
Abstract
Many genes controlling cell proliferation and survival (those most important to cancer biology) are now known to be regulated specifically at the translational (RNA to protein) level. The internal ribosome entry site (IRES) provides a mechanism by which the translational efficiency of an individual or group of mRNAs can be regulated independently of the global controls on general protein synthesis. IRES-mediated translation has been implicated as a significant contributor to the malignant phenotype and chemoresistance, however there has been no effective means by which to interfere with this specialized mode of protein synthesis. A cell-based empirical high-throughput screen was performed in attempt to identify compounds capable of selectively inhibiting translation mediated through the IGF1R IRES. Results obtained using the bicistronic reporter system demonstrate selective inhibition of second cistron translation (IRES-dependent). The lead compound and its structural analogs completely block de novo IGF1R protein synthesis in genetically-unmodified cells, confirming activity against the endogenous IRES. Spectrum of activity extends beyond IGF1R to include the c-myc IRES. The small molecule IRES inhibitor differentially modulates synthesis of the oncogenic (p64) and growth-inhibitory (p67) isoforms of Myc, suggesting that the IRES controls not only translational efficiency, but also choice of initiation codon. Sustained IRES inhibition has profound, detrimental effects on human tumor cells, inducing massive (>99%) cell death and complete loss of clonogenic survival in models of triple-negative breast cancer. The results begin to reveal new insights into the inherent complexity of gene-specific translational regulation, and the importance of IRES-mediated translation to tumor cell biology.
Abbreviations
| 5′-UTR | = | 5′-untranslated region |
| IGF1R | = | type 1 insulin-like growth factor receptor |
| IRES | = | Internal Ribosome Entry Site |
| uORF | = | upstream open reading frame. |
Introduction
There are 2 distinct modes of protein synthesis in the cell. The conventional mechanism for translation initiation involves recruitment of the 40S ribosome to the cap structure at the 5′ end of the mRNA, followed by linear scanning of the 5′-UTR until the initiation codon is reached. Non-canonical translation-regulatory mechanisms such as that mediated by an internal ribosome entry site (IRES) allow the 40S ribosomal subunit to bypass highly structured 5′-untranslated sequences and enter at a point much nearer to, or in the immediate vicinity of the initiation codon.Citation1,2 Importantly, these 2 modes of protein synthesis are not equivalent. The conventional mechanism of cap-dependent ribosomal scanning is utilized for general protein synthesis, whereas IRES-mediated translation appears to be reserved for synthesis of those gene products most critical for cell proliferation and survival, providing a fail-safe or emergency system to ensure that such essential polypeptides can be manufactured whenever needed, particularly under conditions of stress.Citation3-6
Cap-dependent ribosomal scanning is regulated through post-translational modifications to general eukaryotic initiation factors (eIFs) and other molecules which interact directly with these factors. In this manner, the overall rate of protein synthesis can be globally increased or decreased, in response to microenvironmental cues and the physiological status of the cell. IRES-mediated translation, by contrast, utilizes specialized sequence elements within the 5′-untranslated regions of the associated mRNAs, as well as non-canonical translation-regulatory proteins known as IRES trans-acting factors (ITAFs).Citation7,8 In this manner, the IRES provides a mechanism by which the translational efficiency of an individual mRNA or group of mRNAs can be regulated independently of the global controls on general protein synthesis. In fact, one of the defining characteristics of an IRES is its ability to remain operational or even be upregulated under circumstances in which general protein synthesis is severely compromised. Citation9-11
A number of cancer-related proteins are now known to be translated via an IRES, and accumulating evidence from multiple labs suggests that IRES-mediated translation may be of particular importance to malignant cells.Citation12-19 Elegant studies on the IRESs associated with VEGF, FGF-2, c-myc, and XIAP have solidly established the relevance of IRES-mediated translation to cancer.Citation20-25 Furthermore, IRES-mediated translation has been specifically implicated in metastasis and chemotherapeutic drug resistance.Citation26-30 It appears that tumor cells may depend on IRES-mediated translation of key oncogenic proteins to promote their own survival under adverse microenvironmental conditions or exposure to cytotoxic agents.
Our lab has investigated the human IGF1R IRES in considerable detail. The IGF1R mRNA contains an extraordinarily long 5′-untranslated region (1,040 nucleotides, GenBank: NG_009492.1; ) which adopts a highly stable secondary structure (ΔG>-500kcal/mole), with extensive internal base-pairing, serving as a substantial impediment to scanning by the 40S ribosome.Citation31 In addition, an upstream open reading frame (uORF) positioned ∼300 nucleotides upstream of the authentic initiation codon tends to derail many of the scanning ribosomes before they reach the IGF1R coding sequence. The IRES allows the ribosome to bypass the obstacles presented by the complex 5′-UTR. We delimited the core functional IRES to a 90 nucleotide segment of the IGF1R 5′-UTR positioned immediately upstream of the initiation codon.Citation32 Using site-directed mutagenesis to dissect the sequence elements critical for IRES function, we determined that the IGF1R IRES recruits the 40S ribosome at least in part by a Shine-Dalgarno-like (direct mRNA-rRNA base-pairing) interaction between Stem2/Loop2 of the IRES and the G961 loop (helix 23b) of the 18S rRNA.Citation33 We found that translational efficiency through the IGF1R IRES is regulated by dynamic, competitive interactions between sequence-specific RNA-binding proteins which recognize and bind directly to the core functional IRES, among which are hnRNP C (which stimulates IRES activity)Citation31 and HuR (an IRES repressor).Citation32
Figure 1. Approach to identification of small molecule inhibitors of IRES-mediated translation. (A) The 5′-untranslated region of the human IGF1R mRNA. (B) Reporter constructs used to genetically engineer T47D human breast carcinoma cells for use in the high throughput screen to identify compounds that selectively interfere with function of the IGF1R IRES. The bicistronic construct used for the screen contains the full-length IGF1R 5′-untranslated sequence (1,040 nucleotides including the IRES, GenBank: NG_009492.1), positioned between the Renilla and firefly luciferase coding sequences. Cells stably transfected with the reporter construct containing the IGF1R 5′-UTR (in monocistronic context) from which the core functional IRES has been deleted were used for the counterscreen. (C) Scatter plot of representative raw data from one of the pilot HTS assays that preceded the full high-throughput screen / counterscreen. Relative inhibition of IRES activity (firefly bioluminescence signal generated from cells stably transfected with the bicistronic IRES reporter construct) is plotted vs. relative inhibition of signal generated by the cells stably transfected with the monocistronic control (no IRES) construct. (D) Summary of compound progression path for identification of small molecule IRES inhibitors. (E). Structure of IRES inhibitor lead compound P (cpd_P) = N-(4-anilinophenyl)-N′-[2-(4-chlorophenyl)ethyl]thiourea, MW 381; and candidate lead (false positive) cpd_T = N-[5-(5-isopropyl-1,3-benzoxazol-2-yl)-2-methoxyphenyl]butanamide, MW 352. Two closely related analogs of cpd_P are also utilized, in which the chlorine substituent is replaced by either a methoxy group (P-2) or a fluorine atom (P-3).
![Figure 1. Approach to identification of small molecule inhibitors of IRES-mediated translation. (A) The 5′-untranslated region of the human IGF1R mRNA. (B) Reporter constructs used to genetically engineer T47D human breast carcinoma cells for use in the high throughput screen to identify compounds that selectively interfere with function of the IGF1R IRES. The bicistronic construct used for the screen contains the full-length IGF1R 5′-untranslated sequence (1,040 nucleotides including the IRES, GenBank: NG_009492.1), positioned between the Renilla and firefly luciferase coding sequences. Cells stably transfected with the reporter construct containing the IGF1R 5′-UTR (in monocistronic context) from which the core functional IRES has been deleted were used for the counterscreen. (C) Scatter plot of representative raw data from one of the pilot HTS assays that preceded the full high-throughput screen / counterscreen. Relative inhibition of IRES activity (firefly bioluminescence signal generated from cells stably transfected with the bicistronic IRES reporter construct) is plotted vs. relative inhibition of signal generated by the cells stably transfected with the monocistronic control (no IRES) construct. (D) Summary of compound progression path for identification of small molecule IRES inhibitors. (E). Structure of IRES inhibitor lead compound P (cpd_P) = N-(4-anilinophenyl)-N′-[2-(4-chlorophenyl)ethyl]thiourea, MW 381; and candidate lead (false positive) cpd_T = N-[5-(5-isopropyl-1,3-benzoxazol-2-yl)-2-methoxyphenyl]butanamide, MW 352. Two closely related analogs of cpd_P are also utilized, in which the chlorine substituent is replaced by either a methoxy group (P-2) or a fluorine atom (P-3).](/cms/asset/99709bae-baa0-4d1d-906c-583e53232fca/kcbt_a_1071729_f0001_b.gif)
IRES-mediated translation has traditionally been studied through interventions (e.g. polioviral infection) which severely compromise general protein synthesis, leaving only translation initiated through non-canonical mechanisms such as IRES active.Citation34-35 Our objective here was to identify compounds capable of selectively interfering with IRES-mediated translation. The identification of such a small molecule IRES inhibitor would provide the opportunity, for the first time, to selectively perturb this specialized mode of translation and assess the consequences. Although considerable progress has been made toward elucidating the molecules and mechanisms involved in internal ribosome entry, we realized there remains a substantial gap in knowledge with regard to these factors, and therefore elected to employ an empirical screening strategy, rather than attempting a rational drug design approach based on the information currently in hand. We hoped that such a compound would be useful for investigating the contribution of IRES-mediated translation to various physiological processes and pathological states. Furthermore, there was reason to speculate that such a small molecule IRES inhibitor might eventually find clinical utility as well.
A cell-based high-throughput screen of 135,000 compounds was performed to identify compounds capable of inhibiting translation mediated through the IGF1R IRES. From this screen, 3 active lead compounds were identified which consistently and completely block IGF1R protein synthesis in cells in a concentration-dependent manner under a variety of experimental conditions. Here we describe in detail our initial characterization of the first of those compounds. The emphasis of our experiments was on assessing the molecular and phenotypic consequences of IRES inhibition in malignant cells, focusing on IGF1R and c-myc as representatives of 2 very different types of IRESs.
