
Abstract
Colorectal cancer arises via a multistep carcinogenic process and the deregulation of multiple pathways. Thus, the simultaneous targeting of multiple pathways may be a promising therapeutic approach for colorectal treatment. CRM1 is an attractive cancer drug target, because it can regulate multiple pathways and tumor suppressor proteins. In this study, we investigated the anti-tumor activity of a novel reversible CRM1 inhibitor S109 in colorectal cancer. Our data demonstrate that S109 inhibits proliferation and induces cell cycle arrest in colorectal cancer cells. Mechanistically, we demonstrate that the activity of S109 is associated with the nuclear retention of major tumor suppress proteins. Furthermore, the Cys528 mutation of CRM1 prevented the ability of S109 to block nuclear export and inhibit the proliferation of colorectal cancer cells. Interestingly, S109 decreased the CRM1 protein level via proteasomal pathway. We argue that reversible CRM1 inhibitors but not irreversible inhibitors can induce the degradation of CRM1, because the dissociation of reversible inhibitors of CRM1 changes the conformation of CRM1. Taken together, these findings demonstrate that CRM1 is a valid target for the treatment of colorectal cancer and provide a basis for the development of S109 therapies for colorectal cancer.
Abbreviations
| CRM1 | = | chromosomal region maintenance 1 |
| DMSO | = | dimethyl sulfoxide |
| EGFR | = | epidermal growth factor receptor |
| LMB | = | leptomycin B |
| NES | = | leucine-rich nuclear export signal |
| PI3K | = | phosphoinositide 3-kinase |
| RanBP1 | = | Ran-binding protein 1 |
Introduction
Colorectal cancer is the third most frequently diagnosed malignant disease and the fourth highest cause of cancer death worldwide.Citation1 Every year, over one million new cases are diagnosed and more than 600,000 patients die from this disease.Citation2 The standard treatment for colorectal cancer is surgical resection, followed by external radiation and chemotherapy. However, a considerable proportion of patients (40%–50%) experience disease recurrence after surgical resection or develop metastatic disease.Citation3 Chemotherapy is not very effective in the treatment of metastatic colorectal cancer, with 5-year overall survival rates of less than 10%.Citation4 Thus, improved molecular targeted therapies are urgently needed.
Colorectal cancer arises via a multistep carcinogenic process within colon epithelial cells, driving the cells toward immortality.Citation5 This deregulation is caused by genetic and epigenetic alterations that impair essential pathways involving p53,Citation6 epidermal growth factor receptor (EGFR) and PI3KCitation7 signaling pathway. In addition, the mislocalization of tumor suppressor proteins is a driver of colorectal cancer initiation and progression. The negative cell cycle regulator p27 is found in the cytosol of colorectal cancer cells. Patients whose colorectal cancer cells presented an accumulation of cytoplasmic p27 show poorer outcomes for cancer-related relapse and survival.Citation8,9 The mislocalization of p27 may contribute to colorectal tumorigenesis and metastasis. Given the complexity and redundancy of the signaling network, targeting multiple pathways simultaneously may be a promising approach to treat colorectal cancer.
Chromosomal region maintenance 1 (CRM1) is an attractive cancer drug target, because it can regulate multiple pathways and tumor suppressor proteins.Citation10 CRM1 belongs to the karyopherin β superfamily of transport receptors and it is the chief mediator of protein export from the nucleus to the cytoplasm in eukaryotic cells. Protein shuttling between the nucleus and the cytoplasm plays an important role in cell proliferation and survival.Citation11 Many types of proteins, including cell cycle inhibitors and tumor suppressors must localize in the nucleus to perform their functions. The over-expression of CRM1 protein has been found in various tumors, including ovarian cancers,Citation12 gliomas,Citation13 and multiple myelomaCitation14 and is closely associated with poor prognosis. Many studies have demonstrated that CRM1 can recognize the hydrophobic, leucine-rich nuclear export signal (NES) of cargo proteins and transport over 200 proteins, including transcription factors, tumor suppressor proteins and cell cycle regulators such as p53, p21, p27, and Foxos.Citation15 The inhibition of CRM1 function could retain tumor suppressor proteins in the nucleus to induce cancer cell apoptosis or cell cycle arrest. Consequently, CRM1 is deemed to be a promising therapeutic target for anticancer drug development.
Currently, an increasing number of drug-like compounds that target CRM1 have been isolated or synthesized.Citation16-18 Leptomycin B (LMB) was the first CRM1 inhibitor to be identified. The clinical applicability of LMB is limited due to associated toxicity and minimal efficacy.Citation19 However, this limitation did not deter the search for novel low toxicity CRM1 inhibitors. More recently, SINE compounds have recently been reported as novel irreversible inhibitors of CRM1 that can be developed for clinical use.Citation16 The SINE inhibitor KPT-330 is generally well tolerated and can be administered over prolonged periods, as demonstrated in phase I/II clinical trials for several tumors,Citation16,20,21 including breast, pancreatic, and renal cancer as well as acute myeloid leukemia. The preliminary signals of antitumor activity in colorectal cancer patients were observed in a phase I trial of KPT-330.Citation22 We believe that reversible CRM1 inhibitors may be better tolerated and associated with fewer side effects. Furthermore, CBS9106, a novel reversible CRM1 inhibitor, was recently identified and reported to have anti-tumor activity against multiple myeloma.Citation23 However, the effect of CRM1 inhibitors on colorectal cancer cell growth in vitro has not yet been investigated.
