A549DOX11 cells.
5AZA is a DNMT inhibitor that reduces DNA methylation and reactivates silenced genes involved in DNA damage response machinery, apoptosis, signal transduction and drug transport. In the present study, pretreatment of cells with 5AZA before doxorubicin reversed the resistance of A549DOX11 cells without changing the response of A549 cells to doxorubicin. This differential effect could be due to reactivation, by 5AZA, of genes that have been silenced in A549 cells during generation of A549DOX11 cells. Similar results were previously reported by Vijayaraghavalu et al who showed that pretreatment with 5AZA increased the sensitivity of doxorubicin resistant breast cancer cells (MCF7/ADR) more than parental cells (MCF7).Citation26
This postulation has been tested by measurement of global DNA methylation after treatment with 5AZA. The DNA of doxorubicin resistant cells was globally hypomethylated compared to wild type A549 cells and global DNA methylation was reduced in both cell lines after treatment with 5AZA in a concentration-dependent manner. Our finding is in agreement with previous studies showing that chemoresistant cells are globally hypomethylated compared to sensitive cells.Citation27 The degree of global DNA methylation does not reflect activity of specific genes but may give some indications about the effect of 5AZA on the general expression status of genes. It is worth mentioning that gene activation by hypomethylation is not always associated with enhanced response to cytotoxic agents. Activation of genes involved in apoptosis may increase cellular sensitivity whereas activation of genes involved in drug efflux or genes involved in DNA repair may increase resistance to anticancer drugs.
Doxorubicin kills cancer cells by inhibiting DNA topoisomerase II and generation of free radicals inducing DNA double-strand breaks. Hence, we assumed that chromatin relaxation by chromatin modifying agents may enhance accessibility of cellular DNA to doxorubicin and increases the extent of DNA damage. Our results show that doxorubicin induced higher DNA damage in A549 than A549DOX11 cells. Combining TSA, 5AZA or both with doxorubicin increased the extent of DNA damage in both cell lines which may confirm the hypothesis that these agents enhance doxorubicin cell kill by increasing DNA accessibility to doxorubicin. It is noteworthy that 5AZA itself may induce indirect DNA damage through its incorporation during DNA replication which destabilizes the DNA causing its fragmentation.
Analysis of miRNA profile of A549 cells treated with TSA, 5AZA or both revealed the presence of some common miRNAs that are upregulated after this treatment and were also upregulated in the control A549DOX11 cells (). This result confirms the role of these miRNAs as modulators of lung cancer cells response to chemotherapy.
In conclusion, certain cancer cells resistant to DNA damaging anticancer drugs have epigenetic profile different from sensitive cells. Moreover, epigenetic modifiers such as TSA/5AZA may reverse the resistance of NSCLC cells to DNA damaging agents by enhancing their ability to induce DNA damage. In addition, we report some new miRNAs that are involved in the acquired resistance of NSCLC cells to doxorubicin and in mediating their response to epigenetic modifiers. The clinical impact of our results is that epigenetic profile of some cancer cells may be used as a marker for the response to doxorubicin and that response of NSCLC to doxorubicin can be improved by combining low doses of doxorubicin with the epigenetic modifiers TSA/5AZA. This combination allows the use of low doses of doxorubicin and reduces its toxicity. However, confirmation of such finding in other cancer cells is required before drawing clear conclusions.
Materials and methods
Cell lines and culture conditions
Human lung adenocarcinoma epithelial cell line A549 was provided by professor Dr Salem Chouaib (Institute Gustave Roussy – France) and the doxorubicin resistant cell line A549DOX11 was established in our lab (Drug Design and Molecular Pharmacology laboratory, Sharjah Institute for Medical Research, University of Sharjah) by exposing the A549 cells to multiple cycles of 0.1 μM doxorubicin (48 h each) and incubating the cells for 2 d in doxorubicin free medium between every 2 cycles. Resistant clones were selected and cloned after 5 cycles of doxorubicin exposure. Development of resistance was confirmed by the Sulforhodamine-B assay in comparison with the wild-type A549 cells. Moreover, no change in the expression level and the activity of some proteins such as p53, p21, Rb and CDK2 have been observed in the resistant clone over the wild type cells as tested by immune-blot (data not shown). Both cell types were maintained in RPMI supplemented with 10% fetal bovine serum (Gibco, USA) and 1% penicillin / streptomycin (Gibco, USA). Normal fibroblasts, F180 were kindly provided by professor Dr Ekkehard Dikomey (University Cancer Center, Hamburg University, Hamburg, Germany) and were maintained in DMEM supplemented with 15% fetal bovine serum and 1% penicillin/streptomycin. All incubations were done at 37°C in humidified atmosphere of 5% CO2. Resistance phenotype of A549DOX11 cells was maintained by culturing the cells in RPMI (Gibco, USA) medium containing 50 ng/ml doxorubicin. Resistant cells were maintained in drug-free medium in the last 2 passages prior to any experiment.
