
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Bruton's tyrosine kinase (BTK) is a non-receptor tyrosine kinase that has mainly been studied in haematopoietic cells. We have investigated whether BTK is a potential therapeutic target in prostate cancer. We find that BTK is expressed in prostate cells, with the alternate BTK-C isoform predominantly expressed in prostate cancer cells and tumors. This isoform is transcribed from an alternative promoter and results in a protein with an amino-terminal extension. Prostate cancer cell lines and prostate tumors express more BTK-C transcript than the malignant NAMALWA B-cell line or human lymphomas. BTK protein expression is also observed in tumor tissue from prostate cancer patients. Down regulation of this protein with RNAi or inhibition with BTK-specific inhibitors, Ibrutinib, AVL-292 or CGI-1746 decrease cell survival and induce apoptosis in prostate cancer cells. Microarray results show that inhibiting BTK under these conditions increases expression of apoptosis related genes, while overexpression of BTK-C is associated with elevated expression of genes with functions related to cell adhesion, cytoskeletal structure and the extracellular matrix. These results are consistent with studies that show that BTK signaling is important for adhesion and migration of B cells and suggest that BTK-C may confer similar properties to prostate cancer cells. Since BTK-C is a survival factor for these cells, it represents both a potential biomarker and novel therapeutic target for prostate cancer.
Introduction
Therapeutic resistance and metastasis continue to be major challenges in the clinical management of prostate cancer, which is the most commonly diagnosed non-cutaneous cancer in American males and the second leading cause of cancer-related deaths in males in North America.Citation1 Identifying novel mediators that regulate the growth and survival of cancer cells has accelerated the development of therapies that have steadily improved clinical outcomes in cancer patients.Citation2,3 Protein tyrosine kinase inhibitors are among the most promising targeted therapies.Citation4 Dysregulation of tyrosine kinases often occurs through activating mutations or overexpression, and as such members of the family are the most commonly identified dominant oncogenes. Dysregulation of these enzymes causes increases in tumor cell proliferation and abrogation of apoptotic pathways while promoting angiogenesis and metastasis.Citation5-9
Previously, we performed a genome-wide RNAi screen for tyrosine kinase genes whose function is critical for breast cancer cell survival. Bruton's Tyrosine kinase (BTK) was among those genes whose knockdown causes the most significant reduction in survival.Citation10 BTK is a critical regulator of B cell receptor (BCR) signaling.Citation11,12 Mutations in the btk gene lead to B cell deficiency manifested as X-linked agammaglobulinemia in humans and the related but less severe X-linked immunodeficiency (xid) in mice.Citation13 The role of BTK in B-cell development and B-cell malignancies has been extensively studied.Citation14-17 In haematopoietic cells, BTK is involved in multiple signal-transduction pathways regulating survival, activation, proliferation, and differentiation of B-lymphocytes.Citation15,18,Citation19 BTK plays a key role in regulation of osteoclast biology and normal bone homeostasis, and BTK-mediated signaling is disrupted in several bone disorders including osteoporosis and rheumatoid arthritis.Citation18,20 Recently, BTK has emerged as a novel target for the treatment of rheumatoid arthritis and other immune diseases due to its role as a crucial effector in the B-cell antigen receptor (BCR) signaling pathway.Citation21 BTK kinase inhibitors include PCI-32765 (Ibrutinib), AVL-292 and CGI-1746, originally developed as immunosuppressants, this class is among the most promising blood cancer chemotherapeutics in current clinical trials.Citation6 Ibrutinib has already successfully completed phase III clinical testing and is approved for 2 haematopoietic malignancies, mantle cell lymphoma and multiple myeloma.Citation22 Other compounds with BTK-inhibitory properties such as AVL-292, CGI-1746, GDC-0834, HM-71224 and ONO-4059, have progressed through advanced preclinical development to clinical trials (http://clinicaltrials.gov).Citation22,23
We have recently shown that a novel isoform of BTK (BTK-C) is the form of the gene most often expressed in breast cancer cells.Citation10 This isoform is transcribed from an alternative promoter and produces a product that contains an amino-terminal 34 amino acid extension immediately upstream of the pleckstrin homology domain which is critical for BTK signaling in B-cells.Citation10,13 The BTK-C transcript is driven by an alternative promoter located upstream from the BTK-A promoter.Citation10 The transcriptional start site of BTK-C is 255 bp from the start site of ribosomal protein L36a gene which is transcribed in the opposite direction.Citation10
Given the importance of BTK activity in breast cancer cell survivalCitation10 and, as recently reported, prostate cancer,Citation24 we have examined the expression of the novel BTK isoform (BTK-C) in prostate cancer cell lines and tissue microarrays. We show that BTK expression is elevated in a number of prostate cancer cell lines and tumors. Inhibition of BTK with BTK-C siRNA demonstrates that alternative BTK protein isoforms contribute to prostate cancer cell survival. These data indicate that, in addition to its utility in haematopoietic malignancies, targeting BTK may be a potent therapeutic approach for advanced prostate cancer, particularly castration- resistant prostate cancer. These studies provide the framework for clinical development of BTK inhibitors as a novel therapeutic strategy in prostate cancer.
