
Abstract
In a previous study we reported the role of potent bisindole-PBD conjugate as an inclusion in the arsenal of breast cancer therapeutics. In breast cancer cell proliferation, PI3K/AKT/mTOR pathway plays a crucial role by prosurvival mechanism that inhibits programmed cell death. Here, 2 breast cancer cells lines, MCF-7 and MDA-MB-231 were treated with Vorinostat (suberoylanilide hydroxamic acid / SAHA) and bisindole-PBD (5b). We have investigated the effect on PI3K/AKT/mTOR pathway and SIRT expression including epigenetic regulation. There was consistent decrease in the level of PI3K, AKT, mTOR proteins upon treatment of 5b in both MCF-7 and MDA-MB-231 cell lines compared to untreated controls. Treatment with caspase inhibitor (Q-VD-OPH) confirmed that the effect of 5b on PI3K signaling was ahead of apoptosis. Real time PCR and western blot analysis showed profound reduction in the mRNA and protein levels of SIRT1 and SIRT2. Molecular docking studies also supported the interaction of 5b with various amino acids of SIRT2 proteins. Treatment with 5b caused epigenetic changes that include increase of acetylated forms of p53, increase of histone acetylation at p21 promoter as well as decrease in methylation state of p21 gene. Compound 5b thus acts as SIRT inhibitor and cause p53 activation via inhibition of growth factor signaling and activation of p53 dependent apoptotic signaling. This present study focuses bisindole-PBD on epigenetic alteration putting 5b as a promising therapeutic tool in the realm of breast cancer research.
Abbreviations
| ac-p53 | = | acetyl-p53 |
| ChIP | = | chromatin immunoprecipitation |
| mTOR | = | mammalian target of rapamycin |
| PBD | = | Pyrrolo [2, 1-c][1, 4] benzodiazepines |
| PI3K | = | phosphatidylinositide 3-kinases |
| SAHA | = | suberoylanilide hydroxamic acid |
| SIRT | = | Sirtuin. |
Introduction
Breast cancer is the most common form of malignancy diagnosed in women and is the second most common cause of cancer-related deaths among women worldwide. Despite the invention of recent advances in technologies and treatment methodologies, breast cancer still continues to be one of the most dangerous diseases among the women. Therapeutic approach to treat this disease includes a combination of targeted, chemotherapy and hormonal therapy. Yet, most of the patients with breast cancer possess de novo resistance and the remaining patients become drug resistant.Citation1 Drug resistant breast cancer in most cases is associated with poor prognosis,Citation2,3 requiring the need for new therapeutic approach or development of better drugs. Recent studies have indicated that inhibition of cell survival and induction of apoptosis increases the therapeutic efficiency.Citation4 Various growth factors and growth factor mediated signaling pathways play important role in tumorigenesis that is mainly regulated by PI3K/AKt/mTOR signaling cascade. The components of this cascade are aberrantly expressed or mutated in breast cancer. Studies by Carnero A et al. Citation2008 have indicated that a class of HDAC protein SIRT1 is required for PI3K mediated cancer cell growth.Citation5,6 PI3K/Akt /mTOR signaling can reduce p53 response through inhibitory effect on its stability.Citation7
Tumor suppressor gene p53 plays a crucial role in executing anti-proliferative effects such as growth arrest, apoptosis and cell senescence, in response to various types of stress.Citation8 In most cancers, it is found to be mutated. The genomic instability due to loss of p53 function initiates tumor progression.Citation9-11 Chemotherapeutic approach in cancer cells where p53 is mutated does not cause apoptosis; thereby resulting in drug resistance.Citation12,13 p53 stabilization and transcriptional activation are crucial regulatory processes that occur in cancer cells when treated with anti-cancer compounds. Ubiquitination, phosphorylation and acetylation are important post-translational modifications that dictate p53 stability and function.Citation14 Increase in p53 stability is caused by N-terminal phosphorylation at ser15 and ser20. p53 phosphorylation at ser15 position is necessary for its activation.Citation15,16 In addition to phosphorylation, acetylation of p53 at lysine 373 and 382 also plays an important role in p53 activation.Citation17
Epigenetic programming is vital for proper development in mammals. Its stable inheritance is crucial for maintenance of cellular functions. Changes in epigenetic programs cause a change in the accessibility of the chromatin to transcriptional factors.Citation18 Epigenetic modifications along with genetic alterations lead to cancer. Acetylation of histones is related to the relaxation of chromatin structure and transcriptional activation.Citation19 In most cancers, loss of acetylation at lysine 16 and trimethylation at lysine 20 of H4 is observed.Citation20 These modifications at lysine residues affect the expression of genes either by providing binding sites for detector proteins regulating the chromatin access or by altering affinity between DNA and histones. The acetylation state at the N terminal histone tail is controlled by 2 oppositely acting enzymes: histone acetyltransferases (HAT) and histone deacetylases (HDACs).Citation21 In mammals, HDACs are divided into 4 classes: class I HDACs (1– 3 and 8), class IIa HDACs (4, 5, 7 and 9) and Class IIb HDACs (6 and 10), class III HDACs (SIRTs 1–7) and class IV (HDAC11).Citation22 NAD-dependent Class III histone deacetylase SIRT1 is a protein that has multiple functions and has been demonstrated to be critically involved in stress response, cellular metabolism and aging by deacetylation of variety of substrates including p53, forkhead transcription factors, PGC-1α, NF-κB, Ku70 and histones. SIRT2 is also found to be associated with various cellular functions by regulating p53, H3 and H4. Increasing evidences indicate that SIRT1 play a complex role in tumorigenesis with functions in both tumor-promotion and tumor suppression.Citation23 SIRT1 physically interacts with p53 and cause deacetylation of p53 that leads to repression of p53-dependent apoptosis in response to DNA damage.Citation24 In many malignant conditions like breast cancer, colon cancer and leukemia, SIRT1 is found to be over expressed. Recent studies have indicated that inhibition of SIRT proteins lead to an activation and stabilization of p53 via acetylation and induce apoptosis in breast cancer cells.Citation25,26
Epigenetic silencing of tumor suppressor genes and other epigenetic abnormalities also occur due to changes in the methylation pattern of the promoter.Citation27 DNA methylation results in the recruitment of HDACs to the promoter and repressing the expression of the genes. Hypermethylation in CpG-island of promoter region and global hypomethylation are the predominant events frequently found in tumors. DNA hypomethylation in the promoter CpG islands of tumor suppressor genes are associated with histone modifications, such as gain of histone H3, H4 acetylation and loss of H3K9 and H3K27 methylation. These changes in histone lead to activation of tumor suppressor gene p21. Silencing of genes as a result of DNA methylation can be reversed with demethylating agents.Citation28 Reversal of DNA methylation and re expressing the silenced genes lead to a new approach that can be used clinically in malignancies.
Pyrrolo[2,1-c][1,4]benzodiazepines (PBDs), are well known sequence selective DNA interactive agents that bind to the minor groove of DNA and exhibit potent anticancer activity.Citation29 In our previous studies on a small molecule bisindole-pyrrolobenzodiazepine conjugate, we had shown DNA damage induced apoptosis by the compound in breast cancer cells via the involvement of p53 gene.Citation30 Here in this study we have made an attempt to understand the possible role of the compound 5b in regulating the balance between survival and apoptotic factors in breast cancer cells. Studies by Fröjdö S et al. have revealed that a substantial relationship exists between sirtuins and PI3K/AKT pathway.Citation31 Both Sirt1 and Sirt2 play a pivotal role in AKT activation in cancer cells along with constitutive PI3K induction. Present study focus on modulation of growth factor survival proteins such as PI3K/Akt/mTOR and SIRT that regulate the activity of p53 and its dependent apoptotic activity in controlling breast cancer cell proliferation via epigenetic modifications of p21.
Results
Bisindole-PBD (5b) conjugate inhibits breast cancer cell proliferation and migration
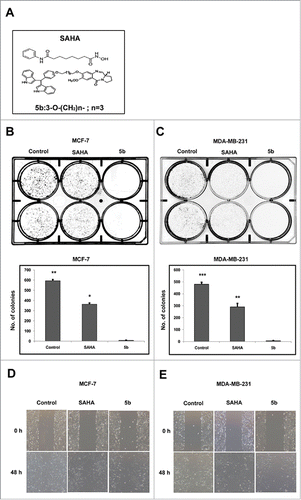
To see the effect of 5b on cell proliferation in vitro clonogenic assay was performed in 2 different breast cancer cell lines MCF-7 and MDA-MB-231. This assay determines the ability of a cell to proliferate indefinitely thereby producing colonies or clones. Cells from actively growing cell culture in monolayer were trypsinized to make a suspension. Approximately 500 cells were seeded per well of a 6 -well plate and treated separately with 4 μM of SAHA (a well known HDAC inhibitor) and 5b for 24 h, followed by PBS wash. Cells were then grown in complete media for 10 d. Number of colonies formed was counted. Simultaneously same numbers of cells were also separately treated with DMSO that serves as control. Compound 5b treated both the breast cancer cell lines exhibited drastic decrease in number compared to DMSO treated control cells (). This observation clearly show that compound 5b produce cell proliferation arrest. We next performed wound healing assay to see the effect of compound 5b on cell migration, an important event in metastasis. Wound healing assay was performed after treatment of both the breast cancer cell lines with 5b and SAHA separately. A clear inhibitory action of 5b on breast cancer cell migration was observed when compared with SAHA (positive control) and DMSO treated cell (negative control) ().