For the first time, we herein report our investigation of the effect of a novel reversible CRM1 inhibitor, S109, on colorectal cancer. S109, a derivative of CBS9106, could block the function of CRM1 followed by the degradation of CRM1. Furthermore, we also found that S109 inhibits cell proliferation and invasion and induces cell cycle arrest in colon cancer cells. These data indicate that S109 is a promising drug for the treatment of colorectal cancer.
Results
S109 inhibits the proliferation and colony formation of colorectal cancer cells
To assess the effects of S109 on growth the inhibition of colon cancer cells, HCT-15 and HT-29 cells were treated with S109, and cell viability was estimated using a CCK8 assay. As shown in , S109 induced a marked decrease in cell viability in a dose-dependent manner compared with the control group. The estimated IC50 values ranged from 1.2 or 0.97 μM in HCT-15 or HT-29 cells. To confirm the anti-proliferative activity of S109, we also tested the rates of cell proliferation by EdU fluorescence staining. S109 treatment resulted in a significant reduction of the mean percentage of proliferating cells compared with the control group ( and ). HCT-15 cells exposure to 2 and 4 μM S109 reduced the proliferation to approximately 59.84% and 32.75%, respectively. These data suggest that S109 can significantly inhibit the viability of colorectal cancer cells.
Figure 1. S109 suppresses cell proliferation and colony formation of colorectal cells. (A) Chemical structure of S109. (B) Cell growth inhibition curves of S109 treatment. HCT-15 and HT-29 cells were treated with vehicle (0.1% DMSO) or different concentrations of S109 for 72 hours. Cell viability was measured by CCK-8 assay. (C) Representative EdU analysis of cell proliferation after S109 treatment. (E) S109 inhibits the colony formation of HCT-15 cells. (G) Representative photographs of invading HCT-15 cells during a 36-hour incubation with S109. (D, F and H) Quantitative results of EdU incorporation assay, clonogenic assay and invading cell numbers, respectively. The percentage of proliferative cells or colony formation were normalized to that of the control group. All data are presented as the mean ± SEM of 3 replicates (*P < 0.05, **P < 0.01).
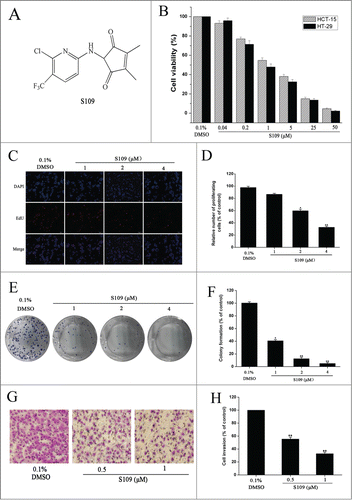
A clonogenic assay was performed to elucidate the long-term effects of S109 on cell proliferation. and 1F show the dose dependent inhibition of clonogenic potential by S109 in HCT-15 cells. Compared with the control group, the colony formation markedly decreased by 58.46%, 83.15% and 91.41% in response 1, 2, and 4 μM treatment, respectively. Taken together, these results provide unequivocal proof of the potential of S109 as a new anticancer drug.
To examine the ability of S109 to prevent the invasion of colorectal cancer cells, we conducted invasion assay. The results showed that S109 induced a dose-dependent decrease in invasion ( and 1H). Exposure of HCT-15 cells to 0.5 and 1 μM S109 decreased the fraction of invading cells by 44.58% and 67.24%, respectively. The results clearly show that S109 treatment decreases the invasiveness of cancer cells compared to the untreated control.
S109-induced G1 arrest is associated with a change in the expression of multiple cell cycle regulators
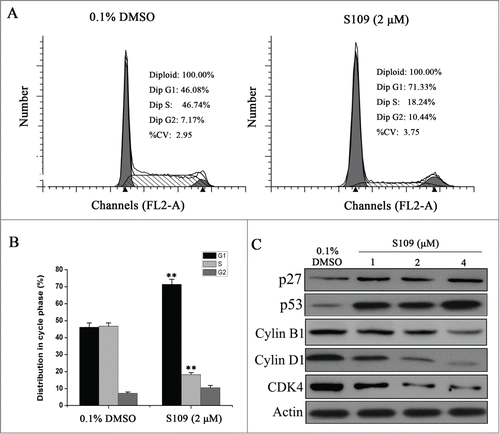
We then analyzed the cell cycle to examine the effect of S109 on colorectal cancer cell cycle progression. The cell cycle distribution of HCT-15 cells was determined by propidium iodide staining after treating cells with either DMSO control or S109 for 24 h. As shown in and 2B, the HCT-15 cells were arrested at G1 phase of the cell cycle in response to treatment with S109, as evidenced by an increase in the G1 fraction from 46.1% in the control cells to 71.3% in S109-treated cells. In addition, a significant decrease in the S phase populations compared with the control group was also observed. We next examined whether S109 modulates cell cycle regulatory proteins to induce G1 arrest. Treatment with S109 enhanced the expression levels of cell cycle inhibitory proteins p27 and p53. Moreover, the expression levels of Cyclin B1, Cyclin D1 and CDK4 were dose-dependently reduced (). Taken together, these results suggest that S109 induces cell cycle arrest by regulating multiple cell cycle regulatory proteins.