Results
BTK expression in prostate cancer cells
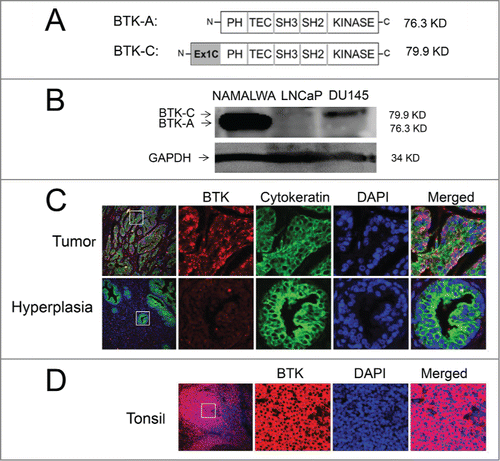
The BTK-C isoform has only recently been described and little is understood regarding its expression. This is due in part to the fact that Affymetrix probes for this region have only been included in exon microarrays very recently. Additionally, the BTK-C isoform encodes the entire B-cell version sequence (BTK-A) and is annotated as a 5′ UTR splice variant of BTK-A.Citation10 An initial exon-level analysis of BTK expression in prostate cancer was carried out using publicly available Affymetrix exon array data in cell intensity format (CEL file) with Affymetrix Expression Console Software. CEL files corresponding to accession number GSE41407 were downloaded from the Gene Expression Omnibus (GEO). BTK-C expression levels were evaluated in several prostate cancer cell lines. These data show that BTK-C represents greater than 50% of the total BTK expression in prostate cancer cells, with the exception of the LAPCA and 22RV1 cell lines. In contrast, BTK-A represents the most prevalent isoform detected in lymphoma cells (Supplementary ). In keeping with these results, we detect BTK-C expression using RT-PCR in both LNCaP and DU145 prostate cancer cells (Supplementary ). Isoform specific qPCR primer sets that discriminate between BTK-A and BTK-C were designed to the heterogeneous regions of the 2 sequences located within the 5′UTRs. As shown in , using the Tissue Scan Cancer Survey Panel, prostate cancer cell lines and the majority of prostate tumors express more BTK-C than BTK-A transcript. In contrast, the NAMALWA B-cell line and leukemia and lymphoma tumors, predominantly express the B-cell isoform, BTK-A. Similar results are seen at the protein level. The BTK-C isoform encodes a product with an amino-terminal 34 amino acid extension that adds 3.6 kDa to the mass of the BTK-A isoform (). A product consistent with this size is observed on immunoblots as an 80 kDa product in prostate cell lines. This agrees with the BTK-C predicted size of 79.9 kDa and is larger than 76.3 kDa BTK-A product in the NAMALWA cells (). Immunofluorescent staining of the prostate cancer tissue microarray (PR8011) shows increased BTK expression in invasive prostate cancers relative to normal and benign prostate tissues using an antibody that recognizes both BTK isoforms (). Supplementary Table 1 summarizes BTK protein expression observed in clinical prostate cancer specimens, including positive staining of a majority of malignant tumors (70%). Staining is less pronounced in prostate cancer epithelial cells compared to tonsil tissue, which contains B-cells and serves as a positive control for BTK expression (). These results confirm the expression of BTK in prostate tumors, although isoform-specific protein level determination is not currently possible since BTK-C specific antibodies are not yet available.
Figure 1. BTK expression in prostate cell lines and tumors. BTK-C message is more abundant than the BTK-A isoform in prostate cancer cells and tumors. cDNA prepared from RNA isolated from prostate cancer cell lines (A) and human prostate normal and tumor tissues. (B) was subjected to qPCR using primers specific for BTK-A and BTK-C isoforms. Expression of each isoform was normalized to an actin control in the respective qPCR reaction. The data represent relative mRNA levels of each BTK isoform as fold increase of the normal tissue sample with the lowest expression. Lymphoma samples are shown for comparison. BTK-C was the predominant isoform in only 3% of lymphomas but 40% of prostate tumors (6 out of 15 cases).
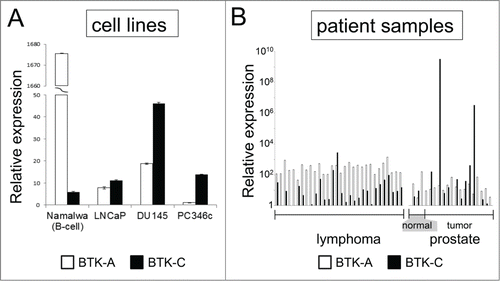
Figure 2. The BTK-(C)isoform is expressed in prostate cancer cell lines and tumors. (A) Schematic representation showing the domains of BTK-A and predicted BTK-C protein. (B) Total lysate from LNCaP, DU145 and Namalwa B-cells subjected to immunoblot analysis blotted and probed with an anti-BTK antibody. (C) Immunohistochemical staining shows increased expression of BTK in malignant prostate cancer tissue compared to hyperplasia prostate tissue. (D) Immunohistochemical staining shows expression of BTK in tonsil tissue using a BTK antibody.
BTK-C is a survival factor in prostate cancer cells
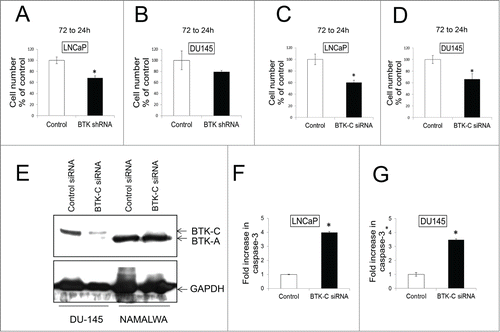
Our previous studies demonstrated that knockdown of BTK has a significant effect on survival of breast cancer cell lines in culture.Citation10 In LNCaP and DU145 prostate cancer cell lines, shRNA-mediated knockdown of BTK results in a 32% and 21% decrease in cell number respectively, although the decrease in DU145 does not reach statistical significance (). However, an siRNA that has been established as specifically silencing the BTK-C isoform transcriptCitation10 leads to a statistically significant decrease in prostate cancer cell number. Transfection of this siRNA into LNCaP and DU145 cells leads to knockdown of BTK-C RNA of 40% and 34% respectively compared with control siRNA after 72 h (). DU145 and NAMALWA cells were transfected with BTK-C specific siRNA and control siRNA for 48h, the immunoblot results show BTK-C siRNA just decreases the BTK-C protein and not BTK-A protein in NAMALWA cells (). These results demonstrate that the BTK-C siRNA is effective at silencing BTK gene expression and that this isoform is critical for prostate cancer cell survival. To establish that the decrease in cell number is the result of apoptosis, caspase-3 cleavage (Asp175) was assessed after transfecting with siRNA specific for BTK-C into LNCaP and DU145 cells. Apoptosis increases by fold4- and 3.fold5- in LNCaP and DU145 cells, respectively, compared to control (). In DU145 cultures, apoptotic cells represented 3.45% of the population in non-silencing siRNA transfected controls and 13.48% in cells transfected with the BTK-C-specific siRNA. Similarly, 3.6% of LNCaP cells transfected with the control non-silencing siRNA and 13.54% transfected with the BTK-C-specific siRNA were apoptotic.