Treatment of cells with epigenetics modifiers (5AZA/TSA)
A549 and its doxorubicin resistant variant A549DOX11 were treated with either 1.0 μM 5AZA (Sigma), 0.1 μM Trichostatin A (Sigma) or their combination for 48 h. Control cells were incubated with the vehicle (DMSO), the DMSO concentration did not exceed 0.1% in all experiments in this study. For combination experiments, cells were treated with TSA or AZA for 24 h followed by doxorubicin or etoposide (both from Sigma Aldrich, USA) for 48 h. This sequence and duration of treatment have been selected because previous studies have demonstrated that DNMT and HDAC levels revert back to normal values at day 2 post-treatment with 5AZA and TSA and that sequential treatment with epigenetics active drugs followed by cytotoxic agents is more effective than concomitant treatment.Citation26
Chemo-sensitivity assay
Cytotoxicity was determined using the Sulphorhodamine-B (SRB) method, as previously described by El-awady et al.Citation28 Cells were seeded in 96-well microtiter plates (104 cells/well) in RPMI medium. After 24 h, cells were incubated for 48 h with either TSA or 5AZA (0.01–100 μM). In combination experiments, cells were treated with either 0.1 μM TSA or 1 μM 5AZA for 24 h followed by either doxorubicin or etoposide (0.01 – 10 μM). After 48 h, cells were fixed with 50% trichloroacetic acid for 1 h at 4°C, washed 5 times with water, stained for 30 min at room temperature with 0.4% SRB dissolved in 1% acetic acid and then washed 4 times with 1% acetic acid. The plates were air-dried, and the dye was solubilized with 100 μl/well of 10 mMTris base (pH 10.5) for 5 min. The optical density (OD) was measured spectrophotometrically at 450 nm with an ELISA micro plate reader (Awareness Technology Chromate, USA). The IC50 values were calculated using sigmoidal concentration–response curve fitting models using the Graph Pad Prizm program (GraphPad Software, San Diego, CA USA).
DNA damage induction
Induction of DNA damage in A549 and A549DOX11 cells was measured by constant-field gel electrophoresis as described previously by Saleh et al.Citation29 Exponentially growing cells were treated with 5AZA, TSA, 5AZA+TSA, doxorubicin alone or their combination for 48 h. In combination experiments, 5AZA or TSA were added 24 h before doxorubicin. Control cells were incubated with the vehicle (DMSO). At the end of the incubation period, cells were detached with trypsin and the resulting cell suspension was mixed with an equal amount of 1.6% low melting point agarose (Bio-Rad, Munich, Germany), pipetted into a 180 µl mold and left on ice to solidify and form agarose cell plugs. The plugs were incubated in lysis buffer (0.4 M ethylene diaminetetraacetic acid, 2% N-lauryl sarcosine and 1 mg/ml proteinase-K; Sigma, Deisenhofen, Germany) for 30 min on ice and then overnight at 37°C. The plugs were washed 3 times in Tris-EDTA buffer, and a portion of the plug containing approximately 105 cells was inserted into a preformed 0.8% agarose gel. The gel was then covered with a thin overlayer of 0.8% agarose to avoid light fraction artifacts upon optical imaging. Electrophoresis was performed for 36 h at 0.6 V/cm in 0.5 x Tris-Borate-EDTA buffer (45 mMTris base, 45 mM boric acid, 2 mM EDTA) in a conventional apparatus (Subcell, Bio- Rad). After completion of electrophoresis, the gel was stained overnight in 0.5 mg /ml ethidium bromide solution and destained overnight in distilled water. The fraction of DNA released (FDR) after each treatment was determined using a gel documentation system (Biometra, Gottingen, Germany).
The FDR released into the gel, which corresponds to fragmented DNA, was calculated according to the equation: FDR = FDR rel/(FDR plug + FDR rel).