Figure 2. S109 induces cell cycle arrest and regulates the expression of cell cycle-related proteins. (A) Representative flow cytometry analysis of cell cycle based on PI staining after S109 treatment for 24 hours. (B) Quantitative analysis of cycle phase distribution in control group and S109-treated group (**P < 0.01). (C) Representative western blot of whole cell lysates isolated from cells treated with 0.1% DMSO or S109 for 24 h using the indicated antibodies.
S109 inhibits CRM1 function and induces nuclear retention of tumor suppress proteins
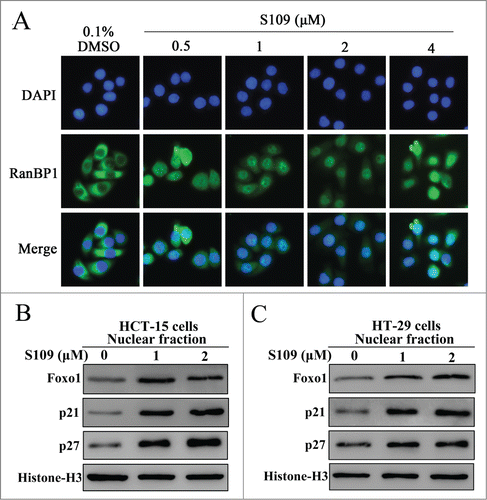
To delineate the molecular mechanisms by which S109 induces cell cycle arrest, we tested the ability of S109 to block CRM1 function. RanBP1, a Ran-binding protein, shuttles between the nucleus and cytoplasm. Previous studies have suggested that the nuclear accumulation of RanBP1 protein is a canonical marker of CRM1 inhibition.Citation24 As shown in , endogenous RanBP1 localized to the cytopasm in DMSO-treated HCT-15 cells. However, S109 treatment induced a clear and rapid shift of RanBP1 from a cytoplasmic localization to the nucleus in a dose-dependent manner. These results indicate that S109 can functionally inhibit CRM1 in colorectal cancer cells. Next, we investigated the ability of S109 treatment to induce the nuclear accumulation of tumor suppressor proteins by subjecting nuclear extracts from S109-treated colorectal cancer cells to Western blotting. As shown in and , S109 treatment resulted in the progressive nuclear accumulation of p21, p27 and Foxo1 in HCT-15 and HT-29 cells compared with the control. These results suggest that S109 can induce the nuclear retention of multiple tumor suppressor proteins and result in the functional inactivation of cell cycle progression.
Figure 3. S109 inhibits CRM1-dependent nuclear export. (A) S109 induces nuclear accumulation of RanBP1 protein. HCT-15 cells were treated with vehicle (0.1% DMSO) and S109 (0.5–4 μM) for 2 hours. Fixed cells were stained for RanBP1 (green) and DAPI (blue) and analyzed by fluorescence microscopy. (B and C) S109 induces nuclear retention of tumor suppressor proteins. HCT-15 and HT-29 cells were treated with S109 at the indicated concentrations for 24 h. Nuclear proteins were then extracted and subjected to Western blot analysis.
S109 decreases CRM1 protein expression via proteasomal-mediated degradation
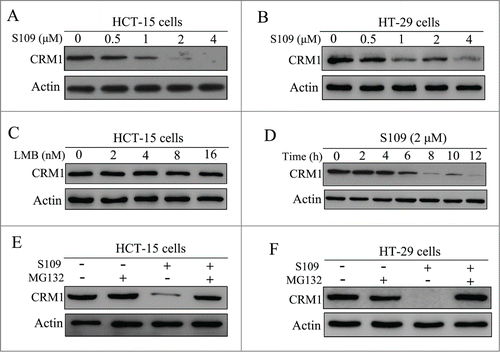
The effects of S109 on the expression of CRM1 protein in colorectal cancer cells were examined by Western blot analysis. As shown in , S109 dose- and time-dependently reduced the levels of CRM1 protein expression. However, LMB treatment did not lead decrease the CRM1 protein levels, indicating that CRM1 is functionally inactivated by LMB without a loss of protein expression (). In addition, S109 treatment did not alter the mRNA expression levels in colorectal cancer cells (data not shown). To investigate the mechanism by which S109 induces CRM1 degradation, we treated HCT-15 and HT-29 cells with S109, alone or in combination with the proteasome inhibitor MG132. While S109 alone decreased the CRM1 protein levels, the combination of S109 and MG132 led stabilized the CRM1 protein (). These results suggest that S109 mediated a reduction in CRM1 protein levels via the ubiquitin/proteasome pathway.
Figure 4. S109 induces proteasomal degradation of CRM1 protein. (A and B) HCT-15 and HT-29 cells were treated with S109 at the indicated concentrations for 24 h, and then subjected to Western blot analyses using anti-CRM1 antibody. (C) HCT-15 cells were treated with LMB at the indicated concentrations for 24 h, and then subjected to Western blot analyses using anti-CRM1 antibody. (D) HCT-15 cells were treated with S109 (2 μM) for the indicated time, and then subjected to Western blot analyses. (E and F) HCT-15 and HT-29 cells were treated with S109 (2 μM) alone, in combination with the proteasomal inhibitor MG132 (10 μM), or with MG132 (10 μM) alone for 12 h and then subjected to Western blot analyses.