Figure 3. BTK shRNA and BTK-(C)specific siRNAs knock down decrease cell survival in prostate cancer cell lines. LNCaP (A) and DU145 (B) were transfected with shRNA and co-transfected with GFP to mark transfected cells. Transfected cells were counted at 24h and 72h and the 72h to 24h ratio was calculated and expressed as % of the control. LNCaP (C) and DU145 (D) were transfected with BTK-C specific siRNA or non-targeting siRNA. Co-transfection with a GFP expressing plasmid marks transfected cells. Transfected cells were counted and the 96h to 24h ratio was calculated and expressed as % of the control. (E) DU145 and NAMALWA cells were transfected with BTK-C specific siRNA and control siRNA for 48h. The cell lysates were prepared for immunoblotting. GAPDH is a loading control; the results show BTK-C siRNA just decreases the BTK-C protein and not BTK-A protein in NAMALWA cells. LNCaP (F) and DU145 (G) were transfected with BTK-C specific siRNA or non-targeting siRNA as a control for 48h. Increased cleaved caspase-3 was detected compared with control. Apoptotic cells for each treatment were calculated as fold increase in Caspase-3 positive cells of control. Mean of triplicate assays ± SD. Student t-test, *p < 0.05.
BTK inhibitors potently inhibit prostate cancer cell viability
In haematopoietic cells, BTK is regulated by membrane recruitment via a PH domain that binds phosphatidylinositol 3,4,5 phosphate (PIP3) followed by auto-phosphorylation of tyrosine residue 551 (Y551) in the activation loop and Y233 in the SH3 domain.Citation11,25 Upon activation, it phosphorylates PLC-, leading to activation of the MAPK, NFκB, and AKT signaling pathways.Citation23,25 Small molecules with BTK-inhibitory properties have emerged as promising therapeutic drugs for the treatment of hematological malignancies and autoimmune disorders.Citation26-28 Ibrutinib (PCI-32765) (Pharmacyclics, Janssen) is an orally bioavailable, highly potent small molecule inhibitor against BTK.Citation29 Ibrutinib selectively binds to Cys481 of BTK, irreversibly blocking its kinase activity.Citation30 AVL-292 (Avila Therapeutic, Celgene) also is an orally bioavailable acrylamide derivative with potent, irreversible anti-BTK activity that abolishes BCR signaling by covalently binding to Cys481 of BTK. This selectively inhibits its auto-phosphorylation as well as activation of PLC
2 and other downstream substrates of BTK in B cells.Citation31 CGI-1746 (Axon Medchem, Reston, VA) is a reversible inhibitor that potently inhibits auto-phosphorylation of BTK. It binds and occupies an SH3 binding pocket within the un-phosphorylated form of BTK, stabilizing the inactive enzyme conformation.Citation32
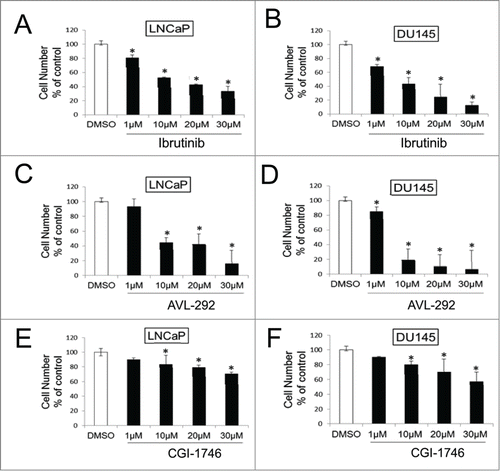
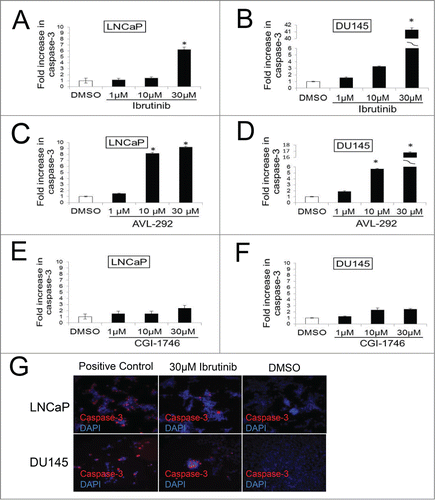
The effect of Ibrutinib, AVL-292 and CGI-1746, was analyzed in LNCaP (AR+) and DU145 (AR-) prostate cancer cells. Both LNCaP and DU145 cells demonstrate a significant dose dependent response to Ibrutinib () and AVL-292 (). Treatment with 10 µM Ibrutinib for 72h reduces cell viability to 57% and 62% for LNCaP and DU145 cells, respectively. Ten µM AVL-292, another irreversible BTK inhibitor, decreases cell viability to 57% and 81% in cultures of LNCaP and DU145 cells, respectively after 72h treatment. In contrast, CGI-1746 does not kill cells as well as the irreversible BTK inhibitors at the same drug concentration ().
Figure 4. BTK inhibitors decrease cell survival in prostate cancer cell lines. LNCaP and DU145 were treated with Ibrutinib (A and B), AVL-292 (C and D) and CGI-1746 (E and F) at the indicated concentrations. Treated cells were counted after 72h of treatment and were presented as % of the control. Mean of triplicate assays ± SD. Student t-test, *p < 0.05.
To establish that the decrease in cell number is the result of apoptosis, caspase-3 cleavage (Asp175) was assessed in LNCaP and DU145 cells 48h after treatment with BTK inhibitors or DMSO. Inhibition of BTK auto-phosphorylation with 30 µM Ibrutinib or AVL-292 induces extensive apoptosis in both cell lines. Apoptosis increases by fold6- in drug treated LNCaP cells and more than 40 fold in DU145 cells compared to control (). Increases in the apoptotic population were also observed in Ibrutinib-treated LNCap cells by Annexin V staining (data not shown). CGI-1746 does not cause significant apoptosis in prostate cancer cells at this concentration (), although it does induce apoptosis in NAMALWA cells (Supplementary Fig. 2). Representative images of apoptotic cells are shown in . Taken together, these data show that the 2 BTK inhibitors that irreversibly abolish BTK activity, Ibrutinib and AVL-292, potently kill prostate cancer cells while CGI-1746, which is a reversible inhibitor, does not.
Figure 5. Inhibition of BTK results in increased apoptosis in prostate cancer cells. DU145 and LNCaP cells were incubated with indicated concentration of Ibrutinib (A and B) and AVL-292 (C and D), CGI (E and F) for 48h results in increased cleaved caspase-3 compared with control cells treated with DMSO. Representative images of apoptotic cells after Ibrutinib or DMSO treatment are shown (G). Apoptotic cells for each treatment were calculated as fold increase of Caspase-3 positive cells of control. Mean of triplicate assays ± SD. Student t-test, *p < 0.05.
