
Abstract
Chewing tobacco is a common practice in certain socio-economic sections of southern Asia, particularly in the Indian subcontinent and has been well associated with head and neck squamous cell carcinoma. The molecular mechanisms of chewing tobacco which leads to malignancy remains unclear. In large majority of studies, short-term exposure to tobacco has been evaluated. From a biological perspective, however, long-term (chronic) exposure to tobacco mimics the pathogenesis of oral cancer more closely. We developed a cell line model to investigate the chronic effects of chewing tobacco. Chronic exposure to tobacco resulted in higher cellular proliferation and invasive ability of the normal oral keratinocytes (OKF6/TERT1). We carried out quantitative proteomic analysis of OKF6/TERT1 cells chronically treated with chewing tobacco compared to the untreated cells. We identified a total of 3,636 proteins among which expression of 408 proteins were found to be significantly altered. Among the overexpressed proteins, stearoyl-CoA desaturase (SCD) was found to be 2.6-fold overexpressed in the tobacco treated cells. Silencing/inhibition of SCD using its specific siRNA or inhibitor led to a decrease in cellular proliferation, invasion and colony forming ability of not only the tobacco treated cells but also in a panel of head and neck cancer cell lines. These findings suggest that chronic exposure to chewing tobacco induced carcinogenesis in non-malignant oral epithelial cells and SCD plays an essential role in this process. The current study provides evidence that SCD can act as a potential therapeutic target in head and neck squamous cell carcinoma, especially in patients who are users of tobacco.
Abbreviations
| iTRAQ | = | Isobaric tags for relative and absolute quantitation |
| SCX | = | Strong cation exchange chromatography |
Introduction
Consumption of tobacco continues to be one of the major established etiological factors in the pathogenesis of head and neck squamous cell carcinoma (HNSCC).Citation1,2 According to the report published by the WHO, every year approximately 6 million people worldwide are killed due to tobacco consumption.Citation3 Tobacco is mainly consumed worldwide in the form of manufactured cigarettes and even smoked as ‘bidis’, cigars and waterpipes.Citation4 Another form in which tobacco is consumed includes smokeless or chewing tobacco. India houses the highest number of chewing tobacco users, which accounts for 80% of global tobacco chewers. In India, more than 50% of oral cancer cases are estimated due to use of chewing tobacco.Citation4
Chewing tobacco is associated with the development of cancers of the oral cavity, esophagus, stomach and pancreas.Citation5,6 In the year 2007, the International Agency for Research on Cancer (IARC) classified chewing tobacco as a human carcinogen.Citation7 Chewing tobacco contains several compounds including tobacco-specific N-nitrosamines (TSNA), N'-nitrosonornicotine (NNN), and 4(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) which are shown to be carcinogenic.Citation8,9 Though several studies have shown the association between chewing tobacco and cancer, the underlying cellular and molecular dynamics that lead to cancer development and metastasis is not well characterized. It is intuitive to consider that the mechanisms of action of chewing tobacco to be similar to that of cigarette smoke. However, it has been reported that there are significant differences in pro-carcinogenic and carcinogenic agents present in chewing tobacco because of which the mode of action might vary significantly.Citation8,10 Current studies have demonstrated the acute effects of chewing tobacco in cells. A study by Wang Y et al., demonstrated the proliferation of normal human epidermal keratinocytes induced by chewing tobacco extract when treated for 3 dCitation11 Studies by other groups have shown that treatment of AMOL-III cells (established from a leukoplakia patient), with chewing tobacco for 48h induced alterations in genes involved in key biological processes such as cell cycle regulation, cell-cell adhesion and DNA methylation.Citation12,13 Validation of these genes on oral pre-malignant lesions (OPLs) and oral squamous cell carcinoma (OSCC) tissues showed similar pattern of expression. The same group also reported activation of nuclear factor-κB (NF-κB) and signal transducer and activator of transcription 3 (STAT3) pathways in chewing tobacco induced oral carcinogenesis.Citation14,15 It is known that carcinogenic effect of chewing tobacco in oral cancer is through chronic exposure and not by acute exposure. Though these studies have identified few of the molecules by which chewing tobacco may exert its effects, but till date, there has been no study on the chronic effects of chewing tobacco in oral keratinocytes even though chewing tobacco is one of the primary risk factors for head and neck cancers.
In this study, we chronically treated normal oral keratinocytes (OKF6/TERT1) with chewing tobacco to model tobacco chewing effects. We found that chronic exposure to chewing tobacco led to increased cellular proliferation and induced invasive ability in the non-invasive oral keratinocytes. To understand the molecular mechanisms of the insult imparted by chewing tobacco, we studied the proteomic profile of the OKF6/TERT1 cells chronically exposed to chewing tobacco compared to untreated cells. Quantitative mass spectrometry-based proteomic analysis resulted in the identification of 408 proteins, which were differentially expressed in the cells chronically exposed to chewing tobacco. The dysregulated proteins included overexpression of proteins involved in the stimulation of cell growth and cell cycle regulation.
Tumor cells exhibit high cellular proliferation rate than the normal cells. This process requires the formation of new membranes which in turn requires the synthesis of lipids/fatty acids to maintain membrane fluidity.Citation16 Saturated fatty acids produced as a result of glycolysis are converted to monounsaturated fatty acids, which serve as the components of cell membrane. The ratio of saturated to monounsaturated fatty acids is tightly regulated in cells. Any imbalance in this ratio affects a wide range of cellular functions leading to either uncontrolled cellular proliferation or cellular senescence and death.Citation17 Stearoy-CoA desaturase (SCD), a delta-9-desaturase, plays a central role in the de novo synthesis of monounsaturated fatty acids from saturated fatty acids. The preferred substrates of SCD include palmitic and stearic acids, which are converted to palmitoleate and oleate, respectively.Citation18 In the absence of SCD, palmitic acid accumulates in the cells, which is toxic to mitochondria and endoplasmic reticulum and induces apoptosis.Citation19,20 Literature evidence suggests SCD as a potential target to block cellular proliferation and invasion in cancer.Citation21-23 However, the role of SCD in HNSCC remains unexplored. In this study, SCD was 2.6-fold overexpressed in cells chronically treated with tobacco and we have assessed the potential of SCD as a novel therapeutic target in head and neck cancer.
Results
Chronic exposure to chewing tobacco increases proliferation and invasive property of oral keratinocytes
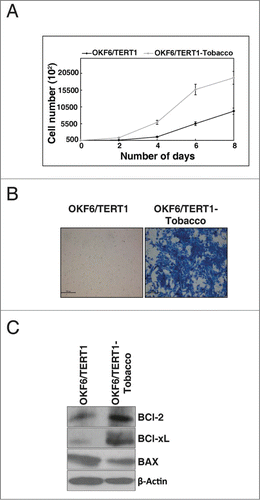
Non-neoplastic oral keratinocytes, OKF6/TERT1, were treated at varying concentrations of chewing tobacco extract ranging from 0–10% to determine the optimum concentration for chronic treatment (data not shown). The highest concentration with which the cells could be treated chronically was 1%. Cells treated at higher concentrations of chewing tobacco (>1 %) underwent apoptosis/necrosis within days of treatment (data not shown). After 3 months of chronic treatment, we observed a change in the invasive property of the oral keratinocytes. The non-invasive cells exhibited signs of invasion (data not shown). The chronic treatment was continued for a total of 6 months before the daughter cells (OKF6/TERT1 cells chronically treated with chewing tobacco, hence forth referred to as OKF6/TERT1-Tobacco) were assessed for proliferation and invasion ability. We observed a significant increase in cellular proliferation of the tobacco treated cells compared to the untreated cells (). In vitro invasion assay using Matrigel showed that the non-invasive OKF6/TERT1 cells had acquired invasive property upon chronic tobacco treatment and more that 80% of the cells had invaded the Matrigel coated PET membrane ().