Results and Discussion
Approach to identification of small molecule inhibitors of IRES-mediated translation
A cell-based functional assay and high-throughput screen of a diverse chemical library (135,000 compounds) were used to identify compounds that selectively inhibit translation mediated by the IGF1R IRES (). We reasoned that such a cell-based assay would provide physiologically-relevant and pharmacologically useful data, increasing the likelihood that a positive scoring compound (and its derivatives) would function in intact biological systems. T47D human breast tumor cells were genetically engineered to express firefly luciferase under control of the IGF1R IRES. The bicistronic IRES-reporter construct contained the full-length human IGF1R 5′-UTR (1,040 nt) including the IRES, positioned between the Renilla and firefly luciferase coding sequences. T47D was selected for this purpose because the IGF1R IRES is particularly active in these cells.Citation31 Control cells stably transfected with a similar reporter construct (monocistronic) containing the IGF1R 5′-UTR but from which the core functional IRES had been deleted (i.e. containing IGF1R 5′-UTR nucleotides 1-959) were used as a counterscreen, to eliminate false positive compounds that exhibit non-specific cytotoxicity, general inhibition of protein synthesis, or inhibit the firefly luciferase enzyme itself. The assay underwent extensive optimization and validation trials, ultimately reaching CVs of 8% and a mean Z’ score of 0.75 (robust).
Of 135,000 compounds screened, ∼6400 scored positive, of which 433 (0.3%) were confirmed active in dose-response titrations. From the pool of 433 confirmed active hits, 60 high priority scaffolds were advanced to laboratory-scale biological assays examining effect on the endogenous target. This selection was based on relative potency, cheminformatic analyses, synthetic accessibility, and chemical tractability. From this group, 3 promising lead compounds (representing distinct chemical scaffolds designated W, P, and V) were identified which consistently and completely block IGF1R protein synthesis, and induce dramatic phenotypic alterations in human breast tumor cells.
Early experiments demonstrated that there was considerable overlap in biological activity for the 3 lead compounds, but that compound P (cpd_P) was consistently intermediate in its properties between those of W and V, and exhibited the broadest spectrum and range of phenotypic outcomes (as described in the results below), and was therefore chosen as the most representative of the group for initial detailed analyses. Compound T (cpd_T) was one of the high priority scaffolds which was ultimately determined to be inactive against the endogenous IRES, and is included as a representative false positive (negative control) compound, as the basis for its ineffectiveness against the endogenous IRES was instructive.
Characterization of small molecule IRES inhibitors by reporter analyses
Detailed analyses of IRES inhibition by cpd_P were performed, employing the same stably transfected IRES reporter cells developed for the high throughput screen and counterscreen (). Three parameters are scored throughout these titrations. Firefly luciferase is indicative of IRES-mediated translation of the second cistron of the bicistronic reporter construct. Renilla luciferase represents translation (via cap-dependent scanning) of the first cistron of the IRES-containing bicistronic construct. The third parameter, designated M, represents translation of the firefly luciferase coding sequence in cells stably transfected with the monocistronic construct containing the IGF1R 5′-UTR from which the IRES had been deleted. The M score serves as an independent control for effects on general translational activity, though it still contains a long, highly structured 5′-untranslated sequence.
Figure 2. Reporter analyses demonstrate selective inhibition of IRES-mediated translation by candidate lead compounds and analogs. T47D breast tumor cells stably transfected with the bicistronic reporter construct containing the IGF1R 5′-UTR / IRES or the monocistronic control construct (no IRES) were seeded (in parallel) in 24-well plates and allowed 48 h to recover and resume proliferation, then treated with increasing concentrations (0 - 20 μg/ml) of IRES inhibitor lead cpd_ P (A, B), analogs P-2 (C, D) or P-3 (E, F), or candidate lead cpd_T (G, H) as indicated, under either Low (0.5%) serum (A,C,E,G) or Full (10%) serum (B, D, F, H) conditions. Following 24 h incubation, cells were harvested, lysates prepared, and firefly and Renilla luciferase activities assayed. Three parameters are assessed: firefly (2nd cistron) translation, as a direct readout of IRES-mediated translation; Renilla (1st cistron) translation, mediated by cap-dependent ribosomal scanning; and M, representing translation of the independent monocistronic control (no IRES). The dotted horizontal line is indicative of the firefly luciferase activity measured following treatment with cycloheximide (100 μg/ml) for 24 h. All data ± standard error.
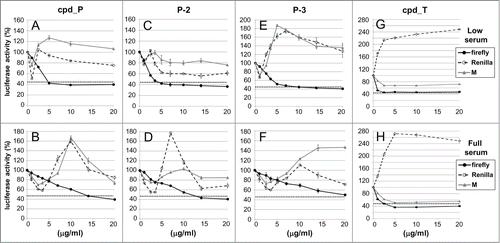
Throughout these experiments, frequent use is made of low serum conditions (acute serum deprivation, an abrupt decrease from 5 or 10% fetal calf serum to 0.5% FCS in media), because the stress of limiting soluble growth / survival factors simulates the suboptimal growth conditions to which tumor cells are exposed in the in vivo microenvironment,Citation36 and increases dependence on IRES-mediated translation, causing cells to become more sensitive to IRES inhibition. Low serum culture conditions have frequently been used to assess response to interventions intended to affect IGF1R function.Citation37 In addition, we have found that cpd_P binds significantly to serum proteins, and thus its bioavailability is actually enhanced in low serum media.
Panel A illustrates the results of the reporter analyses for cpd_P in low serum media. Neither the Renilla curve nor the M curve deviates substantially from 100%, yet the firefly curve decreases dramatically and progressively in a concentration-dependent manner. Note however that the degree of firefly inhibition reaches a plateau with ∼40% firefly luciferase activity remaining. Initially, we interpreted this as indicative of an inherent limitation in the degree of IRES inhibition; however, when the same cells under the same conditions and time period are treated instead with the universal protein synthesis inhibitor cycloheximide (at the standard 100 μg/ml concentration normally utilized for this purpose), firefly luciferase activity reaches the same nadir (∼43%, dotted horizontal line), indicating that the observed limitation relates to the inherent rate of turnover of the reporter enzyme rather than the activity of the IRES inhibitor. (Note that our reporter constructs utilized the second generation recombinant firefly luciferase coding sequence (luc-plus) which was optimized for activity, not the third generation version which was optimized for rapid turnover.) In fact, considering this limitation (equating maximal inhibition achievable with cycloheximide to 100% inhibition), the data indicate that, at sufficient concentration, cpd_P completely blocks firefly luciferase translation (mediated through the IRES).
In full serum (Panel B), the effective concentration range for cpd_P shifts further to the right. Again, cpd_P selectively inhibits firefly (second cistron, IRES-mediated) translation. At sufficient concentration, the degree of firefly inhibition by cpd_P approximates that observed with cycloheximide, indicative of an essentially complete block to IRES-mediated translation. Two closely-related analogs of cpd_P (P-2 and P-3, Panels C, D, E, and F) were also tested, and exhibited reporter outcomes very similar to those of the parent compound, selectively inhibiting IRES-mediated translation of the second cistron.
A second candidate lead compound (T), identified from the same high throughput screen, was evaluated using the same experimental strategy. Firefly luciferase translation decreases dramatically in both low serum and full serum conditions (Panels G and H), reaching a nadir approximately equivalent to that observed with cycloheximide at just 1–2.5 μg/ml, suggesting that cpd_T might be an even more potent IRES inhibitor than cpd_P.
Activity of IRES inhibitors against the endogenous IGF1R IRES
The high throughput screen and dose-response titrations measured the ability of compounds to inhibit firefly luciferase expression in tumor cells genetically-engineered with an IRES reporter construct. It was extremely important that we determine whether these compounds would be capable of interfering with the function of the endogenous IGF1R IRES, effectively blocking translation of the IGF1R mRNA in genetically-unmodified (i.e., untransfected) cells.
The net decrease in IGF1R protein resulting from inhibition of IRES activity, and how this varies as a function of time and concentration of the IRES inhibitor cpd_P were assessed by western blot (). The small molecule IRES inhibitor is intended to interfere with translation (i.e. synthesis) mediated through the internal ribosome entry site, but it is not expected to have any effect on turnover of pre-existing protein molecules. The T47D human breast tumor cells are characterized by a very high baseline expression of IGF1R, and the mature membrane-bound IGF1R protein has a long half-life, therefore an effective block to IGF1R synthesis might not result in a rapid or drastic change in net IGF1R protein under otherwise equilibrium conditions. Indeed this is the case, as a net decrease in mature IGF1R protein in T47D cells treated with cpd_P at concentrations ≥ 5 μg/ml was observed, but was limited to ∼50% over a 72 hour period (). The IGF1R precursor molecule, however, which is characterized by a much more rapid turnover, is effectively depleted in association with cpd_P treatment.
Figure 3. Activity of IRES inhibitors against the endogenous IGF1R IRES. (A) T47D cells were seeded in 6-well plates and allowed 48 h to recover prior to treatment with increasing concentrations of IRES inhibitor cpd_P or vehicle control as indicated. Following 72 h continuous exposure to the compound, cells were harvested, whole cell lysates prepared, equivalent aliquots separated by SDS/PAGE, and analyzed by western blot for IGF1R. (B) Titration of IRES inhibitor cpd_P on regeneration of IGF1R following trypsin catabolism. T47D human breast tumor cells were trypsinized and reseeded (in full serum) into 6-well plates and exposed immediately to increasing concentrations of IRES inhibitor cpd_P. Following 24 h incubation, cells were harvested, whole cell lysates prepared, equivalent aliquots separated by SDS/PAGE, and assayed by western blot for IGF1R. Robust regeneration of trypsin- catabolized IGF1R is observed within 24 h in vehicle (DMSO) treated cells, however, this is completely blocked in the presence of cpd_P at ≥ 5 μg/ml. Trypsin: lysate prepared from cells immediately following trypsinization. The arrow marks the position of intact membrane-bound IGF1R (β subunit). *asterisk marks the position of trypsin-catabolized IGF1R. Results confirm activity of cpd_P against the endogenous IGF1R IRES in native (genetically-unmodified) tumor cells. (C) The trypsin catabolism / regeneration assay was used to titrate IRES inhibition by analog P-3 and candidate lead cpd_T.
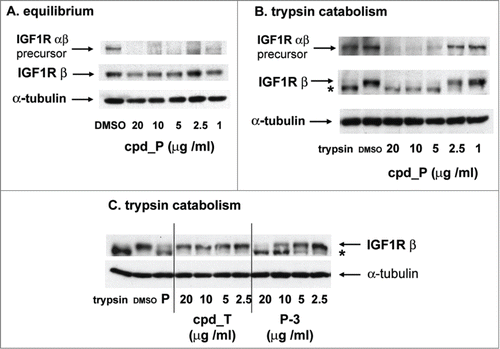
We realized that it would be beneficial to perturb equilibrium so that a block to IGF1R synthesis would be more readily apparent across the backdrop of an inherently low rate of turnover of pre-existing IGF1R molecules. We discovered that trypsinization (the standard protocol for subculturing adherent cells) degrades all pre-existing IGF1R molecules on the surface of the cell, forcing cells to completely regenerate the entire population of mature membrane-bound IGF1R molecules. We made use of this observation to specifically test the ability of cells to re-synthesize IGF1R in the presence of cpd_P (). Note that there is no intact IGF1R remaining after trypsinization when drug treatment is initiated, so the intensity of the band representing full-length IGF1R is a direct reflection of the rate of IGF1R synthesis from that point in time. The results indicate that, at sufficient concentration (≥ 5 μg/ml), IRES inhibitor cpd_P completely blocks de novo synthesis of IGF1R. This assay reflects the activity of the compound against the endogenous IGF1R IRES.