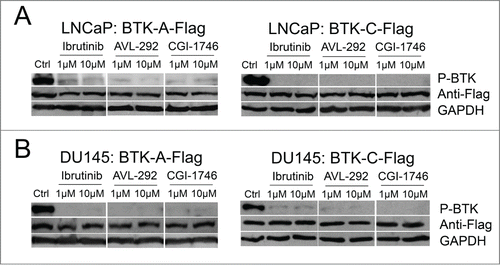
BTK phosphorylation is reduced in prostate cancer cells after treatment with BTK inhibitors
To assess BTK activation in prostate cancer cells, the phosphorylation status of tyrosine 233, which undergoes auto-phosphorylation after activation, was assessed. LNCaP and Du145 cells, stably over-expressing Flag-tagged BTK-A or BTK-C proteins, were treated by BTK inhibitors including Ibrutinib, AVL-292 and CGI-1746 at the indicated concentrations for 24h. Treatment with all 3 BTK inhibitors significantly reduces phosphorylation of both the BTK-A and BTK-C proteins, indicating the auto-phosphorylation of the BTK-C isoform is inhibited in a manner similar to BTK-A (). Notably, even though CGI-1746 does not kill LNCaP or DU145 prostate cancer cells at the same concentrations as Ibrutinib or AVL-292, it demonstrates similar inhibition of BTK phosphorylation at tyrosine 233 in the SH3 domain.
Figure 6. BTK phosphorylation is reduced after treatment with BTK inhibitors in prostate cancer cells. (A) LNCaP or (B) DU145 cells containing the stably integrated BTK-A-Flag, BTK-C-Flag or control vector were treated with Ibrutinib, AVL-292 and CGI-1746 at the indicated concentrations or DMSO as a control for 24h, then cells were lysed, subjected to western blot analysis and probed with the indicated antibodies.
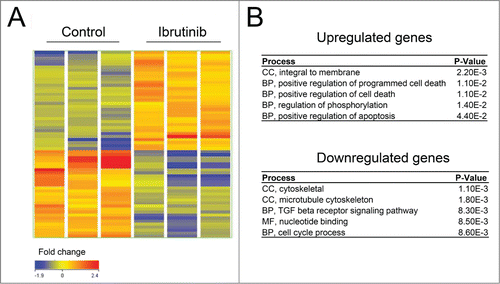
BTK inhibition by Ibrutinib leads to up-regulation of apoptotic genes in prostate cancer cells
Microarray analysis was carried out to explore the potential mechanism of anti-cancer effects of BTK inhibitors in prostate cancer cells. LNCaP cells were treated with 10µM Ibrutinib or vehicle control for 48h. shows a heatmap representation of transcripts exhibiting at least a 1.fold5- change in expression from the control. Ibrutinib treatment of LNCaP cells causes increased expression of 76 genes and decreased expression of 75 genes. Gene ontology (GO) enrichment analysis was performed using the DAVID Bioinformatics Resource Citation33 to identify transcriptional programs regulated in LNCaP cells in response to Ibrutinib. also shows the results of this functional annotation clustering analysis. Up-regulated genes are associated with cellular components including integral membrane (p < 2.2 × 10−3) and biological processes including positive regulation of programmed cell death (p <1.1×10−2), supporting the notion that the induction of apoptosis after BTK inhibition involved selective changes in expression of these ontologies. The top clusters of down-regulated genes are related to cytoskeletal cell components (p <1.1 × 10−3) and biological processes including cell cycle (p <8.6 × 10−3).
Figure 7. BTK inhibition by Ibrutinib leads to upregulation of apoptotic genes. (A) A heatmap representation of transcripts that display at least a 1.5-fold change in expression (B) Results of the Functional Annotation Clustering Analysis after GO-term enrichment of up-regulated and down-regulated genes using the DAVID Bioinformatics Resource. BP; Biological Process, CC; Cellular Component and MF; Molecular Function.
Elevated BTK affects gene expression in prostate cancer cells
As we have shown, the BTK-C isoform is more highly expressed in prostate cancer cells than the BTK-A isoform. It is therefore likely that BTK-C signaling in solid tumor cells will have different downstream consequences than that of the BTK-A isoform in B-cells. To gain a better understanding of how BTK-C acts in prostate cancer cells, we performed microarray analysis on BTK-A or BTK-C overexpressing DU145 cells or an empty vector control to identify potential downstream effectors. shows a heatmap representation of transcripts that display at least a 1.5-fold change in expression. Venn diagram analysis () shows the number of genes that are upregulated or downregulated in BTK-A and BTK-C overexpressing DU145 cells. In BTK-A overexpressing cells, 722 genes (696 + 26 = 722) display increased expression and 320 genes (222 + 98 = 320) show decreased expression. In BTK-C overexpressing cells, 918 genes (696 + 222 = 918) have increased expression and expression of 124 genes (26 + 98 = 124) is decreased. BTK-A and BTK-C isoforms modulate a total of 794 genes in common (696 transcripts are up-regulated 98 transcripts are down-regulated). shows the results of a functional annotation clustering analysis. The most upregulated genes for both isoforms are associated with biological processes such as cell adhesion and cellular components including intermediate filaments and extracellular matrix. Both BTK isoforms up-regulate members of the cadherin family (CDH3, CDH7, CDH15) while CDH11 is only up-regulated by overexpression of the BTK-C isoform. BTK signaling is important for adhesion and migration of B-cells and these gene expression changes may indicate that BTK-C confers similar properties to prostate cancer cells. Although we have observed decreased prostate cell migration and cell invasion after 48h treatment with Ibrutinib (data not shown), we have been unable to unequivocally attribute this effect to changes in migration and invasion rather than to the effects of the drug on cell viability.
Figure 8. Elevated BTK expression affects gene expression in prostate cancer cells. (A) A heatmap representation of transcripts that display at least a 1.fold5- change in expression (B) Venn diagram showing the number of genes those are upregulated or downregulated with overexpression of BTK-A or BTK-C in Du145 cells. (C) Results of the Functional Annotation Clustering Analysis after GO-term enrichment of up-regulated and down-regulated genes using the DAVID Bioinformatics Resource. BP; Biological Process, CC; Cellular Component and MF; Molecular Function.