Figure 1. Chronic exposure to chewing tobacco increases proliferation and invasive property of oral keratinocytes. (A) Growth curve for OKF6/TERT1 and OKF6/TERT1-Tobacco cells. OKF6/TERT1-Tobacco cells showed higher proliferation rate than the parental cells. (B) Invasion assay: OKF6/TERT1 chronically treated with chewing tobacco acquired invasive ability. (C) OKF6/TERT1 chronically treated with chewing tobacco showed an increase in Bcl-xL/Bax ratio.
Chewing tobacco induces the expression of survival proteins
It is established that in the presence of genotypic insult, cancer cells escape cell death by regulating the expression of anti-apoptotic and pro-apoptotic genes.Citation24 As the chewing tobacco treated cells showed enhanced cellular proliferation and invasion compared to the normal oral keratinocytes, we next examined the expression of BCl-2 family proteins in response to chewing tobacco. Western blot analysis showed an increase in expression of both BCl-xL and BCl-2 along with decreased expression of Bax in the OKF6/TERT1-Tobacco cells compared to the parental cells ().
Chronic exposure to chewing tobacco alters the cellular proteome
Once we established that chronic exposure of chewing tobacco induces cellular transformation or leads to progression toward oncogenicity, we sought to study the molecular changes in the tobacco treated cells. We studied the alteration in the cellular proteome of OKF6/TERT1 and OKF6/TERT1-Tobacco cells using isobaric tags for relative and absolute quantitation (iTRAQ)-based quantitative proteomic analysis. The experimental workflow followed is shown in . LC-MS/MS analysis led to the identification of 3,636 proteins among which 174 proteins were overexpressed and 234 proteins were downregulated in the tobacco treated cells compared to the untreated cells (fold change cut-off ≥1.5). The complete list of proteins and their corresponding peptides with their iTRAQ ratios, m/z values and charge state is provided in Supplementary Tables S1 and S2. A partial list of differentially regulated proteins is provided in .
Figure 2. Workflow employed to identify the proteins differentially expressed in response to chewing tobacco. Proteins from OKF6/TERT1 and OKF6/TERT1-Tobacco cells were isolated and quantified. Equal amount of proteins from each condition was subjected to in-solution trypsin digestion. Peptides from OKF6/TERT1 cells were labeled with iTRAQ reagents 114 and 115 and those from OKF6/TERT1-Tobacco cells were labeled with 116 and 117 iTRAQ labels. The samples were pooled and subjected to SCX fractionation, followed by mass spectrometry-based proteomic analysis.
Table 1. Partial list of molecules differentially expressed in OKF6/TERT1 cells chronically treated with chewing tobacco compared to untreated cells.
Chronic exposure to chewing tobacco leads to increased expression of SCD in oral keratinocytes
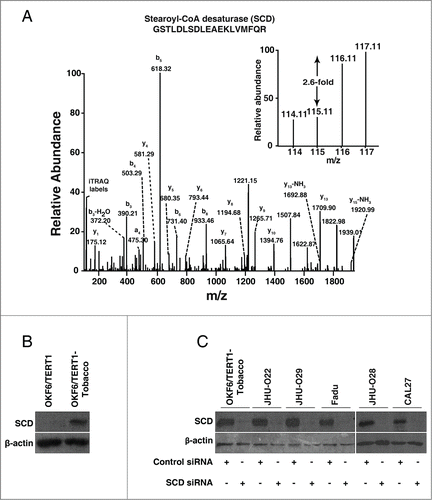
Our proteomics data revealed that SCD was upregulated 2.6-fold in cells chronically treated with chewing tobacco compared to the parental cells. A representative MS/MS spectrum is shown in . In agreement with mass spectrometry data, Western blot analysis revealed a significant increase in the expression of SCD in OKF6/TERT1-Tobacco cells compared to the parental OKF6/TERT1 cells (). We evaluated the expression profile of SCD in a panel of HNSCC cell lines, JHU-O22, JHU-O28, JHU-O29, CAL27 and Fadu. As evident from , all HNSCC cell lines showed an increased expression of SCD compared to normal oral keratinocytes, OKF6/TERT1.
Figure 3. Chronic exposure to chewing tobacco leads to increased expression of SCD in oral keratinocytes. (A) Representative MS/MS spectra of SCD. (B) Western blot analysis showed overexpression of SCD in OKF6/TERT1-Tobacco cells compared to OKF6/TERT1 cells. (C) OKF6/TERT1-Tobacco and a panel of HNSCC cell lines were transfected with SCD siRNA and Western blot was performed 48h post transfection using anti-SCD antibody. β-actin was used as a loading control.
Inhibition of SCD decreases cellular proliferation
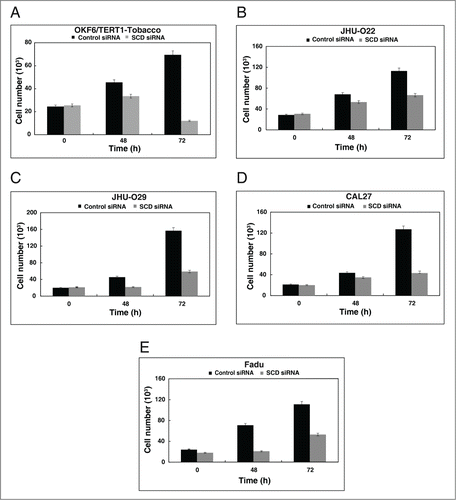
Having observed that SCD was overexpressed both in HNSCC cell lines and OKF6/TERT1-Tobacco cells, we examined the role of SCD in cellular proliferation. Cellular proliferation for the panel of HNSCC cell lines and OKF6/TERT1-Tobacco cells were studied after silencing endogenous expression of SCD using its specific siRNA. Western blot analysis post-transfection with SCD siRNA revealed a successful knockdown of SCD in OKF6/TERT1-Tobacco and HNSCC cell lines (). We observed a decrease in cellular proliferation of the HNSCC cell lines and also in the OKF6/TERT1-Tobacco cells upon siRNA mediated silencing of SCD (). To determine whether inhibition of SCD had any effect on cell survival, the OKF6/TERT1-Tobacco and HNSCC cells were treated with CAY10566, a specific inhibitor of SCDCitation25 and cell proliferation assay was performed. Our results indicated that both the OKF6/TERT1-Tobacco cells and the panel of HNSCC cell lines showed a decrease in cell viability in presence of the inhibitor (Data not shown).