Like the parent compound, analog P-3 also inhibits de novo IGF1R synthesis following trypsin catabolism in a concentration-dependent manner (IC50 ∼7.0 μg/ml, ). However, cpd_T shows no activity against the endogenous IRES, even at concentrations 20-fold greater than that sufficient to inhibit IGF1R IRES function in the context of the bicistronic reporter.
IRES inhibitor cpd_P and its analogs block de novo synthesis of IGF1R in response to acute serum deprivation
The capacity of the small molecule IRES inhibitors to block IGF1R synthesis was further evaluated in a second breast tumor cell line (). SUM159 breast tumor cells express very low IGF1R at baseline under standard culture conditions (5% FCS, 5 μg/ml insulin, lane 1). However, we found that these cells dramatically upregulate IGF1R synthesis in response to acute serum / insulin deprivation (0.5% FCS, no supplemental insulin, lane 2). The increase in IGF1R occurs specifically in response to the decrease in concentration of soluble ligand for IGF1R in the media, as IGF-1 and IGF-2 (the natural ligands for IGF1R) are among the most important growth-promoting components of fetal calf serum, and insulin (at the concentrations commonly used for tissue culture media supplementation) also serves as a ligand for IGF1R. In fact, if the cells are deprived of serum but insulin supplementation is maintained, no increase in IGF1R is observed (data not shown). In the presence of IRES inhibitor cpd_P or analog P-2 or P-3 (lanes 3, 5, 6), this increase in IGF1R was almost completely abrogated. In contrast, cpd_T is completely ineffective in blocking this increase in IGF1R synthesis (lane 4).
Figure 4. IRES inhibitor cpd_P blocks de novo synthesis of IGF1R in response to acute serum deprivation. (A) SUM159 breast tumor cells were seeded in 6-well plates and allowed 48 h in full serum media to recover and resume proliferation. Then, the standard growth media (which includes 5% fetal calf serum and 5 μg/ml insulin) was replaced with media containing only 0.5% FCS and no supplemental insulin, along with IRES inhibitor lead cpd_P, analog P-2 or P-3, candidate lead cpd_T (each at 10 μg/ml), or vehicle (0.1% DMSO) control. After 24 h, cells were harvested, whole cell lysates prepared, equivalent aliquots separated by SDS/PAGE and analyzed by western blot for IGF1R. (B) Trypsin catabolism combined with serum deprivation. SUM159 breast tumor cells were trypsinized and reseeded into 6-well plates and incubated immediately in the presence of compounds as indicated, in low serum media (0.5% FCS, no supplemental insulin). Robust regeneration and upregulation of IGF1R is observed within 24 h in vehicle (DMSO) treated cells, however, this is completely blocked in the presence of IRES inhibitor cpd_P or analogs P-2 or P-3. *asterisk marks the position of trypsin-degraded IGF1R.
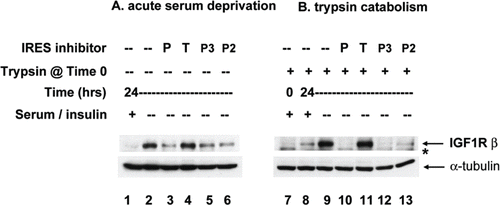
If the cells are simultaneously challenged with trypsinization and acute serum / insulin deprivation, not only is an entirely new population of IGF1R molecules synthesized, but the level of IGF1R is also dramatically increased (lane 9). In the presence of IRES inhibitor cpd_P, regeneration and upregulation of IGF1R is completely blocked (lane 10). Analogs P-2 and P-3 are also effective (lanes 12 and 13). Once again, however, cpd_T exhibits no capacity to block IGF1R translation (lane 11).
These results obtained for IGF1R in SUM159 cells have important implications, demonstrating that sensitivity to IRES inhibition (at the molecular level) does not necessarily correlate with or require high baseline expression of the target protein. In fact, the inability to selectively upregulate synthesis of a key protein when called for, particularly under stressful or adverse microenvironmental conditions, may render the cell vulnerable to drastic phenotypic alteration or cell death (as shown below). Most importantly, the results indicate that cpd_P and its analogs effectively inhibit translation of the endogenous IGF1R mRNA.
Basis for cpd_T as a false positive IRES inhibitor
Although the reporter data indicate that cpd_T has the capacity to interfere with IRES function in the context of the bicistronic mRNA, the western blot data indicate that this compound is completely ineffective against the endogenous IGF1R IRES (naturally monocistronic mRNA). It is important to recognize that the IRES is positioned directly in the path that scanning 40S ribosomes must take to reach the initiation codon. Thus, for a compound to be effective in blocking translation of the endogenous IGF1R mRNA, it must not only interfere with IRES function, but also effectively impede the progress of scanning ribosomes that approach the IRES.
The fundamental difference between the bicistronic reporter and the endogenous monocistronic mRNA is that the artificially-positioned first cistron (Renilla coding sequence) of the bicistronic reporter RNA, functioning essentially as a large upstream open reading frame, essentially eliminates ribosomal scanning through the IGF1R 5′-UTR which is cloned downstream, whereas in the endogenous IGF1R mRNA, in its natural monocistronic context, 40S ribosomes are recruited to the beginning of the 5′-UTR, and proceed to scan, albeit with low efficiency, through this highly structured sequence. In this manner, the bicistronic construct insulates and protects the IRES from scanning ribosomes, so that synthesis of the protein encoded by the second cistron (firefly luciferase) provides a nearly pure read-out of IRES-mediated translation initiation (beneficial to high throughput screening).
It appears that cpd_T effectively blocks translation initiation through the IRES only in the bicistronic context, where there are no 40S ribosomes scanning through the 5′-UTR. Thus, we attribute the failure of this compound to inhibit translation of the endogenous IGF1R mRNA to its apparent susceptibility to disruption by 40S ribosomes concomitantly scanning through the IRES, which takes place on the endogenous monocistronic mRNA but is artificially repressed by the design of the bicistronic construct.
A precedent for such variability in translational inhibition was established by our detailed characterization of the natural IRES-repressor protein HuR.Citation32 HuR when bound alone to the IGF1R IRES, transiently delays translation initiation, but is actively displaced by scanning 40S ribosomes. However, once HuR becomes incorporated into a dysfunctional IRES - RNP complex, it becomes impenetrable to scanning ribosomes, resulting in a perpetual complete block to translation of the coding sequence downstream. It appears that cpd_T is a weak IRES inhibitor, behaving like HuR alone, susceptible to disruption by concomitant ribosomal scanning through the IRES, and fails to effectively block translation of the endogenous monocistronic IGF1R mRNA. In contrast, active lead cpd_P is a strong (true positive) IRES inhibitor, capable of locking the IRES-RNP complex into a non-productive transition state that can withstand oncoming scanning ribosomes, halting IGF1R translation completely. Thus, the bicistronic reporter system is a more sensitive assay for IRES inhibition, while the endogenous mRNA is a more stringent test of an IRES inhibitor.
Spectrum of activity of the IRES inhibitor extends beyond IGF1R and includes Myc
When we began our search for small molecules to interfere with IRES-mediated translation, we could not project how specific such compounds would be for individual or groups of cellular IRESs. We realized that it was possible that the active compound(s) might exhibit: (a) absolute specificity for the IGF1R IRES; (b) pan-IRES activity, taking advantage of a property common to all cellular IRESs; or (c) an intermediate degree of specificity. The cumulative data acquired using a number of experimental approaches clearly indicate that each of the active lead compounds identified in our screen exhibits an intermediate degree of specificity, impacting the translation of a distinct subset of IRES-driven proteins.
We found that cpd_P (in fact, all 3 IRES inhibitor lead compounds) are highly active against Myc, another critical oncogenic protein known to be translated via an IRES. The c-myc IRES allows the Myc protein to be translated under conditions such as apoptosis during which general protein synthesis is largely attenuated.Citation38-39 Relative to IGF1R, c-myc is inherently more amenable to translational modulation because of the considerably shorter half-life of the Myc protein, so that changes in rate of Myc translation (synthesis) are more rapidly reflected as changes in net Myc protein.
The human breast tumor cell line SUM159 expresses Myc at high level at baseline (, lanes 1,2). Treatment of the cells with cpd_P (, lane 5; , 24 hours) brings about a dramatic decrease in p64 (Myc2), the dominant (oncogenic) isoform of Myc, while simultaneously stimulating synthesis of p67 (Myc1), the minor isoform of Myc, which has been attributed potent growth-inhibitory and pro-apoptotic properties.Citation40-41 Thus it appears that the small molecule IRES inhibitor has the capacity to differentially modulate the translation of these 2 functionally distinct Myc protein isoforms.
Figure 5. IRES inhibitor cpd_P is highly active against Myc and is distinguished from other translationally-active drugs. (A) SUM159 breast tumor cells were subjected to acute serum / insulin deprivation (0.5% FCS, no supplemental insulin) and simultaneously treated X 24 h with IRES inhibitor cpd_P (10 μg / ml), rapamycin (100 nM in lanes 3, 6; 200 nM in lane 4), cycloheximide (100 μg / ml), anisomycin (10 μM), or combinations of these reagents as indicated. Whole cell lysates were prepared, equivalent aliquots separated by SDS/PAGE, and analyzed by western blot for IGF1R and c-Myc. Insulin receptor, driven by its own unique IRES,Citation71,72 and mrtl, a Myc-related protein which does not require use of an IRES,Citation73 serve as controls. IRES inhibitor cpd_P differentially modulates translation of the 2 Myc isoforms, decreasing abundance of p64 (oncogenic), while increasing synthesis of p67 (growth-inhibitory). In contrast, rapamycin secondarily stimulates synthesis of both isoforms of Myc, while cycloheximide and anisomycin completely eliminate all Myc protein. (B) Western blot results obtained for Myc after 24, 48, or 72 h treatment of SUM159 breast tumor cells with variable concentrations (1.0 - 10 μg/ml) of cpd_P in full serum.
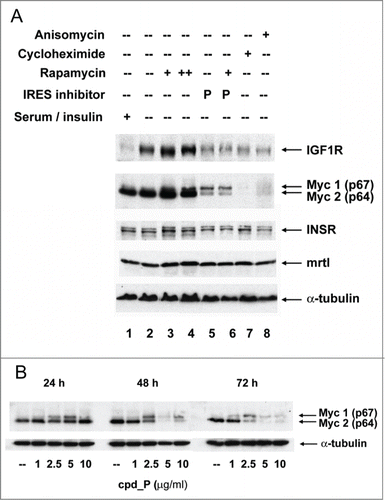
c-myc is known to utilize an IRES, and it is known to have 2 alternative initiation codons (one for p64 and one for p67), but the connection between these 2 translation-regulatory events had not previously been established. Our results suggest that these 2 events are linked, that the IRES controls not only translational efficiency but also choice of initiation codon (and thus which isoform will be synthesized), and the small molecule IRES inhibitor impacts both of these parameters.