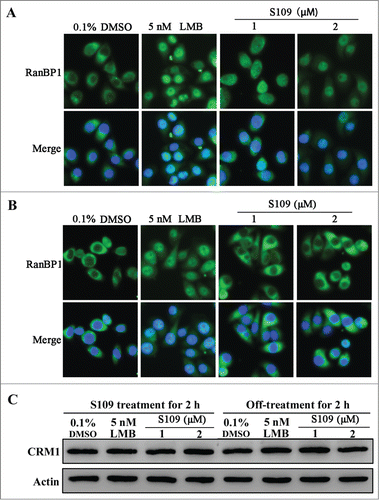
Inhibition of S109 on nucleus export of RanBP1 is reversible
LMB is well known to irreversibly bind to CRM1.Citation25 To investigate whether S109 reversibly binds to CRM1, we analyzed the subcellular localization of RanBP1 after the removal of S109-containing medium. As shown in , treatment with LMB and S109 led to a strong accumulation of RanBP1 in the nucleus. After 2 hours of incubation, S109-containing medium was removed, and fresh medium was added to the wells for another 2 hours. Strikingly, RanBP1 in cells incubated with S109 almost completely translocated to the cytoplasm. However, in the LMB-treated group, RanBP1 remained primarily localized in the nucleus after exchanging the medium (). We then investigated the role of CRM1 degradation in the reversible effect of S109. As shown in , neither S109 nor LMB treatment affected the levels of CRM1 protein. These results preliminarily indicate that S109 can reversibly inhibit the nuclear protein export mediated by CRM1.
Figure 5. The inhibitory effect of CRM1 by S109 is reversible. (A) HCT-15 cells were treated with 0.1% DMSO, LMB or S109 for 2 h, followed by fixation and an immunofluorescence analysis. The localization of RanBP1 was observed. (B) Reversible effect of S109 on the localization of RanBP1. Cells were incubated with the indicated doses of S109 or LMB. After 2 h, the drugs were washed out and fresh medium was added. The cells were further incubated for 2 h and then analyzed by fluorescence microscopy. (C) HCT-15 cells were treated with LMB or S109 at the indicated concentrations for 2 h (left panel). After incubation, the drugs were washed out and fresh medium was added. The cells were further incubated for 2 h (right panel) and then subjected to a Western blot analysis using anti-CRM1 antibody.
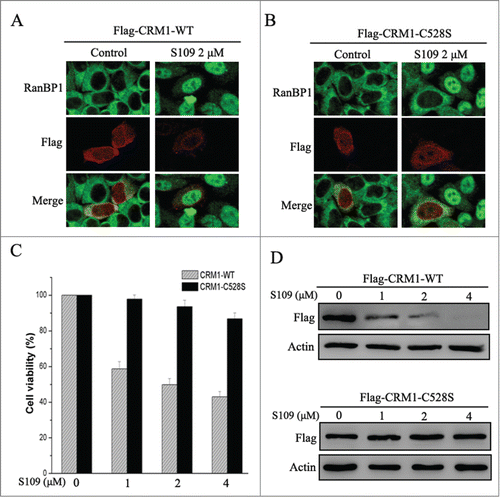
Cys528 mutation of CRM1 abrogates S109 activity in colorectal cancer cells
LMB well known to very selectively bind to Cys528 residue of CRM1.Citation25 Based on these findings, we investigated the ability of CRM1 mutation to abrogate S109 activity. We transiently transfected HCT-15 cells with wild type and Cys528 mutant CRM1 and tested the effect of S109 on RanBP1 nuclear export. Transfection with wild type or mutant CRM1 did not alter the subcellular localization of RanBP1 in control cells. Consistent with our previous results, S109 treatment resulted in the clear nuclear accumulation of RanBP1 in cells expressing wild type CRM1 (). However, cells expressing Cys528 mutant CRM1 were resistant to S109 treatment, because RanBP1 remained in the cytoplasm (). We also evaluated the effect of CRM1 mutation on cell proliferation in response to S109 treatment. As shown in , S109 did not significantly inhibit the growth of cells that stably expressed Cys528 mutant CRM1. Furthermore, Cys528 mutation abrogated the ability of S109 to induce degradation of CRM1 protein (). Taken together, our results show that S109 inhibits CRM1 mediated nuclear export by selectively binding to Cys528 residue of CRM1.
Figure 6. S109 is ineffective in cells expressing Cys528 mutant CRM1. (A and B) HCT-15 cells were transiently transfected with plasmids coding for Flag-tagged wild or C528S mutant CRM1 as indicated. After treatment with 2 μM S109 for 2 h, the cells were fixed and stained with antibodies against RanBP1 and the Flag tag. (C) HCT-15 cells that stably expressed wild type or C528S mutant CRM1 were seeded in 96-well plates and incubated with S109 at the indicated concentrations for 72 h. Growth inhibition was analyzed with the CCK-8 assay. (D) HCT-15 cells stably expressing wild type or C528S mutant CRM1 were treated with S109 at the indicated concentrations for 12 h, and then subjected to a Western blot analyses using anti-Flag antibody
Discussion
Colorectal cancer continues to have the highest incidence and mortality rate among cancers.Citation2 Thus, more effective colorectal cancer therapies are urgently needed. In this study, we report that the novel compound S109, a derivative of CBS9106, could reversibly block CRM1-mediated nuclear export. Furthermore, our data show that S109 significantly inhibits colorectal cells proliferation and invasion and concomitantly induces cell cycle arrest. Most importantly, S109 more easily and cost-effectively synthesized than CBS9106.
