
Abstract
Kinesin-like protein KIFC1, a normally nonessential kinesin motor, plays a critical role in centrosome clustering in cancer cells and is essential for the survival of cancer cells. Herein, we reported that KIFC1 expression is up-regulated in breast cancer, particularly in estrogen receptor negative, progesterone receptor negative and triple negative breast cancer, and is not associated with epidermal growth factor receptor 2 status. In addition, KIFC1 is highly expressed in all 8 tested human breast cancer cell lines, but is absent in normal human mammary epithelial cells and weakly expressed in 2 human lung fibroblast lines. Moreover, KIFC1 silencing significantly reduced breast cancer cell viability. Finally, we found that PJ34, a potent small molecule inhibitor of poly(ADP-ribose) polymerase, suppressed KIFC1 expression and induced multipolar spindle formation in breast cancer cells, and inhibited cell viability and colony formation within the same concentration range, suggesting that KIFC1 suppression by PJ34 contributes to its anti-breast cancer activity. Together, these results suggest that KIFC1 is a novel promising therapeutic target for breast cancer.
Abbreviations
| ER | = | estrogen receptor |
| FBS | = | fetal bovine serum |
| FPKM | = | fragments per kilobase of exon per million fragments mapped |
| HMEC | = | Normal human mammary epithelial cells |
| PARP | = | poly(ADP-ribose) polymerase |
| PR | = | progesterone receptor |
| TCGA | = | The Cancer Genome Atlas. |
Introduction
The centrosome is a cytoplasmic organelle consisting of a pair of centrioles surrounded by an amorphous mass of protein termed the pericentriolar material. Normally, centrosomes undergo duplication precisely once before cell division, and each cell contains one or 2 centrosomes.Citation1-3 During mitosis, the 2 centrosomes form the poles of a bipolar mitotic spindle, a function that is essential for accurate chromosome segregation. However, increased centrosome number, or centrosome amplification, has been reported in a variety of human primary cancers (e.g., breast cancer, lung cancer, bladder cancer, pancreatic cancer, and prostate cancer),Citation4-8 and correlates with aneuploidy, chromosomal instability and tumorigenesis.Citation1-3 In mitosis, supernumerary centrosomes can form multipolar spindles which cause aneuploidy, and ultimately cell death. However, cancer cells can avoid multipolar mitosis by clustering the extra centrosomes into 2 spindle poles thereby enabling bipolar division.Citation1-3 As a cancer cell-specific phenomenon, extra-centrosomes de-clustering emerges as an attractive strategy for cancer therapy.Citation2
KIFC1, also known as HSET, is normally a nonessential kinesin motor protein, and plays a critical role in centrosome clustering in cancer cells.Citation9,10 Knockdown of KIFC1 induced multipolar spindle mitotic defects in cancer cell lines containing extra centrosomes, and induced cancer cell death.Citation9 A recent study further demonstrated that KIFC1 is required for proper spindle assembly, stable pole-focusing and survival of cancer cells irrespective of normal or supernumerary centrosome number, thus indicating a more general and critical role for KIFC1 in cancer cells.Citation10 PJ34 is a potent hydrophilic inhibitor of poly(ADP-ribose) polymerase (PARP). It has been demonstrated that PJ34 was able to prevent the clustering of extra centrosomes in mitosis and exclusively eradicate human cancer cells with supernumerary centrosomes without impairing normal proliferating cells.Citation11 In the present study, we demonstrated that KIFC1 is upregulated in breast cancer tissues and breast cancer cell lines, and that KIFC1 silencing in breast cancer cells significantly reduced cell viability. Furthermore, we found that PJ34 acts as a small molecule KIFC1 inhibitor by suppressing KIFC1 expression in breast cancer cells.
Results
KIFC1 expression in malignant breast cancer tissues
To examine whether KIFC1 is upregulated in human breast cancer, we analyzed KIFC1 expression in breast cancer tissues by quantitative real-time RT-PCR using a Breast Cancer TissueScan Real-Time qPCR Arrays (Origene). This array contains 5 normal control breast tissues and 43 breast cancer tissues representing different clinical stages. All the samples were from female patients with ages ranging from 32 to 84. As seen in , we found that KIFC1 expression was significantly up-regulated in breast tumors.