Hann et al.Citation42 reported a similar shift in p64/p67 balance in avian bursal lymphoma cells in association with high density growth arrest. These investigators were able to attribute this physiological reversal of the p64/p67 ratio specifically to depletion of methionine from the media. Methionine depletion inhibits cap-dependent translation initiation via a decrease in cap methylation (dependent on S-adenosyl methionine as methyl donor), which is required for recognition by the cap-binding protein eIF4E,Citation43 suggesting that the upregulation in p67 synthesis is IRES-mediated. Furthermore, Nanbru et al.Citation44 have demonstrated that the c-myc IRES is capable of initiating translation from both the CUG (p67) and AUG (p64) initiation codons in their native sequence contexts in vitro. In addition, we observe here that p67 synthesis increases upon treatment of cells with rapamycin (lanes 3, 4), which inhibits translation mediated by conventional cap-dependent ribosomal scanning. Together, these observations support the conclusion that p67 synthesis is an alternate outcome of IRES-mediated translation of Myc.
The activity of the small molecule IRES inhibitor can be clearly distinguished from that of other translationally-active drugs. Rapamycin does not impede IGF1R or Myc synthesis, in fact both IGF1R and Myc (both isoforms) are enhanced even further in rapamycin-treated cells (, lanes 3 and 4). This finding is consistent with published data showing that rapamycin secondarily increases IRES-mediated translation, and in fact this secondary increase in IRES-mediated translation is thought to be a mechanism for resistance to rapamycin.Citation45 IRES inhibitor cpd_P not only blocks the increase in IGF1R translation in response to serum deprivation, it also blocks the increase in both IGF1R and Myc in response to rapamycin treatment (lane 6). Thus, the IRES inhibitor functions in a manner which is essentially reciprocal to, and dominant over, rapamycin. The effects of the IRES inhibitor are also distinguished from those of cycloheximide and anisomycin, 2 universal inhibitors of protein synthesis, which eliminate all Myc protein (lanes 7, 8).
Additional experiments were performed to evaluate the time and dose-dependent changes taking place in Myc in cells continuously exposed to cpd_P (). At 24 hours there is first evidence for an increase in the p67 isoform. This shift in translation favoring synthesis of p67 continues through 48 hours and is associated with a progressive decline in p64. By the 72 hour time point, there is nearly complete disappearance of both Myc protein isoforms from cells exposed to ≥ 5 μg/ml cpd_P. This dynamic shift in the balance between the oncogenic and growth inhibitory isoforms, culminating in a complete shutdown of Myc translation, would be expected to have a drastic impact on cell phenotype (see below).
Phenotypic consequences of sustained IRES inhibition
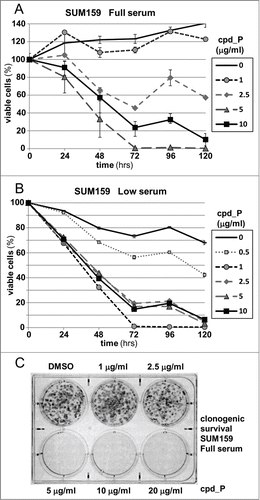
Next, cell viability was assessed as a function of concentration and duration of exposure to the IRES inhibitor (). Continuous exposure of SUM159 cells to 5-10 μg/ml cpd_P for ≥72 hours results in loss of viability affecting 75 to >99% of the breast tumor cell population. (An in-depth characterization of 2 distinct modes of cell death induced by cpd_P is the subject of an ongoing investigation.) When the viability assays are repeated under low serum conditions (0.5% FCS, ), precisely the same pattern of response to cpd_P is observed. Furthermore, beyond a critical threshold concentration, clonogenic survival of the breast tumor cells is completely eliminated (). Note that the concentrations of cpd_P required for effective inhibition of IRES-mediated translation initiation are remarkably consistent between the different types of assays utilized, with concordant reporter and western (endogenous target) outcomes (IC50 in the range of 5 - 10 μg/ml in full serum), and this same effective dose range also applies to cytotoxicity and clonogenic survival. Together, these results suggest that IRES-mediated translation is of critical importance to the survival of malignant cells.
Figure 6. Phenotypic consequences of IRES inhibition. (A) Viability of SUM159 breast tumor cells assessed following treatment with increasing concentrations of cpd_P for periods of 24 to 120 h. Cell survival is presented relative to the cell number at initiation of treatment (time 0 = 100%) ± standard error. (B) The viability time course titrations of IRES inhibitor cpd_P in SUM159 breast tumor cells were repeated under Low serum (0.5%) conditions (all data ± standard error). (C) Clonogenic survival assay. SUM159 breast tumor cells were seeded at low density, allowed 48 h to recover, then treated with increasing concentrations of cpd_P in full serum for 72 h. Media was then changed, compound removed, and cells allowed an additional 120 h to recover and form colonies. Cultures were stained with MTT to enhance visualization of colonies.
RNase protection analyses confirm that the changes in Myc protein associated with IRES inhibition are not accompanied by any discernible changes at the mRNA level
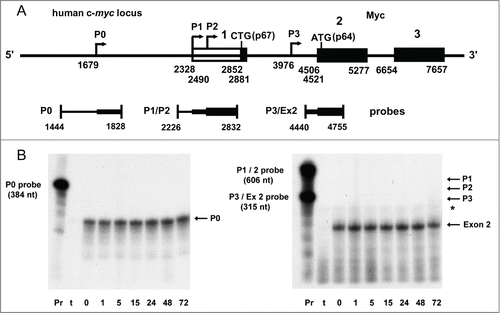
A series of RNase protection assays were performed to examine the status of the c-myc mRNA, and to place the results into context with the dramatic alterations in Myc protein induced by the small molecule IRES inhibitor. The human c-myc locus is extraordinarily complex, with 4 distinct transcription start sites generating 4 mRNA isoforms (P0, P1, P2, P3) which differ in extent of the 5′-untranslated sequence.Citation46,47 The relative translational efficiency of each of the 4 c-myc transcripts, the degree to which each utilizes the IRES, and the propensity for synthesis of p67 versus p64 in vivo have not been established, except that the P3 transcript, which initiates within the first intron, does not include the IRES and is incapable of encoding p67.
A series of antisense probes bracketing each of the transcription start sites was used to distinguish and quantify the relative abundance of each of the 4 c-myc mRNA isoforms. In addition, a probe overlapping the intron 1 – exon 2 boundary was used to measure the cumulative abundance of all Myc-encoding mRNAs. For the SUM159 breast tumor cells at baseline, we found that the P0 transcript accounted for the overwhelming majority of c-myc transcripts, with the intensity of the P0-protected band nearly equal to that produced by the exon 2 probe (representing total c-myc mRNA). We were consistently unable to detect the presence of P1 or P2 initiated transcripts, in spite of having tested 8 different probes (both PCR-generated and plasmid-derived) covering this sequence, and having amplified the template for these probes directly from the genomic DNA of the cells from which the RNA was isolated (to control for possible sequence polymorphisms). Protection attributable to the P3 start site was modest in intensity and equivocal. Thus, our findings suggest that in SUM159 cells, the c-myc P0 transcript is responsible for IRES-mediated translation of Myc, as well as generation of the p67 isoform. This conclusion is consistent with results reported by Nanbru et al.Citation44, who demonstrated that the c-myc IRES is active in the context of the P0 5′-UTR in transfected cells. This predominance of the P0 transcript in malignant cells is not without precedent, as c-myc is expressed exclusively from the P0 transcription start site in the majority (70%) of multiple myelomas.Citation48.
The pattern of c-myc transcription was established in the mid-1980's in work focused primarily on hematological models (particularly Burkitt's lymphomas). There is a paucity of experimental data directly addressing this issue in other cell types. However, there is considerable evidence that the P0 promoter is regulated independently of the P1 and P2 promoters, and that P0 transcription or changes in chromatin structure surrounding the P0 promoter often correlate more precisely with c-myc exon 2 levels and cell phenotype than do the other c-myc promoters (e.g., downregulation of Myc accompanying differentiation of HL-60 cells),Citation47,49-51 suggesting that the P0 transcript may be of major significance in c-myc function even in cells in which the P1 and P2 transcripts appear to dominate. In breast cancer, the sole difference in c-myc chromatin structure between ER-positive and ER-negative tumor cells is the significantly greater accessibility of DNase I hypersensitive site II-2 associated with the P0 promoter in the ER-negative cells.Citation52
A time course assay was performed to assess whether any changes occur in c-myc mRNA during treatment with cpd_P, when dramatic alterations to Myc protein are taking place (). SUM159 cells were treated with cpd_P for various periods of time (ranging from 1 hour to 72 hours), RNA recovered, and equivalent aliquots hybridized to the P0, P1/2, P3, and exon2 probes. The probes were prepared simultaneously, labeled to equivalent specific activity, and the sizes of the protected fragments are comparable, so that band intensities accurately reflect the relative abundance of each of the mRNA species. Analysis of these samples revealed no discernible alterations to the P0 transcript or to exon 2 (representing total c-myc mRNA). Furthermore, there was no evidence for utilization of the P1 or P2 start sites, nor did the data suggest any change in P3 (minimal protection of full-length exon 2 probe, and additional data not shown). These findings indicate that the dramatic alterations in Myc protein induced by the IRES inhibitor are not accompanied by any corresponding changes in c-myc mRNA, i.e., no apparent alteration in start site utilization, and no significant increase or decrease in transcriptional activity (or mRNA stability), confirming that these changes take place exclusively at the translational level.
Figure 7. RNase protection analysis of c-myc mRNA in human breast tumor cells treated with IRES inhibitor cpd_P. (A) Diagram of the human c-myc locus and the series of antisense probes designed to assess the abundance of each of the 4 c-myc mRNA isoforms. Sequence coordinates are as described in Gazin et alCitation74 (GenBank: X00364). (B) RNase protection time course assay. SUM159 breast tumor cells were treated with IRES inhibitor cpd_P (10 μg/ml in full serum) and harvested at selected time points. 5 μg of total RNA recovered from each sample (or tRNA as negative control) was hybridized with each of the probes overnight at 42°C. An extensive series of pilot experiments had indicated that P0 is the predominant c-myc mRNA isoform present in these cells, and that good resolution could be obtained by combining the P1/P2 and P3/exon 2 probes while analyzing the P0 probe separately. Following hybridization, samples were digested with RNase A / T1, recovered by ethanol precipitation, and separated on 5% acrylamide, 8M urea denaturing gels. Only 1/20th of the amount of probe included in each digested sample was loaded in the positive control (undigested) lanes. The P0 transcript is expected to protect a fragment of 149 nucleotides. Protected fragments of 504 and 342 nt are expected for the P1 and P2 transcripts. P3-initiated transcripts would be contiguous with exon 2, and should protect the full-length intron 1 – exon 2 probe. With exon 2 beginning at 4506, the spliced c-myc mRNAs (cumulatively) are expected to protect a fragment of 249 nt. The asterisk marks the position of a faint band which maps to the 3′ end of exon 1 and apparently represents a rare splicing event for the P0 transcript (additional data not shown). The two gels were run in parallel and exposed to autoradiography for the same amount of time. Pr = full-length probe (undigested). t = tRNA (negative control).