Figure 4. Inhibition of SCD decreases cellular proliferation. Silencing of SCD led to a decrease in cell proliferation of (A) OKF6/TERT1-Tobacco, (B) JHU-O22, (C) JHU-O29, (D) CAL27 and (E) Fadu cells.
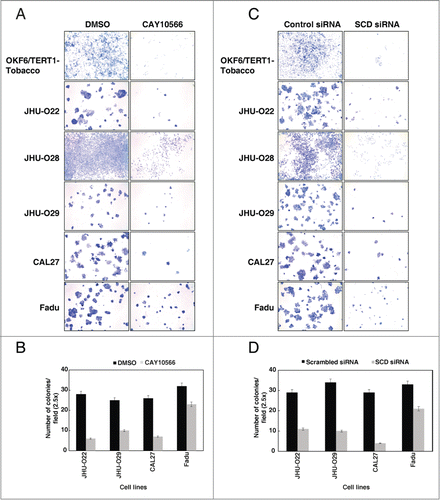
Inhibition of SCD reduces the colony formation ability of oral keratinocytes chronically exposed to chewing tobacco
Colony formation assay is often performed to understand the oncogenic potential or infinite growth potential of cells.Citation26 Having observed that SCD plays an essential role in cellular proliferation, we next studied the effect of SCD ablation in the colony forming ability of the cells. HNSCC cell lines, including the OKF6/TERT1-Tobacco cells showed a decrease in their colony forming ability in the presence of SCD inhibitor, CAY10566 (). Using an alternative strategy to suppress the expression of SCD in the HNSCC cell lines, we silenced SCD expression using its specific siRNA. In agreement to the results of SCD inhibitor, CAY10566, over the colony formation ability of the HNSCC cell lines, siRNA-mediated silencing of SCD in HNSCC cell lines resulted in a significant decrease in the colony formation ability of the cells (). It is to be noted that, the OKF6/TERT1-Tobacco and JHU-O28 cells did not form discrete colonies. However, a significant decrease in cell population was observed both in the presence of SCD siRNA and CAY10566. Taken together, our results indicate that SCD plays an essential role in the early cellular changes induced by chewing tobacco that lead to malignancy.
Figure 5. Inhibition of SCD reduces the colony formation ability of oral keratinocytes chronically exposed to chewing tobacco and HNSCC cells. (A) Colony formation ability of the OKF6/TERT1-Tobacco and HNSCC cells were decreased after inhibition of SCD activity using CAY10566 (10 µM). (B) A graphical representation of the same. (C) siRNA-mediated silencing of SCD resulted in decreased colony forming ability of OKF6/TERT1-Tobacco and HNSCC cells. (D) A graphical representation of the same.
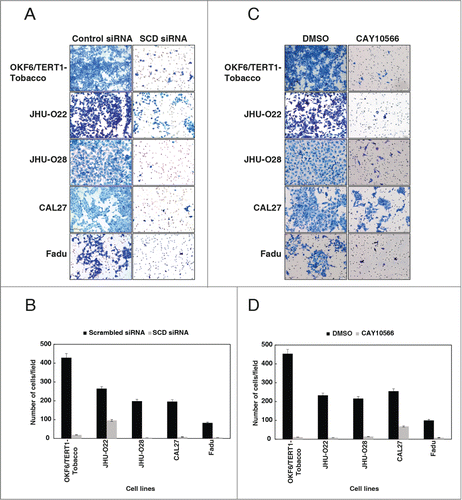
Inhibition of SCD reduces the invasive property of cells exposed to chewing tobacco
Decreased colony formation potential is often associated with loss of invasion capabilities in cancer cells.Citation27 Since invasion is a critical property for metastasis of tumor cells, we further investigated the in vitro invasive capabilities of the HNSCC cells using Matrigel coated transwell chambers. Silencing of SCD using siRNA in OKF6/TERT1-Tobacco and a panel of HNSCC cell lines decreased the invasive property of the cells (). In concordance with the siRNA results, inhibition of SCD with its inhibitor, CAY10566, resulted in a significant decrease in the invasive property of both the HNSCC and OKF6/TERT1-Tobacco cells (). Taken together, these results indicate that silencing of SCD decreases the invasive properties of HNSCC cells.
Figure 6. Inhibition of SCD reduces the invasive property of cells exposed to chewing tobacco. (A) OKF6/TERT1-Tobacco and HNSCC cells were transfected with SCD specific siRNA and control siRNA. Silencing of SCD led to a decrease in the invasive property of the cells. (B) A graphical representation of the same. (C) Treatment of OKF6/TERT1-Tobacco and HNSCC cells with SCD inhibitor, CAY10566 (10 µM) for 48h resulted in a decrease in the invasive ability of the cells. (D) A graphical representation of the same.
Discussion
Earlier studies have indicated that chronic exposure to cigarette smoke provides a better model in vitro than acute exposure to cigarette smoke.Citation28 Number of cellular models have been developed and employed to understand the mechanisms of cellular transformation from normal to tumorigenic phenotype in response to chronic exposure to cigarette smoke.Citation28-32 All the above studies have shown that chronic cigarette smoke exposure/treatment induces selection of clones that have incorporated the molecular level changes necessary for cancer progression and resistance to apoptosis.
Smokeless tobacco or chewing tobacco is a known risk factor in the development of oral cancer. Although the tumor inducing property of chewing tobacco was proven in the year 1964, the molecular events that lead to tumor growth and progression upon its consumption are not known.Citation33 It is important to understand the molecular mechanisms and pathobiology of oral cancer resulting from chewing tobacco as it differs significantly from cigarette smoking because of differences in composition. Chewing tobacco contains several compounds such as nicotine, tobacco-specific N-nitrosamines and polycyclic aromatic hydrocarbons which are known to be carcinogenic. As mentioned earlier tobacco demonstrates its carcinogenic effect through long-term (chronic) exposure and not acute. The goal of this study was to understand the total effects of chewing tobacco and not its individual components in inducing cellular transformation of normal oral keratinocytes upon chronic exposure. Toward this end, we developed a cellular model that mimics the in vivo system of chronic habit of chewing tobacco.
