
Abstract
To avoid cell cycle arrest or apoptosis, rapidly proliferating cancer cells have to promote DNA double strand break (DSB) repair to fix replication stress induced DSBs. Therefore, developing drugs blocking homologous recombination (HR) and nonhomologous end joining (NHEJ) – 2 major DSB repair pathways – holds great potential for cancer therapy. Over the last few decades, much attention has been paid to explore drugs targeting DSB repair pathways for cancer therapy. Here, using 2 well-established reporters for analyzing HR and NHEJ efficiency, we found that both HR and NHEJ are elevated in hepatoma cell lines Hep3B and HuH7 compared with normal liver cell lines Chang liver and QSG-7701. Our further study found that Harmine, a natural compound, negatively regulates HR but not NHEJ by interfering Rad51 recruitment, resulting in severe cytotoxicity in hepatoma cells. Furthermore, NHEJ inhibitor Nu7441 markedly sensitizes Hep3B cells to the anti-proliferative effects of Harmine. Taken together, our study suggested that Harmine holds great promise as an oncologic drug and combination of Harmine with a NHEJ inhibitor might be an effective strategy for anti-cancer treatment.
Abbreviations
| DSB | = | double strand break |
| HR | = | homologous recombination |
| NHEJ | = | nonhomologous end joining |
| DDR | = | DNA damage response |
Introduction
Sustaining proliferation is one of the major hallmarks of cancer.Citation1 High speed of cancer cell division is associated with increasing rates of DNA replication, which inevitably leads to more collapsed DNA replication forks in cancer cells than those in slowly growing or quiescent normal cells.Citation2 However, collapsed DNA replication forks are one of the intrinsic causes of DNA DSBs, which are considered the most detrimental type among all sorts of DNA damages. Improperly repaired or unrepaired DSBs are extremely toxic to cancer cells as they may result in cell death. Therefore, it is essential for cancer cells to activate DNA DSB repair pathways to relieve replication stress, thereafter avoiding apoptosis. DSBs are predominantly repaired by HR or NHEJ.Citation3,4 HR is an error-free repair pathway that requires a non-damaged sister chromatid to serve as a template for repair. Factors such as the MRN complex, CtIP, RPA, BRCA1, BRCA2 and Rad51 are involved in HR pathway. NHEJ is an error-prone repair pathway that is mediated by the direct joining of the 2 broken ends. Factors involved in NHEJ include the Ku70/Ku80 complex, DNA-PKcs, Artemis, XLF, XRCC4 and DNA ligase IV.
Indeed, previous studies indicate that in several types of cancer cells, NHEJ and HR are aberrantly activated.Citation5,6 For instance, compared with normal breast epithelial cells, HR efficiency is greatly increased in breast cancer cells. These findings suggest that targeting DNA DSB repair pathways might be a promising cancer therapeutic method. Many inhibitors targeting NHEJ have been developed and tested for their potential to enhance DNA damage-induced tumor cell killing in preclinical studies and clinical trials. For instances, inhibitors of DNA-PKcs, including Nu7026 and Nu7441, were found to induce extreme sensitivity to ionizing radiation and DNA-damaging agents in preclinical studies.Citation7,8 SCR7, a small molecule inhibitor of human DNA ligase IV, reduces cell proliferation in a DNA ligase IV-dependent manner and increases the tumor-inhibitory effects of agents that cause DSBs.Citation9 Likewise, many inhibitors targeting HR pathway have been developed over recent years.Citation10,11 B02, A03 and A10 were demonstrated to inhibit Rad51 to sensitize cancer cells to DNA-damaging agents and overcome imatinib resistance in CML cells.Citation12,13
Harmine is a tricyclic β-carboline alkaloid that was originally isolated from seeds of Peganum harmala and it is widely distributed in nature, such as various plants, marine creatures, insects and mammals. Harmine has been reported to possess anxiolytic, behavioral effects both in vitro and in vivo. Several potential molecular targets have been identified for the central pharmacological effects of Harmine, including dual-specificity tyrosine-phosphorylation regulated kinase (DYRK), monoamine oxidase A (MAO-A), 5-HT2A receptor and imidazoline receptors.Citation14-16 In addition, interestingly, Harmine suppresses the growth of several types of cancer cells, hinting its potential use in cancer therapy.Citation17 It inhibits breast cancer resistance protein (BCRP) in a BCRP overexpressing breast cancer cell line and reverse drug resistance.Citation18 Alone or in combination with ATRA and G-CSF, Harmine reduces proliferation of HL60 cells in a dose and time dependent manner.Citation19 Although previous studies indicate that Harmine might suppress cell growth by activating intrinsic and extrinsic pathways of apoptosis and regulating transcription factors such as p53 and Bax,Citation20 the mechanisms of anti-tumor effect by Harmine remain unelucidated.