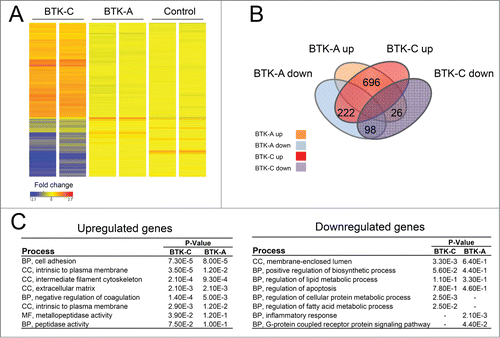
The sets of genes that are downregulated in response to BTK-A or BTK-C overexpression differ between the isoforms. Down-regulated genes in BTK-C overexpressing DU145 cells are associated with regulation of cellular protein metabolism and fatty acid metabolism, while downregulated genes in response to BTK-A overexpression are associated primarily with inflammatory responses. The down-regulated genes include GPNMB and pro-apoptotic genes such as BAK1 and CID for both BTK isoforms. Gene Spring analysis of cells overexpressing BTK-C indicates that many of the genes that are downregulated are involved in fatty acid degradation and cholesterol biosynthesis (Suppl. ). These results indicate that BTK-C may play a role in the alternative energy metabolism that operates in breast and prostate cancer cellsCitation34,35 and that it may have similar effects on glucose metabolism previously associated with BTK-C overexpression.Citation10
Discussion
BTK has attracted interest as a specific therapeutic target in B-cell malignancies and autoimmune diseases.Citation14 In this study, we have shown that BTK is expressed in prostate cancer cells and tumors predominantly as the BTK-C form. Immunofluorescent staining of human prostate cancers shows increased BTK expression in invasive prostate cancers relative to normal and benign prostate tissues. Our current data do not support a correlation between BTK staining and Gleason score. BTK-C specific siRNA causes a significant increase in cell death indicating that expression of BTK (BTK-C) in prostate cancer is critical for cell survival. Microarray experiments show inhibition of BTK, specifically by the irreversible inhibitors Ibrutinib and AVL-292, leads to upregulation of apoptosis related genes. In addition, overexpression of BTK-C is associated with elevated expression of genes with functions related to cell adhesion, cytoskeletal structure and the extracellular matrix, demonstrating that BTK-C related signaling is critical for the survival of prostate cancer cells. These data suggest that the novel BTK-C isoform is an attractive new prostate cancer drug target.
While the study of BTK has mainly been carried out in haematopoietic cells, recent work has indicated that it may be important in solid tumors. BTK-C isoform activity has been shown to be critical for the survival of breast cancer cells.Citation10 A recent study has reported that BTK is found in prostate cancer cells.Citation24 Transcription of BTK-C initiates in the ∼200bp RPL36A-BTK-C intergenic region. CHIP-SEQ data from the ENCODE project indicates that more than 20 transcription factors bind within this region, suggesting that differences in transcriptional regulation of BTK-A and BTK-C isoforms are important for the expression and function of BTK isoforms (http://www.genome.ucsc.edu/ENCODE/).Citation36 The additional 34 amino acid C-terminal extension in BTK-C is likely to impact the enzyme such that the subcellular localization and function of this isoform in solid tumors may be different from BTK-A in haematopoietic cells (X. Wang et al, unpublished). This may significantly influence the response of prostate cancer cell to BTK isoform selective inhibitors.
The BTK inhibitors, Ibrutinib and AVL-292, demonstrate better efficacy in killing prostate cancer cells than CGI-1746, although all 3 inhibitors block phosphorylation of BTK at a similar concentration. That CGI-1746 is not as effective is not surprising since it has the highest IC50 in terms of growth inhibition of B cells of the 3 compounds. Reports have shown that the irreversible BTK inhibitors, Ibrutinib (proliferation IC50 = 8nM)Citation37 and AVL-292 (proliferation IC50 = 3nM)Citation38 are more effective at blocking B cell proliferation in vitro than the reversible inhibitor CGI-1746 (proliferation IC50 = 42nM).Citation32 This may also explain why AVL-292 kills more prostate cancer cells than ibrutinib at 10µM (). At present, the discrepancy between the effect of CGI-1746 on inhibiting the kinase and its effect on B cell proliferation has not been addressed. In prostate cells, residual kinase activity that escapes the reversible inhibitor, differences in the metabolic clearance of the drug, or differences in the subcellular localization of the proteins involved in the survival signaling pathway in which BTK operates may be relevant. For example, CGI-1746 may bind to cytosolic BTK and block its activation, but have low affinity for the active membrane-bound BTK which can potentiate pro-survival signaling downstream. It is also possible that the increased efficacy of Ibrutinib and AVL-292 over CGI-1746 is explained by these drugs having off-target effects due to binding to cysteine residues in kinase domains of the EGFR family or other targets (X. Wang et al, unpublished), even though the RNA interference experiments indicate that BTK itself is required for survival.
Changes in gene expression identified by microarray analysis in prostate cancer cells indicate that BTK may affect transcription of specific targets. Introduction of BTK overexpression constructs in DU145 cells resulted in a considerable increase in the levels of either isoform to levels in the range seen in the NAMALWA cells. Changes in expression of 40 2 genes are found in common between the ibrutinib treated cells and BTK-A or BTK-C overexpressing cells. Supplementary Table 2 shows the results of the functional annotation clustering analysis of transcript changes that supports the induction of apoptosis after BTK inhibition in prostate cancer cells and the suppression of apoptosis in BTK-A and BTK-C overexpressing cells.
A number of both upregulated and down-regulated genes are associated with overexpression of the BTK isoforms. These experiments focused on the effect of overexpression as there were basal levels of expression of both isoforms. For example, BTK-C is expressed in the BTK-A overexpressing DU145 cells, although the amount of intrinsic BTK-C expression is relatively low in comparison to BTK-A expression in BTK-A overexpressing DU145 cells. Expression of cadherins (CDH3, CDH7 and CDH15) is elevated in cells overexpressing either BTK Isoform, whereas cadherin-11 (CDH11) is elevated uniquely in cells overexpressing the BTK-C isoform. Cadherins are a large family of calcium dependent cell adhesion proteins whose differential expression is associated with tumor aggressiveness.Citation39,40 CDH11 is also involved in the metastasis of prostate cancer cells to bone.Citation41 In haematopoietic cells, BTK provides signals that stimulate the proliferation, suppress apoptosis and increase mobility of maturing B-cells.Citation42 These findings suggest that BTK-C confers these same attributes to prostate cancer cells although additional experimentation will be required to test this hypothesis.