Figure 1. Bisindole-PBD conjugate chemical structure, Cell proliferation studies and wound healing assays. (A) Chemical structure of SAHA and bisindole-PBD conjugate (5b). (B-C) Effects of SAHA and 5b on MCF-7 and MDA-MB-231 cell proliferation at 4 μM and the images of the colonies along with untreated controls. The numbers of colonies were counted from 3 independent results and plotted to compare the effect of 5b. 5b was more efficient in inhibiting cell proliferation. Histograms were drawn from 3 independent experiments. Error bars with *** represents P<0.001 and ** represents p<0.01. (D, E) Scratched monolayer of cells was treated with SAHA and 5b at 4 μM. Cell migration was photographed after stimulation with drug at 48 hour time point.
Bisindole-PBD (5b) induces the expression of p53-dependent genes involved in cell cycle and apoptosis
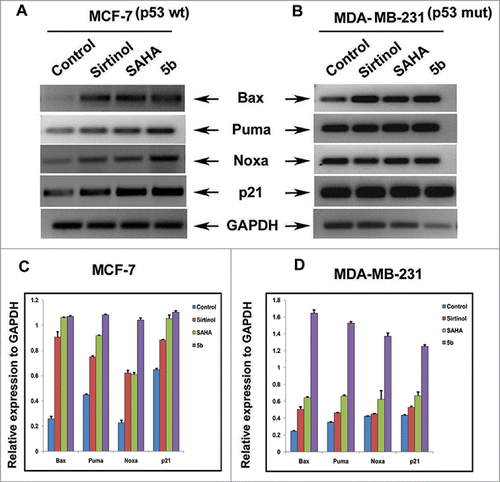
p53 is an essential transcription factor involved in expression of genes in response to cellular stress.Citation32 It regulates Bcl-2 family genes such as Bax, Puma and Noxa via intrinsic form of apoptosis.Citation33 p21 another p53 dependent gene plays a crucial regulatory role in cell cycle by binding and inhibiting cyclin dependent kinases (CDKs). Therefore we estimated the expression of the proapoptotic genes as well p21 in both the breast cancer cell lines upon compound treatment. Cells were incubated with compounds (i.e. 5b, Sirtinol and SAHA) for 24 hours. Total cellular RNA was isolated and Reverse transcription PCR for Bax, Puma, Noxa, and p21 was performed using gene specific primers to examine the level of expression (Table S1). We observed marked increase in mRNA expression of Bax, Puma, Noxa and p21 genes in compound 5b treated cells when compared to untreated control. Interestingly compound 5b treated cells showed prominent increase in the transcript level of these genes than standard HDAC inhibitor SAHA or Sirtinol in both MCF-7 and MDA-MB-231 cells ().
Figure 2. mRNA expression level of proapoptotic Bcl-2 family members. Cells were treated with 50 μM of Sirtinol, 4 μM of SAHA and 4 μM of 5b and were incubated for 24 h. (A, B) RT-PCR analysis of different members of the family indicating the level of mRNA expression after treatment. (C, D) Histograms represent relative expression of gene levels to internal control GAPDH. Plots are mean values from 3 independent experiments. Error bars represent standard deviation.
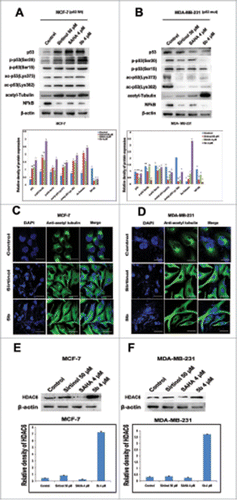
Bisindole-PBD (5b) inhibits PI3K/AKT/mTOR pathway
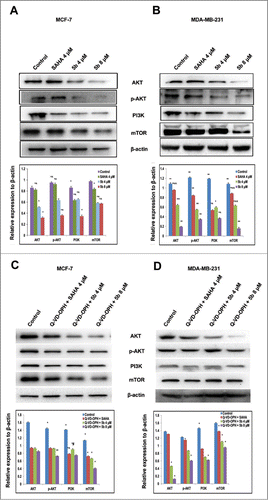
Interactions between the p53 and PI3K/AKT pathways play a significant role in the regulation of cell death and survival signaling.Citation34 PI3K/AKT/mTOR is an intracellular signaling pathway that is often deregulated in human cancers. It inhibits p53 tumor suppressor activity resulting in tumor progression and drug resistance.Citation35-37 To uncover the effect of 5b on PI3K/AKT/mTOR alterations and see its effect on tumorigenesis, we treated both the breast cancer cell lines with the compound 5b and estimated the expression of the proteins by performing protein gel blot analysis. Both the cell lines (MCF-7 and MDA-MB-231) were treated with 5b for 24 hours before isolation of proteins. Western blot analysis was performed using antibodies against PI3K, AKT, phospho-AKT (ser 473), and mTOR proteins. There was a significant reduction in PI3K, AKT, phospho-AKT (ser 473), and mTOR protein expression in 5b treated samples compared to untreated control. There was 2- to 3-fold decrease in protein levels as compared to untreated control () in MCF-7 cells whereas in MDA-MB-231, there was 4- to 5 -fold decrease in protein levels after 5b treatment () Treatment of the same cell lines with 4 μM of SAHA (positive control) also showed reduction of PI3K, AKT, phospho-AKT (ser 473) and mTOR protein expression. This study clearly demonstrated that the compound 5b was capable of inhibiting PI3K/AKT/mTOR pathway leading to growth arrest and inhibition of tumor cell progression. We further wanted to verify that the down regulation of PI3K/AKT/mTOR signaling cascade occurred before or prior to apoptosis. So, we treated the cells with 500 nM of Q-VD-OPH, a caspase inhibitor one hour prior to treatment of 5b and SAHA. We observed similar change in expression of PI3K, AKT and mTOR proteins confirming that the reduction in their expression might be direct and were not due to the induction of apoptosis ().
Figure 3. Western blot analysis showing that Bisindole PBD (5b) dampens the prosurvival PI3K/AKT/mTOR signaling pathway in breast cancer cells. (A, B) Proteins extracted from MCF-7 and MDA-MB-231 cells incubated with 4 μM of SAHA and 4, 8 μM of 5b for 24 h along with DMSO (0.1%) treated control cells were immunoblotted and hybridized with antibodies against AKT, p-AKT, PI3K and mTOR to check the level of expressions. β-Actin was used as gel loading control. The relative expression of proteins from 3 independent blots was plotted in histograms. Bars with * are significantly different from other groups p ≤ 0.01 except that they bear same symbol. In case of MDA-MB-231 histogram, bars with ** are significantly different from other groups p ≤ 0.01 and Bars with * are significantly different from other groups p ≤ 0.05 except that they bear same symbol. (C, D) Co- treatment with Q-VD-OPH 500 nm, SAHA 4 μM and 5b 4 μM was given to examine the expression pattern of AKT, p-AKT, PI3K and mTOR protein levels. β-Actin was used as loading control. Histograms represent data from 3 independent experiments. Data are represented as the mean ± SD. *P < 0.05.