In the present study, using a sensitive and quantitative assay of measuring HR and NHEJ efficiencies, we identified Harmine as a putative inhibitor of HR pathway. Harmine reduces HR by impairing Rad51 recruitment, thereby leading to accumulation of DSBs and significant S or G2/M phase arrest within hepatoma cells, culminating into cytotoxicity. The combination of Harmine and DNA-PKcs inhibitor Nu7441 has a cooperative anti-proliferative and cell cycle arrest effect on Hep3B cells. Collectively, our results showed that Harmine is a newly developed inhibitor targeting HR pathway, contributing to converting spontaneous DNA lesions into fatal replication lesions that specifically kill cancer cells.
Results
Both HR and NHEJ efficiencies are markedly elevated in Hep3B and HuH7 cells
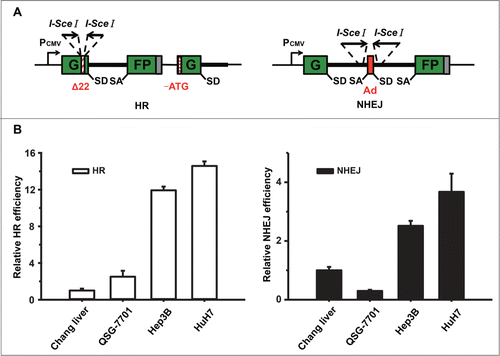
The sustaining proliferation signaling is hyperactivated in cancer cells in comparison to normal cells. The phenomenon of fast growing results in increasing number of collapsed DNA replication forks, which causes more DNA DSBs. To examine whether hepatoma cells grow faster than normal liver cells, we compared the growth rate of 2 normal liver cell lines Chang liver and QSG-7701 to that of the hepatocellular carcinoma cell line Hep3B. Cells were collected and counted at indicated time points. Among all the cell lines, Hep3B exhibits a strikingly higher proliferation rate (Fig. S1), indicating they probably undergo high replication stress. To compare HR and NHEJ efficiencies between different cell lines in a quantitative manner, we used fluorescent reporter constructs in which a functional GFP gene is reconstituted following an HR or NHEJ event as described previously ().Citation21-26 Notably, since the HR reporter lacks a second copy of GFP second exon, a single strand annealing event can only result in first exon of GFP, which does not turn cells green fluorescence. Instead, only gene conversion, the major pathway of homologous recombination repair, can restore the GFP expression. We transfected linearized HR or NHEJ reporter by I-SceI enzyme into different cell lines. A plasmid expressing DsRed was co-transfected to normalize for differences in transfection efficiency between the cell lines. Seventy-two hours post transfection, the numbers of GFP+ and DsRed+ cells were determined by flow cytometry and the ratio between GFP+ and DsRed+ cells was used as a measure of DSB repair efficiency. The analysis showed that in Hep3B and HuH7 cells both HR and NHEJ efficiencies are much higher than that in Chang liver and QSG-7701 (), suggesting that targeting HR and NHEJ might be a feasible strategy for cancer therapy.
Figure 1. DSB repair pathways are hyperactivated in Hep3B and HuH7 cells. (A) Reporter constructs for analysis of DSB repair. The HR reporter comprises 2 inactivated copies of GFP-Pem1. In the first one, a 22 nt and an insertion of 2 I-SceI recognition sites in inverted orientation are inserted into the first exon of GFP. In the second copy, both the ATG and second exon of GFP are deleted. Upon induction of DSBs by I-SceI, only gene conversion restores GFP expression. The starting NHEJ reporter is also GFP negative as it contains a GFP gene with an intron from the rat Pem1 gene and an adenoviral exon (Ad). Two I-SceI recognition sites flank the Ad exon in an inverted orientation. Upon induction of a DSB, only NHEJ can turn cells GFP+. SA, splice acceptor; SD, splice donor. (B) Analysis of HR or NHEJ efficiency in 4 liver cell lines. At 48 h post splitting, cells were co-electroporated with I-SceI linearized 0.6 μg HR or 0.2 μg NHEJ reporter plasmids and 0.005 μg of the DsRed expression vectors for normalizing transfection efficiency. Three days later, cells were harvested for FACS analysis. The ratio of GFP+ versus DsRed+ cells was used as a measure of repair efficiency. All the experiments were repeated more than 3 times and all results were normalized with that of Chang liver. Error bars stand for standard deviation (SD).