Among downregulated genes in either BTK isoform overexpressing cells, the glycoprotein transmembrane nmbgene (GPNMB) expression decreased by18.fold6- and 16.fold7- in BTK-A and BTK-C overexpressing cells, respectively. GPNMB may be involved in growth delay and reduction of metastatic potential in cancer, one study shows GPNMB gene is an anti-tumor gene for prostate cancer.Citation43 Interestingly, genes implicated in fatty acid degradation, cholesterol biosynthesis and statin pathways are also down-regulated in BTK-C overexpressing cells. A recent report shows inhibition of fatty acid degradation plays a role in elevating glucose metabolism in prostate cancer.Citation44 Fatty acids are essential for cancer cell proliferation and limiting their availability can kill cancer cells.Citation45 BTK-C overexpression in prostate cancer may be associated with decreased fatty acid degradation, leading to more fatty acids for anabolic processes required for membrane biogenesis while simultaneously instigating elevated glucose uptake to support central carbon metabolism. Importantly, previous work has shown that BTK-C activity is correlated with glucose uptake in breast cancer cells.Citation10
In summary, our work has shown BTK-C isoform is expressed in prostate cancer cells and tumors and that FDA approved therapeutics targeting BTK-C such as Ibrutinib and drugs in development such as AVL-292 are efficacious in killing prostate cancer cells. These results suggest that drugs targeting BTK warrant additional study as a novel treatment strategy for prostate cancer.
Materials and Methods
Cell culture
Cell lines, NAMALWA, LNCaP, DU145 and Phoenix AMPHO, were obtained from the American Type Culture Collection (Manassas, VA). HEK293FT cells were obtained from Invitrogen. NAMALWA and LNCaP cells were cultured in RPMI-1640 medium (Hyclone, Logan, UT) supplemented with 10% FBS and 100 U/mL of penicillin- streptomycin. DU145, HEK293FT and Phoenix cells were cultured in DMEM (Hyclone, Logan, UT) supplemented with 10% FBS and 100 U/mL of penicillin- streptomycin. PC346c cells were cultured in DME/F12 supplemented with 2% FCS, 0.01% BSA, 10 ng/ml EGF, 1% ITS-G, 1.4µM hydrocortisone, 0.1 nM R188, 100 U/ml of penicillin and 100 µg/ml streptomycin.
RNAi methods
BTK shRNAs from the p-SHAG-MAGIC 2 (pSM2) shRNA libraryCitation46 were used for targeting BTK-signaling pathway kinases. Plasmid DNA was isolated from bacterial stocks containing each of shRNAs using a plasmid midi kit (Qiagen, Valencia CA). Transfection efficiency was monitored by co-transfection with a modified MSCV-Puro vector expressing green fluorescent protein (GFP)(Clontech). BTK was also targeted using siGENOME SMART pool duplex (Dharmacon, Lafayette, CO) transfected with Xtreme GENE siRNA transfection reagent (Roche) according to the manufacturer's instructions. A BTK-C specific siRNA was custom ordered (Dharmacon, Lafayette, CO), siRNA sense: GGUUAUUGGAUGCCCAUUAUU, antisense: UAAUGGGCAUCCAAUAACCUUCitation10
Cell viability assays
5×103 DU145 cells or 104 LNCaP cells per well, grown on 96 well plates for 24h, were treated with 1 to 30 µM BTK inhibitors. Cells were fixed after 72h with 2.5% formaldehyde, and stained with Hoechst 33342 (Molecular Probes® Invitrogen). Control cells were treated with DMSO. Cell images were acquired using an IN Cell Analyzer 2200 (GE Healthcare) high content imaging system, with a 20X objective. At least 9 fields were imaged per single well of each experiment. Cell numbers were determined and statistics performed using IN Cell Investigator 3.4 high content image analysis software (GE Healthcare). Each experiment was replicated 3 times, and data are presented as mean ± SD. Results were considered significant if p < 0.05.
Apoptosis assays
Apoptosis was detected by cleavage of caspase-3 after 48h after siRNA transfection or treatment with the BTK inhibitors. For the detection of cleaved caspase-3, LNCaP and DU145 cells were treated with BTK-C specific siRNA and with BTK inhibitors at the indicated concentrations. Control cells were treated with non-target siRNA or DMSO, and staurosporine or thapsagargine was used as a positive control. After treatment, cells were fixed with 2.5% formaldehyde, washed 3 times with 1X DPBS, permeabilized with 0.1% Triton-X 100, incubated overnight (4˚C) with 1:200 dilution of cleaved caspase-3 antibody (Asp175, #9579; Cell Signaling), washed 2 times with 1X DPBS, incubated 1h with a 1:200 secondary antibody (Alexia Fluor 568 goat anti-rabbit IgG, #A-11011; Invitrogen), washed again 2 times with 1X PBS and finally stained with Hoechst 33342. Cell images were acquired using an IN Cell Analyzer 2200 (GE Healthcare) high content imaging system, with a 20X objective. At least 9 fields were imaged per single well of each experiment. ImageJ was used to normalize Caspase-3 staining to DAPI staining. The mean area of Caspase-3 divided by the mean area of DAPI for each treatment. The ratio for the control was obtained and set at 1. The Y-axis values for each treatment or BTK-C siRNA knockdown refer to fold increase Caspase-3 positive cells compared to normal. The data are presented as mean ± SD. Results were considered significant if p < 0.05. Annexin V staining was performed after 48h of Ibrutinib or DMSO treatment of LNCaP cells. 105 cells/ml was stained with Muse (TM) Annexin V & Dead Cell Reagent (Millipore) at 1:1 (v/v) ratio for 20 min at room temperature in the dark. Samples were immediately analyzed by the Muse (TM) cell analyzer after staining. A minimum of 5,000 events was analyzed for each experimental condition.
Transfection, infection and selection
Phoenix AMPHO packaging cells were transfected with BTK-A Flag-tagged MarxIV, BTK-C Flag-tagged MarxIV and Empty MarxIV vectors for generating retroviruses.Citation10 LNCaP and DU145 cells were infected with retroviruses for 48h in the presence of 8µg/ml of polybrene. Stable cell lines expressing these constructs were selected with 100 µg/ml and 200 µg/ml hygromycin B for LNCaP and DU145 cells, respectively. BTK-A and BTK-C overexpression was confirmed by immunoblot and RT-PCR analysis. Quantitative RT-PCR overexpression of BTK-C showed a ˜1400-fold increase compared to empty vector control in DU145 cells. Overexpression of BTK-A showed a ˜3500-fold increase in BTK-A mRNA compared to empty vector control in DU145 cells (data not shown).