Figure 1. KIFC1 is upregulated in human breast cancer. (A) Breast cancer TissueScan Real-Time qPCR array was analyzed for KIFC1 expression by real-time PCR. The relative levels of KIFC1 expression are plotted with clinical status. (B) KIFC1 expression in human normal mammary epithelial cells (HMEC), 8 breast cancer cell lines and 2 human lung fibroblast cell lines LL24 and LL47 was examined by Western blotting. Samples were also probed with the anti-actin antibody to verify equal loading.
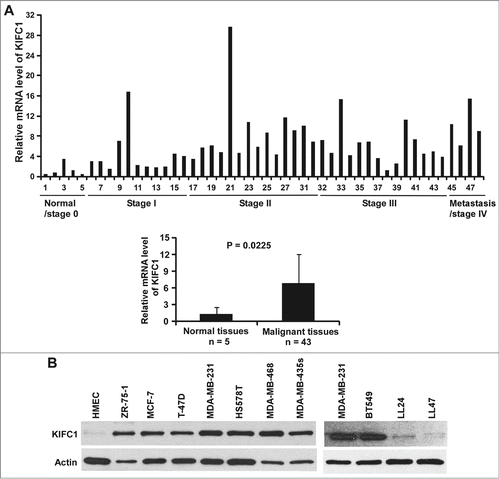
TCGA is a national collaborative program for a comprehensive genomic characterization of a large number of clinical samples of different tumor types (http://cancergenome.nih.gov). Through the TCGA Data Portal, we further characterized KIFC1 expression in breast cancer patients. Comparison of tumor N stages (N2, 3, 4 vs. N1) and metastasis M stages (M1 vs. M0) did not show any significant differences in KIFC1 expression (data not shown). However, we found that the level of KIFC1 expression is higher in estrogen receptor (ER) negative and progesterone receptor (PR) negative than in corresponding ER positive and PR positive breast tumors (). Although KIFC1 expression is not associated with human epidermal growth factor receptor 2 (HER2) status, the level of KIFC1 expression is higher in triple-negative breast cancer (TNBC; ER, PR, and HER2-negative breast cancer) than in non-TNBC (). Together, these results indicate that KIFC1 expression is upregulated in human breast cancer, particularly in ER negative breast cancer, PR negative breast cancer and TNBC when all 3 markers are taken into account.
Table 1. KIFC1 expression as related to TNBC markersFootnote*
KIFC1 expression in breast cancer cell lines
To further characterize KIFC1 expression in breast cancer, we performed Western blotting to examine KIFC1 expression in HMEC and breast cancer cell lines. As seen in , KIFC1 was highly expressed in all 8 tested breast cancer cell lines, but was undetectable in HMEC. Moreover, we also found that the levels of KIFC1 expression were very low in human lung fibroblast lines LL24 and LL47.
Knockdown of KIFC1 expression in breast cancer cells inhibits cell viability
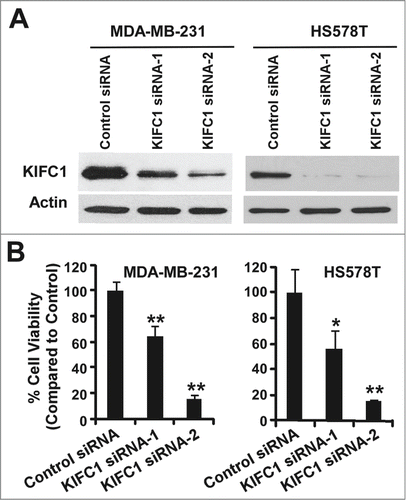
We next examined the effects of modulating KIFC1 expression on breast cancer growth. Using two independent siRNAs targeting distinct regions of KIFC1, we knocked down KIFC1 expression in TNBC MDA-MB-231 and HS578T cells (), and found that cell viability was significantly inhibited when KIFC1 expression was knocked down in breast cancer cells (). It was noted that while both siRNAs efficiently knocked down KIFC1 expression in HS578T cells, KIFC1 siRNA-2 was more potent in killing HS578T cells than KIFC1 siRNA-1, suggesting that there are some off-target effects of KIFC1 siRNA-2 in HS578T cells. Nevertheless, these results indicate that inhibition of KIFC1 might be an effective strategy to suppress the growth of breast cancer.