Bisindole-PBD conjugate (5b) down regulates the SIRT expression both at transcriptional and translational level
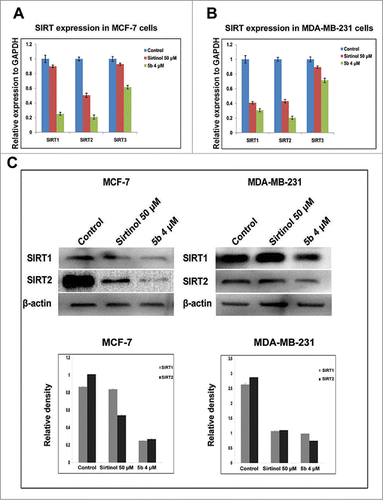
Sirtuins (SIRTs) belong to class III group of histone deacetylases and play important role in survival of cell and control of cell division by regulating transcription, microtubule organization and responses to DNA damaging agents. Earlier work by Trapp et al. had shown bisindolylmaleimide to be a specific inhibitor of class III HDACs - SIRTs.Citation38 Therefore we explored the possible regulatory role of the compound 5b on the mRNA expression of SIRT-1, 2, 3 in both MCF-7 and MDA-MB-231 cells. We first performed a time-dependent study for SIRT mRNA expression after treating the cell with 5b at 4 μM for 4, 8, 12 h and with Sirtinol at 50 μM for 12 h. There was moderate down regulation in expression of SIRT mRNAs in both the cell lines at 8 and 12 h (Fig. S1). We also performed simultaneously SIRT1, 2, 3 mRNA expression study by real time PCR after treating the cell lines with 5b at 4 µM and Sirtinol at 50 μM concentration. In both the cell lines, relative quantification showed 4- to 5-fold decrease in SIRT1 and SIRT2 mRNA expression level upon 5b treatment whereas SIRT3 showed a lesser down regulation (). We further estimated the expression of SIRT1 and SIRT2 protein after treating both the breast cancer cell lines with either Sirtinol (50 μM) or 5b (4 μM). There was profound suppression in the expression of both the proteins in both the cell lines (). In case of MCF-7, SIRT1 showed a 4-fold down regulation where as in case of SIRT2 the expression was still lower (5-fold) in 5b treated cells. MDA-MB-231 cells showed a 2- fold decrease in SIRT1 expression and 3-fold decrease in SIRT2 expression. In case of both the cell lines the compound showed a much lower expression of both the SIRT proteins compared to Sirtinol treated condition. These results proved that compound 5b inhibited the SIRT protein expression in both breast cancer cell lines MCF-7 and MDA-MB-231 having wild-type p53 or lacking functional p53 respectively.Citation25,26
Figure 4. 5b down modulates levels of SIRTs both at transcriptional level and post transcriptional level. (A, B) Cells treated with Sirtinol and 5b was subjected to qRTPCR with SIRT primers taking GAPDH as endogeneous control. Relative expression has been shown with respect to GAPDH. (C) Cells were pretreated with Sirtinol 50 μM and 5b 4 μM for 24 h. Downregulation of SIRT protein levels were assessed by western blot. β- actin was used as loading control. Data are shown as the mean + SD. All these data were obtained from 3 independent experiments and a representative figure has been shown from 3 replicates.
Immunocytochemical expressions of SIRT1 and SIRT2
In normal cells, SIRT 1 is localized in the nucleus. But in cancer cells its localization is partially delocalized to the cytoplasm.Citation39 On the contrary, SIRT2 is predominantly localized in the cytoplasm. In order to visualize the effect of 5b on SIRT localization we performed immunocytochemistry study after treating the cells with compound 5b/SAHA/Sirtinol for 24 hours. The cells were subsequently subjected to fixation followed by immunostaining with anti-SIRT1 and anti-SIRT2 antibodies. There was drastic delocalization of the SIRT1 protein from the nucleus to the cytoplasm in 5b treated MCF-7 and MDA-MB-231 cells thereby increasing SIRT1 localization in cytoplasm () whereas the untreated cells showed a perfect localization in the nucleus, indicating a clear movement of the cells toward apoptosis or CRM1 mediated nuclear export of SIRT1.Citation39 In case of SIRT2, the delocalization was moderate after incubating the cells with 5b or SAHA ().
Figure 5. Immunocytochemical expression of SIRT1 and SIRT2 in breast cancer cells after compound treatment and docking study between SIRT2 and 5b. MCF-7 and MDA-MB-231 cells were treated with 50 μM of Sirtinol and 4 μM of 5b for 24 h and fixed for immunocytochemical analysis of SIRT1 and SIRT2. (A-B) Reduced expression of cytoplasmic localized SIRT1 was observed after 5b treatment. Images were taken by using 60X oil objective under confocal microscope. Bar scale represents here 10 µM. (C-D) Diminished level of SIRT2 was observed after incubation with Sirtinol and 5b. Bars represent 10 µM. (E) Molecular modeling showed binding between 5b and SIRT2 (with different amino acids residues). Figure showed receptor-ligand Hydrogen bonds (Green) and Receptor-ligand bumps of 5b with active site residues of SIRT2 protein.
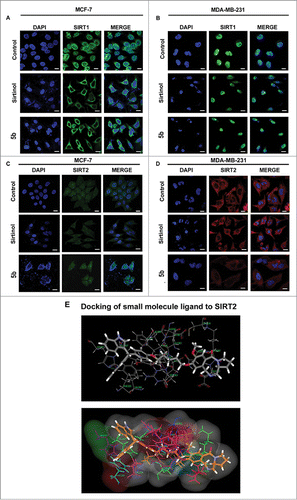
Bisindole-PBD conjugate (5b) as SIRT2 inhibitor
As the compound 5b inhibited SIRT-1 and SIRT-2 both at mRNA and protein level we investigated the binding modes of the 5b in the active sites of human SIRT2 by performing molecular docking studies using C DOCKER software. Dock score was used to estimate the ligand-binding energies. Apart from these, other input parameters for docking were also considered for evaluating the 5b docking efficacy with the protein. Different conformations were generated of which we focused on the conformation having maximal c docker interaction. In this study, 5b was screened for SIRT2 inhibitory role in order to find the receptor ligand binding orientation, affinity, free energies of the 5b against the selected targets. Therefore, re-scoring of best docked based on their interaction energies with respective protein active site residues was carried out using different scoring function. Hydrogen bond interaction plays crucial role in docking study and considered as major parameter. Here Ligand molecule has exhibited a strong Hydrogen bonding interaction with aminoacid His187 of SIRT2 ().
Bisindole-PBD conjugate (5b) targets downstream players of SIRT1 and SIRT2
Increased cancer cell death is associated with SIRT1 inhibition.Citation40 SIRTs are protein deacetylases that modulates target genes that includes histones as well as non-histones such as p53 and α tubulin. In general, it is well known that DNA damaging agents modulate expression of p53 protein as well as its post-translational modifications such as acetylation and phosphorylation and cause cellular apoptosis.Citation41 The stability and transcriptional activity of p53 depends upon the acetylation status at lysine residues. SIRT1 is known to destabilize p53 by deacetylating p53 at lysine 382 position.Citation42,43 We therefore examined the acetylation pattern of p53 after treatment with the compound 5b in MCF-7 and MDA-MB-231 cancer cell lines. Here Sirtinol, the known SIRT inhibitor was used as positive control and untreated cells as negative control. An increased acetylation at 2 distinct lysine residues (K382 and K373) in MCF-7 was observed. But in case of MDA-MB-231 there was decrease in the level of acetylated p53. Next we checked the status of cleaved caspase-3 as an indicator of cell death/apoptosis. There was activation of caspase-3, after treatment with 5b in MDA-MB-231 cells indicating apoptosis independent of p53 (Fig. S2).Citation26 Further we have also examined the phosphorylation of p53 (p53 ser 15 and p53 Ser 20) in 5b treated cells. Interestingly, MCF-7 cells showed increased phosphorylated forms of p53 at ser15 and ser20 positions. But in case of MDA-MB-231 cells, phosphorylated p53 levels were suppressed probably due to the presence of mutation in p53 in these cells (). These results indicate that compound 5b functions as transcriptional activators as well as stabilizers of p53 though further study is required to understand the modifications in mutant p53 in MDA-MB-231 cells. Next to confirm the effect of 5b on SIRT2, the level and expression of acetylated tubulin, a specific target for SIRT2 was examined.Citation44 There was a significant elevation in acetylated tubulin in 5b treated cells compared to untreated controls. The level of expression was higher than Sirtinol treated cells. A similar pattern of elevation in acetylated tubulin in 5b treated cells compared to untreated controls was observed from immunofluorescence study (). To rule out the involvement of HDAC-6 and to confirm the specific effect of compound as SIRT2 inhibitor by inducing acetylated tubulin in compound 5b treated cells we performed western blot analysis. Interestingly there was increase in the level of HDAC-6 protein after incubation with 5b () confirming induction of acetylated tubulin was due to inhibition of SIRT2 but not for HDAC-6. Tubulin polymerization assay also confirmed that there was no depolymerization of tubulin in cells treated with 5b at 4 μM concentration as shown in earlier study (Fig. S3).Citation29
Figure 6. Effect of 5b on post translational modifications of p53 and tubulin acetylation. (A, B) Proteins extracted from cells treated with indicated doses of Sirtinol, SAHA and 5b for 24 h were immunoblotted with antibody specific to acetylated tubulin, p53, acetylated p53(Lys373), acetylated p53(Lys382), phospho p53(Ser20), phospho p53(Ser15)and NF-kB. β-actin was taken as loading control. ‘*’ p < 0.05; bars with same symbols are dissimilar statistically. (C, D) Immunofluorescence studies with anti-acetylated tubulin of cells after treatment with 5b. Images were observed under confocal microscope (Olympus FV1000) and processed with flow view version 1.7c software program. Bar represents 10 µM scales. (E, F) Pretreated cells were subjected to protein gel blot to decipher the role of 5b upon HDAC6 expression. Relative density has been calculated taking β-actin as loading control. Bars represent mean + SD.