Myc translation-regulatory status varies with cell context
The degree to which Myc translation relies on the IRES has not been clearly defined, and apparently conflicting data on this point exist in the literature. Evidence has been published supporting a conclusion that Myc is translated via the conventional cap-dependent ribosomal scanning mechanism (enhanced by overexpression of eIF4ECitation53), while other investigators have demonstrated clearly that Myc is translated via an IRES (finding that c-myc is among the small fraction of mRNAs which remain associated with polysomes during polioviral infection, that the IRES is responsible for Myc translation during apoptosis, and having characterized biologically and clinically-relevant ITAFs and point mutationsCitation34,54,55). We addressed this question, using the IRES inhibitor cpd_P as a probe, and find that the status of Myc translational regulation varies with cell context.
MDA-MB-231 is another human triple-negative breast tumor cell line which expresses Myc at a relatively high level. However, the western blot data appear to indicate that Myc translation is IRES-independent in these cells (). The first clue in this regard is that Myc is decreased rather than increased by serum deprivation, suggesting that c-myc is not relied on as an acute stress response gene in these cells. Furthermore, Myc actually increases rather than decreasing when treated with IRES inhibitor cpd_P. Importantly, IGF1R is quite sensitive to cpd_P in these cells, as a significant decline in IGF1R is evident even within 24 hours. Apparently the turnover of pre-existing IGF1R is quite rapid (likely a function of the extraordinarily high rate of cell division exhibited by these cells). Thus IRES-mediated translation is active in these cells, but Myc translation is IRES-independent.
Figure 8. Myc translation-regulatory status evaluated on the basis of sensitivity to IRES inhibition. (A) MDA-MB-231 or (B) ZR-75-1 breast tumor cells were treated for 24 h with IRES inhibitor cpd_P (10 μg/ml), rapamycin (100 nM), cycloheximide (100 μg / ml), anisomycin (10 μM), or combinations of these reagents as indicated, in either Full serum (10% for MDA-MB-231, 20% for ZR-75-1) or Low serum (0.5%) conditions. Cells were harvested, whole cell lysates prepared, equivalent aliquots separated by SDS/PAGE, and analyzed by western blot for IGF1R and c-Myc. (C) ZR-75-1 breast tumor cells were seeded in 8-well chamber slides, treated with IRES inhibitor cpd_P (10 μg/ml) or vehicle control (DMSO 0.1%) for 24 h in low (0.5%) serum, then stained for Myc, following PFA (2% × 15 min) fixation and low (0.2% × 10 min) Triton X-100 permeabilization. Confocal imaging demonstrates dramatic loss of Myc from cpd_P-treated cells. DAPI staining in the associated panels marks the locations of all nuclei in each field. (D) MDA-MB-231 breast tumor cells were treated with cpd_P (0 - 10 μg/ml in low serum) for 72 or 144 h, or treated for 72 h followed by 72 h incubation in absence of compound (washout). The graph displays viability outcomes for the cpd_P-treated cells relative to DMSO (vehicle)-treated controls (100%) ± standard error. (E) Clonogenic survival assay. MDA-MB-231 cells were seeded at low density, allowed 48 h to recover, then treated with increasing concentrations of cpd_P in full (10%) serum for 96 h. Media was then changed, compound removed, and cells allowed an additional 96 h to recover and form colonies. Cultures were stained with MTT to enhance visualization of colonies.
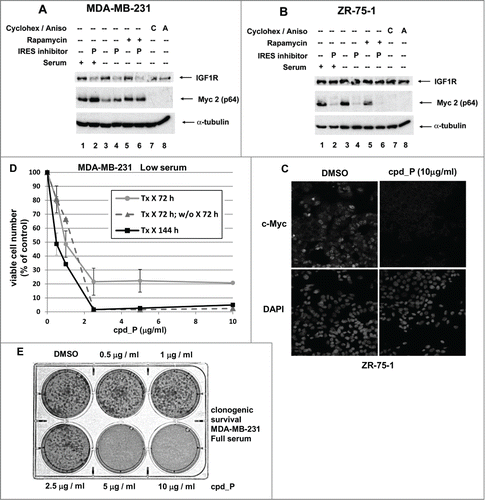
A contrasting set of results is provided by the ZR-75-1 human breast tumor cell line (). Myc is highly sensitive to IRES inhibition, with a dramatic decline in p64 noted following 24 hours exposure to cpd_P under either full or low serum conditions. No significant decrease in IGF1R is appreciated with 24 hours exposure to cpd_P, consistent with an inherently low turnover of pre-existing IGF1R molecules, commensurate with the low rate of proliferation of these cells (similar to what was seen in T47D cells in ).
Myc is only modestly sensitive to IRES inhibition in T47D cells (data not shown). Thus, of our 4 breast tumor models, we have 2 (SUM159 and ZR-75-1) in which Myc is IRES-dependent and highly sensitive to IRES inhibition, and 2 in which Myc appears to be either partially (T47D) or completely (MDA-MB-231) IRES-independent. We were curious to test whether Myc translational status in the different breast tumor cell lines could be correlated with differences in relative abundance of the 4 c-myc transcripts. Using the RNase protection strategy described above, we consistently observed that the P0 transcript was highest in the SUM159 and ZR-75-1 cell lines, intermediate in T47D, and considerably lower in MDA-MB-231 cells. The P3 transcript was detected at roughly equivalent levels, and there was no evidence for P1 or P2 initiated transcripts (data not shown). Although further experimentation in a larger number of cell types would be required to arrive at a definitive conclusion, these results suggest that Myc IRES-dependence and sensitivity of Myc to IRES inhibition correlate positively with relative abundance of the P0 transcript.
Dramatic loss of Myc from cells treated with IRES inhibitor cpd_P
To confirm and extend these results, indirect immunofluorescence staining and confocal imaging were used to assess changes in abundance and intracellular localization of Myc protein in response to IRES inhibition (). The ZR-75-1 breast tumor cells express moderately high level of Myc protein at baseline, localized predominantly to the nucleus. In cells treated for 24 hours with cpd_P, a dramatic decrease in the intensity of Myc staining is observed, consistent with the western blot data showing near complete disappearance of p64 under these same conditions. ZR75-1 cells are uniquely well-suited for this assay because these cells are not conducive to synthesis of the alternative isoform (p67) of Myc, which is transiently stimulated by cpd_P in SUM159 cells. Thus the unilateral decrease in p64 allows for a more straightforward evaluation by immunofluorescence staining (where the antibody recognizes both isoforms).
MDA-MB-231 cells are highly dependent on IRES-mediated translation for their survival, even though Myc in these cells is IRES-independent
Since we learned that Myc was IRES-independent in MDA-MB-231 cells, we wondered whether this would cause these cells to be resistant to cpd_P, averting the cytotoxicity that had been observed in association with sustained IRES inhibition in SUM159 cells, in which Myc was IRES-dependent (). The results indicate that 72 hours exposure to cpd_P at ≥ 2.5 μg/ml is sufficient to trigger irreversible commitment to cell death in a very large proportion of the cells. Furthermore, beyond a critical threshold in cpd_P concentration, clonogenic survival of MDA-MB-231 cells is completely eliminated (). Together, the results indicate that Myc translation status being IRES-independent in a particular cell line does not mean that those cells do not utilize the IRES mechanism for translation of other proteins, or that they are not vulnerable to IRES inhibition.
Therapeutic modalities designed to specifically inhibit IGF1R function (blocking antibodies, kinase inhibitors) when used as single agents typically induce growth inhibition, not cell death, and combination with other agents is required to elicit a cytotoxic response.Citation37,56,57 Thus it is very likely that additional IRES-driven proteins beyond IGF1R are impacted by cpd_P and contribute to the loss of viability observed in the MDA-MB-231 cells.
IGF1R and Myc as representatives of 2 distinct categories of IRESs
This investigation has focused on 2 oncogenic proteins, IGF1R and Myc, both of which are unequivocally implicated in the pathogenesis of a large proportion of human malignancies,Citation58-67 and both of which are translated through an IRES. The results obtained for IGF1R and c-myc clearly indicate that there are fundamental differences in the way these 2 IRESs are organized, how they operate, and their potential for modulation by small molecule IRES inhibitors.
The IGF1R IRES is positioned immediately adjacent to the initiation codon. cpd_P and its analogs are capable of completely blocking IGF1R synthesis. The architecture of the human c-myc 5′-untranslated region and IRES is considerably more complex, with 2 alternative initiation codons, and the IRES ribosome entry window positioned ∼100-150 nucleotides further upstream.Citation23,68 The transient stimulation of p67 in association with progressive down-modulation of p64 by cpd_P eventually squelches all Myc translation and results in complete disappearance of both Myc protein isoforms. Thus the c-myc IRES might be thought of as operating more like a faucet than a switch, i.e. a control that can be turned or twisted from “hot” to “cold” and then to “off.” In contrast, the IGF1R IRES appears to operate more like a binary (on – off) toggle switch. It will be interesting to determine how many other IRESs resemble either IGF1R or c-myc in organization, operation, and response to small molecule IRES inhibitors, and how many other such categories of cellular IRESs may exist.
The phenotypic outcomes observed with sustained IRES inhibition likely depend on combinatorial effects on multiple IRES-driven proteins, which almost certainly extend beyond IGF1R and Myc. Elucidation of the full spectrum of IRES targets impacted by cpd_P will require a genome-scale approach, which is currently underway.
Precise molecular target of the small molecule IRES inhibitor
IRES-mediated translation initiation is a complex process involving the 5′-untranslated region of the mRNA, a diverse group of sequence-specific translation-regulatory proteins (ITAFs), and the 40S ribosomal subunit itself. Each of these components could potentially be impacted by a small molecule IRES inhibitor. While additional experimentation will be required to define the precise molecular target of cpd_P and its analogs, the results may be most readily reconciled if the compound interacts with the 40S ribosomal subunit rather than the IRES (5′-UTR) or an individual ITAF(s). The fact that cpd_P, which was identified on the basis of its ability to interfere with function of the IGF1R IRES, was found also to dramatically impact the c-myc IRES, suggests that this compound targets a feature or component which these IRESs have in common. The primary sequences and secondary structures of the IGF1R and c-myc IRESs do not exhibit any notable similarity, the ITAFs identified for each of these IRESs are distinct, and the physiological conditions to which each of these IRESs responds and the purposes of their respective gene products are highly dissimilar (except that both are implicated in cell proliferation and malignant transformation).