Figure 2. Depletion of KIFC1 in breast cancer cells results in loss of cell viability. (A) Western blots showing the depletion of KIFC1 after 2 d of transiently transfection of 50 nM KIFC1 siRNA (Dharmacon) in MDA-MB-231 and HS578T cells. (B) Breast cancer cells in 96-well plates were transiently transfected with KIFC1 siRNAs. After 7 d of culture, cell viability was measured by the CellTiter-Glo assay. All the values are the average of quadruple determinations with the s.d. indicated by error bars. *P < 0.05 verse cells treated with transfected with control siRNA. **P < 0.01 verse cells treated with transfected with control siRNA.
Small molecule PJ34 suppresses KIFC1 expression in breast cancer cells
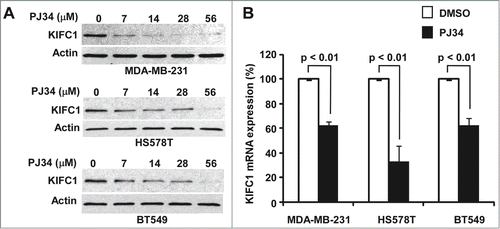
PJ34 is a potent small molecule inhibitor of poly(ADP-ribose) polymerase (PARP).Citation12 Recently, it has been found that PJ34 is a centrosome declustering agent exclusively eradicating human cancer cells.Citation11 However the molecular mechanism underlying PJ34-induced centrosome declustering was unclear. Interestingly, we found that PJ34 at 7–56 µM was able to markedly suppress KIFC1 expression in breast cancer MDA-MB-231, HS578T and BT549 cells (). To test whether PJ34 regulates KIFC1 expression at the transcription level too, we examined KIFC1 mRNA levels by real-time RT-PCR. As shown in , KIFC1 mRNA levels were also significantly decreased after PJ34 treatment in MDA-MB-231, HS578T and BT549 cells.
Figure 3. PJ34 suppresses KIFC1 expression in breast cancer cells. (A) Breast cancer MDA-MB-231, HS578T and BT549 cells in 6-well plates were treated with PJ34 at the indicated concentrations for 24 h. The levels of KIFC1 were analyzed with Western blotting. Samples were also probed with the anti-actin antibody to verify equal loading. (B) Breast cancer MDA-MB-231, HS578T and BT549 cells in 96-well plates were treated with PJ34 (56 µM) in quadruplicate for 24 h. At the end of treatment, total RNA was extracted and KIFC1 mRNA was determined by real-time RT-PCR and normalized to the message levels of 18s rRNA. All the values are the average of 3 independent experiments with the SD indicated by error bars.
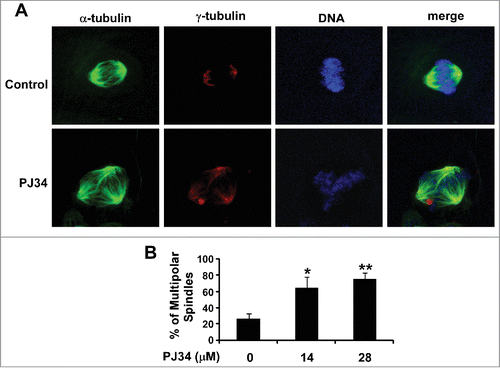
It has been demonstrated that PJ34 at 10–30 µM markedly induced multipolar spindle formation in MDA-MB-231 cells.Citation11 To confirm PJ34 acting as an extra-centrosome declustering agent, we performed multipolar spindle formation assay in BT549 cells. As shown in , PJ34 at 14 and 28 µM significantly enhanced the percentage of BT549 cells containing multipolar spindles, suggesting that PJ34 causes declustering of extra centrosomes by suppressing KIFC1 expression in breast cancer cells.