We demonstrate that chronic exposure to chewing tobacco induces molecular changes in a cellular system that is a pre-requisite for progression to malignancy. This is evident by the increased cellular proliferation and invasion ability of the tobacco treated cells. Apoptotic signaling by Bax and Bak can be sequestered by Bcl-xL, leading to cellular survival.Citation34 Our results indicated an increase in the expression of Bcl-xL and Bcl-2 in the chewing tobacco treated cells compared to the parental cells. Bcl-2 has been shown to be overexpressed in chewing tobacco-induced oral cancer.Citation35 In addition, Bcl-xL has been found to be upregulated in other cancers and is considered to be a marker for increased tumorigenesis.Citation36 An increase in the Bcl-xL/Bax ratio provides resistance to DNA damage and aids in survival of cancer cells.Citation34 The increase in the Bcl-xL/Bax ratio in the tobacco treated cells probably leads to an increase in cellular survival due to selective pressure of the tumor microenvironment which enables them to acquire oncogenic property. This was supported by mass spectrometry-based analysis of the cell line pair (OKF6/TERT1-Parental and OKF6/TERT1-Tobacco). A wide array of proteins known to play an essential role in increased cellular proliferation were found to be overexpressed in the cells chronically treated with tobacco. These proteins included TIMP metallopeptidase inhibitor 1 (TIMP1) (2.7-fold), the antigen identified by monoclonal antibody Ki-67 (MKI67) (2.8-fold) and filamin B (FLNB) (2.0-fold).Citation37-39 These observations provide a possible explanation for enhanced cellular proliferation in the tobacco treated cells. We also observed a decrease/loss of expression of multiple tumor suppressor genes. This includes, programmed cell death protein 4 (PDCD4) (0.3-fold) and S100 calcium binding protein A2 (S100A2) (0.4-fold).Citation40,41
Our proteomics data also showed an increased expression of proteins such as epidermal growth factor receptor pathway substrate 8 (EPS8) (2.0-fold), S100 calcium binding protein A14 (S100A14) (1.9-fold) and heat shock 70kDa protein 5 (HSPA5) (1.6-fold), which are known to be aberrantly expressed in head and neck cancer.Citation42-44 This data supports our hypothesis that chronic exposure to chewing tobacco induces malignancy and potentiates the induction of head and neck carcinoma. Apart from the known proteins in literature, we also identified few novel proteins (enhancer of rudimentary homolog (ERH) (7.1-fold) and teneurin transmembrane protein 2 (TENM2) (0.3-fold), whose role in HNSCC remains to be elucidated, which is beyond the scope of this manuscript.
Our observation that SCD is overexpressed in HNSCC is supported by studies in other cancer types.Citation17,21,Citation45 SCD is known to induce epithelial to mesenchymal transition and cancer progression.Citation46 SCD mediates its effects through activation of various signaling events such as activation of PI3K/AKT pathway, Wnt/β-catenin and through the activation of MAP kinases (MAPK1/3).Citation45-47 Here, we show a direct effect of chewing tobacco on normal oral cells and provide evidence that increased expression of SCD in presence of chewing tobacco leads to oncogenic transformation of oral cells. We further demonstrate that inhibition of SCD results in loss of oncogenic and metastatic potential of head and neck cancer cells. Taken together, our results indicate that SCD plays an essential role in HNSCC progression and can serve as a potential therapeutic target in head and neck cancer. Our current study does not rule out the role of other proteins in the progression of HNSCC. In addition, the precise mechanisms of how SCD acts as a therapeutic agent in HNSCC have not yet been clearly defined. However, a detailed understanding of the role of SCD in HNSCC progression is needed and requires both in vivo and in vitro models. In this study, we report the linkage of overexpression of SCD to chewing tobacco mediated oncogenic transformation in oral cells that has not been previously reported.
Materials and Methods
Preparation of chewing tobacco extract
Chewing tobacco was procured from local supplier. 10 g of tobacco was finely powdered and homogenized in 100 ml of 1X phosphate buffer saline (PBS). The mixture was stirred at 37°C for 24h, followed by centrifugation at 2000 g for 15 min. The supernatant was collected and sterilized using 0.22 µm filter.Citation12 This was considered as 100% extract of chewing tobacco. The chewing tobacco extract was aliquoted and stored at −80°C until further use.
Cell culture
Normal human oral keratinocytes, OKF6/TERT1, were a generous gift from Dr. James Rheinwald (Brigham and Women's Hospital, Boston, MA). OKF6/TERT1 were cultured and maintained in keratinocyte serum free medium (KSFM) supplemented with bovine pituitary extract (25 µg/ml), epidermal growth factor (EGF) (0.2 ng/ml), 1% penicillin/streptomycin and CaCl2 (0.4 mM). Fadu and CAL27 cells were procured from American Type Culture Collection (ATCC, Manassas, VA). JHU-O22, JHU-O28, JHU-O29 and Fadu cells were grown in RPMI media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin mixture. CAL27 cells were grown and maintained in DMEM containing 10% FBS and 1% penicillin/streptomycin. All cells were grown at 37°C in a humidified 5% CO2 incubator.
Treatment of OKF6/TERT1 cells with chewing tobacco
OKF6/TERT1 cells were chronically treated with chewing tobacco extract at 1% concentration for 6 months. Henceforth, OKF6/TERT1 cells chronically treated with chewing tobacco extract will be termed as “OKF6/TERT1-Tobacco” and the parental cells will be referred to as “OKF6/TERT1.” Both these cells were grown at 37°C in a humidified 5% CO2 incubator.
siRNA transfection
ON-TARGETplus SMARTpool control siRNA (catalog # D-001810–10–05, Invitrogen, Carlsbad, CA) and SCD siRNA (catalog # L-005061–00–0005, Invitrogen, Carlsbad, CA) were purchased from Dharmacon (Lafayette, CO). Cells were transiently transfected with scrambled or SCD siRNA using Lipofectamine RNAiMAX reagent (catalog # 13778075, Invitrogen, Carlsbad, CA) as described previously.Citation30
Cell proliferation assays
OKF6/TERT1-Tobacco cells and HNSCC cell lines were seeded at a density of 12 ×103 cells per well in 12-well plates. The cells were transfected with either SCD siRNA or control siRNA. The cellular proliferation was monitored for 3 d post-transfection where the cells were counted using trypan blue exclusion method. All assays were carried out in triplicate.
Sample preparation
OKF6/TERT1 and OKF6/TERT1-Tobacco cells were grown to 80% confluence. The cells were starved for 12h. Then, the cells were washed with 1X PBS and harvested in 1X ice-cold PBS. Cells were lysed in 0.5% SDS buffer followed by sonication (Branson Sonifier, Danbury, CT) and centrifugation. The cleared supernatant was used for protein estimation using the bicinchoninic acid assay (BCA).Citation48
In-solution digestion and iTRAQ labeling
In-solution trypsin digestion and iTRAQ labeling of samples from both conditions was carried out as described previously.Citation49 Equal amounts of cell lysate (200 μg) from both OKF6/TERT1 and OKF6/TERT1-Tobacco cells were reduced with (tris(2-carboxyethyl) phosphine (TCEP)) at 60°C for 1h. This was followed by alkylation with cysteine blocking reagent, methyl methanethiosulfonate (MMTS) for 10 min at room temperature. The samples were then subjected to trypsin digestion at 37°C for 12–16 h using sequencing grade trypsin (catalog # V5111, Promega, Madison, WI) at enzyme to substrate ratio of 1:20. Following trypsin digestion, the peptides were labeled with iTRAQ reagents as follows, peptides from OKF6/TERT1 cells were labeled with reagents containing the reporter ions of m/Z 114 and 115 and those from OKF6/TERT1-Tobacco cells were labeled with reagents containing reporter ions of m/Z 116 and 117. The samples from both the conditions were pooled and subjected to fractionation.