Harmine inhibits HR, resulting in selective cytotoxicity to Hep3B and HuH7 cells
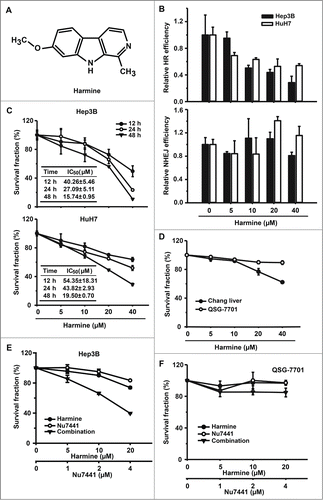
Harmine is a tricyclic β-carboline alkaloid (). It has been suggested to be involved in suppressing the growth of cancer cells, but the molecular mechanism has not been well characterized. To test if it inhibits DNA DSB repair by HR or NHEJ, we used the same assay for analysis of HR and NHEJ in Hep3B and HuH7 cells. Intriguingly, upon the treatment of Harmine, a dose dependent decrease of HR rather than NHEJ was observed (). We then further tested if Harmine suppresses the proliferation of Hep3B and HuH7. In the presence of 0–40 μM Harmine for 12–48 h, the proliferation rate of Hep3B and HuH7 cells reduced in a dose and time-dependent fashion with an IC50 of 40.26±5.46 and 54.35±18.31, 27.09±5.11 and 43.82±2.93, 15.74±0.95 and 19.50±0.70 μM, respectively (). In contrast, Harmine-mediated cytotoxicity is relatively mild to normal liver cell lines Chang liver and QSG-7701 (), suggesting that Harmine is an ideal drug candidate for cancer therapy as it is selectively harmful to cancer cells but has minimal side effects to normal cells. NHEJ efficiency was also dramatically elevated in Hep3B cells (). We therefore proposed that inhibiting NHEJ might sensitize Hep3B cells to Harmine. To test this hypothesis, DNA-PKcs inhibitor Nu7441 was supplemented to the Harmine treated Hep3B cells. We found that at the concentration of 10 μM, Harmine displays very moderate cytotoxicity to Hep3B cells whereas the combination of Nu7441 markedly increased the cytotoxicity (). In addition, the combination of the 2 drugs exhibits very mild toxic effect on normal cells ()
Figure 2. Harmine inhibits the efficiency of HR and suppresses the proliferation in Hep3B and HuH7 cells. (A) Chemical structure of Harmine. (B) Effect of Harmine on HR or NHEJ efficiency. Hep3B and HuH7 cells were pretreated with indicated concentrations of Harmine for 6 h, followed by co-transfection with linearized HR or NHEJ reporter constructs and DsRed expression plasmid. Twenty-four h later, cells were analyzed by flow cytometry. All results were normalized with that of control. (C) Effects of Harmine on the proliferation of Hep3B and HuH7. After treated with Harmine for 12 h, 24 h and 48 h, the numbers of cells were determined by MTT assay. The IC50 value at each time point was calculated using Dose-Effect Analysis software. (D) Cytotoxicity of Harmine on Chang liver or QSG-7701 cells lines. The procedure was the same as described in Figure 2C. (E and F) The combinatory treatment of Harmine and Nu7441. Hep3B or QSG-7701 cells were plated in 96-well plate and exposed to serial concentrations treatments of Nu7441 and Harmine either alone or in combination for 24 h. Cell viability was then determined by MTT assay. All experiments were repeated at least 3 times. Error bars represent SD
Harmine disrupts HR by inhibiting Rad51 recruitment, leading to generation of unrepaired DSBs
To reveal the underlying molecular mechanism of inhibiting HR by Harmine, we first examined the expression levels of several HR related proteins by Western blot. No significant differences were observed in expression levels of these proteins in irradiated or non-irradiated Hep3B cells in the presence or absence of Harmine treatment (). Previous researches have indicated that Rad51 is localized in both cytoplasm and nucleus, and in response to DNA damage Rad51 is re-localized to the sites of DNA damage to form distinct repair foci.Citation27,28 We then assessed if Harmine affects the recruitment of Rad51 to damage sites. We found that upon the induction of DNA DSBs by ionizing radiation, Harmine treatment reduced the number of Rad51 foci ( and Fig. S2A), suggesting that Harmine inhibits HR by blocking the recruitment of Rad51.