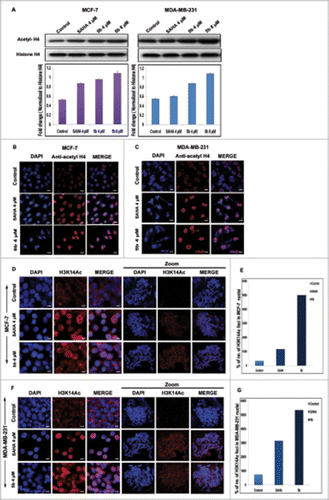
Bisindole-PBD conjugate leads to the acetylation of histones
Sirtuins are a family of histone deacetylases that functions in chromatin regulation. As the compound 5b has shown to affect SIRT1 and SIRT2, we investigated its role in chromatin regulation by directly modifying the histones. Earlier studies have shown that changes in the expression of SIRTs produce changes in the acetylation pattern of the histones globally.Citation45 Therefore the acetylation pattern of H4 was studied by incubating the cells with 5b/SAHA (HDAC inhibitor). There was elevated accumulation of acetylated-H4 both in MCF-7 and MDA-MB-231 cells at dose dependent manner. The level of accumulation was 1.5-fold at 4 μM concentration and 2-folds at 8 μM concentration in MDA-MB-231 cells. The level of accumulation in SAHA treated cells were lower compared to 5b treated ones. In case of MCF-7 cells, the increase in acetylated H4 was 1.5 to 2-fold higher at 4 and 8 μM concentrations respectively as compared to untreated control cells (). Here non-acetylated form of histone H4 served as loading control. To further confirm the efficacy of 5b, we performed immunofluorescence staining with anti-acetyl histone H4 (). This study revealed distinct and significant enrichment in acetyl histone H4 (histone acetylation) level in interphase nuclei in 5b treated cells. A significant enrichment of H3acK14 in the chromatin of metaphase spreads in compound treated cells was observed (), leading to the conclusion that the compound 5b functions as modifier of chromatin organization.
Figure 7. Effect of 5b on histone acetylation and methylation. (A) Treatment with 5b at 4, 8 μM concentrations for 24 h in MCF-7 and MDA-MB-231 cells significantly induced the level of acetylated histone H4 as compared to DMSO (0.1%) control. (B, C) Immunofluorescence studies with anti-acetyl histone H4 antibody on cells treated with 5b showed profound expression of acetylated histone H4. (D, F) Metaphase spreads isolated from MCF-7 and MDA-MB-231 cells treated with SAHA and 5b at 4 μM concentration for 24 h and control untreated cells hybridized with histone H3K14ac antibody. (E, G) Percentage of H3K14ac foci is shown. Error bar represents standard deviation from 3 different experiments. Bars with * are significantly different from all the other groups at p ≤ 0.01.
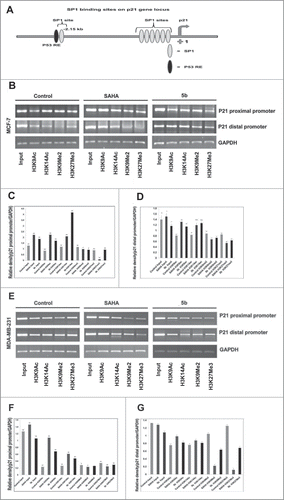
Epigenetic regulation of the p21WAF1 gene locus
Suppression of p21 is observed in the progression of various cancers. Among various epigenetic modifications histone acetylation and methylation forms an important epigenetic modification in regulating gene silencing associated with cancers. In our previous study we have shown that induction of apoptosis by the compound 5b was caused by an increase in Bax and p21 levels. Here in order to understand the cause of increase in the p21 levels after treatment with 5b, we have studied the differential methylation and acetylation of N-terminal histone tails on increased p21 transcription in breast cancer cells. To understand the relation between induction of p21 and chromatin organization with respect to acetylated and methylated histone H3 we performed chromatin immunoprecipitation studies (ChIP) at the p21 promoter. We found that the induction of p21 was independent of p53 status in case of MDA-MB-231 cells. So, we focused our study at the 2 sp1 sites at the distal (−2476 to −2175) and proximal promoter (−389 to +44) of p21 (). ChIP assay was performed against H3K9ac, H3K14ac, H3K9me2 and H3K27me3 at both the mentioned sites of p21 promoter. There was significant enrichment of H3K9ac and H3K14ac in SAHA and 5b treated MCF-7 cells. H3K14ac was more enriched in 5b treated cells as compared to SAHA treated. In case of MDA-MB-231, binding of H3K9ac, H3K14ac was significantly higher in the distal region of p21 promoter whereas not much change was observed with proximal promoter primer. A moderate change in methylation pattern was observed for H3K9me2 and H3K27me3 with p21 promoter (sp1 binding site) specific primers in MCF-7 cells treated with 5b (). On the total the results indicated that bisindole PBD conjugate (5b) affected both histone acetylation and methylation pattern at p21 promoter.
Figure 8. Effect of 5b on histone modifications and sp1 at p21 promoter in MCF-7 and MDA-MB-231 cells. (A) Schematic diagram of the human p21WAF1 promoter illustrating regulatory factor binding sites;−2260 (p53), −2080 (sp1-distal) and −100 (sp1 proximal). Arrow indicates transcriptional start site at +1. (B) Two different binding sites (distal and proximal) of sp1 in the p21 promoter were considered for ChIP analysis after treating the cells with SAHA and 5b at 4 μM concentration for 24 h. H3K9ac, H3K14ac, H3K9Me2 and H3K27me3 antibodies were used for the study. (B-G) Representation of gel pictures of chromatin DNA amplified with distal and proximal promoter specific primers and their respective histograms. Each experiment was performed thrice from which standard deviation derived and error bars were plotted. Bars with * and ** are significantly different from all the other groups at p ≤ 0.05 and p ≤ 0.01 respectively; bars with ˆ superscript are indifferent from each other in Oneway-ANOVA, posthoc-LSD in MCF-7. In MDA-MB-231 bars with * and ** are significantly different from all the other groups at p ≤ 0.05 and p ≤ 0.01 respectively in Oneway-ANOVA, posthoc-LSD.
Reversal of p21 promoter DNA methylation by 5b in Breast cancer cells
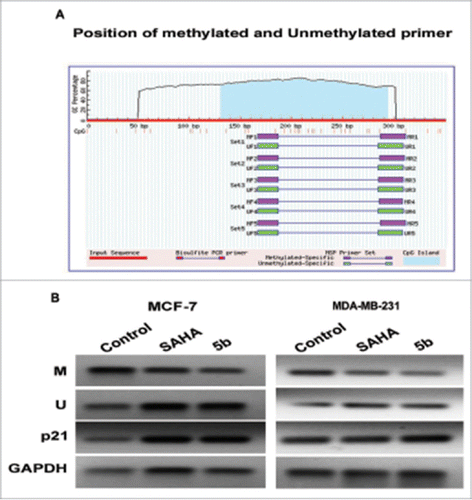
We further studied the methylation pattern at p21 promoter with and without compound treatment by performing methylation specific PCR. Here the breast cancer cells were incubated with SAHA/5b at 4 µM concentration for 24 h followed by PCR studies using methylation specific primers (primer sequences are provided in the methods/supplement). There was considerable decrease in the methylation of p21 promoter in both the cell lines (). Hence it can be concluded that p21 down regulation may be associated with hypermethylation, which can be reversed by 5b. This study might support the fact that certain HDAC inhibitors can also affect the DNA methylation such as Azacytidine. Primers used (Methylated M, Unmethylated U) are listed in supplementary Table S1.
Figure 9. Methylation analysis at the p21 promoter by MSP in MCF-7 and MDA-MB-231 cells. (A) Genomic sequence of the p21 promoter (www.urogene.org/methprimer) revealed the presence of a high content of GC in CpG islands (blue). Methylated and unmethylated primers for p21 were desingned from Methprimer software. (B) Expression of the methylated and unmethylated DNA estimated by methylated and unmethylated primers obtainded from methprimer software. M primer anneal only to methylated sequences and U primer anneal unmethylated sequences. We took primer set 1 from the software output.
Discussion
Anti-cancer properties of bisindoles have already been established in most of the cell lines.Citation46 In this present study, it has been shown that bisindole conjugate 5b exhibited profound control over growth factor signaling pathway mediated by PI3K/AKT/mTOR and modulate the balance between survival signals as well as apoptotic signals via SIRT and p53 axis in breast cancer cell- lines. Till date not many SIRT inhibitors have been studied in metabolic disorders, nor their regulation of balance between growth factor signaling and p53 mediated apoptotic pathway in breast cancer cells is understood.