Figure 4. PJ34 induced multiple spindle formation in breast cancer BT549 cells. (A) BT549 cells were treated with PJ34 at 14 µM. After 24 h incubation, the cells were fixed, permeabilized and immunolabeled for α- and γ-tubulin for the detection of spindles and centrosomes (green and red fluorescent labeling, respectively). DNA was labeled with NucRed647 (blue). (B) The percentage of mitotic cells with multipolar spindles was calculated in BT549 cells treated with DMSO control or PJ34 at 14 and 28 µM. All the values are the average of triplicate determinations with the s.d. indicated by error bars. *P < 0.05, **P < 0.01 versus corresponding BT549 cells treated with DMSO control.
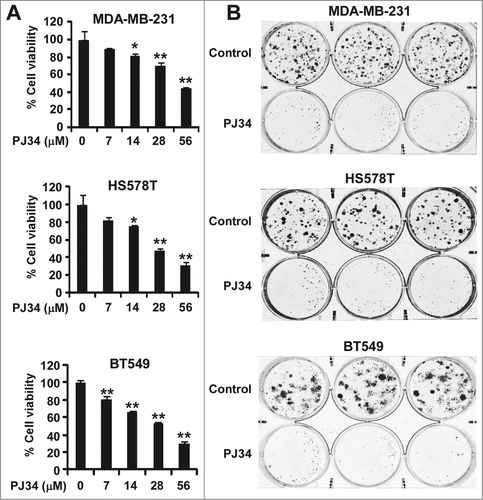
PJ34 inhibits breast cancer cell growth
Given that PJ34 can suppress KIFC1 expression in breast cancer cells, we then examined the effect of PJ34 on cell viability of breast cancer cells. As shown in , PJ34 inhibited breast cancer cell viability at concentrations shown to suppress KIFC1 expression and induce multipolar spindle formation in breast cancer cells. Colony formation is generally considered as the most accurate measure of viability after drug treatment. Therefore, we performed colony formation assays, and found that PJ34 at 14 µM significantly suppressed colony formation in breast cancer cells (). It is interesting to note that the effects of PJ34 at 14 µM on breast cancer cell colony formation were greater than on cell viability. Together, these results indicate that suppression of KIFC1 expression by PJ34 contributes to its anti-breast cancer activity.
Figure 5. PJ34 inhibits breast cancer cell growth. (A) Breast cancer MDA-MB-231 and HS578T cells in 96-well plates were treated with PJ34 at the indicated concentrations for 72 h. Cell viability was measured by the CellTiter-Glo assay. All the values are the average of triplicate determinations with the s.d. indicated by error bars. **P < 0.01 vs. corresponding control value. (B) Cancer cells in 6-well plates were treated with PJ34 (14 µM) for 10–12 d The media were changed every 3 d Colonies were fixed with formaldehyde and stained with crystal violet. All the values are the average of triplicate determinations with the s.d. indicated by error bars. *P < 0.05, **P < 0.01 compared to cells treated with DMSO.
Discussion
In cancer cells with supernumerary centrosomes, KIFC1 functions as an important force to cluster amplified centrosomes into 2 functional spindle poles for bipolar mitotic division, and is essential for the survival of cancer cells, but not normal cells.Citation9,10 It was reported that KIFC1 expression was significantly elevated in a broad panel of cancer tissues,Citation10 and that KIFC1 expression in primary non-small cell lung cancer was associated with metastatic spread to the brain.Citation13 Moreover, overexpression of KIFC1 mediates the resistance of docetaxel, a clinically well-established chemotherapeutic agent for cancer, in breast cancer MDA-MB-231 and MDA-MB 468 cells.Citation14 In the current study, we found that KIFC1 expression is up-regulated in human breast cancer, particularly in TNBC. This is consistent with the finding from a recent study showing that KIFC1 expression is high in triple negative subtype of breast cancer.Citation15 Importantly, we also demonstrated for the first time that KIFC1 expression was higher in ER-negative and PR-negative breast cancer than in corresponding ER positive and PR positive breast cancer, but was not associated with HER2 status. Furthermore, in the present study we demonstrated that KIFC1 was expressed in breast cancer cell lines, but was absent in HMEC. Finally, we confirmed that cell viability was significantly inhibited when KIFC1 expression was knocked down in breast cancer cells. All together, these findings suggest that KIFC1 is a novel therapeutic target for breast cancer.