Strong cation exchange chromatography (SCX)
SCX fractionation of the iTRAQ labeled peptides was performed as described previously.Citation50 Pooled sample was diluted to 1ml with solvent A (10mM KH2PO4, 25% (v/v) ACN, pH 2.7). The pH of sample was adjusted to 2.7 using ortho-phosphoric acid. The peptides were loaded on PolySULFOETHYL A column (PolyLC, Columbia, MD) (5 µm, 200Å, 200×2.1mm) using an Agilent 1290 Infinity series HPLC system (Agilent Technologies, Santa Clara, CA). Peptides were fractionated using a 50 min gradient from 0% to 40% solvent B (350 mM KCl in solvent A). A total of 96 fractions were collected and further pooled into 23 fractions based on chromatographic peaks. The pooled fractions were dried and desalted using C18 StageTips and stored at −20°C till further analysis.
LC-MS/MS analysis
LTQ-Orbitrap Velos mass spectrometer (Thermo Fischer Scientific, Bermen, Germany) interfaced with Proxeon Easy nLC system (Thermo Scientific, Bremen, Germany) was used for proteomic analysis. Enrichment of the peptides was carried on a trap column (75 µm × 2cm) packed in-house using C18 material (Magic C18AQ, 5 um, 100A, Michrom Biosciences Inc.) with a flow rate of 3 µl/min using solvent A (0.1% formic acid) and resolved on an analytical column (75 µm × 10 cm, Magic C18AQ, 3 µm, 100A, Michrom Biosciences Inc.) at a flow rate of 350 nL/min using a linear gradient of 7- 30% solvent B (95% acetonitrile, 0.1% formic acid) over 70 min. MS and MS/MS scans were acquired with a mass resolution of 60,000 and 15,000 at 400 m/z using the Orbitrap mass analyzer. Precursor MS scan was set to m/z 350–1,800. In each duty cycle 20 most intense monoisotopic precursors were selected for MS/MS fragmentation using higher-energy collision dissociation (HCD) mode at 41% normalized collision energy. Isolation width was set to 1.9 m/z. Singly charged and unassigned charge precursor ions were rejected. Dynamic exclusion setting was enabled and acquired ions were excluded for 45s. The automatic gain control for full MS and MS/MS were set to 1 × 106 and 5 × 104 ions, respectively. The maximum ion injection time was set to 100 ms for MS and 250 ms for MS/MS scans. The lock mass option was enabled using polydimethylcyclosiloxane ions (m/z, 445.120025) for internal calibration.
Data analysis
The raw data obtained was searched using Sequest and Mascot (version 2.2.0, Matrix Science, London, UK) search algorithms through Proteome Discoverer (version 1.4.0.288, Thermo Scientific, Bremen, Germany) suite against NCBI RefSeq human protein database (version 65 containing 34,453 protein sequences and known contaminants). The search parameters included trypsin as proteolytic enzyme with 1 missed cleavage and oxidation of methionine as variable modification. The static modifications included alkylation (methylthio) at cysteine and iTRAQ modification at N-terminus of the peptide and lysine. Mass tolerances of precursor and fragment ions were set to 20 ppm and 0.1 Da, respectively. False discovery rate of 1% was considered to report identification. The average of reporter ion intensities from technical replicates was used for iTRAQ quantitation. Quantitation of identified peptide was normalized on protein median.
Western blotting
Cells were grown to 80% confluence and the proteins were harvested in RIPA lysis buffer (10 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton-X-100, 0.1% SDS containing protease and phosphatase inhibitor cocktails) and sonicated. Western blot analysis was carried out as described previously.Citation30 Briefly, 30 µg of the cell lysate was resolved by SDS-PAGE and transferred onto nitrocellulose membrane. The membrane was blocked with 5% non-fat dry milk in PBS-T and incubated overnight with monoclonal mouse anti-SCD antibody (catalog # ab19862, Abcam, Cambridge, MA). Anti-mouse IgG bound to the HRP conjugate was used as a secondary antibody. Proteins on the membrane were visualized using enhanced chemiluminescence detection kit as per manufacturer's instructions. β-Actin was used as loading control for all Western blots.
Cell invasion assays
Invasion assay was performed in a transwell system (BD Biosciences, San Jose, CA) with Matrigel coated filters and cellular invasion was evaluated after 48h. Briefly, invasiveness of the cells was assayed in the membrane invasion culture system using polyethylene terephthalate (PET) membrane (8-µm pore size) (Catalog # 353097, BD Biosciences, San Jose, CA) in the upper compartment of a transwell coated with Matrigel (Catalog # 354234, BD Biosciences, San Jose, CA). The cells were seeded at 2.0 × 104 cells in 500 µl of media on the Matrigel-coated PET membrane in the upper compartment. The lower compartment was filled with complete growth media and the plates were incubated at 37°C for 48 h. At the end of the incubation time, the upper surface of the membrane was wiped with a cotton-tip applicator to remove nonmigratory cells. Cells that migrated to bottom side of membrane were fixed and stained using 4% methylene blue. Each measurement was performed in duplicate. All experiments were repeated thrice.
Colony formation assays
OKF6/TERT1-Tobacco and the HNSCC cell lines were transfected with either SCD siRNA or control siRNA. Post-transfection 3×103 cells were seeded into 6-well plates with complete media. Cell colonies were allowed to grow for 10–14 days, before the colonies were fixed with methanol and stained with 4% methylene blue. The number of colonies per well was counted. Similarly, colony formation ability of the OKF6/TERT1-Tobacco and HNSCC cells was monitored in presence of SCD inhibitor, CAY10566 (3-[4-(2-chloro-5-fluorophenoxy)-1-piperidinyl]-6-(5-methyl-1,3,4-oxadiazol-2-yl)-pyridazine) (Catalog # CAY10566, Cayman Chemicals Ann Arbor, USA). All experiments were performed in triplicate.
Data availability
The mass spectrometry data files generated in this study can be accessed freely available at ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE public data repository.Citation51 The data can be downloaded using dataset identifier PXD001554. Alternatively, the peptides and their associated MS/MS spectra can be downloaded from Human Proteinpedia at http://www.humanproteinpedia.org/data_display?exp_id=00804.Citation52
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Acknowledgments
We thank the Department of Biotechnology (DBT), Government of India for research support to the Institute of Bioinformatics (IOB), Bangalore. We thank the “Infosys Foundation” for research support to the Institute of Bioinformatics. IOB is supported by DBT Program Support on Neuroproteomics and infrastructure for proteomic data analysis (BT/01/COE/08/05). This work was supported by the Science and Engineering Research Board, Department of Science and Technology, Government of India grant “miRNAs in chronic tobacco-induced oral cancer (SR/S0/HS-02081/2012);” NCI's Clinical Proteomic Tumor Analysis Consortium initiative (U24CA160036), and FAMRI-funded 072017_YCSA. Gajanan J. Sathe and Aneesha Radhakrishnan are recipients of Senior Research Fellowship from the Council of Scientific and Industrial Research (CSIR), New Delhi, India. Santosh Renuse and Nazia Syed are recipients of Senior Research Fellowship from University Grants Commission (UGC), India. Remya Raja is a recipient of Research Associateship from DBT, Government of India. Harsha Gowda is a Wellcome Trust/DBT India Alliance Early Career Fellow. We thank Dr. S. K. Shankar of National Institute of Mental Health and Neurological Sciences (NIMHANS), for providing access to the microscopy imaging facility. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Hashibe M, Brennan P, Benhamou S, Castellsague X, Chen C, Curado MP, Dal Maso L, Daudt AW, Fabianova E, Fernandez L, et al. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. J Natl Cancer Inst 2007; 99:777-89; PMID:17505073; http://dx.doi.org/10.1093/jnci/djk179
- Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer 2011; 11:9-22; PMID:21160525; http://dx.doi.org/10.1038/nrc2982
- http://www.who.int/mediacentre/factsheets/fs339/en/.