There are numerous precedents for highly efficacious drugs targeting the translational machinery. Many of these drugs are antibacterial agents which exploit subtle differences between the prokaryotic and eukaryotic ribosomes.Citation69,70 We have identified a compound that selectively interferes with IRES-mediated translation, apparently exploiting the subtle distinctions between the conventional mechanism for translation initiation (cap-dependent ribosomal scanning) and the encounter taking place when the 40S ribosome engages the mRNA through the IRES.
Materials and Methods
Chemical Reagents
Compounds P, P-2, P-3, and T were solubilized in 100% DMSO to a concentration of 10 mg/ml and used fresh or stored at −20C. Stock solutions were diluted a minimum of 1:500 in media such that final DMSO concentration did not exceed 0.2%, which was matched in vehicle-only control samples. Compounds were thoroughly dispersed in media before adding to cells and incubating for up to 72 hours. For incubations extending beyond 72 hours, media was changed and compound re-added.
Cycloheximide and anisomycin were obtained from Sigma. Rapamycin was obtained from Calbiochem.
Cells, constructs, and stable transfections
T47D human breast tumor cells were stably transfected with the bicistronic IGF1R 5′-UTR / IRES reporter construct (pDualIGF1R(1-1040)), or the control (IRES-deleted) construct (plucIGF1R(1-959)), together with a linear puromycin resistance cassette (ClonTech). The derivation of pDualIGF1R(1-1040) and plucIGF1R(1-959) has been previously described, and these constructs have been utilized extensively for characterization of the IGF1R IRES and its cognate IRES-regulatory proteins.Citation31,32 Stable transfectants were selected with puromycin (2 μg/ml). Once established, it was not necessary to maintain the cells in puromycin, and cells utilized for experiments had not been exposed to puromycin for at least 8 passages.
T47D (ER-positive) human breast tumor cells were obtained from ATCC and propagated in RPMI1640 supplemented with 10% FCS and 10 μg/ml insulin. SUM159PT (triple-negative) human breast tumor cells were obtained from Asterand and propagated in Ham's F-12 supplemented with 5% FCS, 10 mM Hepes, 5 μg/ml insulin, and 1 μg/ml hydrocortisone. ZR-75-1 (ER-positive) human breast tumor cells were a generous gift from Dr. Patsy Oliver and propagated in RPMI1640 with 20% FCS. MDA-MB-231 (triple-negative) human breast tumor cells were a generous gift from Dr. Dan Welch and propagated in DMEM with 10% FCS.
Antibodies
Rabbit polyclonal anti-IGF1R (C-20, Santa Cruz) recognizes both the β subunit of the mature membrane-bound IGF1R (∼90 kDa), as well as the αβ precursor molecule (∼200 kDa). Other antibodies included c-Myc N262 for western blot, c-Myc C33 for immunofluorescence, insulin receptor N-20 (all from Santa Cruz), mrtl 131-5-2,73 and α−tubulin (B-5-1-2, Sigma). Secondary antibody for indirect immunofluorescence staining was Alexafluor 488-conjugated goat anti-mouse IgG (Invitrogen). DAPI (4′,6-Diamidino-2-phenylindole dihydrochloride) was from Sigma.
High-throughput screen
The genetically-engineered IRES-reporter and control (IRES-deleted) cells described above were used in a high-throughput screen / counterscreen to identify compounds capable of selectively inhibiting translation mediated through the IGF1R IRES. The assay was adapted to high-throughput (384-well, robotics)-compatible format, undergoing extensive optimization and validation trials, which led to establishment of a non-homogeneous yet robust protocol with a mean Z’ score of 0.75 and CV of 8%. Two replicate pilot screens (10,000 compounds) were performed on 2 separate occasions to confirm quality and reproducibility of the data, prior to initiation of the full screen. The full screen / counterscreen was performed on a diverse collection of 135,000 compounds, the bulk of which were obtained from Chembridge, but which also included focused libraries enriched for compounds known to be active on the translational machinery as well as compounds capable of binding RNA or serving as RNA / nucleoside analogs. Cells were incubated with compounds (10 µg/ml final concentration) for 24 hours prior to measurement of firefly luminescence. Each plate included wells treated with a potent inhibitor of the luciferase enzyme which had been validated for use as a positive control. Compounds were scored positive if they exhibited a statistically significant degree of inhibition of IRES activity (firefly luciferase activity ≥ 3 standard deviations below median of control) and no more than 20% decrease in general protein synthesis (as measured using the counterscreen). Compounds scoring positive in the initial screen / counterscreen were re-tested using a 10-point 2-fold dilution dose-response titration, to confirm activity and assess relative potency.
Reporter assays
Stably transfected IRES reporter and control (IRES-deleted) cells were treated in parallel with IRES inhibitors as indicated for 24 hours. Lysates were prepared (1X passive lysis buffer) and firefly and Renilla luciferase activities were measured using the dual luciferase system as recommended by the manufacturer (Promega).
Western blot assays
Following incubation with IRES inhibitors or other agents as indicated (see figure legends for details), whole cell lysates were rapidly prepared by adding lysis buffer (containing 4% SDS and 720 mM 2-mercaptoethanol, preheated to 100°C) directly to cell monolayer, recovery and addition of glycerol to 10%, and heating for an additional 5 minutes. For instances in which cell adhesion was compromised (e.g. trypsinization followed immediately by exposure to IRES inhibitor), care was taken (using centrifugation) to ensure recovery of loosely adherent or floating cells. Equivalent aliquots (by protein content) were separated on 10% SDS/PAGE gels, transferred to 0.2 μm nitrocellulose membranes, and subjected to standard immunoblotting procedures followed by chemiluminescence image capture.
RNase protection
Probe templates were amplified from normal human placental DNA or genomic DNA recovered from breast tumor cell lines, incorporating the T7 promoter into the 5′-tail of the reverse primer. Radiolabeled probes were transcribed in vitro in antisense orientation using T7 RNA polymerase (Promega) in the presence of [α-32P]-UTP. RNA was recovered from cells using Qiazol reagent. RNase protection was performed using the RPA III kit (Ambion) and following precisely the manufacturer's protocol. Briefly, following hybridization of probe with cellular RNA overnight at 42°C, samples were digested with RNase A / T1 for 30 min at 25°C, then precipitated, separated on 5% acrylamide / 8M urea denaturing gels, and results obtained by autoradiography.
Cell viability assays
Cell viability was assayed using standard MTT ((3-[4,5-dimethylthiazol-2-yl]-2,5- diphenyltetrazolium bromide), Sigma) protocol. Briefly, at selected time points, sterile MTT solution was added directly to cell cultures (final 250 μg/ml), incubated for 3 hours, after which media was aspirated, formazan dye solubilized in DMSO, and absorbance at 570 nm recorded (correcting for background at 670 nm).
Immunofluorescence staining and confocal imaging
Cells were seeded in 8-well chamber slides (Nunc) and allowed 48 hours to recover and resume proliferation prior to treatment with cpd_P (10 μg/ml) or vehicle control (0.1% DMSO) for 24 hours. Cells were fixed with freshly prepared 2% paraformaldehyde for 15 minutes, followed by permeabilization with 0.2% Triton X-100 for 10 minutes at room temperature. After blocking with 10% normal goat serum in PBS for 45 minutes, cells were incubated with monoclonal anti-c-Myc (C33, Santa Cruz) at 1:100 dilution in blocking solution for 1 hour at room temperature. After two consecutive washes in PBS and re-blocking, secondary antibody (AlexaFluor 488-conjugated goat anti-mouse IgG, highly cross-adsorbed, Life Technologies) was added for 45 minutes at room temperature. Following two additional PBS washes, nuclei were stained with DAPI (0.2 μg/ml), and mounted using ProLong Gold (Life Technologies). Images were captured using a Nikon A1 confocal instrument with 40X 1.3NA objective. Paired images of control and experimental wells were acquired sequentially and all settings including laser power, PMT voltage, and pinhole were held constant between samples. Prior to the definitive experiments, a series of trial assays had been performed in which concentration and duration of exposure to paraformaldehyde and Triton X-100 were varied and optimal conditions for staining determined. Controls incubated with secondary antibody alone were used to document specificity of staining.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors wish to express their grateful appreciation to Drs. Edward E. Partridge, Richard J. Whitley, Maaike Everts, John A. (Jack) Secrist, and the late W. Blaine Knight, whose vision and administrative support were critical for the initiation and advancement of this project. The authors wish to gratefully acknowledge Drs. E. Lucile White, Lynn Rasmussen, Joseph A. Maddry, Miranda Nebane-Akah, Krister Wennerberg, Omar Moukha-Chafiq, Nameta Bansal, and Cecil Kwong of Southern Research Institute for their outstanding contributions to the high throughput screening and medicinal chemistry aspects of the project.
Funding
This work was supported by grants from the NIH / NCI (R01CA108886); Department of Defense Breast Cancer Research Program (W81XWH-09-1-0183); the American Society for Clinical Oncology Young Investigator Award (to C.V.); the UAB Comprehensive Cancer Center Drug Discovery and Development Program; and the Alabama Drug Discovery Alliance.