Cancer is caused due to abnormalities in cell cycle progression. Hallmark of tumorigenesis is associated with abnormal gene expression. The activities of cyclins and cdks regulate the expression of mammalian cell cycle progression. The fate of progression toward cell cycle or induction of cancer of a cell depends on the correlation between the suppression and activation of cyclin and cdk inhibitors.Citation47 Regulation of aberrantly expressed gene as a consequence of alteration of chromatin state provides us a tool to target specific therapeutics that in turn enhances the importance of HDAC inhibitors in cancer treatment.Citation48 SIRT inhibitors perform various cellular functions such as induction of apoptosis and cell cycle arrest.Citation49 Our previous study showed DNA damage induced apoptosis by 5b. Here we have further shown that compound 5b at 4 μM concentration induced the expression of proapoptotic members of Bcl-2 family that are p53 dependent such as Bax, Noxa and Puma. There was significant upregulation of these factors upon treatment with 5b. Both p53 dependent and independent mechanisms were observed in MCF-7 (wt p53) and MDA-MB-231(mut p53) cells simultaneously. In MCF-7 cells p53 mediated transcriptional activation of proapototic genes Bax, Noxa and Puma was observed. In addition 5b induced the level of phosphorylated p53 at ser15 and ser20 as well as acetylated forms of p53 resulted in transcriptional activation and stability thus revealing the p53 activating nature of the compound in MCF-7 cells. But in case of MDA-MB-231 cells, induction of proapoptotic genes was found to be independent of mutant p53 expression. The phosphorylation as well as acetylation status of p53 in MDA-MB-231 cells was downregulated. The suppressed level of mutant p53 did not interfere with the cell death due to activation of cleaved caspase - 3. Aberrantly expressed pathways in cancerous condition lead to the development of new therapeutic approaches. Different genetic and epigenetic modifications stimulate the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway, that induce cancer development.Citation50 This PI3K/AKT/mTOR pathway regulates the growth, proliferation, cellular differentiation, and survival mechanism. In PI3K pathway Akt, serine / threonine kinase, acts as central mediator. This Akt increases the protein synthesis and growth of cancer cell. This process in turn activates mTOR (mammalian target of rapamycin) which is considered to be the key switch of different cell growth regulatory factors.Citation51 mTOR influences PI3K signaling via a feedback loop of S6K-IRS1. Influence on PI3K occurs through mTORC2 directed Akt-Ser473.Citation52,53 Targeting this prosurvival pathway along with apoptosis inducing property of 5b, may put more insight toward targeted therapy for breast cancer.
Earlier study by Kamal et.al on HDAC inhibitory role of 5b in breast cancer cells has shown it to be classic Class I HDAC inhibitor.Citation29 We observed more potent inhibitory action of 5b on SIRT gene expression. As member of histone deacetylation the SIRT protein is found to regulate the histone proteins (i.e., H3K9 Ac) as well non-histone proteins such as p53. Immunofluorescene study showed down regulation of SIRT1 and SIRT2 expressions in MCF-7 and MDA-MB-231 cells. In this study, for the first time we have deciphered the SIRT inhibitory role of bisindole-PBD (5b), its mechanism of action and cellular targets in MCF-7 and MDA-MB-231 cells. Docking study also further supported the interaction of 5b with human SIRT2 protein in silico. In vitro studies on acetylation of tubulin also support the results obtained from docking studies. This was further strengthened by lack of inhibition on HDAC-6 by 5b. Recent studies on neuroblastoma cells have shown that many compounds that inhibit HDAC-6 also effect directly by increasing the status of tubulin acetylation. Taken together, these findings confirm that the cytotoxicity of the compound 5b occurs through the inhibition of SIRT1 and SIRT2.
Epigenetic alterations are prevalent in breast cancers. Silencing of tumor suppressor gene by promoter hypermethylation is a common phenomenon in cancer progression. The agents targeting these alterations have got immense importance in medical biology. Rational combination of epigenetic modifiers with existing drug for cancer treatment may provide easier way to treat cancer. As an extension of our experiments, we explored the possible role of 5b as a demethylating agent in context to reactivate p21 gene expression. It is well known that DNA methylation at the promoter region of a gene leads to silencing /transcriptional repression.Citation54 As a putative tumor suppressor gene, mutation in p21 gene has been implicated in different cancers. Epigenetic modifications at the p21 promoter render complete suppression of its expression and thereby induce breast cancer development. By performing methylation-specific PCR (MSP), we differentiated between methylated and unmethylated cytosine upon sodium bisulfite treatment of DNA. 5b treated cells showed decrease in methylation level in both MCF-7 and MDA-MB-231 cells. Reversal of epigenetic alteration and reactivating silenced gene expression by small molecule 5b may provide new strategy to treat breast cancer.
In recent years, HDAC inhibitors and SIRT inhibitors have got immense importance as anti-cancer drugs as they can modulate epigenetic state within a cell and reactivate the silenced state of a gene by regulating cellular differentiation, cell growth and apoptosis. Here in this study we clearly established compound 5b as a typical SIRT inhibitor. There is a well-known concept of apoptosis inducing property by SIRT inhibitors that may be p53 dependent or independent.Citation26 Sirtuins are NAD-dependent deacetylases commonly associated with the regulation of various vital biological functions. SIRTs are known to target and regulate the activity of important tumor suppressor proteins such as p53, p73, E2F1, and FOXO3a.Citation55 p53 deacetylation by SIRT1 indicates that SIRT1 functions as an oncogene by inhibiting tumor suppressor activity of p53.Citation56 However some studies also suggest that tumor suppressor activity is also shown by SIRT1. SIRT1 has the ability to deacetylase histones H1, H3 and H4 Citation57,58 as well as non-histone substrates.Citation59
p53, the known tumor suppressor gene is often mutated in human solid tumors.Citation60 Regulation of p53 expression is mediated by both transcriptionally and post-transcriptionally. Post-translational modification of p53 such as acetylation initiates the transactivation of many specific genes, induces apoptotic cell death and regulates cell cycle arrest. SIRT1 is known to target p53, responsible for its deacetylation and attenuation of p53-mediated transcriptional activity. Hence SIRT-p53 axis is considered as a well established and targetable pathway to regulate tumor suppressive activity of p53. Different studies have shown that overexpression of wild-type SIRT1 is linked to the removal or reduction of p53 acetylation. On the contrary, catalytically in active SIRT1 overexpression did not seem to cause deacetylation. These findings suggest that SIRT1 is a key regulator of p53 transcriptional activity.Citation56 So, SIRT1 inhibition leads to transactivation with induced p53 acetylation resulting apoptotic cell death.
Various small molecules and endogenous factors have been found to target and regulate SIRT1-p53 pathway. These molecules work as SIRT activators (AROS) as well as SIRT inhibitors such as Sirtinol, Splitomicin, Tenovin etc. A well known Indole, EX-527 known to regulate and inhibit SIRT1. Similarly, sirtinol and salermide are known as dual SIRT1/2 inhibitors which have the ability to enhance p53 acetylation and stabilization in MCF-7 cells.Citation26 Recent studies by Gildon Choi et al., Citation2013 has shown the SIRT1/2 inhibitory role of Taxoflavins and its role in increased p53 acetylation.Citation61 We have also investigated the level of acetyl histone H4, which was elevated upon treatment with 5b and showed even higher expression when compared to SAHA in a dose dependent manner. Moreover immunocytochemical studies with acetyl histone H4 also showed hyperacetylation in the nucleus of breast cancer cells after treatment with SAHA and 5b. Similar to a typical HDAC inhibitor, bisindole-PBD enhances the p21WAF1 transcription. Compound 5b also enhanced the accumulation of acetylated histones at the SP1 binding site in p21 promoter. Hyperacetylation of histones after treatment with 5b in concert with reduced expression of methyaltion in SP1 sequence may recognize non-histone proteins for transcriptional activation of p21. Histone acetylation and methylation modulation at different sites in p21 promoter are necessary for p21 transcriptional gene regulation. Methylation at the lysine residue of histone (H3K4) is found to be associated with active genes.Citation62 But methylation at lysine residue H3K9 is linked with heterochromatin formation.Citation63 So, from this observation it can be summarized that chromatin modification induced by compound 5b such as methylation and acetylation which are the hallmarks of epigenetic mechanism in the histone tails, controls different transcription factors from binding to the gene.
SIRT1/2 is considered as the potent emerging therapeutic targets for cancer. Identification of SIRT inhibitors will show a new avenue in the development of anticancer agent. As discussed earlier, targeting of both SIRT1 and SIRT2 by SIRT inhibitors caused induction of p53 acetylation and effective cell death.Citation26 Studies by Solomon et al., Citation2006 have shown the SIRT inhibitory role of EX-527 and its efficacy in increasing the hypothalamic acetyl-p53 levels by inhibiting SIRT1 activity of hypothalamus.Citation64 Another class of compounds, Tenovins (Tenovin-1, Tenovin-6) were found to show inhibitory role on SIRT deacetylating activities along with enhanced p53 activity that decreased tumor growth in xenografts at low dose of drug. Thus both Tenovin-1 (water insoluble) and Tenovin-6 (water soluble) had the ability to block SIRT1 mediated p53 deacetylation. Further studies on Inauhzin, a SIRT inhibitor has shown that it has the efficacy in reducing xenograft tumor size in a p53-dependent manner.Citation65 Various literatures have strongly indicated that Indoles are the key class of anti cancer agents which can be useful for clinical use in specifically in activating SIRT1. Thus employing bisindole-PBD in xenograft studies (in vivo) would provide more therapeutic benefits against breast cancer. Hence compound 5b can be used alone or in combination with other HDAC/SIRT inhibitors and demethylating agent for the better prospect of breast cancer treatment.