PARP inhibitors are currently being clinically developed either as single agents in BRCA1/2 deficient cancers and cancers with “BRCAness,” or in combination therapy with other DNA damaging agents to derive additional therapeutic benefit from DNA damage.Citation16,17 PJ34 is a potent hydrophilic PARP inhibitor.Citation12 Castiel et al. reported that PJ34 was able to prevent clustering of extra centrosomes in mitosis and exclusively eradicate human cancer cells with supernumerary centrosomes without impairing normal proliferating cells.Citation11 It has been found that several PARP members including PARP-1 and PARP-3 can be localized to the centrosome of cancer cells, and that correct regulation of poly(ADP-ribosyl)ation levels in vivo is important for maintenance of proper centrosome duplication and chromosomal stability.Citation18-21 However, recent studies indicate that PJ34 displays some PARP-1-independent effects in cancer cells.Citation11,22,23 It was demonstrated that, despite acting as a PARP inhibitor, the action of PJ34 on centrosome clustering in cancer cells was not attributed to PARP inhibition.Citation11 In the present study, we found that PJ34 was able to suppress KIFC1 expression in breast cancer cells, and that PJ34 inhibited breast cancer cell viability at concentrations shown to suppress KIFC1 expression. Therefore, our results present a novel mechanism underlying PJ34-mediated breast cancer cell death.
An ideal cancer treatment should selectively kill cancer cells without harming normal cells. However, standard chemotherapy not only destroys cancer cells but also unavoidably damage normal cells. KIFC1 is essential for bipolar mitotic division of cancer cells, but is not required for normal cells,Citation9,10 suggesting that targeting KIFC1 is a promising strategy to develop highly selective anti-cancer agents. AZ82 and CW069 are 2 small molecule KIFC1 inhibitors reported so far, which inhibit microtubule-stimulated KIFC1 ATPase activity.Citation15,24 In the present study, we have identified PJ34 as a KIFC1 inhibitor by suppressing KIFC1 expressing in breast cancer cells. This finding supports the notion that PJ34 may be developed into an effective agent in treating breast cancer.
Materials and Methods
Materials
PJ34 was purchased from Tocris. Rabbit polyclonal antibody to KIFC1 (ab72452) was purchased from Abcam. Peroxidase labeled anti-mouse antibody and ECL system were purchased from Amersham Life Science. Tissue culture media, fetal bovine serum (FBS), and plastic-ware were obtained from Life Technologies, Inc. Proteinase inhibitor cocktail Complete™ was obtained from Boehringer Mannheim.
Cell Culture
All breast cancer cell lines and lung fibroblast lines were obtained from ATCC, and cultured in cultured in DMEM medium containing 10% fetal bovine serum, 2 mM of L-glutamine, 100 units/ml of penicillin, and 100 μg/ml of streptomycin. Normal human mammary epithelial cells (HMEC) were purchased from Lonza, and cultured with mammary epithelial growth medium (Lonza). Cells were grown under standard cell culture conditions at 37°C in a humidified atmosphere with 5% CO2.