The development of colorectal cancer is a multi-step process and that involves the deregulation of multiple pathways.Citation26 Thus, targeting multiple pathways simultaneously may be an effective therapeutic approach for colorectal cancer treatment. Overexpression of CRM1 protein has been observed and correlated with poor prognosis in several cancer types.Citation18 Most importantly, this increased expression of CRM1 leads to mislocalization of major tumor suppressor proteins and deregulation of multiple growth-regulatory pathways. Therefore, inhibiting CRM1 may be a potential treatment option. For example, the PI3K/AKT oncogenic signaling pathway is activated in colorectal cancer cells, resulting in the inhibition of apoptosis and promotion of cell proliferation.Citation7 Foxo1, a key downstream target gene of PI3K/AKT pathway, is inactivated by cytoplasmic mislocalization in cancer cells.Citation27 Our data show that CRM1 inhibition can induce nuclear retention of Foxo1. A Recent study reported that cytoplasmic localization inactivated the negative cell cycle regulator p27 in colorectal cancer.Citation8 Our results show that S109 increases the expression of p27 and blocks its nuclear export. Given the key roles of CRM1 in the proliferation and survival of cancer cells, CRM1 may be a novel therapeutic target in colorectal cancer treatment.
An increasing number of CRM1 inhibitors have been isolated or synthesized over the past 2 decades. Although the canonical CRM1 inhibitor LMB shows strong antitumor activity in vitro, phase 1 trials of LMB were discontinued due to its toxicity.Citation19 Recently, several novel inhibitors of CRM1, known as SINE compounds, have been developed.Citation16 Of these compounds, KPT-330 has been administered to patients with several tumors in phase I/II clinical trials. The phase I clinical trial of KPT-330 on colorectal cancer patients showed that most patients had stable disease and KPT-330 is well tolerated.Citation22 Further evaluations are ongoing; however, the effects of CRM1 inhibitors on the proliferation of colorectal cancer cells in vitro have not yet been reported. CBS9106, a small molecule reversible inhibitor of CRM1, can induce cell cycle arrest in various cancer cell types.Citation23 S109 is more easily and cost-effectively synthesized derivative of CBS9106. Thus, it should be more suitable for commercial production. Our results demonstrate that S109 is also a reversible inhibitor of CRM1. We believe that the future development of low toxicity, small molecule CRM1 inhibitors may provide a new approach to treat colorectal cancer.
Our results demonstrate that S109 significantly inhibits the proliferation and invasion of colorectal cancer cells. Colorectal cancer development primarily results in the deregulation of cell cycle control and/or suppressed apoptosis.Citation5 Our results show significant G1-phase arrest in the S109-treated group. Cyclin D1 and CDK4 are well known key negative regulatory proteins for cell cycle progression in G1.Citation28 In our study, we also observed a marked reduction in the levels of Cyclin D1 and CDK4 in S109-treated colorectal cancer cells. These results strongly correlated with the altered cell cycle distribution phenotype.
Our results indicate that S109 can induce the proteasome-mediated degradation of CRM1. Interestingly, only the SINE compounds,Citation29 CBS9106Citation23 and S109 are currently known to degrade CRM1. Although several studies have reported that SINE compounds are irreversible inhibitors of CRM1, this conclusion is as of yet unsupported by evidence. The structure of these SINE compounds similar to that of PKF050-638, a reversible small molecule CRM1 inhibitor.Citation30 Thus, we believe that SINE compounds are also reversible or semi-reversible CRM1 inhibitors. We argued that although reversible inhibitors bind to the same residue in CRM1 as LMB, reversible inhibitors can likely change the conformation of CRM1 such that it is recognized by the proteasomal degradation machinery and thereby degraded. The levels of CRM1 protein were not changed after washout of S109-containing medium. Furthermore, cells must be exposed to S109 for 6 hours to induce CRM1 degradation (). Therefore, the reversible effect of S109 is not due to new CRM1 protein production.
In this study, we investigated the effect of a novel reversible CRM1 inhibitor S109 on colorectal cancer. S109 treatment induces potent cell cycle arrest in colorectal cancer cells. Mechanistically, S109 reduces the levels of CRM1 protein and induces significant nuclear accumulation of tumor suppress proteins. However, further studies are needed to confirm the anti-tumor activity of S109 in vivo. In conclusion, our study identifies CRM1 as a novel target in colorectal cancer and demonstrates that S109 can act as a promising anti-tumor agents.