Microarray
LNCaP cells were incubated for 48h with 10 µM Ibrutinib or vehicle control. BTK-A and BTK-C overexpressing prostate cancer cells were also used for microarray analysis. Total RNA was extracted using TRizol (Invitrogen) from cells, followed by the addition of DNaseI (Roche) for 20 min at room temperature and purified using the RNAeasy column (Qiagen). The quality and concentrations of total RNAs were assessed using the Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA). Total RNA (100 ng) deemed to be of good quality (RNA integrity number (RIN) greater than 8) was processed according to the standard Affymetrix Whole Transcript Sense Target labeling protocol (Affymetrix, Santa Clara, CA). The fragmented biotin labeled cDNA from 3 independent biological replicates was hybridized over 16h to Affymetrix Gene 1.0 ST arrays and scanned on an Affymetrix Scanner 3000 &G using AGCC software. The resulting CEL files were analyzed for quality using Affymetrix Expression Console software and were imported into GeneSpring GX12.6 (Agilent Technologies) where the data was quantile normalized using PLIER and baseline transformed to the median of control samples. The probe sets were further filtered to exclude the bottom 20th percentile across all samples as well as probe sets with expression levels with CV > 20% across all replicates in a condition. The resulting entity list was then subjected to a t-test with Benjamini-Hochberg False Discovery RateCitation47 or Bonferroni correctionCitation48. Gene ontology (GO) enrichment analysis, performed using the DAVID Bioinformatics Resource.Citation33
Immunoblot analysis
Cells were washed with cold PBS containing 0.9 mM CaCl2 and 0.5 mM MgCl2 and lysed in Laemmli buffer supplemented with 5 % β-mercaptoethanol. The cell lysates were incubated at 95°C for 5 min and sonicated 3 times for 10 s. Total cell lysates were separated by SDS-PAGE and transferred to PVDF membranes (EMD Milipore, Billerica, MA) using wet transfer. Blots were imaged using STORM Scanner (GE Healthcare, Piscataway, NJ). The following primary antibodies were used; BTK (#611116, BD Bioscience), BTK (#8547, Cell Signaling, Danvers, MA), P-BTK (Y223, #5082, Cell Signaling), anti-Flag M2 (#080M6035, Sigma-Aldrich, St. Louis, MO), GAPDH (#5174, Cell Signaling). Secondary antibodies included anti-rabbit IgG-HRP (#7074, Cell Signaling) and anti-mouse IgG-HRP (#7076, Cell Signaling).
RNA Isolation, RT-PCR and quantitative RT-PCR
Total RNA was extracted from cells using Trizol (Invitrogen), followed by the addition of DNaseI (Roche) for 20 min at room temperature and purified using the RNAeasy column (Qiagen) clean up protocol. cDNA was synthesized using DyNAmo cDNA synthesis kit (Thermo Scientific). PCR was performed using AccuStart II GelTrack PCR SuperMix (Quanta Bioscience). The PCR reaction consisted of an initial denaturation step (95˚C for 5 min), 45 cycles of amplification (95˚C for 45 sec, 55˚C for 40 sec and 72˚C for 40 sec), followed by a final extension step (72˚C for 10 min). Aliquots of each PCR reaction were electrophoresed on a 1% gel. Quantitative RT-PCR reactions using SYBR Green Master Mix (Applied Biosystems) or TaqMan qPCR using TaqMan Gene Expression Master Mix (Applied Biosystems) were performed on ABI PRISM 7900HT Fast Real Time PCR system (Applied Biosystems). The primers and probes are shown in supplementary Table 3. After the initial denaturation step, 95˚C for 10 min, PCR reactions consisted of 45 cycles of a 95˚C for 15 sec step and a 60˚C for 1 min. Analysis was conducted using Applied Biosystem Real-Time Analysis software. Measurement of BTK-A and BTK-C mRNA expression in prostate tumor tissues was performed using an array of first strand cDNA (cDNA) from human prostate cancer tissues and lymphoma samples contained in the TissueScan Cancer Survey Panel (CSRT302) in 384-well plates from OriGene (Rockville, MD).
BTK-Immunolocalization
Cells were fixed and immunostained on cover slips with anti-BTK antibody (#8547, Cell Signaling), anti-phospho-BTK (Y223), (#5082, Cell Signaling) and anti-Flag M2 (#080M6035, Sigma-Aldrich). Primary antibodies detected with Cy3 donkey anti-rabbit and Cy5 donkey anti-mouse. Prostate cancer tissue arrays (PR8011) and multiple organ tissue arrays (UNC241) were obtained from US Biomax (Rockville, MD). UNC241 has tonsil tissue which serves as a positive control for BTK. PR8011 contained 34 prostate cancer cases with a range of disease stages and patients' ages, 26 cases with hyperplasia, 6 cases with chronic inflammation, 6 cancer adjacent normal cases and 8 normal prostate cases. Arrays were processed with standard immunohistochemical (IHC) procedures. Slides were baked at 65˚C for 1h, de-paraffinizedin HistoChoice clearing agent, and rehydrated through a series of decreasing concentrations of ethanol (100, 75, and 50%) and placed in 1X PBS. Slides were permeabilized with 0.1% Triton-X 100 for 10 min and epitopes retrieved in a pressure cooker for 20 min. Sections were incubated overnight at 4˚C with BTK antibody (1:100 dilution in 3% BSA/PBS), washed in 1X PBS and primary antibody detected with Cy3 donkey anti-rabbit secondary antibody (1:200) dilution in 3% BSA/FBS). Slides were washed, stained with Hoechst 33342 to visualize the nuclei, rinsed, and mounted with DABCO anti-fade in 90% glycerol. TMA cores were imaged using an Olympus inverted fluorescence microscopy platform.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Authors' contributions
LK, CS, WW, XW, LC and SC performed the experiments, data analysis and computational studies. LK, XW, MK, MT and DSC conceived and designed the study, interpreted the data and drafted the article.
Supplemental Material
Download MS Word (291.1 KB)Acknowledgments
We thank Dr. Yan Sun, Jason Wong, Marcy Kuentzel and Dr. Jan Baumann for their help.
Funding
This work was supported by U.S. Army Medical Research Acquisition Activity grant DAMD17–02–1–0729 and NCI R01CA136658 to DSC.