References
- Baird SD, Turcotte M, Korneluk RG, Holcik M. Searching for IRES. RNA 2006; 12: 1755-85; PMID:16957278; http://dx.doi.org/10.1261/rna.157806
- Prats AC, Prats H. Translational control of gene expression: role of IRESs and consequences for cell transformation and angiogenesis. Prog. Nucleic Acid Res Mol Biol 2002; 72: 367-413; http://dx.doi.org/10.1016/S0079-6603(02)72075-8
- Marash L, Liberman N, Henis-Korenblit S, Sivan G, Reem E, Elroy-Stein O, Kimchi A. DAP5 promotes cap-independent translation of Bcl-2 and CDK1 to facilitate cell survival during mitosis. Mol Cell 2008; 30: 447-59; PMID:18450493; http://dx.doi.org/10.1016/j.molcel.2008.03.018
- Majumder M, Yaman I, Gaccioli F, Zeenko VV, Wang C, Caprara MG, Venema RC, Komar AA, Snider MD, Hatzoglou M. The hnRNA-binding proteins hnRNP L and PTB are required for efficient translation of the Cat-1 arginine/lysine transporter mRNA during amino acid starvation. Mol Cell Biol 2009; 29:2899-912; PMID:19273590; http://dx.doi.org/10.1128/MCB.01774-08
- Lang KJ, Kappel A, Goodall GJ. Hypoxia-inducible factor-1alpha mRNA contains an internal ribosome entry site that allows efficient translation during normoxia and hypoxia. Mol Biol Cell 2002; 13:1792-1801; PMID:12006670; http://dx.doi.org/10.1091/mbc.02-02-0017
- Riley A, Jordan LE, Holcik M. Distinct 5′ UTRs regulate XIAP expression under normal growth conditions and during cellular stress. Nucleic Acids Res 2010; 38:4665-74; PMID:20385593; http://dx.doi.org/10.1093/nar/gkq241
- Komar AA, Hatzoglou M. Cellular IRES-mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle 2011; 10:229-40; PMID:21220943; http://dx.doi.org/10.4161/cc.10.2.14472
- Komar AA, Mazumder B, Merrick WC. A new framework for understanding IRES-mediated translation. Gene 2012; 502:75-86; PMID:22555019; http://dx.doi.org/10.1016/j.gene.2012.04.039
- Fernandez J, Yaman I, Mishra R, Merrick WC, Snider MD, Lamers WH, Hatzoglou M. Internal ribosome entry site-mediated translation of a mammalian mRNA is regulated by amino acid availability. J Biol Chem 2001; 276:12285-91; PMID:11114306; http://dx.doi.org/10.1074/jbc.M009714200
- Morrish BC, Rumsby MG. The 5′ untranslated region of protein kinase Cdelta directs translation by an internal ribosome entry segment that is most active in densely growing cells and during apoptosis. Mol Cell Biol 2002; 22:6089-99; PMID:12167703; http://dx.doi.org/10.1128/MCB.22.17.6089-6099.2002
- Sherrill KW, Byrd MP, Van Eden ME, Lloyd RE. BCL-2 translation is mediated via internal ribosome entry during cell stress. J Biol Chem 2004; 279:29066-74; PMID:15123638; http://dx.doi.org/10.1074/jbc.M402727200
- Eto I. Upstream molecular signaling pathways of p27(Kip1) expression: effects of 4-hydroxytamoxifen, dexamethasone, and retinoic acids. Cancer Cell Int 2010; 10:3; PMID:20170512; http://dx.doi.org/10.1186/1475-2867-10-3
- Jimenez J, Jang GM, Semler BL, Waterman ML. An internal ribosome entry site mediates translation of lymphoid enhancer factor-1. RNA 2005; 11:1385-99; PMID:16120831; http://dx.doi.org/10.1261/rna.7226105
- Petz M, Them N, Huber H, Beug H, Mikulits W. La enhances IRES-mediated translation of laminin B1 during malignant epithelial to mesenchymal transition. Nucleic Acids Res 2012; 40: 290-302; PMID:21896617; http://dx.doi.org/10.1093/nar/gkr717
- Pozner A, Goldenberg D, Negreanu V, Le SY, Elroy-Stein O, Levanon D, Groner Y. Transcription-coupled translation control of AML1/RUNX1 is mediated by cap- and internal ribosome entry site-dependent mechanisms. Mol Cell Biol 2000; 20:2297-307; PMID:10713153; http://dx.doi.org/10.1128/MCB.20.7.2297-2307.2000
- Yoon A, Peng G, Brandenburger Y, Zollo O, Xu W, Rego E, Ruggero D. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 2006; 312:902-6; PMID:16690864; http://dx.doi.org/10.1126/science.1123835
- Zheng Y, Miskimins WK. Far upstream element binding protein 1 activates translation of p27Kip1 mRNA through its internal ribosomal entry site. Int J Biochem Cell Biol 2011; 43:1641-8; PMID:21855647; http://dx.doi.org/10.1016/j.biocel.2011.08.001
- Nagamachi A, Htun PW, Ma F, Miyazaki K, Yamasaki N, Kanno M, Inaba T, Honda Z, Okuda T, Oda H, Tsuji K, Honda H. A 5′ untranslated region containing the IRES element in the Runx1 gene is required for angiogenesis, hematopoiesis and leukemogenesis in a knock-in mouse model. Dev Biol 2010; 345:226-36; PMID:20647008; http://dx.doi.org/10.1016/j.ydbio.2010.07.015
- Galy B, Maret A, Prats AC, Prats H. Cell transformation results in the loss of the density-dependent translational regulation of the expression of fibroblast growth factor 2 isoforms. Cancer Res 1999; 59:165-71; PMID:9892202
- Bonnal S, Schaeffer C, Créancier L, Clamens S, Moine H, Prats AC, Vagner S. A single internal ribosome entry site containing a G quartet RNA structure drives fibroblast growth factor 2 gene expression at four alternative translation initiation codons. J Biol Chem 2003; 278:39330-6; PMID:12857733; http://dx.doi.org/10.1074/jbc.M305580200
- Durie D, Lewis SM, Liwak U, Kisilewicz M, Gorospe M, Holcik M. RNA-binding protein HuR mediates cytoprotection through stimulation of XIAP translation. Oncogene 2011; 30:1460-9; PMID:21102524; http://dx.doi.org/10.1038/onc.2010.527
- Huez I, Créancier L, Audigier S, Gensac MC, Prats AC, Prats H. Two independent internal ribosome entry sites are involved in translation initiation of vascular endothelial growth factor mRNA. Mol Cell Biol 1998; 18:6178-90; PMID:9774635
- Le Quesne JP, Stoneley M, Fraser GA, Willis AE. Derivation of a structural model for the c-myc IRES. J Mol Biol 2001; 310:111-26; PMID:11419940; http://dx.doi.org/10.1006/jmbi.2001.4745
- Riley A, Jordan LE, Holcik M. Distinct 5′ UTRs regulate XIAP expression under normal growth conditions and during cellular stress. Nucleic Acids Res 2010; 38:4665-74; PMID:20385593; http://dx.doi.org/10.1093/nar/gkq241
- Shi Y, Frost P, Hoang B, Benavides A, Gera J, Lichtenstein A. IL-6-induced enhancement of c-Myc translation in multiple myeloma cells: critical role of cytoplasmic localization of the rna-binding protein hnRNP A1. J Biol Chem 2011; 286:67-78; PMID:20974848; http://dx.doi.org/10.1074/jbc.M110.153221
- Bellodi C, Krasnykh O, Haynes N, Theodoropoulou M, Peng G, Montanaro L, Ruggero D. Loss of function of the tumor suppressor DKC1 perturbs p27 translation control and contributes to pituitary tumorigenesis. Cancer Res 2010; 70:6026-35; PMID:20587522; http://dx.doi.org/10.1158/0008-5472.CAN-09-4730
- Blau L, Knirsh R, Ben-Dror I, Oren S, Kuphal S, Hau P, Proescholdt M, Bosserhoff AK, Vardimon L. Aberrant expression of c-Jun in glioblastoma by internal ribosome entry site (IRES)-mediated translational activation. Proc Natl Acad Sci USA 2012; 109:E2875-84; PMID:23027969; http://dx.doi.org/10.1073/pnas.1203659109
- Blume SW, Jackson NL, Frost AR, Grizzle WE, Shcherbakov OD, Choi H, Meng Z. Northwestern profiling of potential translation-regulatory proteins in human breast epithelial cells and malignant breast tissues: evidence for pathological activation of the IGF1R IRES. Exp Mol Pathol 2010; 88:341-52; PMID:20233590; http://dx.doi.org/10.1016/j.yexmp.2010.03.006
- Dobson T, Chen J, Krushel LA. Dysregulating IRES-dependent translation contributes to over-expression of the Aurora A kinase onco-protein. Mol Cancer Res 2013; 11:887-900; PMID:23661421; http://dx.doi.org/10.1158/1541-7786.MCR-12-0707
- Warnakulasuriyarachchi D, Cerquozzi S, Cheung HH, Holcík M. Translational induction of the inhibitor of apoptosis protein HIAP2 during endoplasmic reticulum stress attenuates cell death and is mediated via an inducible internal ribosome entry site element. J Biol Chem 2004; 279:17148-57; PMID:14960583; http://dx.doi.org/10.1074/jbc.M308737200
- Meng Z, Jackson NL, Choi H, King PH, Emanuel PD, Blume SW. Alterations in RNA-binding activities of IRES-regulatory proteins as a mechanism for physiological variability and pathological dysregulation of IGF-IR translational control in human breast tumor cells. J Cell Physiol 2008; 217:172-83; PMID:18452152; http://dx.doi.org/10.1002/jcp.21486
- Meng Z, King PH, Nabors LB, Jackson NL, Chen C-Y, Emanuel PD, Blume SW. The ELAV RNA-stability factor HuR binds the 5′-Untranslated region of the human IGF1R transcript and differentially represses cap-dependent and IRES-mediated translation. Nucleic Acids Res 2005; 33:2962-79; PMID:15914670; http://dx.doi.org/10.1093/nar/gki603
- Meng Z, Jackson NL, Shcherbakov OD, Choi H, Blume SW. The human IGF1R IRES likely operates through a Shine-Dalgarno-like interaction with the G961 loop (E-site) of the 18S rRNA and is kinetically modulated by a naturally-polymorphic polyU loop. J Cell Biochem 2010; 110:531-44; PMID:20432247; http://dx.doi.org/10.1002/jcb.22569
- Johannes G, Carter MS, Eisen MB, Brown PO, Sarnow P. Identification of eukaryotic mRNAs that are translated at reduced cap binding complex eIF4F concentrations using a cDNA microarray. Proc Natl Acad Sci U S A 1999; 96:13118-23; PMID:10557283; http://dx.doi.org/10.1073/pnas.96.23.13118
- Spriggs KA, Stoneley M, Bushell M, Willis AE. Re-programming of translation following cell stress allows IRES-mediated translation to predominate. Biol Cell 2008; 100:27-38; PMID:18072942; http://dx.doi.org/10.1042/BC20070098
- Beltran PJ, Mitchell P, Chung YA, Cajulis E, Lu J, Belmontes B, Ho J, Tsai MM, Zhu M, Vonderfecht S, Baserga R, Kendall R, Radinsky R, Calzone FJ. AMG 479, a fully human anti-insulin-like growth factor receptor type I monoclonal antibody, inhibits the growth and survival of pancreatic carcinoma cells. Mol Cancer Ther 2009; 8:1095-105; PMID:19366899; http://dx.doi.org/10.1158/1535-7163.MCT-08-1171