Figure 3. Harmine blocks HR by inhibiting Rad51 recruitment. (A and B) Harmine does not affect the expression levels of HR related proteins. Hep3B cells were treated with indicated concentrations of Harmine, followed by x-ray irradiation. The expression levels of HR related proteins were analyzed by Western blot. (C) Harmine disrupts the formation of Rad51 foci after DNA damage. Hep3B cells pretreated with 40 μM Harmine for 2 h were irradiated with 8 Gy X-ray. At 6 h post irradiation, Rad51 foci formation was examined by immunofluorescence. The percentage of cells containing Rad51 foci (>5 ) was displayed. **, p < 0 .01. All experiments were repeated 3 times and at least 50 nuclei were examined and quantified for each time. (D) Harmine upregulates γ-H2Ax level in Hep3B cells. Cells were treated with Harmine at various concentrations for 6 hours before whole cells lysates were extracted and analyzed by Western blot. (E) The p-Chk1 level is upregulated in Hep3B cells following Harmine treatment. The experimental procedure is as described in . (F) Harmine treatment leads to a persistent DNA damage signaling after x-ray irradiation. Hep3B cells were exposed to X-ray at 8 Gy, followed by treatment with 40 μM Harmine for 2 h. Then γ-H2Ax foci were immune-stained. The percentage of cells positive for γ-H2Ax foci (>10 ) was shown. **, p < 0 .01. All experiments were repeated 3 times and at least 50 nuclei were examined and quantified for each time.
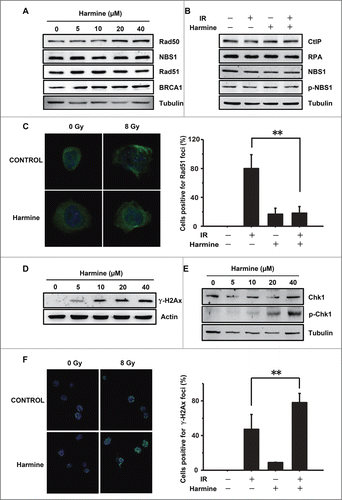
We thereafter hypothesized that the inhibition of HR by Harmine may result in the accumulation of unrepaired DSBs in genomes. To test this, we first examined the level of γ-H2Ax, a marker of DNA DSBs, in the presence of Harmine by Western blot. We found that the γ-H2Ax level is increased by Harmine in a dose dependent manner (). Replication stress generated DNA DSBs triggers the activation of ATR, which phosphorylates Chk1. We therefore further examined the effect of Harmine on Chk1 phosphorylation.Citation29 The Western blot analysis indicates that similar to H2Ax phosphorylation, Chk1 phosphorylation level also increases upon Harmine treatment in a dose dependent manner (), suggesting that the inhibition of clearance of replication stress induced DNA DSBs by Harmine leads to accumulated DNA damages. Furthermore, when treated with ionizing radiation, in the presence of Harmine, Hep3B cells failed to repair DNA DSBs, leaving more γ-H2Ax foci formed at the damaged sites ( and Fig. S2B), further confirming that Harmine mediates the repression of DNA DSB repair by HR.
Harmine induces S and G2/M phase arrest in Hep3B cells
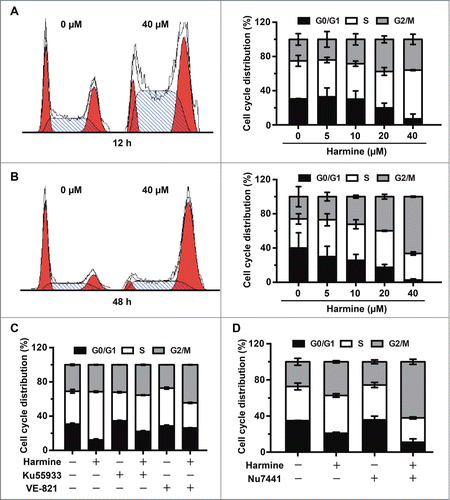
Unrepaired DSBs cause persistent DNA damage response (DDR), resulting in either cell death or cell cycle arrest.Citation30 To determine whether Harmine induces cell death or cell cycle arrest by inhibiting HR repair, we first analyzed the apoptotic and necrotic rate in the presence of Harmine. We did not observe significant change of cell death rate in Hep3B cells with Harmine treatment even at the concentration of 40 μM for 48 h (Fig. S3). Instead, cell cycle distribution analysis revealed that Harmine induces significant S and G2/M phase arrest after 12 h and 48 h treatment, indicating that the inhibition of cell survival by Harmine is through inducing cell cycle arrest rather than triggering cell death in Hep3B cells (). To further demonstrate that Harmine treatment blocks cell cycle progression through inhibiting DNA repair, we analyzed cell cycle distribution in the presence of inhibitors of ATM or ATR, 2 major sensors of DNA DSBs. We observed that in Hep3B cells the pretreatment of Ku55933, an inhibitor of ATM, or VE-821, an inhibitor of ATR, rescued the cell cycle arrest in S phase (), strongly indicating that the induction of cell cycle arrest by Harmine is mediated by its effect on blocking DNA repair pathways. Moreover, the combinatory treatment of Harmine and Nu7441 greatly stimulated the S and G2/M phase arrest to the level of 90%. In contrast, monotherapy with either agent only arrested 79% and 65% of cells in the 2 phases (). Therefore, we conclude that Harmine together with Nu7441 cooperatively triggers cell cycle arrest in Hep3B cells.