FDR rel: fraction of DNA released outside the well. FDR plug: fraction of DNA remaining in the well.
miRNA microarray expression analysis
After treatment of A549 and A549DOX11 cells with either 0.1 μM TSA or 1 μM 5AZA or their combination for 48h, total RNA was extracted by using trizol (sigma) reagent according to the manufacturer's instructions. One microliter was used for determination of RNA concentration using a Nanodrop spectrophotometer (Thermo Fisher Scientific). Quality of RNA-preparations was further assessed by Agilent Bioanalyzer 2000 technology (Agilent technologies), based on the 28S/18S rRNAs ratio. MicroRNA expression analysis was performed with Agilent® Unrestricted Human miRNA V16.0 8 × 60 K Microarray (Agilent Technologies, AMADID 31181) with the following design: each sample was hybridized on separate arrays. Agilent miRNA Microarray System with miRNA complete labeling and hybridization kit was used for Cy3 labeling. Briefly, isolated total RNA was dephosphorylated, labeled with pCp-Cy3 and hybridized on arrays for 20h at 55°C in a rotating oven (Robbins Scientific) at 20 rpm. Slides were washed and scanned by using an Agilent G2565C DNA microarray scanner. Microarray images were analyzed by using Feature Extraction software version (10.7.3.1) from Agilent technologies.
First, control spots were systematically removed, followed by flagged spots. Then, quantile normalization was performed on the MedianSignal values of each sample using the normalizeBetweenArray function from LIMMA (Smyth, 2004, Statistical applications in Genetics and molecular biology, vol3, N°1, article3). To summarize the miRNA data, the median of all probes for a given miRNA was computed, and the corresponding value was assigned to the miRNA.
To assess differentially expressed miRNA, the fold changes between 2 groups of samples were determined. Differentially detected signals were those with fold changes 1.5 or more. To extract a table of the top-ranked miRNA or genes from the linear model, the topTable function from LIMMA was used. The results were saved in table file format.
RT-PCR
For validation of the miRNA microarray results, real time PCR was used. miRNA was extracted using a column based method, miRNeasy mini kit (Qiagen, cat no. 217004). cDNA was synthesized using miscript II RT kit (Qiagen, cat no. 218161). According to the manufacturer's instructions, 2 µg of extracted RNA was incubated in 37°C with the miScript reverse transcriptase mix and the enzyme was inactivated by heating to 95°C for 10 min. The cDNA was used for relative quantification with real time PCR. For U6 and mir494, miScript syber green PCR kit was used (Qiagen, cat no. 218073). This kit includes the syber green hot-start mastermix in addition to a universal miRNA primer. The reaction was run in 20 µl that included 50 ng of cDNA and 0.3 nM of the corresponding primers. The U6 and mir-494 primers sequences were obtained from Zhou et al.Citation30 For U6 forward primer sequence was ATTGGAACGATACAGAGAAGATT and the reverse was GGAACGCTTCACGAATTTG. For mir-494, the forward primer sequence was TGACCTGAAACATACACGGGA while the reverse was TATCGTTGTACTCCACTCCTTGAC. Cycling conditions included initial heating at 95°C for 15 min followed by 40 cycles of 94°C for 12 sec, 62°C for 40 sec and 72°C for 30 sec. For miR-29b and miR-4286, predesigned assays were obtained (Applied Biosystems, cat no. 4427976 and 4427978). The data was obtained as cycle threshold, corrected to the expression of U6 and the quantification was performed using 2−ΔΔCt method.Citation31 The results represent the mean and standard deviation of 3 replicates.
DNA methylation quantification
DNA isolation: A549 and A549DOX11 cells were treated with 5AZA at 2 different concentrations for 48 h (0.1 and 1μM). Control cells were incubated with the vehicle (DMSO). Genomic DNA was extracted from pelleted cells using Wizard® Genomic DNA Purification Kit (Promega) according to the manufacturer's protocol. The concentration and purity were determined by measuring the absorbance at 230, 260 and 280 nm.
Global DNA quantification was detected using EpiSeeker methylated DNA Colormetric Quantification Kit (Abcam). Briefly, isolated DNA was bound to the assay wells and captured by a methyl–cytosine specific antibody. Antibodies were detected by using enhancer solution. After 10 minutes, absorbance was read on ELISA microplate reader (Awareness Technology Chromate, USA). Absolute and relative methyl-cytosine content was then calculated using the provided formula.