Materials and Methods
Cell culture, antibodies and reagents
The human colon cancer cell lines HCT-15 and HT-29 were cultured in RPMI-1640 medium supplemented with 10% FBS and grown in a humidified incubator containing 5% CO2 at 37°C. The antibodies to CRM1 and RanBP1 were purchased from Santa Cruz Biotechnology. Antibodies against Cyclin D1, Cyclin B1, CDK4, p27, foxo1 and Histone-H3 were obtained from Cell Signaling Technology. Mouse anti-p53 was obtained from Becton, Dickinson and Company. Compound S109 was synthesized house. All other chemicals were obtained from Sigma-Aldrich. S109 was dissolved in dimethyl sulfoxide (DMSO) to make a 10 mM stock solution, which was then diluted with culture medium to different concentrations before use.
Cell viability assay
The cell viability was assessed with a Cell Counting Kit-8 (CCK8) assay. Briefly, Cells (2 × 103) were seeded on 96-well plates and incubated overnight to allow cell attachment. The cells were treated with different concentrations of S109 (0.04, 0.2, 1, 5, 25 and 50 μM) and then cultured for 72 h. Ten microliters of CCK8 was added to each well, and the 96-well plates were then incubated for 3 hours at 37 °C. The absorbance was measured using a microplate reader at a wavelength of 450 nm.
EdU cell proliferation assay
Cell proliferation was assessed with 5-ethynyl-2′-deoxyuridine (EdU) fluorescence staining using the Cell-Light™ EdU DNA Cell Proliferation Kit (Ruibo Biotech, Guangzhou, China) according to the manufacturer's instructions. The HCT-15 cells were seeded in 96-well culture plates and incubated overnight. The cells were then treated with 0.1% DMSO (vehicle) or various concentrations of S109 (1, 2 and 4 μM) for 12 h and incubated with 50 µM EdU for 4 h at 37 °C. Subsequently, the cells were fixed with 4% paraformaldehyde for 15 min and then treated with 0.5% Triton X-100 for 20 min. The cells were then incubated with 100 µL of 1 × Apollo® reaction cocktail for 30 min and then stained with DAPI for 15 min. After three washs with phosphate-buffered saline (PBS), the cells were examined with fluorescence microscopy and photographed (Olympus, Japan).
Cell clonogenic assay
To determine long-term effects, HCT-15 cells seeded in 6-well plates (600 cells/well) were treated with 0.1% DMSO (vehicle) or S109 (1, 2 and 4 μM) for 12 h. After treatment, the drug-containing medium was removed, and fresh medium was added to the wells. The medium was exchanged every 4 d for 10–14 d to allow for colony formation. The cells were then fixed with 4% formaldehyde and stained with 0.1% crystal violet solution. Finally, colony formation was confirmed by manual counting.
Cell cycle analysis
HCT-15 cells were seeded in 6-well plates at a density of 2 × 106 cells per well and treated with S109 for 24 h. After treatment, the cells were collected, fixed in 70% ethanol, and incubated overnight. The cells were then washed twice with PBS and stained in the dark for 30 min with PI solution that contained 50 µg/mL PtdIns and 25 µg/mL Rnase. Subsequently, the cells were analyzed by flow cytometry (FACSCalibur, Becton-Dickinson) and the CellQuest Pro software (Becton-Dickinson).
Invasion assay
Cell invasion assays were performed using a transwell system (Corning, NY) as described previously.Citation31 Approximately, 2 × 104 HCT-15 cells in serum-free RPMI-1640 media with 0.1% DMSO and different concentrations of S109 (0.5 and 1 μM) were added to the top chamber, and the bottom chamber was filled with RPMI-1640 containing 10% FBS. After 36 hours of incubation, invading cells were fixed with 4% paraformaldehyde and stained with 1% crystal violet. Five fields of adherent cells in each well were randomly photographed under an inverted microscope and counted.
Immunofluorescence microscopy
Approximately 1 × 104 cells were seeded onto 96-well culture plates and grown overnight at 37 °C. The cells were treated with S109 or 0.1% DMSO (vehicle) for 2 hours. The cells were then fixed with 4% paraformaldehyde in PBS for 20 minutes at room temperature and permeabilized with 0.3% Triton X-100 in PBS for 20 minutes. After blocking with 1% bovine serum albumin (BSA) in PBS for 1 h, the cells were treated with primary and fluorescent secondary antibody. The cellular nucleus was stained with DAPI. The stained cells were visualized using fluorescence microscopy and photographed (Olympus, Japan).
Western blot analysis
Cells were seeded into 6-well plates and then treated with different concentrations of S109. After incubation, whole or nuclear cell extracts from treated or untreated cells were subjected to Western blot analyses as described previously.Citation32
Production of lentiviral vector and establishment of stable cell lines
The wild type or C528S mutant human CRM1 was cloned into a pWPXL lentiviral vector containing a sequence that codes for a flag tag. The overexpression lentiviral vector constructs were co-transfected with pSPXA2 and pMD2.G plasmids into 293FT packaging cells using lipofectamine 2000 and a standard protocol (Invitrogen). Forty-eight hours after transfection, the supernatants were harvested and filtered through a 0.45-μm filter. The supernatants were then concentrated using ultracentrifugation. The cells were seeded into 6-well plates and infected with CRM1-WT or CRM1-C528S lentivirus. Forty-eight hours after infection, the cells were continuously cultured in selecting medium containing 2.5 µg/mL puromycin. The surviving cells were then cultured to generate cell lines that stably expressed CRM1-WT or CRM1-C528S.