Materials and Methods
Cell lines
Human breast cancer cell lines MDA-MB-231(p53 mutant) and MCF-7(p53 wild type) were purchased from American Type Culture collection and routinely maintained in RPMI-1640 and Dulbecco's Modified Eagle's Medium (DMEM) (Sigma) respectively, supplemented with 10% fetal bovine serum and 100U/ml Penicillin and 100 mg/ml streptomycin sulfate (Invitrogen) at 37 °C with 5% CO2.
Reagents
Vorinostat (suberoylanilide hydroxamic acid, cat# SML0061), Sirtinol (cat# S7942), Q-VD-OPH(cat# SML0063) and DMSO(cat# D2650) were obtained from Sigma. TRIzol® Reagent (cat#15596-018) was purchased from Life Technologies. Compound 5b was obtained from Dr. Ahmed Kamal'S Lab CSIR-IICT; India.Citation29 A 10 mM stock was prepared in DMSO and stored at −20°C. Anti-Histone H4 (1:1000, cat# 07-108), anti-acetyl Histone H4 (1:1000, cat# 06-598), anti-SIRT1 (1:1000,cat# 04-1557), anti-acetyl-Histone H3 (Lys9) (cat# 07-352), anti-Dimethyl Histone H3 (Lys9) Antibody, clone CMA307 (cat# 05-1249), anti-trimethyl-Histone H3 (Lys27) Antibody(cat# 07-449), anti-HDAC6 (1:500, cat# 07-732) and anti-SIRT2 (1:1000, cat# 04-1124), anti- ac-p53(Lys373)(1:1000, cat# 06-916), Anti-PI3 Kinase Antibody (1:500, cat# 04-399) and anti- ac-p53(Lys382)(1:1000, 04-1146) antibodies were purchased from Merck Millipore. Anti -mTOR(1:1000, 2972), anti-Akt(1:1000, cat# 4691), anti-Phospho-Akt (Ser473) (1:500, cat# 4060), anti-phospho p53(ser15) (1:1000, cat# 9284) and anti-phospho p53(ser20) (1:1000, cat# 9287)were purchased from Cell Signaling technology. Anti- acetylated tubulin (1:2000, cat# T7451) was purchased from Sigma. Anti-NF-kB (1: 500, cat# 250769) and anti-p53 (1:500, cat# 251786) antibody were bought from Abbiotec. Loading control β-actin (1:1000, cat# ab8226) was obtained from Abcam. Rabbit and Mouse polyclonal secondary antibodies were purchased from Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA).
Clonogenic survival assays
Cells were seeded into 6 well plate at a density of 500 cells per well. Thereafter they were treated vorinostat (SAHA) or 5b at 4 μM concentration for 24 hours followed by 2 washes with PBS. The cells were then allowed to grow in fresh media for 10 d in a humidified incubator at 37°C with 5% CO2. Colonies were washed in PBS then fixed with methanol. Fixed colonies were stained with crystal violet (0.5% crystal violet) and counted. Every experiment was performed in triplicate and from individual experiment colony pictures were taken in Gel Doc XR system.
Wound - Healing assay in vitro
Both MCF-7 and MDA-MB-231 cells were plated in 6-well plate at a density of 1 × 105. After the cells have reached almost 80% confluency, monolayer of cells was scratched with 200μL pipette tip, washed with PBS and fresh 2 ml media was added. There after it was treated with SAHA or 5b at desired concentration and incubated for 48 h to see the wound healing process. Images were captured using OLYMPUS CKX41 inverted microscope in 4X magnification.
Confocal microscopy
Cells were seeded at a density of 2.5 × 105 on cover slips. After 24 h, cells were incubated with SAHA 4 μM, Sirtinol 50 μM and 5b 4 μM for 24 h. After the desired incubation time, the media was removed and cells were washed with PBS. Cells were fixed in 4% paraformaldehyde for 20 minutes followed by incubation with 0.2% triton -X 100 in PBS for 5 min. Cells were washed twice with PBS and blocked with 1% BSA in PBS for 1 hour. Cells were incubated with anti-acetyl tubulin, anti- acetyl Histone H4, anti-SIRT1 and anti-SIRT2 antibodies at a dilution of 1:100 for 2 h in PBST at room temperature, followed by 3 washes of 10 min each in PBS. For detection the cells were incubated in dark with FITC/cy3-conjugated anti-rabbit secondary antibody (1:50 dilution) (Jackson Immuno Research Laboratories Inc., Pennsylvania, USA) at room temperature for 1 h. After washes for 3 times (10 min each in PBS), the coverslips were mounted with DAPI and observed under confocal microscope (Olympus FV1000). Images were processed with flow view version 1.7c software program at 60X objective.
Immunostaining of metaphase chromosomes
Accumulation of modified histones on the chromosomal arms was observed by indirect Immuno-fluorescence. Metaphase cell spreads on the slides were incubated for 1 h at 37°C in a humidifier chamber with serial dilutions with either primary Lys-14 acetyl H3 (1:75; # 06-911) anti-sera followed by wash in KCM (120 mM KCl, 20 mM NaCl, 10 mM Tris-Cl− pH 8.0, 0.5 M EDTA, 0.1% Triton). Cy3- conjugated, affinity-purified, donkey anti-rabbit IgG antibody (Jackson Immuno-Research) diluted 1:100 in KCM, was incubated with the mixture for 30 min at room temperature. Chromosomes were further washed with KCM and fixed in 4% formaldehyde for 10 min at room temperature. After a wash in sterile water, chromosomes were counterstained with DAPI, mounted with anti-fade media (Vectashield) and viewed under confocal microscope (Olympus FV1000). Images were processed with flow view version 1.7c software program.
Molecular modeling
The high resolution X-ray crystal structure of human SIR2 is retrieved and the pdb code is 3ZGV with a resolution of 2.27 A° and the method of incorporation is X-ray diffraction method. The ligand and crystallographic water molecules are removed from the protein; and the chemistry of the protein is corrected for missing hydrogen. Crystallographic disorders and unfilled valence atoms are connected using alternate conformations and valence monitor options. Following the above steps of preparation, the protein is subjected to energy minimization using the CHARMM Force field. Ligand structure of 5b was sketched using ACD/ChemSketch (12.0) software and saved in Mol2 format. The saved ligand compounds are later imported in to DS and hydrogen bonds are added and the energy is minimized using CHARMM force field. The individual compounds are finally saved in mol file format for further binding studies. The Dock Ligands (CDOCKER) protocol is an implementation of the CDOCKER algorithm. It allows you to run a refinement docking of any number of ligands with a single protein receptor. CDOCKER is a grid-based molecular docking method that employs CHARMM. The receptor is held rigid while the ligands are allowed to flex during the refinement. For pre-docked ligands, prior knowledge of the binding site is not required. It is possible, however, to specify the ligand placement in the active site using a binding site sphere. Random ligand conformations are generated from the initial ligand structure through high temperature molecular dynamics, followed by random rotations. The random conformations are refined by grid-based (GRID 1) simulated annealing and a final grid-based or full forcefield minimization.
Protein extraction and Immunoblot analysis
Total cell lysates were obtained by lysing the cells with ice cold RIPA buffer (Sigma R 0278). Protease inhibitor (Roche) was added to RIPA buffer prior to lysis. Lysates were centrifuged at 12,000 rpm for 15 min at 4 °C. The protein obtained from supernatant was quantified by Bradford method (BIO-RAD) using Multimode Varioskan instrument (Thermo-Fischer Scientifics). Proteins were separated by SDS-PAGE and transferred onto PVDF membrane. Blots were developed in BioRad Molecular Imager Chemidoc XRS with Image Lab software. Intensity of bands were observed and analyzed with Image J software.
Gene expression analysis by Semi-quantitative reverse transcription PCR (RT-PCR)
Total RNA from compound (5b) treated, SAHA treated and untreated cells was extracted using Trizol reagent obtained from Invitrogen (Life Technologies) and reverse transcribed into cDNA using RNA to cDNA EcoDry™ Premix kit (Double Primed, Clontech USA). PCR was carried out using specific primers in Eppendorf Mastercycler Gradient PCR machine. PCR products were electrophoresed on agarose gel (1.2%) and visualized under U.V. light. Respective band signal was measured by Quantity one version 4.1.1 software (CA, USA). Primer sets used for PCR amplification are listed in supplementary Table S1.