Western blotting
The expression level of different HDACs and the level of histone acetylation in A549 and A549DOX11 cells have been investigated by Western blot analysis. The A549 and A549DOX11 cells were treated with the indicated concentration of TSA (0.1, 0.2 μM) for 48 h. Control cells were treated with the vehicle (DMSO). The histone deacetylase antibody sampler kit and the acetyl-Histone antibody sampler kit from cell Signaling Technology were used. Adherent cells were washed in 1 X PBS and lysed in plates with lysis buffer (1 mM Tris-HCl [pH 6.8], 2% w/v SDS, 10% glycerol, 2 mM PMSF), and a commercial protease inhibitor mixture (Complete Protease Inhibitor Mixture; Roche Molecular Biochemicals). Lysates were sonicated on ice, and the concentration was analyzed using Bicinchoninic Acid (BCA) protein assay kit (Pierce). Fourteen μg of total cell lysate was loaded and resolved in SDS PAGE and electro-transferred to nitrocellulose membranes. The membranes were blocked in TBS-Tween 20 buffer containing 5% nonfat dried milk. The primary antibody solutions total H4, acetyl-Histone H4 (Lys8), acetyl-Histone H2A (Lys5), HDACs 1,2,3,4,6,phospho-HDACs 4,5and7 and DNMT1 (1:1000; all Cell Signaling Technology) in 5% BSA TBS-T was added and incubated overnight at 4°C. Anti-species secondary antibody binding was carried out at room temperature for 1 h. The membranes were washed 3 times for 10 min each with TBS-T. Chemiluminescence detection was carried out with the ECL plus Western Blotting Detection System (Pierce). Quantification of the relative intensity of individual bands was done using the image studio™ Lite-Western blot analysis Program (LI-COR Biosciences, Lincoln, NE) and the given band intensity was normalized to the corresponding actin level
Immunofluorescence detection of γ–H2AX
Immunofluorescence detection of γ-H2AX foci was used for detection of apoptosis. Two × 105 exponentially growing cells were seeded on glass slides placed in 10 cm Petri dishes and treated 16 h later with 0.05 μM doxorubicin for 48 h. At the end of the incubation period, slides were removed, fixed in 2% formaldehyde in 1× PBS solution for 15 min at room temperature and washed 3 times for 10 min each in PBS. The cells were permeabilized in 0.2% Triton-X100/PBS/1% BSA for 5 min on ice and blocked for 1 h at room temperature in PBS/3% BSA. 50 μl (1:100) anti-phospho-H2AX (Ser139) antibody (Upstate, Lake Placid, NY USA) in PBS/1% BSA was dispersed on each slide followed by incubation in humidified atmosphere for 1 h at room temperature. Slides were washed 3 times for 10 min each in PBS/1%BSA, incubated for 1 h with the secondary
antibody (1:600, Alexa Fluor-594 goat anti-mouse, Molecular Probes-Invitrogen, Karlsruhe, Germany) and washed again 4 times in PBS/0.5%Tween 20. Cells were stained in a DAPI antifade mixture and formation of γ-H2AX foci was observed using a fluorescence microscope (Axioplan 2, Zeiss, Göttingen, Germany). The number of cells showing enlarged nucleus and more than 50 γ–H2AX foci were counted in both cell lines.
Measurement of caspase3 activity
Activity of caspase-3 enzyme was measured colorimetrically using the CaspACE assay system purchased from Promega (Madison, WI USA). The assay was done according to the manufacturer's instructions. Briefly, cells were seeded in T175 cm2 flasks, treated after 24 h with 0.05 µM doxorubicin for 48 h, collected by trypsinization and cell density was adjustedto106 cells/ml. Cell pellets were resuspended in cell lysis buffer (supplied with the kit). The supernatant was collected by centrifugation at 4°C. This cell extract was used to measure caspase activity by mixing in a 96-multiwell plate with the reagents supplied with the kit and the absorbance was measured at 405 nm in an ELISA microplate reader (Meter tech. Σ960, USA) and caspase specific activity was calculated.
Statistics
Each experiment was repeated at least 3 times and the data are presented as the mean ±SEM. Multiple comparisons were carried out using a one-way analysis of variance (ANOVA) followed by LSD (least significant difference) for post hoc analysis. Statistical significance was acceptable to a level of P < 0.05. Statistical analysis, data fitting and graphics were performed by the Prism computer program (GraphPad Software, San Diego, CA USA).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the KEF-HOLDING-FAIZAL and Shabana Foundation and the Boehringer Ingelheim Pharmaceutical company.