Figure 4. Harmine induces S and G2/M phase arrest in Hep3B cells in an ATM and ATR dependent manner. (A and B) Harmine arrests cells in S and G2/M phases. Hep3B cells were grown in the absence or presence of Harmine for 12 h or 48 h, respectively. Cell cycle distributions were examined by flow cytometry after PI staining. Results were representative of 3 independent experiments. (C) ATM or ATR inhibition abrogates the Harmine induced cell cycle arrest. Hep3B cells pretreated with 10 μM Ku55933 or VE-821 for 2 h were further treated with 40 μM Harmine for another 12 h. Cell cycle profiles of Hep3B cells were then detected by PtdIns staining and flow cytometry. (D) The combinatory treatment of Harmine and Nu7441 promotes cell cycle arrest of Hep3B cells. Cell cycle distributions of Hep3B cells after treatment with 4 μM Nu7441 and 20 μM Harmine either alone or in combination for 24 h were determined by PI staining followed by flow cytometry.
Discussion
Although efficient DNA repair pathways are evolved to protect normal cells from exogenous or endogenous assault to the genetic carriers, cancer cells ironically take advantage of these pathways to secure themselves from exogenously or endogenously aroused DNA damages, such as chemo- and radio- therapy, or replication stress.Citation5 Here, with the unique reporters for analyzing HR and NHEJ efficiency, we first found that HR and NHEJ efficiencies are both markedly elevated in Hep3B and HuH7 cells compared with normal cell lines. Given the fact that accumulated DNA damages by inhibiting DNA repair eventually leads to cell death or cell cycle arrest,Citation31 our finding strongly hints that targeting DNA DSB repair pathways is an ideal strategy for selectively killing cancer cells.
Great effort has been exerted to screen small molecules for targeting DNA repair pathways. For example, olaparib, a potent inhibitor of PARP1, which is mainly involved in base excision repair and single strand break repair, has been approved by European regulators and FDA.Citation32-34 As successful repair of DNA DSBs is essential to the survival and rapid proliferation of cancer cells, much progress has been made toward developing novel drugs targeting HR or NHEJ pathways for cancer therapy. Here, Harmine was identified as a potent HR inhibitor by disrupting Rad51 recruitment, leading to unrepaired DSBs, culminating into severe cytotoxicity in Hep3B and HuH7 cells. Interestingly, previous studies have suggested that Harmine induces DNA damages including DNA DSBs in various species, such as yeast and Chinese hamster lung fibroblasts V79.Citation35,36 Their data could be explained by 2 possibilities. One is that impaired HR pathway by Harmine results in DNA damage in these cells. The other is that Harmine is a dual-target drug candidate integrating DNA damage and repair inhibition. Since Harmine shows slight cytotoxicity in normal cells in our experiments, it is highly likely that the induced DNA damages result from unrepaired DNA DSBs, although more experiments are needed to further clarify if Harmine directly induces DNA damages.
Although Harmine was characterized as an anti-tumor molecule long ago,Citation37 its undefined mechanisms of regulating HR, and moderate anti-tumor effects, hamper it from being further tested in clinic. Here, we found that in Hep3B and HuH7 cells not only HR are elevated but also NHEJ are greatly stimulated, hinting that merely blocking one of the 2 pathways is insufficient to stop cancer cells from prevailing. Our data indicates that blocking both pathways with Harmine and Nu7441 has an impressive synergistic anti-tumor effect. This finding not only demonstrates that Harmine is a potent HR inhibitor, but also hints that a complete analysis of the DNA repair capacities of a specific tumor might provide physicians better clues of personized therapy.