Statistical analysis
Data are expressed as the mean ± SEM. Comparisons between untreated and S109-treated groups were evaluated with Student's t test. P < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
The research was supported by National Natural Science Foundation of China (No. 81402074; No. 81400167); Natural Science Foundation of Jiangsu province (No. BK20140224; No. BK20140227); Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 14KJB320022).
References
- Fang Y, Yu H, Liang X, Xu J, Cai X. Chk1-induced CCNB1 overexpression promotes cell proliferation and tumor growth in human colorectal cancer. Cancer Biol Ther 2014; 15:1268-79; PMID:24971465; http://dx.doi.org/10.4161/cbt.29691
- Tong JH, Lung RW, Sin FM, Law PP, Kang W, Chan AW, Ma BB, Mak TW, Ng SS, To KF. Characterization of rare transforming KRAS mutations in sporadic colorectal cancer. Cancer Biol Ther 2014; 15:768-76; PMID:24642870; http://dx.doi.org/10.4161/cbt.28550
- Sugarbaker PH, Ryan DP. Cytoreductive surgery plus hyperthermic perioperative chemotherapy to treat peritoneal metastases from colorectal cancer: standard of care or an experimental approach? Lancet Oncol 2012; 13:e362-9; PMID:22846841; http://dx.doi.org/10.1016/S1470-2045(12)70210-3
- Gallant JN, Allen JE, Smith CD, Dicker DT, Wang W, Dolloff NG, Navaraj A, El-Deiry WS. Quinacrine synergizes with 5-fluorouracil and other therapies in colorectal cancer. Cancer Biol Ther 2011; 12:239-51; PMID:21725213; http://dx.doi.org/10.4161/cbt.12.3.17034
- Bogaert J, Prenen H. Molecular genetics of colorectal cancer. Ann Gastroenterol 2014; 27:9-14; PMID:24714764
- Cooks T, Pateras IS, Tarcic O, Solomon H, Schetter AJ, Wilder S, Lozano G, Pikarsky E, Forshew T, Rosenfeld N, et al. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013; 23:634-46; PMID:23680148; http://dx.doi.org/10.1016/j.ccr.2013.03.022
- Berg M, Soreide K. EGFR and downstream genetic alterations in KRAS/BRAF and PI3K/AKT pathways in colorectal cancer: implications for targeted therapy. Discov Med 2012; 14:207-14; PMID:23021375
- Bottini C, Platini F, Rinaldi M, Leutner M, Alabiso O, Garavoglia M, Tessitore L. p27Kip1 is inactivated in human colorectal cancer by cytoplasmic localization associated with activation of Akt/PKB. Int J Oncol 2009; 34:69-77; PMID:19082479
- Cheah PY, Choo PH, Yao J, Eu KW, Seow-Choen F. A survival-stratification model of human colorectal carcinomas with beta-catenin and p27kip1. Cancer 2002; 95:2479-86; PMID:12467060; http://dx.doi.org/10.1002/cncr.10986
- Nguyen KT, Holloway MP, Altura RA. The CRM1 nuclear export protein in normal development and disease. Int J Biochem Mol Biol 2012; 3:137-51; PMID:22773955
- Mor A, White MA, Fontoura BM. Nuclear trafficking in health and disease. Curr Opin Cell Biol 2014; 28:28-35; PMID:24530809; http://dx.doi.org/10.1016/j.ceb.2014.01.007
- Wang Y, Xiang J, Ji F, Deng Y, Tang C, Yang S, Xi Q, Liu R, Di W. Knockdown of CRM1 inhibits the nuclear export of p27(Kip1) phosphorylated at serine 10 and plays a role in the pathogenesis of epithelial ovarian cancer. Cancer Lett 2014; 343:6-13; PMID:24018641; http://dx.doi.org/10.1016/j.canlet.2013.09.002
- Shen A, Wang Y, Zhao Y, Zou L, Sun L, Cheng C. Expression of CRM1 in human gliomas and its significance in p27 expression and clinical prognosis. Neurosurgery 2009; 65:153-9; discussion 9–60; PMID:19574837; http://dx.doi.org/10.1227/01.NEU.0000348550.47441.4B
- Zhang K, Wang M, Tamayo AT, Shacham S, Kauffman M, Lee J, Zhang L, Ou Z, Li C, Sun L, et al. Novel selective inhibitors of nuclear export CRM1 antagonists for therapy in mantle cell lymphoma. Exp Hematol 2013; 41:67-78 e4; PMID:22986101; http://dx.doi.org/10.1016/j.exphem.2012.09.002
- Tan DS, Bedard PL, Kuruvilla J, Siu LL, Razak AR. Promising SINEs for embargoing nuclear-cytoplasmic export as an anticancer strategy. Cancer Discov 2014; 4:527-37; PMID:24743138; http://dx.doi.org/10.1158/2159-8290.CD-13-1005
- Parikh K, Cang S, Sekhri A, Liu D. Selective inhibitors of nuclear export (SINE)–a novel class of anti-cancer agents. J Hematol Oncol 2014; 7:78; PMID:25316614; http://dx.doi.org/10.1186/s13045-014-0078-0