References
- Dong Z, Meller J, Succop P, Wang J, Wikenheiser-Brokamp K, Starnes S, Lu S. Secretory phospholipase A2-IIa upregulates HER/HER2-elicited signaling in lung cancer cells. Int J Oncol 2014; 45:978-84; PMID:24913497
- Chang A. Lung Cancer Chemotherapy, Chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer 2011; 71:3-10; PMID:20951465; http://dx.doi.org/10.1016/j.lungcan.2010.08.022
- Pronzato P, Vigani A, Tognoni A, Vaira F, Canessa P. Anthracyclines in non small cell lung cancer. Lung Cancer 2011; 34:57-9; http://dx.doi.org/10.1016/S0169-5002(01)00394-4
- Singhal S, Wickramarachchi D, Singhal J, Yadav S, Awasthi Y, Awasthi S. Determinants of differential doxorubicin sensitivity between SCLC and NSCLC. FEBS Lett 2006; 580:2258-64; PMID:16579994; http://dx.doi.org/10.1016/j.febslet.2006.03.038
- Yang F, Teves S, Kemp C, Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim Biophys Acta 2014; 1845:84-9; PMID:24361676
- Li L, Wu P, Lee J, Li P, Hsieh W, Ho C, Ho C, Chen W, Wang C, Yen M. Hinokitiol induces DNA damage and autophagy followed by cell cycle arrest and senescence in gefitinib-resistant lung adenocarcinoma cells. PLoS One 2014; 9:e104203; PMID:25105411; http://dx.doi.org/10.1371/journal.pone.0104203
- Rishabh K. MicroRNAs: an emerging science in cancer epigenetics. J Clin Bioinforma 2013; 3:1-8; PMID:23317455; http://dx.doi.org/10.1186/2043-9113-3-1
- Robertson K. DNA methylation, methyltransferases, and cancer. Oncogene 2001; 20:3139-55; PMID:11420731; http://dx.doi.org/10.1038/sj.onc.1204341
- Glasspool R, Teodoridis J, Brown R. Epigenetics as a mechanism driving polygenic clinical drug resistance. Br J cancer 2006; 94:1087-92; PMID:16495912; http://dx.doi.org/10.1038/sj.bjc.6603024
- Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, Jones PA. specific activation of microRNA-127 with downregulation of the Proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer cell 2006; 9:435-43; PMID:16766263; http://dx.doi.org/10.1016/j.ccr.2006.04.020
- Taylor S, Jones P. Multiple new phenotypes induced in 10T1/2 and 3T3 cells treated with 5-azacytidine. Cell 1979; 17:771-9; PMID:90553; http://dx.doi.org/10.1016/0092-8674(79)90317-9
- Vigushin D, Ali S, Pace P, Mirsaidi N, Ito K, Adcock I, Coombes R. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin Cancer Res 2001; 1:971-6; PMID:11309348
- Hajji N, Wallenborg K, Vlachos P, Nyman U, Hermanson O, Joseph B. Combinatorial action of the HDAC inhibitor trichostatin A and etoposide induces caspase -mediated AIF-dependent apoptotic cell death in non-small cell lung carcinoma cells. Oncogene 2008; 27:3134-44; PMID:18071312; http://dx.doi.org/10.1038/sj.onc.1210976
- Heller G, Weinzierl M, Noll C, Babinsky V, Ziegler B, Altenberger C, Minichsdorfer C, Lang G, Döme B, End-Pfützenreuter A. Genome-wide miRNA expression profiling identifies miR-9-3 and miR-193a as targets for DNA methylation in non − small cell lung cancers. Clin Cancer Res 2012; 15:1619-29; PMID:22282464; http://dx.doi.org/10.1158/1078-0432.CCR-11-2450
- Pang B, Qiao X, Janssen L, Velds A, Groothuis T, Kerkhoven R, Nieuwland M, Ovaa H, Rottenberg S, van Tellingen O. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat Commun 2013; 4:1-13; PMID:23715267
- Basile V, Mantovani R, Imbriano C. DNA damage promotes histone deacetylase 4 nuclear localization and repression of G2/M promoters, via p53 C-terminal lysines. J Biol Chem 2006; 28:2347-57; PMID:16293626; http://dx.doi.org/10.1074/jbc.M507712200
- Calcagno A, Fostel J, To K, Salcido C, Martin S, Chewning K, Wu CP, Varticovski L, Bates S, Caplen N. Single-step doxorubicin-selected cancer cells overexpress the ABCG2 drug transporter through epigenetic changes. Br J Cancer 2008; 98:1515-24; PMID:18382425; http://dx.doi.org/10.1038/sj.bjc.6604334