Unrepaired DNA damages cause cell cycle arrest or cell death. It is not fully understood which of these 2 responses is chosen in a given cell. Cell type appears to be one of the factors involved in this process. In addition, several cellular and viral proteins contribute to the choice of the 2 responses. Moreover, previous studies suggest that p53 level is a determining factor.Citation38 Upon DNA damage, cells with low expression of p53 show slowed or arrested growth, whereas high expression level of p53 causes cell death.Citation38,39 In our case, Hep3B cells undergo S and G2/M phase arrest rather than apoptosis in response to Harmine treatment. This could be due to that Hep3B is a p53 deficient cell line.Citation40
In conclusion, we demonstrated that inhibition of HR by Harmine results in accumulated unrepaired DSBs, leading to severe cytotoxicity in hepatoma cells. In addition, our work indicates Harmine targets rapidly dividing cells while has very limited side effect on normal cells. More importantly, our findings demonstrate a concept of suppressing both pathways of DNA DSBs repair could be a potentially efficient way of curing cancer.
Materials and Methods
Chemicals and antibodies
Harmine was obtained from Abcam. Ku55933, VE-821 and Nu7441 were purchased from Selleckchem. All the compounds were dissolved in DMSO.
The γ-H2Ax (Ser139) (20E3) Rabbit mAb was obtained from Cell Signaling Technology. Rad51 (A6268), NBS1 (A0783), Phospho-NBS1 (AP0413), RPA (A2189), Rad50 (A3078), BRCA1 (A0212), Chk1 (A0700), Phospho-Chk1 (AP0018), Actin (AC006) and Tubulin (AC007) polyclonal antibodies were purchased from ABclonal Technology. Secondary antibodies used for Western blot were purchased from Bio-Rad Laboratories and for immunofluorescence were obtained from Abcam.
Cell culture
Human hepatocellular carcinoma cell lines Hep3B and HuH7 were grown in complete DMEM medium. Human normal liver cell lines Chang liver and QSG-7701 were grown in complete RPMI-1640 medium. All media were supplemented with 10% fetal bovine serum (Gibco, Cat. # 16000) plus penicillin (100 U/ml) and streptomycin (100 U/ml). All cells were incubated at 37 °C in a 5% CO2 atmosphere.
Transfections
All cells were seeded on a 10 cm dish 24 h before electroporation. Transfections were performed with the indicated program on a Lonza 4D machine.
Immunofluorescence
Hep3B cells were washed twice with PBS, fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and blocked with 1×goat serum. Cells were then incubated overnight at 4 °C with anti-γ-H2Ax or anti-Rad51 antibody diluted 1:50 in 1×goat serum. After washed twice with PBS, cells were incubated with FITC-conjugated secondary antibody (1:100 dilution) for 1 h at room temperature in the dark. The probed cells were then washed 3 times followed by staining with DAPI. The cells were observed using a confocal scanning laser microscopy (Leica).
Protein extraction and Western blot
Cells were lysed in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1, and protease inhibitors). The protein concentration was determined using the DC Protein Assay kit (Bio-Rad Laboratories). After protein quantification and normalization, equivalent amounts of proteins were electrophoresed on 8–15% SDS-PAGE gels and transferred to PVDF membranes (Millipore). Nonspecific binding sites were blocked by incubation in TBS containing 0.1% Tween-20 and 5% dry milk. After incubation with primary antibodies, the proteins were visualized by incubation with HRP-conjugated secondary antibodies followed by enhanced chemiluminescence detection (Thermo Fisher).
Apoptosis assay
Apoptosis of Hep3B cells caused by Harmine were analyzed by Annexin V-FITC Apoptosis Detection Kit (Biotool). Briefly, cells (1–5×105 per sample) were collected, washed twice with cold PBS, centrifuged, and resuspended in 100 μl 1×binding buffer. Annexin V-FITC and PI staining solution were then added. After incubation for 10 min in the dark, fluorescence was analyzed by FACSCalibur flow cytometer (BD Biosciences).
Cell cycle analysis
Cells were collected, washed with PBS twice and fixed with 75% alcohol overnight. Cells were then washed with cold PBS and resuspended. After elimination the interference of RNA with RNase, the cells were treated with PtdIns for 15 min. Cells were washed and the DNA contents were analyzed on FACSCalibur.
MTT assay
Cells were cultured in 96-well plates and the cytotoxic activity of Harmine was detected using the MTT assay. After treatment with various concentrations of Harmine for 12 h, 24 h and 48 h, cells were added 5 mg/ml MTT solution and incubated for an additional 4 h at 37 °C. The purple formazan crystals were then dissolved in 100 μl DMSO. Absorbance in each well was detected on a reader (Biotek) at 570 nm. The IC50 values were calculated using Dose-Effect Analysis software.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
KCBT_A_1078021_Fig_S3.docx
Download MS Word (78.8 KB)KCBT_A_1078021_Fig_S2.docx
Download MS Word (115.9 KB)KCBT_A_1078021_Fig_S1.docx
Download MS Word (100 KB)Acknowledgments
Shanghai Municipal Natural Science Foundation (Grant No. 15ZR1442600), the Cultural Program for Outstanding Young Talents in Tongji University to Y.J.; and the National Science Foundation of China (Grant No. 81402543), the Cultural Program for Outstanding Young Talents in Tongji University, the China Postdoctoral Science Foundation (Grant No. 2014M551446) to L.Z.