Quantitative Real-Time PCR
Human KIFC1 real-time primer set (PPH08277A) and 18s rRNA real-time primer set (PPH05666E) were from Qiagen. TissueScan Breast Cancer cDNA Array II was purchased from Origene. This product contains first-strand cDNAs prepared from 48 human breast tissues including both malignant and normal controls. These 48 cDNAs have been normalized against β-actin by RT-PCR, and arrayed onto PCR plates. KIFC1 expression was quantitatively measured by real-time PCR using SYBR Green (Qiagen) in 96-well format in a total volume of 30 µl over 42 2-step cycles using the following temperature protocol: 95°C for 15 seconds and 55°C for 60 seconds. For analysis KIFC1 mRNA in breast cancer cell lines, 1–2 ×104 breast cancer cells were seeded into 96-well plates. After overnight culture, the cells were treated with PJ34 for 24 h. Total RNA was extracted from 96-well cultured cells using RNeasy 96 kit (Qiagen), and One-step RT-PCR amplification was performed using QuantiFast SYBR Green RT-PCR kit (Qiagen) in 384-well format. The relative amount of KIFC1 mRNA was normalized to 18s rRNA level as a housekeeping gene, and the data were analyzed according to the 2−ΔΔCT method.
Analysis of KIFC1 Expression with Breast Cancer Dataset Obtained from The Cancer Genome Atlas (TCGA)
TCGA Data Portal (https://tcga-data.nci.nih.gov/tcga) was used to download expression data of breast invasive carcinoma samples (n = 1, 100). The RNAseqV2 level 3 data that includes fragments per kilobase of exon per million fragments mapped (FPKM)-normalized gene level data were used before statistics. In addition, idf file and sdrf file were also downloaded for sample mapping and annotation. The clinical outcome data was downloaded for correlation and model building. Group wise comparison as well as ANOVA was used to select genes of significance. False Discovery Rate (FDR)-corrected p values were used for multiple hypothesis testing purpose.
siRNA depletion of KIFC1
Two different oligos of siRNAs against human KIFC1were purchased from Dharmacon: siRNA KIFC1–1: sense, 5′-UAACUGACCCUUUAAGUCCUU-3′; siRNA KIFC1–2, 5′-UGGUCCAACGUUUGAGUCCUU-3′. Non-specific scrambled siRNA was used as control (Ambion). Cells were transfected with Lipofectamine RNAiMAX (Invitrogen) with a final siRNA concentration of 50 nM according to the manufacturer's instructions.
Western Blotting
Cells in 6-well plates were lysed in 0.5 ml of phosphate-buffered saline containing 1% Triton X-100, 1 mM PMSF and 1 X protease inhibitor cocktail (lysis buffer) at 4°C for 10 min. Equal quantities of protein were subjected to SDS-PAGE under reducing conditions, and transferred to immobilon-P transfer membrane. Membranes were blocked with 5% nonfat milk in PBS containing 0.1% Tween 20 (blocking buffer) for 0.5 h at room temperature, and incubated overnight at 4°C with anti-KIFC1 (Abcam) or anti-actin (Sigma-Aldrich) antibody diluted in the blocking buffer. After several washes with PBS containing 0.1% Tween 20, membranes were incubated in the blocking buffer with horseradish peroxide conjugated secondary antibody for 60 min at room temperature. After washing, the immunoreactive proteins were then detected using the ECL system. Films showing immunoreactive bands were scanned by Kodak Digital Science DC120 Zoom Digital Camera.
Confocal microscopy
BT549 cells were grown on glass coverslips in 24-well plates. After 24 h treatment of PJ34, the cells were fixed with 4% paraformaldehyde in phosphate-buffered saline for 20 min, and permeabilized with 0.2% Triton X-100 for 10 min at room temperature. The cells were then incubated with anti-α-tubulin-FITC antibody (Sigma) and anti-γ-tubulin Cy3 conjugate (Sigma) for 45 min, and nuclei were stained with NucRed647 (Life Technologies) for 10 min. The cells were then examined by a laser-scan confocal microscope (Leica DMI 4000 B), and the percentage of mitotic cells with multipolar spindles was calculated out of a total number of a minimum of 20 mitotic cells per coverslip that were detected in different and randomly chosen microscopic fields. Images were processed and analyzed using Leica's LAS Image Analysis software.
Cell viability assay
Cells were seeded into 96-well tissue culture treated microtiter plates at a density of 5000 cells/well. After overnight incubation, the cells were treated with PJ34 for 72 h. Cell viability was measured by the CellTiter-Glo Assay (Promega).