- Bao L, Hazari S, Mehra S, Kaushal D, Moroz K, Dash S. Increased expression of P-glycoprotein and doxorubicin chemoresistance of metastatic breast cancer is regulated by miR-298. Am J Pathol 2012; 180:2490-503; PMID:22521303; http://dx.doi.org/10.1016/j.ajpath.2012.02.024
- Zhu X, Li H, Long L, Hui L, Chen H, Wang X, Shen H, Xu W. miR-126 enhances the sensitivity of non-small cell lung cancer cells to anticancer agents by targeting vascular endothelial growth factor A. Acta Biochim Biophys Sin (Shanghai) 2012; 44:519-26; PMID:22510476; http://dx.doi.org/10.1093/abbs/gms026
- Tsuchiya Y, Nakajima M, Takagi S, Taniya T, Yokoi T. MicroRNA regulates the expression of human cytochrome P450 1B1. Cancer Res 2006; 66:9090-8; PMID:16982751; http://dx.doi.org/10.1158/0008-5472.CAN-06-1403
- Zhao A, Zeng Q, Xie X, Zhou J, Yue W, Li Y, Pei X. MicroRNA-125b induces cancer cell apoptosis through suppression of Bcl-2 expression. J Genet Genomics 2012; 39:29-35; PMID:22293115; http://dx.doi.org/10.1016/j.jgg.2011.12.003
- Fang L, Deng Z, Shatseva T, Yang J, Peng C, Du WW, Yee AJ, Ang LC, He C, Shan SW. MicroRNA miR-93 promotes tumor growth and angiogenesis by targeting integrin-beta8. Oncogene 2011; 30:806-21; PMID:20956944; http://dx.doi.org/10.1038/onc.2010.465
- Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007; 131:1109-23; PMID:18083101; http://dx.doi.org/10.1016/j.cell.2007.10.054
- Romano G, Acunzo M, Garofalo M, Di Leva G, Cascione L, Zanca C, Bolon B, Condorelli G, Croce C. MiR-494 is regulated by ERK1/2 and modulates TRAIL-induced apoptosis in non – small-cell Lung cancer through BIM down-regulation. PNAS 2012; 109:16570-5; PMID:23012423; http://dx.doi.org/10.1073/pnas.1207917109
- Newbold A, Salmon J, Martin B, Stanley K, Johnstone R. The role of p21waf1/cip1 and p27kip1 in HDACi-mediated tumor cell death and cell cycle arrest in the Eµ-myc model of B-cell lymphoma. Oncogene 2014; 33:5415-23; PMID:24292681; http://dx.doi.org/10.1038/onc.2013.482
- Vijayaraghavalau S, Dermawan J, Venugopalan C, Labhasetwar V. Highly synergistic effect of sequential treatment with epigenetic and anticancer drugs to overcome drug resistance in breast cancer cells is mediated via activation of p21 gene expression leading to G2/M cycle arrest. Mol Pharm 2013; 10:337-52; PMID:23215027; http://dx.doi.org/10.1021/mp3004622
- Robertson K, Jones P. DNA methylation: past, present and future directions. Carcinogenesis 2000; 21:461-7; PMID:10688866; http://dx.doi.org/10.1093/carcin/21.3.461
- El-Awady R, Saleh E, Ezz M, El-Sayed A. Interaction of celecoxib with different anti-cancer drugs is antagonistic in breast but not in other cancer cells. Toxicol Appl Pharmacol 2011; 255:271-86; PMID:21763710; http://dx.doi.org/10.1016/j.taap.2011.06.019
- Saleh, M, El-Awady R, Eissa N, Abdel-Rahman W. Antagonism between curcumin and the topoisomerase II inhibitor etoposide: A study of DNA damage, cell cycle regulation and death pathways. Cancer Biol Ther 2012; 13:1058-71; PMID:22895066; http://dx.doi.org/10.4161/cbt.21078
- Zhou R, Chen G, Shen Z, Pan L. Cinobufacin suppresses cell proliferation via miR-494 in BGC- 823 gastric cancer cells. Asian Pac J Cancer Prev 2014; 15:1241-5; PMID:24606447; http://dx.doi.org/10.7314/APJCP.2014.15.3.1241
- Livak K, Schmittgen T. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402-8; PMID:11846609; http://dx.doi.org/10.1006/meth.2001.1262
- Hsu SD, Tseng YT, Shrestha S, Lin YL, Khaleel A, Chou CH, Chu CF, Huang HY, Lin CM, Ho SY, et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res 2014 Jan; 42(Database issue):D78-85; PMID:24304892; http://dx.doi.org/10.1093/nar/gkt1266