- Giovino GA, Mirza SA, Samet JM, Gupta PC, Jarvis MJ, Bhala N, Peto R, Zatonski W, Hsia J, Morton J, et al. Tobacco use in 3 billion individuals from 16 countries: an analysis of nationally representative cross-sectional household surveys. Lancet 2012; 380:668-79; PMID:22901888; http://dx.doi.org/10.1016/S0140-6736(12)61085-X
- Boffetta P, Aagnes B, Weiderpass E, Andersen A. Smokeless tobacco use and risk of cancer of the pancreas and other organs. Int J Cancer 2005; 114:992-5; PMID:15645430; http://dx.doi.org/10.1002/ijc.20811
- Boffetta P, Hecht S, Gray N, Gupta P, Straif K. Smokeless tobacco and cancer. Lancet Oncol 2008; 9:667-75; PMID:18598931; http://dx.doi.org/10.1016/S1470-2045(08)70173-6
- http://monographs.iarc.fr/ENG/Monographs/vol89/mono89.pdf.
- Hoffmann D, Djordjevic MV. Chemical composition and carcinogenicity of smokeless tobacco. Adv Dent Res 1997; 11:322-9; PMID:9524432; http://dx.doi.org/10.1177/08959374970110030301
- Hecht SS, Carmella SG, Murphy SE, Riley WT, Le C, Luo X, Mooney M, Hatsukami DK. Similar exposure to a tobacco-specific carcinogen in smokeless tobacco users and cigarette smokers. Cancer Epidemiol Biomarkers Prev 2007; 16:1567-72; PMID:17684130; http://dx.doi.org/10.1158/1055-9965.EPI-07-0227
- Hoffmann D, Hoffmann I, El-Bayoumy K. The less harmful cigarette: a controversial issue. a tribute to Ernst L. Wynder. Chem Res Toxicol 2001; 14:767-90; PMID:11453723; http://dx.doi.org/10.1021/tx000260u
- Wang Y, Rotem E, Andriani F, Garlick JA. Smokeless tobacco extracts modulate keratinocyte and fibroblast growth in organotypic culture. J Dent Res 2001; 80:1862-6; PMID:11926249; http://dx.doi.org/10.1177/00220345010800091801
- Rohatgi N, Kaur J, Srivastava A, Ralhan R. Smokeless tobacco (khaini) extracts modulate gene expression in epithelial cell culture from an oral hyperplasia. Oral Oncol 2005; 41:806-20; PMID:15979382; http://dx.doi.org/10.1016/j.oraloncology.2005.04.010
- Rohatgi N, Matta A, Kaur J, Srivastava A, Ralhan R. Novel molecular targets of smokeless tobacco (khaini) in cell culture from oral hyperplasia. Toxicology 2006; 224:1-13; PMID:16730401; http://dx.doi.org/10.1016/j.tox.2006.03.014
- Macha MA, Matta A, Chauhan SS, Siu KW, Ralhan R. Guggulsterone (GS) inhibits smokeless tobacco and nicotine-induced NF-kappaB and STAT3 pathways in head and neck cancer cells. Carcinogenesis 2011; 32:368-80; PMID:21177768; http://dx.doi.org/10.1093/carcin/bgq278
- Sawhney M, Rohatgi N, Kaur J, Shishodia S, Sethi G, Gupta SD, Deo SV, Shukla NK, Aggarwal BB, Ralhan R. Expression of NF-kappaB parallels COX-2 expression in oral precancer and cancer: association with smokeless tobacco. Int J Cancer 2007; 120:2545-56; PMID:17354234; http://dx.doi.org/10.1002/ijc.22657
- Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J 2012; 279:2610-23; PMID:22621751; http://dx.doi.org/10.1111/j.1742-4658.2012.08644.x
- Igal RA. Stearoyl-CoA desaturase-1: a novel key player in the mechanisms of cell proliferation, programmed cell death and transformation to cancer. Carcinogenesis 2010; 31:1509-15; PMID:20595235; http://dx.doi.org/10.1093/carcin/bgq131
- Enoch HG, Catala A, Strittmatter P. Mechanism of rat liver microsomal stearyl-CoA desaturase. Studies of the substrate specificity, enzyme-substrate interactions, and the function of lipid. J Biol Chem 1976; 251:5095-103; PMID:8453
- Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr., Ory DS, Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 2003; 100:3077-82; PMID:12629214; http://dx.doi.org/10.1073/pnas.0630588100
- Park EJ, Lee AY, Park S, Kim JH, Cho MH. Multiple pathways are involved in palmitic acid-induced toxicity. Food Chem Toxicol 2014; 67:26-34; PMID:24486139; http://dx.doi.org/10.1016/j.fct.2014.01.027
- Du X, Wang QR, Chan E, Merchant M, Liu J, French D, Ashkenazi A, Qing J. FGFR3 stimulates stearoyl CoA desaturase 1 activity to promote bladder tumor growth. Cancer Res 2012; 72:5843-55; PMID:23019225; http://dx.doi.org/10.1158/0008-5472.CAN-12-1329
- Hess D, Chisholm JW, Igal RA. Inhibition of stearoylCoA desaturase activity blocks cell cycle progression and induces programmed cell death in lung cancer cells. PLoS One 2010; 5:e11394
- Scaglia N, Igal RA. Inhibition of Stearoyl-CoA Desaturase 1 expression in human lung adenocarcinoma cells impairs tumorigenesis. Int J Oncol 2008; 33:839-50; PMID:18813799
- Jaattela M. Escaping cell death: survival proteins in cancer. Exp Cell Res 1999; 248:30-43; PMID:10094811; http://dx.doi.org/10.1006/excr.1999.4455
- Liu G, Lynch JK, Freeman J, Liu B, Xin Z, Zhao H, Serby MD, Kym PR, Suhar TS, Smith HT, et al. Discovery of potent, selective, orally bioavailable stearoyl-CoA desaturase 1 inhibitors. J Med Chem 2007; 50:3086-100; PMID:17530838; http://dx.doi.org/10.1021/jm070219p
- Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc 2006; 1:2315-9; PMID:17406473; http://dx.doi.org/10.1038/nprot.2006.339
- Subramani R, Lopez-Valdez R, Arumugam A, Nandy S, Boopalan T, Lakshmanaswamy R. Targeting insulin-like growth factor 1 receptor inhibits pancreatic cancer growth and metastasis. PLoS One 2014; 9:e97016; PMID:24809702; http://dx.doi.org/10.1371/journal.pone.0097016
- Chang SS, Jiang WW, Smith I, Glazer C, Sun WY, Mithani S, Califano JA. Chronic cigarette smoke extract treatment selects for apoptotic dysfunction and mitochondrial mutations in minimally transformed oral keratinocytes. Int J Cancer 2010; 126:19-27; PMID:19634139; http://dx.doi.org/10.1002/ijc.24777