Gene expression analysis by Real time PCR
Total cellular RNA was isolated by Trizol and RNase-Free Turbo DNase treatment was carried out to remove DNA contaminants. RNA was purified by RNeasy Mini Kit (Qiagen, Germany). Two micrograms of RNA were used for first strand cDNA synthesis using SuperScriptTM (Invitrogen, USA). To quantify the level of mRNA, real-time RT-PCR was conducted with a TaqMan Universal Master Mix (Applied Biosystems) using reverse transcribed cDNA as a template, in triplicate and the amplification was performed using an ABI 7900. Conditions used were 50°C for 2 min and 95°C for 10 min, followed by 40 cycles (95°C for 15 s and 60°C for 1 min). GAPDH levels were used for normalization. Primers for amplification were purchased from Eurofins. Primer list has been provided in supplementary Table S1.
DNA extraction and methylation-specific polymerase chain reaction (MSP)
Genomic DNA from compound (5b) treated, SAHA treated and untreated cells was extracted using NucleoSpin® Tissue obtained from MACHEREY-NAGEL. Bisulfite conversion of DNA was carried out by EpiTect Bisulfite Kits (Qiagen). PCR was carried out using specific primers in Eppendorf Mastercycler Gradient PCR machine with EpiTect MSP Kit (Qiagen). PCR products were electrophoresed on agarose gel (1.2%) and visualized under U.V. light. Respective band signal was measured by Quantity one version 4.1.1 software (CA, USA). Primer sets used for PCR amplification are listed in supplementary Table S1.
ChIP assay
Chromatin immunoprecipitation assay was conducted as per company protocol by EZ-ChIP (Millipore). The optimal reaction conditions for PCR were determined for each primer pair. Parameters were denaturation at 95°C for 1 min and annealing at 60°C for 1 min, followed by elongation at 72°C for 1 min. PCR products were analyzed by 2.5% agarose/ethidium bromide gel electrophoresis. Different primer pairs used for p21WAF1 promoter ChIP analysis are provided in supplementary Table S1.
Statistical analysis
Densitometry analysis was done by Image J software. The region (area) of interest of the gel picture was selected and the densitometry value was estimated by Image J. Relative densitometry values were calculated considering β-actin (in case of protein gel blot) and GAPDH (in PCR) as an internal control and were plotted in Microsoft Office Excel. All variables were tested in 3 independent experiments. The results were reported as mean ± SD. Statistics performed by SPSS 17.0 One-Way ANOVA, Bonferroni.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Data
Download MS Word (16.2 KB)Acknowledgments
PS thanks CSIR for providing the SRF (ID 8169). IB thanks CSIR for providing her post doctoral fellowship. All the authors thank P. Devender for maintaining the cell culture facility.
Funding
The authors acknowledge CSIR 12th FYP (CSC0111) for the financial support.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Rivera E, Gomez H. Chemotherapy resistance in metastatic breast cancer: the evolving role of ixabepilone. Breast Cancer Res 2010; 12 Suppl 2:S2; http://dx.doi.org/10.1186/bcr2573
- Yardley DA. Drug resistance and the role of combination chemotherapy in improving patient outcomes. Int J Breast Cancer 2013:137414; PMID:23864953; http://dx.doi.org/10.1155/2013/137414
- van Agthoven T, Sieuwerts AM, Meijer-van Gelder ME, Look MP, Smid M, Veldscholte J, Sleijfer S, Foekens JA, Dorssers LC. Relevance of breast cancer antiestrogen resistance genes in human breast cancer progression and tamoxifen resistance. J Clin Oncol 2009; 1; 27(4):542-9; PMID:19075277; http://dx.doi.org/10.1200/JCO.2008.17.1462
- X Sui, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q, Wang X, He C, Pan H. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death and Disease 2013; Oct 10; 4:e838; PMID:24113172; http://dx.doi.org/10.1038/cddis.2013.350
- Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets 2008; 8(3):187-98; PMID:18473732; http://dx.doi.org/10.2174/156800908784293659
- German S, Hafiz Muhammad Aslam, Shafaq Saleem, Aisha Raees, Alvi AA. Carcinogenesis of PIK3CA. Hered Cancer Clin Pract 2013; 11:5; PMID:23768168; http://dx.doi.org/10.1186/1897-4287-11-5
- Astle MV, Hannan KM, Ng PY, Lee RS, George AJ, Hsu AK, Haupt Y, Hannan RD, Pearson RB. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: implications for targeting mTOR during malignancy. Oncogene 2012; 31(15):1949-1962; PMID:21909130; http://dx.doi.org/10.1038/onc.2011.394
- Saretzki G. Cellular senescence in the development and treatment of cancer. Curr Pharm Des 2010 Jan; 16(1):79-100; PMID:20214620; http://dx.doi.org/10.2174/138161210789941874
- Lane DP. Cancer. p53, guardian of the genome. Nature 1992; 2; 358(6381):15-6; PMID:1614522; http://dx.doi.org/10.1038/358015a0
- Gottlieb TM, Oren M. p53 in growth control and neoplasia. Biochim Biophys Acta 1996; 1287:77-102; PMID:8672531; http://dx.doi.org/10.1016/0304-419X(95)00019-C
- Ko LJ, Prives C. p53: puzzle and paradigm. Genes Dev 1996; 10:1054-1072; PMID:8654922; http://dx.doi.org/10.1101/gad.10.9.1054
- Fan S, Smith ML, Rivet DJ 2nd, Duba D, Zhan Q, Kohn KW, Fornace AJ Jr, O'Connor PM. Disruption of p53 function sensitizes breast cancer MCF-7 cells to cisplatin and pentoxifylline. Cancer Res 1995; 55:1649-1654; PMID:7712469
- Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell 2002; 108(2):153-64; PMID:11832206; http://dx.doi.org/10.1016/S0092-8674(02)00625-6
- Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol 2003; 15(2):164-71; PMID:12648672; http://dx.doi.org/10.1016/S0955-0674(03)00003-6
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998; 281:1677-1679; PMID:9733515; http://dx.doi.org/10.1126/science.281.5383.1677
- Amano T, Nakamizo A, Mishra SK, Gumin J, Shinojima N, Sawaya R, Lang FF. Simultaneous phosphorylation of p53 at serine 15 and 20 induces apoptosis in human glioma cells by increasing expression of pro-apoptotic genes. J Neurooncol 2009; 92(3):357-71; PMID:19357962; http://dx.doi.org/10.1007/s11060-009-9844-1
- Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci USA 2004; Feb 24; 101(8):2259-64; PMID:14982997; http://dx.doi.org/10.1073/pnas.0308762101
- Sabnis GJ, Goloubeva O, Chumsri S, Nguyen N, Sukumar S, Brodie AM. Functional activation of the estrogen receptor-alpha and aromatase by the HDAC inhibitor entinostat sensitizes ER-negative tumors to letrozole. Cancer Res 2011; 71(5):1893-903; PMID:21245100; http://dx.doi.org/10.1158/0008-5472.CAN-10-2458
- Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell 2007; 128(4):721-33; PMID:17320509; http://dx.doi.org/10.1016/j.cell.2007.01.030
- Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 2005; 37(4):391-400; PMID:15765097; http://dx.doi.org/10.1038/ng1531
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of Histone deacetylase inhibitors. Nat Rev Drug Discov 2006; 5(9):769-84; PMID:16955068; http://dx.doi.org/10.1038/nrd2133
- Woan KV, Sahakian E, Sotomayor EM, Seto E, Villagra A. Modulation of antigen presenting cells by HDAC inhibitors: implications in autoimmunity and cancer. Immunol Cell Biol 2012; Jan; 90(1):55-65; PMID:22105512; http://dx.doi.org/10.1038/icb.2011.96
- Kai Li, Jianyuan Luo. The role of SIRT1 in tumorigenesis. N Am J Med Sci 2011; 4(2):104-106; PMID:22180829; http://dx.doi.org/10.7156/v4i2p104
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001;107(2):137-148; PMID:11672522; http://dx.doi.org/10.1016/S0092-8674(01)00524-4
- Botta G, De Santis LP, Saladino R. Current advances in the synthesis and antitumoral activity of SIRT1-2 inhibitors by modulation of p53 and pro-apoptotic proteins. Curr Med Chem 2012; 19(34):5871-84; PMID:22998567; http://dx.doi.org/10.2174/092986712804143303