- Liu X, Niu M, Xu X, Cai W, Zeng L, Zhou X, Yu R, Xu K. CRM1 is a direct cellular target of the natural anti-cancer agent plumbagin. J Pharmacol Sci 2014; 124:486-93; PMID:24739265; http://dx.doi.org/10.1254/jphs.13240FP
- Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol 2012; 83:1021-32; PMID:22209898; http://dx.doi.org/10.1016/j.bcp.2011.12.016
- Newlands ES, Rustin GJ, Brampton MH. Phase I trial of elactocin. Br J Cancer 1996; 74:648-9; PMID:8761384; http://dx.doi.org/10.1038/bjc.1996.415
- Senapedis WT, Baloglu E, Landesman Y. Clinical translation of nuclear export inhibitors in cancer. Semin Cancer Biol 2014; 27:74-86; PMID:24755012; http://dx.doi.org/10.1016/j.semcancer.2014.04.005
- Turner JG, Dawson J, Cubitt CL, Baz R, Sullivan DM. Inhibition of CRM1-dependent nuclear export sensitizes malignant cells to cytotoxic and targeted agents. Semin Cancer Biol 2014; 27:62-73; PMID:24631834; http://dx.doi.org/10.1016/j.semcancer.2014.03.001
- Mau-Soerensen M, Razak ARA, Mahipal A, Mahaseth H, Gerecitano JF, Shacham S, Yau CYF, Lassen U, Shields AF, McCauley D, et al. Safety and antitumor activity of selinexor (KPT-330), a first-in -class, oral XPO1 selective inhibitor of nuclear export: A phase I study expanded with colon cancer cohort. J Clin Oncol 2014; 32:482; abstract; PMID:24395844; http://dx.doi.org/10.1200/JCO.2013.53.3448
- Sakakibara K, Saito N, Sato T, Suzuki A, Hasegawa Y, Friedman JM, Kufe DW, Vonhoff DD, Iwami T, Kawabe T. CBS9106 is a novel reversible oral CRM1 inhibitor with CRM1 degrading activity. Blood 2011; 118:3922-31; PMID:21841164; http://dx.doi.org/10.1182/blood-2011-01-333138
- Hilliard M, Frohnert C, Spillner C, Marcone S, Nath A, Lampe T, Fitzgerald DJ, Kehlenbach RH. The anti-inflammatory prostaglandin 15-deoxy-delta(12,14)-PGJ2 inhibits CRM1-dependent nuclear protein export. J Biol Chem 2010; 285:22202-10; PMID:20457605; http://dx.doi.org/10.1074/jbc.M110.131821
- Sun Q, Carrasco YP, Hu Y, Guo X, Mirzaei H, Macmillan J, Chook YM. Nuclear export inhibition through covalent conjugation and hydrolysis of Leptomycin B by CRM1. Proc Natl Acad Sci U S A 2013; 110:1303-8; PMID:23297231; http://dx.doi.org/10.1073/pnas.1217203110
- Ewing I, Hurley JJ, Josephides E, Millar A. The molecular genetics of colorectal cancer. Frontline Gastroenterol 2014; 5:26-30; PMID:24416503; http://dx.doi.org/10.1136/flgastro-2013-100329
- Kau TR, Schroeder F, Ramaswamy S, Wojciechowski CL, Zhao JJ, Roberts TM, Clardy J, Sellers WR, Silver PA. A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell 2003; 4:463-76; PMID:14706338; http://dx.doi.org/10.1016/S1535-6108(03)00303-9
- Jirawatnotai S, Sharma S, Michowski W, Suktitipat B, Geng Y, Quackenbush J, Elias JE, Gygi SP, Wang YE, Sicinski P. The cyclin D1-CDK4 oncogenic interactome enables identification of potential novel oncogenes and clinical prognosis. Cell Cycle 2014; 13:2889-900; PMID:25486477; http://dx.doi.org/10.4161/15384101.2014.946850
- Mendonca J, Sharma A, Kim HS, Hammers H, Meeker A, De Marzo A, Carducci M, Kauffman M, Shacham S, Kachhap S. Selective inhibitors of nuclear export (SINE) as novel therapeutics for prostate cancer. Oncotarget 2014; 5:6102-12; PMID:25026284
- Daelemans D, Afonina E, Nilsson J, Werner G, Kjems J, De Clercq E, Pavlakis GN, Vandamme AM. A synthetic HIV-1 Rev inhibitor interfering with the CRM1-mediated nuclear export. Proc Natl Acad Sci U S A 2002; 99:14440-5; PMID:12374846; http://dx.doi.org/10.1073/pnas.212285299
- Niu M, Sun Y, Liu X, Tang L, Qiu R. Tautomycetin induces apoptosis by inactivating Akt through a PP1-independent signaling pathway in human breast cancer cells. J Pharmacol Sci 2013; 121:17-24; PMID:23269238; http://dx.doi.org/10.1254/jphs.12206FP
- Liu X, Cai W, Niu M, Chong Y, Liu H, Hu W, Wang D, Gao S, Shi Q, Hu J, et al. Plumbagin induces growth inhibition of human glioma cells by downregulating the expression and activity of FOXM1. J Neurooncol 2015; 121:469-77; PMID:25528634; http://dx.doi.org/10.1007/s11060-014-1664-2