- Brait M, Munari E, LeBron C, Noordhuis MG, Begum S, Michailidi C, Gonzalez-Roibon N, Maldonado L, Sen T, Guerrero-Preston R, et al. Genome-wide methylation profiling and the PI3K-AKT pathway analysis associated with smoking in urothelial cell carcinoma. Cell Cycle 2013; 12:1058-70; PMID:23435205; http://dx.doi.org/10.4161/cc.24050
- Chang X, Ravi R, Pham V, Bedi A, Chatterjee A, Sidransky D. Adenylate kinase 3 sensitizes cells to cigarette smoke condensate vapor induced cisplatin resistance. PLoS One 2011; 6:e20806; PMID:21698293; http://dx.doi.org/10.1371/journal.pone.0020806
- Kim MS, Huang Y, Lee J, Zhong X, Jiang WW, Ratovitski EA, Sidransky D. Cellular transformation by cigarette smoke extract involves alteration of glycolysis and mitochondrial function in esophageal epithelial cells. Int J Cancer 2010; 127:269-81; PMID:19937795
- Sun W, Chang SS, Fu Y, Liu Y, Califano JA. Chronic CSE treatment induces the growth of normal oral keratinocytes via PDK2 upregulation, increased glycolysis and HIF1alpha stabilization. PLoS One 2011; 6:e16207; PMID:21283817; http://dx.doi.org/10.1371/journal.pone.0016207
- Bock FG, Moore GE, Crouch SK. Tumor-Promoting Activity of Extracts of Unburned Tobacco. Science 1964; 145:831-3; PMID:14163329; http://dx.doi.org/10.1126/science.145.3634.831
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ. BCL−2, BCL−X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 2001; 8:705-11; PMID:11583631; http://dx.doi.org/10.1016/S1097-2765(01)00320-3
- Teni T, Pawar S, Sanghvi V, Saranath D. Expression of bcl−2 and bax in chewing tobacco-induced oral cancers and oral lesions from India. Pathol Oncol Res 2002; 8:109-14; PMID:12172574; http://dx.doi.org/10.1007/BF03033719
- Olopade OI, Adeyanju MO, Safa AR, Hagos F, Mick R, Thompson CB, Recant WM. Overexpression of BCL−x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J Sci Am 1997; 3:230-7; PMID:9263629
- Avalos BR, Kaufman SE, Tomonaga M, Williams RE, Golde DW, Gasson JC. K562 cells produce and respond to human erythroid-potentiating activity. Blood 1988; 71:1720-5; PMID:2836003
- Hu J, Lu J, Lian G, Zhang J, Hecht JL, Sheen VL. Filamin B regulates chondrocyte proliferation and differentiation through Cdk1 signaling. PLoS One 2014; 9:e89352; PMID:24551245; http://dx.doi.org/10.1371/journal.pone.0089352
- Schluter C, Duchrow M, Wohlenberg C, Becker MH, Key G, Flad HD, Gerdes J. The cell proliferation-associated antigen of antibody Ki-67: a very large, ubiquitous nuclear protein with numerous repeated elements, representing a new kind of cell cycle-maintaining proteins. J Cell Biol 1993; 123:513-22; PMID:8227122; http://dx.doi.org/10.1083/jcb.123.3.513
- Goke R, Gregel C, Goke A, Arnold R, Schmidt H, Lankat-Buttgereit B. Programmed cell death protein 4 (PDCD4) acts as a tumor suppressor in neuroendocrine tumor cells. Ann N Y Acad Sci 2004; 1014:220-1; PMID:15153438; http://dx.doi.org/10.1196/annals.1294.024
- Nagy N, Brenner C, Markadieu N, Chaboteaux C, Camby I, Schafer BW, Pochet R, Heizmann CW, Salmon I, Kiss R, et al. S100A2, a putative tumor suppressor gene, regulates in vitro squamous cell carcinoma migration. Lab Invest 2001; 81:599-612; PMID:11304580; http://dx.doi.org/10.1038/labinvest.3780269
- Chiu CC, Lin CY, Lee LY, Chen YJ, Kuo TF, Chang JT, Liao CT, Wang HM, Yen TC, Shen CR, et al. Glucose-regulated protein 78 regulates multiple malignant phenotypes in head and neck cancer and may serve as a molecular target of therapeutic intervention. Mol Cancer Ther 2008; 7:2788-97; PMID:18790759; http://dx.doi.org/10.1158/1535-7163.MCT-08-0172
- Sapkota D, Bruland O, Costea DE, Haugen H, Vasstrand EN, Ibrahim SO. S100A14 regulates the invasive potential of oral squamous cell carcinoma derived cell-lines in vitro by modulating expression of matrix metalloproteinases, MMP1 and MMP9. Eur J Cancer 2011; 47:600-10; PMID:21074410; http://dx.doi.org/10.1016/j.ejca.2010.10.012
- Xia F, Xu JC, Zhang P, Zhang YY, Zhang QW, Chao ZH, Wang F. Glucose-regulated protein 78 and heparanase expression in oral squamous cell carcinoma: correlations and prognostic significance. World J Surg Oncol 2014; 12:121; PMID:24766948; http://dx.doi.org/10.1186/1477-7819-12-121
- Fritz V, Benfodda Z, Rodier G, Henriquet C, Iborra F, Avances C, Allory Y, de la Taille A, Culine S, Blancou H, et al. Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Mol Cancer Ther 2010; 9:1740-54; PMID:20530718; http://dx.doi.org/10.1158/1535-7163.MCT-09-1064
- Mauvoisin D, Charfi C, Lounis AM, Rassart E, Mounier C. Decreasing stearoyl-CoA desaturase-1 expression inhibits β-catenin signaling in breast cancer cells. Cancer Sci 2013; 104:36-42; PMID:23013158; http://dx.doi.org/10.1111/cas.12032
- Scaglia N, Chisholm JW, Igal RA. Inhibition of stearoylCoA desaturase-1 inactivates acetyl-CoA carboxylase and impairs proliferation in cancer cells: role of AMPK. PLoS One 2009; 4:e6812; PMID:19710915; http://dx.doi.org/10.1371/journal.pone.0006812
- Walker JM. The bicinchoninic acid (BCA) assay for protein quantitation. Methods Mol Biol 1994; 32:5-8; PMID:7951748
- Marimuthu A, Chavan S, Sathe G, Sahasrabuddhe NA, Srikanth SM, Renuse S, Ahmad S, Radhakrishnan A, Barbhuiya MA, Kumar RV, et al. Identification of head and neck squamous cell carcinoma biomarker candidates through proteomic analysis of cancer cell secretome. Biochim Biophys Acta 2013; 1834:2308-16; PMID:23665456; http://dx.doi.org/10.1016/j.bbapap.2013.04.029