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science 2008; 319:1352-5; PMID:18323444; http://dx.doi.org/10.1126/science.1140735
- Chu G. Double strand break repair. J Biol Chem 1997; 272:24097-100; PMID:9305850; http://dx.doi.org/10.1074/jbc.272.39.24097
- Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res 2008; 18:134-47; PMID:18157161; http://dx.doi.org/10.1038/cr.2007.111
- Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 2008; 8:193-204; PMID:18256616; http://dx.doi.org/10.1038/nrc2342
- Mao Z, Jiang Y, Liu X, Seluanov A, Gorbunova V. DNA repair by homologous recombination, but not by nonhomologous end joining, is elevated in breast cancer cells. Neoplasia 2009; 11:683-91; PMID:19568413; http://dx.doi.org/10.1593/neo.09312
- Willmore E, de Caux S, Sunter NJ, Tilby MJ, Jackson GH, Austin CA, Durkacz BW. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood 2004; 103:4659-65; PMID:15010369; http://dx.doi.org/10.1182/blood-2003-07-2527
- Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, Griffin RJ, Calvert AH, Newell DR, Smith GC, Curtin NJ. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res 2006; 66:5354-62; PMID:16707462; http://dx.doi.org/10.1158/0008-5472.CAN-05-4275
- Srivastava M, Nambiar M, Sharma S, Karki SS, Goldsmith G, Hegde M, Kumar S, Pandey M, Singh RK, Ray P, et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell 2012; 151:1474-87; PMID:23260137; http://dx.doi.org/10.1016/j.cell.2012.11.054
- Budke B, Kalin JH, Pawlowski M, Zelivianskaia AS, Wu M, Kozikowski AP, Connell PP. An optimized RAD51 inhibitor that disrupts homologous recombination without requiring Michael acceptor reactivity. J Med Chem 2013; 56:254-63; PMID:23231413; http://dx.doi.org/10.1021/jm301565b
- Budke B, Logan HL, Kalin JH, Zelivianskaia AS, Cameron McGuire W, Miller LL, Stark JM, Kozikowski AP, Bishop DK, Connell PP. RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res 2012; 40:7347-57
- Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem Biol 2011; 6:628-35; PMID:21428443; http://dx.doi.org/10.1021/cb100428c
- Hosoya N, Miyagawa K. Targeting DNA damage response in cancer therapy. Cancer Sci 2014; 105:370-88; PMID:24484288; http://dx.doi.org/10.1111/cas.12366
- Gockler N, Jofre G, Papadopoulos C, Soppa U, Tejedor FJ, Becker W. Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS J 2009; 276:6324-37; PMID:19796173; http://dx.doi.org/10.1111/j.1742-4658.2009.07346.x
- Bergstrom M, Westerberg G, Langstrom B. 11C-harmine as a tracer for monoamine oxidase A (MAO-A): in vitro and in vivo studies. Nucl Med Biol 1997; 24:287-93; PMID:9257326; http://dx.doi.org/10.1016/S0969-8051(97)00013-9
- Li Y, Sattler R, Yang EJ, Nunes A, Ayukawa Y, Akhtar S, Ji G, Zhang PW, Rothstein JD. Harmine, a natural β-carboline alkaloid, upregulates astroglial glutamate transporter expression. Neuropharmacol 2011; 60:1168-75; PMID:21034752; http://dx.doi.org/10.1016/j.neuropharm.2010.10.016
- Song Y, Kesuma D, Wang J, Deng Y, Duan J, Wang JH, Qi RZ. Specific inhibition of cyclin-dependent kinases and cell proliferation by harmine. Biochem Biophys Res Commun 2004; 317:128-32; PMID:15047157; http://dx.doi.org/10.1016/j.bbrc.2004.03.019
- Ma Y, Wink M. The β-carboline alkaloid harmine inhibits BCRP and can reverse resistance to the anticancer drugs mitoxantrone and camptothecin in breast cancer cells. Phytother Res 2010; 24:146-9; PMID:19548284; http://dx.doi.org/10.1002/ptr.2860
- Zaker F, Oody A, Arjmand A. A study on the antitumoral and differentiation effects of peganum harmala derivatives in combination with ATRA on leukaemic cells. Arch Pharm Res 2007; 30:844-9; PMID:17703736; http://dx.doi.org/10.1007/BF02978835
- Hamsa TP, Kuttan G. Harmine activates intrinsic and extrinsic pathways of apoptosis in B16F-10 melanoma. Chinese Med 2011; 6:11; PMID:21429205; http://dx.doi.org/10.1186/1749-8546-6-11