- Peck B, Chen CY, Ho KK, Di Fruscia P, Myatt SS, Coombes RC, Fuchter MJ, Hsiao CD, Lam EW. SIRT inhibitors induce cell death and p53 acetylation through targeting SIRT1 and SIRT2. Mol Cancer Ther 2010; Apr; 9(4):844-55; PMID:20371709; http://dx.doi.org/10.1158/1535-7163.MCT-09-0971
- Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128(4):683-92; PMID:17320506; http://dx.doi.org/10.1016/j.cell.2007.01.029
- Plumb JA, Strathdee G, Sludden J, Kaye SB, Brown R. Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res 2000; 60(21):6039-44; PMID:11085525
- Kamal A, Srikanth YV, Ramaiah MJ, Khan MN, Kashi Reddy M, Ashraf M, Lavanya A, Pushpavalli SN, Pal-Bhadra M. Synthesis, anticancer activity and apoptosis inducing ability of bisindole linked pyrrolo[2,1-c][1,4]benzodiazepine conjugates. Bioorg Med Chem Lett 2012; 22:571-8; PMID:22104151; http://dx.doi.org/10.1016/j.bmcl.2011.10.080
- Sarma P, Ramaiah MJ, Kamal A, Bhadra U, Bhadra MP. A novel bisindole-PBD conjugate causes DNA damage induced apoptosis via inhibition of DNA repair pathway. Cancer Biol Ther 2014; 15(10):1320-32; PMID:25010292; http://dx.doi.org/10.4161/cbt.29705
- Fröjdö S, Durand C, Molin L, Carey AL, El-Osta A, Kingwell BA, Febbraio MA, Solari F, Vidal H, Pirola L. Phosphoinositide 3-kinase as a novel functional target for the regulation of the insulin signaling pathway by SIRT1. Mol Cell Endocrinol 2011; Mar 30; 335(2):166-76; PMID:21241768; http://dx.doi.org/10.1016/j.mce.2011.01.008
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; Nov 16; 408(6810):307-10; PMID:11099028; http://dx.doi.org/10.1038/35042675
- Igata E, Inoue T, Ohtani-Fujita N, Sowa Y, Tsujimoto Y, Sakai T. Molecular cloning and functional analysis of the murine bax gene promoter. Gene 1999; 238(2):407-15; PMID:10570968; http://dx.doi.org/10.1016/S0378-1119(99)00348-0
- Bhuvanesh S, Reddy PG, Goberdhan A, Walsh C, Dao S, Ngai I, Chou TC, O-Charoenrat P, Levine AJ, Rao PH, et al. p53 regulates cell survival by inhibiting PIK3CA in squamous cell carcinomas. Genes Dev 2002; 16(8):984-993; PMID:11959846; http://dx.doi.org/10.1101/gad.973602
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci 2005; 102:8204-8209; PMID:15928081; http://dx.doi.org/10.1073/pnas.0502857102
- Brünner-Kubath C, Shabbir W, Saferding V, Wagner R, Singer CF, Valent P, Berger W, Marian B, Zielinski CC, Grusch M, et al. The PI3 kinase/mTOR blocker NVPBEZ235 overrides resistance against irreversible ErbB inhibitors in breast cancer cells. Breast Cancer Res Treat 2011; 129(2):387-400; PMID:21046231; http://dx.doi.org/10.1007/s10549-010-1232-1
- McAuliffe PF, Meric-Bernstam F, Mills GB, Gonzalez-Angulo AM. Deciphering the role of PI3K/Akt/mTOR pathway in breast cancer biology and pathogenesis. Clin Breast Cancer 2010; 10 Suppl. 3:S59-65; PMID:21115423; http://dx.doi.org/10.3816/CBC.2010.s.013
- Trapp J, Jochum A, Meier R, Saunders L, Marshall B, Kunick C, Verdin E, Goekjian P, Sippl W, Jung M, et al. Adenosine mimetics as inhibitors of NAD+-dependent histone deacetylases, from kinase to sirtuin inhibition. J Med Chem 2006; 49(25):7307-16; PMID:17149860; http://dx.doi.org/10.1021/jm060118b
- Jin Q, Yan T, Ge X, Sun C, Shi X, Zhai Q. Cytoplasm-localized SIRT1 enhances apoptosis. J Cell Physiol 2007; 213(1):88-97; PMID:17516504; http://dx.doi.org/10.1177/1947601912475079
- Zhenghong L, Deyu F. The roles of SIRT1 in cancer. Genes Cancer 2013; 4(3-4):97-104; PMID:24020000; http://dx.doi.org/10.1177/1947601912475079
- Deng CX. SIRT1, is it a tumor promoter or tumor suppressor? Int J Biol Sci 2009; 5(2):147-52; PMID:19173036; http://dx.doi.org/10.7150/ijbs.5.147
- Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, Kouzarides T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J 2002; 21:2383-2396; PMID:12006491; http://dx.doi.org/10.1093/emboj/21.10.2383
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001; 107(2):137-48; PMID:11672522; http://dx.doi.org/10.1016/S0092-8674(01)00524-4
- North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell 2003; 11:437-44; PMID:12620231; http://dx.doi.org/10.1016/S1097-2765(03)00038-8
- Brian JN, Eric V. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol 2004; 5(5):224; PMID:15128440; http://dx.doi.org/10.1186/gb-2004-5-5-224
- Andreani A, Burnelli S, Granaiola M, Leoni A, Locatelli A, Morigi R, Rambaldi M, Varoli L, Landi L, Prata C, Berridge MV, Grasso C Fiebig HH, Kelter G, Burger AM, Kunkel MW. Antitumor activity of bis-indole derivatives. J Med Chem 2008; 51(15):4563-70; PMID:18598018; http://dx.doi.org/10.1021/jm800194k
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev 1995; 9(10):1149-63; PMID:7758941; http://dx.doi.org/10.1101/gad.9.10.1149
- Pandolfi PP. Transcription therapy for cancer. Oncogene 2001; 20(24):3116-27; PMID:11420728; http://dx.doi.org/10.1038/sj.onc.1204299
- Minakshi N, Ahmad N, Wood GS. SIRT1 is upregulated in cutaneous T-cell lymphoma, and its inhibition induces growth arrest and apoptosis. Cell Cycle 2014; 13 (4); PMID:24343700; http://dx.doi.org/10.4161/cc.27523
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov 2005; 4(12):988-1004; PMID:16341064; http://dx.doi.org/10.1038/nrd1902
- Chiang GG and Abraham RT. Targeting the mTOR signaling network in cancer. J Clin Oncol 2009; 27(13):2278-87; PMID:17905659; http://dx.doi.org/10.1200/JCO.2008.20.0766
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307:1098-1101; PMID:15718470; http://dx.doi.org/10.1126/science.1106148
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell 2007; 12:9-22; PMID:17613433; http://dx.doi.org/10.1016/j.ccr.2007.05.008
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3(6):415-28; PMID:12042769; http://dx.doi.org/10.1038/nrg816
- Olmos Y, Brosens JJ, Lam EW. Interplay between SIRT proteins and tumour suppressor transcription factors in chemotherapeutic resistance of cancer. Drug Resist Updat 2011 Feb; 14 (1):35-44; PMID:21195657; http://dx.doi.org/10.1016/j.drup.2010.12.001
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2 (SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001 Oct 19; 107 (2):149-59; PMID:11672523; http://dx.doi.org/10.3410/f.1001553.2395457
- Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine56. Nature 2009 May 7; 459 (7243):113-7; PMID:19270680; http://dx.doi.org/10.1038/nature07861
- Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell 2004 Oct 8; 16 (1):93-105; PMID:15469825; http://dx.doi.org/10.1016/j.molcel.2004.08.031
- Lavu S, Boss O, Elliott PJ, Lambert PD. Sirtuins - novel therapeutic targets to treat age associated diseases. Nat Rev Drug Discov 2008 Oct; 7(10):841-53; PMID:18827827; http://dx.doi.org/10.1038/nrd2665
- Lain S, Lane D. Improving cancer therapy by non-genotoxic activation of p53. Eur J Cancer 2003 May; 39(8):1053-60; PMID:12736103, http://dx.doi.org/10.1016/S0959-8049(03)00063-7
- Choi G, Lee J, Ji JY, Woo J, Kang NS, Cho SY, Kim HR, Ha JD, Han SY. Discovery of a potent small molecule SIRT1/2 inhibitor with anticancer effects. Int J Oncol 2013 Oct; 43(4):1205-11; PMID:23900402; http://dx.doi.org/10.3892/ijo.2013.2035;
- Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell 2007; 25:15-30; PMID:17218268; http://dx.doi.org/10.1016/j.molcel.2006.12.014
- Grewal SI, Jia S. Heterochromatin revisited. Nat Rev Genet 2007; 8(1):35-46; PMID:17173056; http://dx.doi.org/10.1038/nrg2008
- Solomon JM, Pasupuleti R, Xu L, McDonagh T, Curtis R, DiStefano PS, Huber LJ. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol Cell Biol 2006 Jan; 26(1):28-38; PMID:16354677; http://dx.doi.org/10.1128/MCB.26.1.28-38.2006
- Zhang Q, Zeng SX, Zhang Y, Zhang Y, Ding D, Ye Q, Meroueh SO, Lu H. A small molecule Inauhzin inhibits SIRT1 activity and suppresses tumour growth through activation of p53. EMBO Mol Med 2012 Apr; 4(4):298-312; PMID:22331558; http://dx.doi.org/10.1002/emmm.201100211