Colony formation assay
Cancer cells were seeded at a density of 500 cells/well into 6-well plates. Sixteen hours after the plates had been set up, PJ34 was added, and media were replenished every 3 d After being incubated for 10–14 days, colonies were fixed with 4% formaldehyde, stained with 0.5 mg/mL crystal violet, and imaged on a FluorChem HD2 Imager System (Alpha Innotech).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The results presented in this study are in part based upon data generated by the TCGA Research (https://tcga-data.nci.nih.gov/tcga). We are very grateful to all specimen donors and research groups that have made the data publicly available.
Funding
This work was partially supported by grants from the National Institutes of Health R21CA182056 and R03DA033983 and Alabama Innovation Fund.
References
- Fukasawa K. Oncogenes and tumour suppressors take on centrosomes. Nature Rev Cancer 2007; 7:911-24; http://dx.doi.org/10.1038/nrc2249.
- Ogden A, Rida PC, Aneja R. Let's huddle to prevent a muddle: Centrosome declustering as an attractive anticancer strategy. Cell Death Differ 2012; 19:1255-67; PMID:22653338; http://dx.doi.org/10.1038/cdd.2012.61.
- Anderhub SJ, Kramer A, Maier B. Centrosome amplification in tumorigenesis. Cancer Lett 2012; 322:8-17; PMID:22342684; http://dx.doi.org/10.1016/j.canlet.2012.02.006.
- Pihan GA, Purohit A, Wallace J, Knecht H, Woda B, Quesenberry P, Doxsey SJ. Centrosome defects and genetic instability in malignant tumors. Cancer Res 1998; 58:3974-85; PMID:9731511.
- Sato N, Mizumoto K, Nakamura M, Nakamura K, Kusumoto M, Niiyama H, Ogawa T, Tanaka M. Centrosome abnormalities in pancreatic ductal carcinoma. Clin Cancer Res 1999; 5:963-70; PMID:10353727.
- Pihan GA, Purohit A, Wallace J, Malhotra R, Liotta L, Doxsey SJ. Centrosome defects can account for cellular and genetic changes that characterize prostate cancer progression. Cancer Res 2001; 61:2212-9; PMID:11280789.
- Kawamura K, Izumi H, Ma Z, Ikeda R, Moriyama M, Tanaka T, Nojima T, Levin LS, Fujikawa-Yamamoto K, Suzuki K, et al. Induction of centrosome amplification and chromosome instability in human bladder cancer cells by p53 mutation and cyclin e overexpression. Cancer Res 2004; 64:4800-9; PMID:15256449; http://dx.doi.org/10.1158/0008-5472.CAN-03-3908.
- Zyss D, Gergely F. Centrosome function in cancer: Guilty or innocent? Trends Cell Biol 2009; 19:334-46; PMID:19570677; http://dx.doi.org/10.1016/j.tcb.2009.04.001.
- Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, Pellman D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev 2008; 22:2189-203; PMID:18662975; http://dx.doi.org/10.1101/gad.1700908.
- Kleylein-Sohn J, Pollinger B, Ohmer M, Hofmann F, Nigg EA, Hemmings BA, Wartmann M. Acentrosomal spindle organization renders cancer cells dependent on the kinesin hset. J Cell Sci 2012; 125:5391-402; PMID:22946058; http://dx.doi.org/10.1242/jcs.107474.
- Castiel A, Visochek L, Mittelman L, Dantzer F, Izraeli S, Cohen-Armon M. A phenanthrene derived parp inhibitor is an extra-centrosomes de-clustering agent exclusively eradicating human cancer cells. BMC Cancer 2011; 11:412; PMID:21943092; http://dx.doi.org/10.1186/1471-2407-11-412.
- Antolin AA, Jalencas X, Yelamos J, Mestres J. Identification of pim kinases as novel targets for pj34 with confounding effects in parp biology. ACS Chem Biol 2012; 7:1962-7; PMID:23025350; http://dx.doi.org/10.1021/cb300317y.