- Seluanov A, Mao Z, Gorbunova V. Analysis of DNA double-strand break (DSB) repair in mammalian cells. J Vis Exp 2010; 8(43). pii: 2002. doi: 10.3791/2002
- Seluanov A, Mittelman D, Pereira-Smith OM, Wilson JH, Gorbunova V. DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc Natl Acad Sci USA 2004; 101:7624-9
- Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011; 332:1443-6; PMID:21680843; http://dx.doi.org/10.1126/science.1202723
- Mao Z, Seluanov A, Jiang Y, Gorbunova V. TRF2 is required for repair of nontelomeric DNA double-strand breaks by homologous recombination. Proc Natl Acad Sci USA 2007; 104:13068-73
- Mao Z, Tian X, Van Meter M, Ke Z, Gorbunova V, Seluanov A. Sirtuin 6 (SIRT6) rescues the decline of homologous recombination repair during replicative senescence. Proc Natl Acad Sci USA 2012; 109:11800-5
- Chen S, Wang C, Sun L, Wang DL, Chen L, Huang Z, Yang Q, Gao J, Yang XB, Chang JF, et al. RAD6 promotes homologous recombination repair by activating the autophagy-mediated degradation of heterochromatin protein HP1. Mol Cell Biol 2015; 35:406-16; PMID:25384975; http://dx.doi.org/10.1128/MCB.01044-14
- Essers J, Houtsmuller AB, van Veelen L, Paulusma C, Nigg AL, Pastink A, Vermeulen W, Hoeijmakers JH, Kanaar R. Nuclear dynamics of RAD52 group homologous recombination proteins in response to DNA damage. EMBO J 2002; 21:2030-7; PMID:11953322; http://dx.doi.org/10.1093/emboj/21.8.2030
- Davies AA, Masson JY, McIlwraith MJ, Stasiak AZ, Stasiak A, Venkitaraman AR, West SC. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell 2001; 7:273-82; PMID:11239456; http://dx.doi.org/10.1016/S1097-2765(01)00175-7
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 2000; 14:1448-59; PMID:10859164; http://dx.doi.org/10.1101/gad.840500
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071-8; PMID:19847258; http://dx.doi.org/10.1038/nature08467
- Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408:433-9; PMID:11100718; http://dx.doi.org/10.1038/35044005
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. New Engl J Med 2009; 361:123-34; PMID:19553641; http://dx.doi.org/10.1056/NEJMoa0900212
- Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 2010; 376:245-51; PMID:20609468; http://dx.doi.org/10.1016/S0140-6736(10)60893-8
- Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 2010; 376:235-44; PMID:20609467; http://dx.doi.org/10.1016/S0140-6736(10)60892-6
- Boeira JM, Viana AF, Picada JN, Henriques JA. Genotoxic and recombinogenic activities of the two β-carboline alkaloids harman and harmine in Saccharomyces cerevisiae. Mutat Res 2002; 500:39-48; PMID:11890933; http://dx.doi.org/10.1016/S0027-5107(01)00294-9
- Boeira JM, da Silva J, Erdtmann B, Henriques JA. Genotoxic effects of the alkaloids harman and harmine assessed by comet assay and chromosome aberration test in mammalian cells in vitro. Pharmacol Toxicol 2001; 89:287-94; PMID:11903953; http://dx.doi.org/10.1034/j.1600-0773.2001.d01-162.x
- Ishida J, Wang HK, Bastow KF, Hu CQ, Lee KH. Antitumor agents 201. Cytotoxicity of harmine and β-carboline analogs. Bioorg Med Chem Lett 1999; 9:3319-24; PMID:10612592; http://dx.doi.org/10.1016/S0960-894X(99)00598-3
- Chen X, Ko LJ, Jayaraman L, Prives C. p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev 1996; 10:2438-51; PMID:8843196; http://dx.doi.org/10.1101/gad.10.19.2438
- Ryan KM, Vousden KH. Characterization of structural p53 mutants which show selective defects in apoptosis but not cell cycle arrest. Mol Cell Biol 1998; 18:3692-8; PMID:9632751
- Stahler F, Roemer K. Mutant p53 can provoke apoptosis in p53-deficient Hep3B cells with delayed kinetics relative to wild-type p53. Oncogene 1998; 17:3507-12; PMID:10030675; http://dx.doi.org/10.1038/sj.onc.1202245