- Marimuthu A, Subbannayya Y, Sahasrabuddhe NA, Balakrishnan L, Syed N, Sekhar NR, Katte TV, Pinto SM, Srikanth SM, Kumar P, et al. SILAC-based quantitative proteomic analysis of gastric cancer secretome. Proteomics Clin Appl 2013; 7:355-66; PMID:23161554; http://dx.doi.org/10.1002/prca.201200069
- Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res 2013; 41:D1063-9; PMID:23203882; http://dx.doi.org/10.1093/nar/gks1262
- Muthusamy B, Thomas JK, Prasad TS, Pandey A. Access guide to human proteinpedia. Curr Protoc Bioinformatics 2013; Chapter 1:Unit 1 21; PMID:23504933
- Fujimura A, Kishimoto H, Yanagisawa J, Kimura K. Enhancer of rudimentary homolog (ERH) plays an essential role in the progression of mitosis by promoting mitotic chromosome alignment. Biochem Biophys Res Commun 2012; 423:588-92; PMID:22704934; http://dx.doi.org/10.1016/j.bbrc.2012.06.018
- Nishikawa M, Kira Y, Yabunaka Y, Inoue M. Identification and characterization of endoplasmic reticulum-associated protein, ERp43. Gene 2007; 386:42-51; PMID:17020792; http://dx.doi.org/10.1016/j.gene.2006.06.030
- Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol 2000; 182:311-22; PMID:10653597; http://dx.doi.org/10.1002/(SICI)1097-4652(200003)182:3%3c311::AID-JCP1%3e3.0.CO;2-9
- Gasson JC, Golde DW, Kaufman SE, Westbrook CA, Hewick RM, Kaufman RJ, Wong GG, Temple PA, Leary AC, Brown EL, et al. Molecular characterization and expression of the gene encoding human erythroid-potentiating activity. Nature 1985; 315:768-71; PMID:3839290; http://dx.doi.org/10.1038/315768a0
- Seo WY, Jeong BC, Yu EJ, Kim HJ, Kim SH, Lim JE, Kwon GY, Lee HM, Kim JH. CCAR1 promotes chromatin loading of androgen receptor (AR) transcription complex by stabilizing the association between AR and GATA2. Nucleic Acids Res 2013; 41:8526-36; PMID:23887938; http://dx.doi.org/10.1093/nar/gkt644
- Fazioli F, Minichiello L, Matoska V, Castagnino P, Miki T, Wong WT, Di Fiore PP. Eps8, a substrate for the epidermal growth factor receptor kinase, enhances EGF-dependent mitogenic signals. EMBO J 1993; 12:3799-808; PMID:8404850
- Wolf R, Howard OM, Dong HF, Voscopoulos C, Boeshans K, Winston J, Divi R, Gunsior M, Goldsmith P, Ahvazi B, et al. Chemotactic activity of S100A7 (Psoriasin) is mediated by the receptor for advanced glycation end products and potentiates inflammation with highly homologous but functionally distinct S100A15. J Immunol 2008; 181:1499-506; PMID:18606705; http://dx.doi.org/10.4049/jimmunol.181.2.1499
- Miyake H, Hara I, Arakawa S, Kamidono S. Stress protein GRP78 prevents apoptosis induced by calcium ionophore, ionomycin, but not by glycosylation inhibitor, tunicamycin, in human prostate cancer cells. J Cell Biochem 2000; 77:396-408; PMID:10760948; http://dx.doi.org/10.1002/(SICI)1097-4644(20000601)77:3%3c396::AID-JCB5%3e3.0.CO;2-5
- Ghosh AK, Steele R, Ray RB. Functional domains of c-myc promoter binding protein 1 involved in transcriptional repression and cell growth regulation. Mol Cell Biol 1999; 19:2880-6; PMID:10082554
- Yang Z, Chang YJ, Miyamoto H, Ni J, Niu Y, Chen Z, Chen YL, Yao JL, di Sant'Agnese PA, Chang C. Transgelin functions as a suppressor via inhibition of ARA54-enhanced androgen receptor transactivation and prostate cancer cell growth. Mol Endocrinol 2007; 21:343-58; PMID:17082327; http://dx.doi.org/10.1210/me.2006-0104
- Diaz E, Pfeffer SR. TIP47: a cargo selection device for mannose 6-phosphate receptor trafficking. Cell 1998; 93:433-43; PMID:9590177; http://dx.doi.org/10.1016/S0092-8674(00)81171-X
- Sagona AP, Nezis IP, Pedersen NM, Liestol K, Poulton J, Rusten TE, Skotheim RI, Raiborg C, Stenmark H. PtdIns(3)P controls cytokinesis through KIF13A-mediated recruitment of FYVE-CENT to the midbody. Nat Cell Biol 2010; 12:362-71; PMID:20208530; http://dx.doi.org/10.1038/ncb2036
- Takeuchi T, Misaki A, Liang SB, Tachibana A, Hayashi N, Sonobe H, Ohtsuki Y. Expression of T-cadherin (CDH13, H-Cadherin) in human brain and its characteristics as a negative growth regulator of epidermal growth factor in neuroblastoma cells. J Neurochem 2000; 74:1489-97; PMID:10737605; http://dx.doi.org/10.1046/j.1471-4159.2000.0741489.x
- Stetak A, Veress R, Ovadi J, Csermely P, Keri G, Ullrich A. Nuclear translocation of the tumor marker pyruvate kinase M2 induces programmed cell death. Cancer Res 2007; 67:1602-8; PMID:17308100; http://dx.doi.org/10.1158/0008-5472.CAN-06-2870
- Ohuchida K, Mizumoto K, Ohhashi S, Yamaguchi H, Konomi H, Nagai E, Yamaguchi K, Tsuneyoshi M, Tanaka M. S100A11, a putative tumor suppressor gene, is overexpressed in pancreatic carcinogenesis. Clin Cancer Res 2006; 12:5417-22; PMID:17000675; http://dx.doi.org/10.1158/1078-0432.CCR-06-0222
- Silva JP, Lelianova VG, Ermolyuk YS, Vysokov N, Hitchen PG, Berninghausen O, Rahman MA, Zangrandi A, Fidalgo S, Tonevitsky AG, et al. Latrophilin 1 and its endogenous ligand Lasso/teneurin-2 form a high-affinity transsynaptic receptor pair with signaling capabilities. Proc Natl Acad Sci U S A 2011; 108:12113-8; PMID:21724987; http://dx.doi.org/10.1073/pnas.1019434108
- Koya RC, Fujita H, Shimizu S, Ohtsu M, Takimoto M, Tsujimoto Y, Kuzumaki N. Gelsolin inhibits apoptosis by blocking mitochondrial membrane potential loss and cytochrome c release. J Biol Chem 2000; 275:15343-9; PMID:10809769; http://dx.doi.org/10.1074/jbc.275.20.15343