- Grinberg-Rashi H, Ofek E, Perelman M, Skarda J, Yaron P, Hajduch M, Jacob-Hirsch J, Amariglio N, Krupsky M, Simansky DA, et al. The expression of three genes in primary non-small cell lung cancer is associated with metastatic spread to the brain. Clin Cancer Res 2009; 15:1755-61; PMID:19190132; http://dx.doi.org/10.1158/1078-0432.CCR-08-2124.
- De S, Cipriano R, Jackson MW, Stark GR. Overexpression of kinesins mediates docetaxel resistance in breast cancer cells. Cancer Res 2009; 69:8035-42; PMID:19789344; http://dx.doi.org/10.1158/0008-5472.CAN-09-1224.
- Wu J, Mikule K, Wang W, Su N, Petteruti P, Gharahdaghi F, Code E, Zhu X, Jacques K, Lai Z, et al. Discovery and mechanistic study of a small molecule inhibitor for motor protein kifc1. ACS Chem Biol 2013; 8:2201-8; PMID:23895133; http://dx.doi.org/10.1021/cb400186w.
- Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. Parp inhibition: Parp1 and beyond. Nature Rev Cancer 2010; 10:293-301; http://dx.doi.org/10.1038/nrc2812.
- Davar D, Beumer JH, Hamieh L, Tawbi H. Role of parp inhibitors in cancer biology and therapy. Curr Med Chem 2012; 19:3907-21; PMID:22788767; http://dx.doi.org/10.2174/092986712802002464.
- Kanai M, Uchida M, Hanai S, Uematsu N, Uchida K, Miwa M. Poly(adp-ribose) polymerase localizes to the centrosomes and chromosomes. Biochem Biophys Res Commun 2000; 278:385-9; PMID:11097846; http://dx.doi.org/10.1006/bbrc.2000.3801.
- Kanai M, Tong WM, Sugihara E, Wang ZQ, Fukasawa K, Miwa M. Involvement of poly(adp-ribose) polymerase 1 and poly(adp-ribosyl)ation in regulation of centrosome function. Mol Cell Biol 2003; 23:2451-62; PMID:12640128; http://dx.doi.org/10.1128/MCB.23.7.2451-2462.2003.
- Kanai M, Tong WM, Wang ZQ, Miwa M. Haploinsufficiency of poly(adp-ribose) polymerase-1-mediated poly(adp-ribosyl)ation for centrosome duplication. Biochem Biophys Res Commun 2007; 359:426-30; PMID:17553458; http://dx.doi.org/10.1016/j.bbrc.2007.05.108.
- Augustin A, Spenlehauer C, Dumond H, Menissier-De Murcia J, Piel M, Schmit AC, Apiou F, Vonesch JL, Kock M, Bornens M, et al. Parp-3 localizes preferentially to the daughter centriole and interferes with the g1/s cell cycle progression. J Cell Sci 2003; 116:1551-62; PMID:12640039; http://dx.doi.org/10.1242/jcs.00341.
- Toller IM, Altmeyer M, Kohler E, Hottiger MO, Muller A. Inhibition of adp ribosylation prevents and cures helicobacter-induced gastric preneoplasia. Cancer Res 2010; 70:5912-22; PMID:20634404; http://dx.doi.org/10.1158/0008-5472.CAN-10-0528.
- Madison DL, Stauffer D, Lundblad JR. The parp inhibitor pj34 causes a parp1-independent, p21 dependent mitotic arrest. DNA Repair 2011; 10:1003-13; PMID:21840268; http://dx.doi.org/10.1016/j.dnarep.2011.07.006.
- Watts CA, Richards FM, Bender A, Bond PJ, Korb O, Kern O, Riddick M, Owen P, Myers RM, Raff J, et al. Design, synthesis, and biological evaluation of an allosteric inhibitor of hset that targets cancer cells with supernumerary centrosomes. Chem Biol 2013; 20:1399-410; PMID:24210220; http://dx.doi.org/10.1016/j.chembiol.2013.09.012.