- Zeng X, Sachdev D, Zhang H, Gaillard-Kelly M, Yee D. Sequencing of type I insulin-like growth factor receptor inhibition affects chemotherapy response in vitro and in vivo. Clin Cancer Res 2009; 15:2840-9; PMID:19351773; http://dx.doi.org/10.1158/1078-0432.CCR-08-1401
- Stoneley M, Chappell SA, Jopling CL, Dickens M, MacFarlane M, Willis AE. c-Myc protein synthesis is initiated from the internal ribosome entry segment during apoptosis. Mol Cell Biol 2000; 20:1162-9; PMID:10648601; http://dx.doi.org/10.1128/MCB.20.4.1162-1169.2000
- Créancier L, Mercier P, Prats AC, Morello D. c-myc Internal ribosome entry site activity is developmentally controlled and subjected to a strong translational repression in adult transgenic mice. Mol Cell Biol 2001; 21:1833-40; http://dx.doi.org/10.1128/MCB.21.5.1833-1840.2001
- Benassayag C, Montero L, Colombié N, Gallant P, Cribbs D, Morello D. Human c-Myc isoforms differentially regulate cell growth and apoptosis in Drosophila melanogaster. Mol Cell Biol 2005; 25:9897-909; PMID:16260605; http://dx.doi.org/10.1128/MCB.25.22.9897-9909.2005
- Hann SR, King MW, Bentley DL, Anderson CW, Eisenman RN. A non-AUG translational initiation in c-myc exon 1 generates an N-terminally distinct protein whose synthesis is disrupted in Burkitt's lymphomas. Cell 1988; 52:185-95; PMID:3277717; http://dx.doi.org/10.1016/0092-8674(88)90507-7
- Hann SR, Sloan-Brown K, Spotts GD. Translational activation of the non-AUG-initiated c-myc 1 protein at high cell densities due to methionine deprivation. Genes Dev 1992; 6:1229-40; PMID:1628829; http://dx.doi.org/10.1101/gad.6.7.1229
- Cowling VH. Regulation of mRNA cap methylation. Biochem J 2009; 425:295-302; PMID:20025612; http://dx.doi.org/10.1042/BJ20091352
- Nanbru C, Lafon I, Audigier S, Gensac MC, Vagner S, Huez G, Prats AC. Alternative translation of the proto-oncogene c-myc by an internal ribosome entry site. J Biol Chem 1997; 272:32061-6; PMID:9405401; http://dx.doi.org/10.1074/jbc.272.51.32061
- Shi Y, Sharma A, Wu H, Lichtenstein A, Gera J. Cyclin D1 and c-myc internal ribosome entry site (IRES)-dependent translation is regulated by AKT activity and enhanced by rapamycin through a p38 MAPK- and ERK-dependent pathway. J Biol Chem 2005; 280:10964-73; PMID:15634685; http://dx.doi.org/10.1074/jbc.M407874200
- Bentley DL, Groudine M. Novel promoter upstream of the human c-myc gene and regulation of c-myc expression in B-cell lymphomas. Mol Cell Biol 1986; 6:3481-9; PMID:3540591
- Eick D, Polack A, Kofler E, Lenoir GM, Rickinson AB, Bornkamm GW. Expression of P0- and P3-RNA from the normal and translocated c-myc allele in Burkitt's lymphoma cells. Oncogene 1990; 5:1397-402; PMID:2216463
- Hoover RG, Kaushal V, Lary C, Travis P, Sneed T. c-myc transcription is initiated from P0 in 70% of patients with multiple myeloma. Curr Top Microbiol Immunol 1995; 194:257-64; PMID:7895497
- Bentley DL, Groudine M. A block to elongation is largely responsible for decreased transcription of c-myc in differentiated HL60 cells. Nature 1986; 321:702-6; PMID:3520340; http://dx.doi.org/10.1038/321702a0
- Lang JC, Wilkie NM, Clark AM, Chudleigh A, Talbot S, Whitelaw B, Frame MC. Regulatory domains within the P0 promoter of human c-myc. Oncogene 1991; 6:2067-75; PMID:1945411
- Pullner A, Mautner J, Albert T, Eick D. Nucleosomal structure of active and inactive c-myc genes. J Biol Chem 1996; 271:31452-7; PMID:8940157; http://dx.doi.org/10.1074/jbc.271.49.31452
- Miller TL, Huzel NJ, Davie JR, Murphy LC. C-myc gene chromatin of estrogen receptor positive and negative breast cancer cells. Mol Cell Endocrinol 1993; 91:83-9; PMID:8472857; http://dx.doi.org/10.1016/0303-7207(93)90258-L
- De Benedetti A, Harris AL. eIF4E expression in tumors: its possible role in progression of malignancies. Int J Biochem Cell Biol 1999; 31:59-72; PMID:10216944; http://dx.doi.org/10.1016/S1357-2725(98)00132-0
- Chappell SA, LeQuesne JP, Paulin FE, deSchoolmeester ML, Stoneley M, Soutar RL, Ralston SH, Helfrich MH, Willis AE. A mutation in the c-myc-IRES leads to enhanced internal ribosome entry in multiple myeloma: a novel mechanism of oncogene de-regulation. Oncogene 2000; 19:4437-40; PMID:10980620; http://dx.doi.org/10.1038/sj.onc.1203791
- Cobbold LC, Spriggs KA, Haines SJ, Dobbyn HC, Hayes C, de Moor CH, Lilley KS, Bushell M, Willis AE. Identification of internal ribosome entry segment (IRES)-trans-acting factors for the Myc family of IRESs. Mol Cell Biol 2008; 28:40-9; PMID:17967896; http://dx.doi.org/10.1128/MCB.01298-07
- Carboni JM, Wittman M, Yang Z, Lee F, Greer A, Hurlburt W, Hillerman S, Cao C, Cantor GH, Dell-John J, et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol Cancer Ther 2009; 8:3341-9; PMID:19996272; http://dx.doi.org/10.1158/1535-7163.MCT-09-0499
- Litzenburger BC, Creighton CJ, Tsimelzon A, Chan BT, Hilsenbeck SG, Wang T, Carboni JM, Gottardis MM, Huang F, Chang JC, et al. High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy. Clin Cancer Res 2011; 17:2314-27; PMID:21177763; http://dx.doi.org/10.1158/1078-0432.CCR-10-1903
- Turner BC, Haffty BG, Narayanan L, Yuan J, Havre PA, Gumbs AA, Kaplan L, Burgaud JL, Carter D, Baserga R. et al. Insulin-like growth factor-I receptor overexpression mediates cellular radioresistance and local breast cancer recurrence after lumpectomy and radiation. Cancer Res 1997; 57:3079-83; PMID:9242428
- Scotlandi K, Avnet S, Benini S, Manara MC, Serra M, Cerisano V, Perdichizzi S, Lollini PL, De Giovanni C, Landuzzi L. et al.. Expression of an IGF-I receptor dominant negative mutant induces apoptosis, inhibits tumorigenesis and enhances chemosensitivity in Ewing's sarcoma cells. Int J Cancer 2002; 101:11-16; PMID:12209582; http://dx.doi.org/10.1002/ijc.10537
- Sachdev D, Hartell JS, Lee AV, Zhang X, Yee D. A dominant negative type I insulin-like growth factor receptor inhibits metastasis of human cancer cells. J Biol Chem 2004; 279: 5017-24; PMID:14615489; http://dx.doi.org/10.1074/jbc.M305403200
- Yanochko GM, Eckhart W. Type I insulin-like growth factor receptor over-expression induces proliferation and anti-apoptotic signaling in a three-dimensional culture model of breast epithelial cells. Breast Cancer Res 2006: 8:R18; PMID:16584539; http://dx.doi.org/10.1186/bcr1392
- Jones RA, Campbell CI, Gunther EJ, Chodosh LA, Petrik JJ, Khokha R, Moorehead RA. Transgenic overexpression of IGF-IR disrupts mammary ductal morphogenesis and induces tumor formation. Oncogene 2007; 26:1636-44; PMID:16953219; http://dx.doi.org/10.1038/sj.onc.1209955
- Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, Weinberg RA. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev 2001; 15:50-65; PMID:11156605; http://dx.doi.org/10.1101/gad.828901
- Notari M, Neviani P, Santhanam R, Blaser BW, Chang JS, Galietta A, Willis AE, Roy DC, Caligiuri MA, Marcucci G, Perrotti D. A MAPK/HNRPK pathway controls BCR/ABL oncogenic potential by regulating MYC mRNA translation. Blood 2006; 107:2507-16; PMID:16293596; http://dx.doi.org/10.1182/blood-2005-09-3732
- Alles MC, Gardiner-Garden M, Nott DJ, Wang Y, Foekens JA, Sutherland RL, Musgrove EA, Ormandy CJ. Meta-analysis and gene set enrichment relative to er status reveal elevated activity of MYC and E2F in the “basal” breast cancer subgroup. PLoS One 2009; 4:e4710; PMID:19270750; http://dx.doi.org/10.1371/journal.pone.0004710
- Chandriani S, Frengen E, Cowling VH, Pendergrass SA, Perou CM, Whitfield ML, Cole MD. A core MYC gene expression signature is prominent in basal-like breast cancer but only partially overlaps the core serum response. PLoS One 2009; 4:e6693; PMID:19690609; http://dx.doi.org/10.1371/journal.pone.0006693
- Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, Hjelmeland AB, Rich JN. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One 2008; 3:e3769; PMID:19020659; http://dx.doi.org/10.1371/journal.pone.0003769
- Cencig S, Nanbru C, Le SY, Gueydan C, Huez G, Kruys V. Mapping and characterization of the minimal internal ribosome entry segment in the human c-myc mRNA 5′ untranslated region. Oncogene 2004; 23:267-77; PMID:14712232; http://dx.doi.org/10.1038/sj.onc.1207017
- Lynch SR, Puglisi JD. Structural origins of aminoglycoside specificity for prokaryotic ribosomes. J Mol Biol 2001; 306:1037-58; PMID:11237617; http://dx.doi.org/10.1006/jmbi.2000.4420
- Hansen JL, Moore PB, Steitz TA. Structures of five antibiotics bound to the peptidyl transferase center of the large ribosomal subunit. J Mol Biol 2003; 330:1061-75; PMID:12860128; http://dx.doi.org/10.1016/S0022-2836(03)00668-5
- Spriggs KA, Cobbold LC, Ridley SH, Coldwell M, Bottley A, Bushell M, Willis AE, Siddle K. The human insulin receptor mRNA contains a functional internal ribosome entry segment. Nucleic Acids Res 2009; 37:5881-93; PMID:19654240; http://dx.doi.org/10.1093/nar/gkp623
- Olson CM, Donovan MR, Spellberg MJ, Marr MT 2nd. The insulin receptor cellular IRES confers resistance to eIF4A inhibition. Elife 2013; 2:e00542; PMID:23878722
- Choi H, Jackson NL, Shaw DR, Emanuel PD, Liu YL, Tousson A, Meng Z, Blume SW. mrtl – a translation / localization regulatory protein encoded within the human c-myc locus and distributed throughout the endoplasmic and nucleoplasmic reticular network. J Cell Biochem 2008; 105:1092-108; also GenBank: BK006467; PMID:18816594; http://dx.doi.org/10.1002/jcb.21909
- Gazin C, Dupont de Dinechin S, Hampe A, Masson JM, Martin P, Stehelin D, Galibert F. Nucleotide sequence of the human c-myc locus: provocative open reading frame within the first exon. EMBO J 1984; 3:383-7; PMID:6714223