References
- Altekruse SF, Huang L, Cucinelli JE, McNeel TS, Wells KM, Oliver MN. Spatial patterns of localized-stage prostate cancer incidence among white and black men in the southeastern United States, 1999-2001. Cancer Epidemiol Biomarkers Prev 2010; 19:1460-7; http://dx.doi.org/10.1158/1055-9965.EPI-09-1310
- Akinleye A, Chen Y, Mukhi N, Song Y, Liu D. Ibrutinib and novel BTK inhibitors in clinical development. J Hematol Oncol 2013; 6:59; PMID:23958373; http://dx.doi.org/10.1186/1756-8722-6-59
- Lee B, Mukhi N, Liu DL. Current management and novel agents for malignant melanoma. J Hematol Oncol 2012; 5:3; http://dx.doi.org/10.1186/1756-8722-5-3
- Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007; 318:287-90; PMID:17872411; http://dx.doi.org/10.1126/science.1142946
- Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature 2001; 411:355-65; PMID:11357143; http://dx.doi.org/10.1038/35077225
- Vassilev AO, Uckun FM. Therapeutic potential of inhibiting Bruton's tyrosine kinase, (BTK). Curr Pharm Des 2004; 10:1757-66; PMID:15180538; http://dx.doi.org/10.2174/1381612043384475
- Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Eng J Med 2005; 353:172-87; PMID:16014887; http://dx.doi.org/10.1056/NEJMra044389
- Baselga J. Targeting tyrosine kinases in cancer: the second wave. Science 2006; 312:1175-8; PMID:16728632; http://dx.doi.org/10.1126/science.1125951
- Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther 2005; 315:971-9; PMID:16002463; http://dx.doi.org/10.1124/jpet.105.084145
- Eifert C, Wang X, Kokabee L, Kourtidis A, Jain R, Gerdes MJ, Conklin DS. A novel isoform of the B cell tyrosine kinase BTK protects breast cancer cells from apoptosis. Genes Chromosomes Cancer 2013; 52:961-75; http://dx.doi.org/10.1002/gcc.22091
- Smith CI, Baskin B, Humire-Greiff P, Zhou JN, Olsson PG, Maniar HS, Kjellén P, Lambris JD, Christensson B, Hammarström L. Expression of Bruton's agammaglobulinemia tyrosine kinase gene, BTK, is selectively downregulated in T lymphocytes and plasma cells. J Immunol 1994; 152:557-65
- de Weers M, Verschuren MC, Kraakman ME, Mensink RG, Schuurman RK, van Dongen JJ, Hendriks RW. The Bruton's tyrosine kinase gene is expressed throughout B cell differentiation, from early precursor B cell stages preceding immunoglobulin gene rearrangement up to mature B cell stages. Eur J Immunol 1993; 23:3109-14; PMID:8258324; http://dx.doi.org/10.1002/eji.1830231210
- Mohamed AJ, Yu L, Backesjo CM, Vargas L, Faryal R, Aints A, Christensson B, Berglöf A, Vihinen M, Nore BF, et al. Bruton's tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev 2009; 228:58-73; PMID:19290921; http://dx.doi.org/10.1111/j.1600-065X.2008.00741.x
- Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol 2012; 31:119-32; PMID:22449073; http://dx.doi.org/10.3109/08830185.2012.664797
- Kurosaki T, Tsukada S. BLNK: connecting Syk and Btk to calcium signals. Immunity 2000; 12:1-5; PMID:10661400; http://dx.doi.org/10.1016/S1074-7613(00)80153-3
- Kurosaki T. Regulation of B-cell signal transduction by adaptor proteins. Nat Rev Immunol 2002; 2:354-63; PMID:12033741; http://dx.doi.org/10.1038/nri801
- Schmidt U, Boucheron N, Unger B, Ellmeier W. The role of Tec family kinases in myeloid cells. Int Arch Allergy Immunol 2004; 134:65-78; PMID:15133303; http://dx.doi.org/10.1159/000078339
- D'Cruz OJ, Uckun FM. Novel Bruton's tyrosine kinase inhibitors currently in development. Onco Targets Ther 2013; 6:161-76; PMID:23493945
- Rawlings DJ. Bruton's tyrosine kinase controls a sustained calcium signal essential for B lineage development and function. Clin Immunol 1999; 91:243-53; PMID:10370369; http://dx.doi.org/10.1006/clim.1999.4732
- Lee SH, Kim T, Jeong D, Kim N, Choi Y. The tec family tyrosine kinase Btk Regulates RANKL-induced osteoclast maturation. J Biol Chem 2008; 283:11526-34; PMID:18281276; http://dx.doi.org/10.1074/jbc.M708935200
- Shinohara M, Chang BY, Buggy JJ, Nagai Y, Kodama T, Asahara H, Takayanagi H. The orally available Btk inhibitor ibrutinib (PCI-32765) protects against osteoclast-mediated bone loss. Bone 2014; 60:8-15; PMID:24316417; http://dx.doi.org/10.1016/j.bone.2013.11.025
- Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, Flynn J, Jones J, Blum KA, Buggy JJ, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood 2011; 117:6287-96; PMID:21422473; http://dx.doi.org/10.1182/blood-2011-01-328484
- Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton's tyrosine kinase in B cell malignancies. Nat Rev Cancer 2014; 14:219-32; PMID:24658273; http://dx.doi.org/10.1038/nrc3702
- Guo W, Liu R, Bhardwaj G, Yang JC, Changou C, Ma AH, Mazloom A, Chintapalli S, Xiao K, Xiao W, et al. Targeting Btk/Etk of prostate cancer cells by a novel dual inhibitor. Cell Death Dis 2014; 5:e1409; PMID:25188519; http://dx.doi.org/10.1038/cddis.2014.343
- Tai YT, Chang BY, Kong SY, Fulciniti M, Yang G, Calle Y, Hu Y, Lin J, Zhao JJ, Cagnetta A, et al. Bruton tyrosine kinase inhibition is a novel therapeutic strategy targeting tumor in the bone marrow microenvironment in multiple myeloma. Blood 2012; 120:1877-87; PMID:22689860; http://dx.doi.org/10.1182/blood-2011-12-396853
- Burger JA, Buggy JJ. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765). Leuk Lymphoma 2013; 54:2385-91; PMID:23425038; http://dx.doi.org/10.3109/10428194.2013.777837
- Hutcheson J, Vanarsa K, Bashmakov A, Grewal S, Sajitharan D, Chang BY, Buggy JJ, Zhou XJ, Du Y, Satterthwaite AB, et al. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end-organ disease in murine lupus. Arthritis Res Ther 2012; 14:R243; PMID:23136880; http://dx.doi.org/10.1186/ar4086
- Ruderman EM, Pope RM. More than just B-cell inhibition. Arthritis Res Ther 2011; 13:125; PMID:21878134; http://dx.doi.org/10.1186/ar3439
- Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A 2010; 107:13075-80; PMID:20615965; http://dx.doi.org/10.1073/pnas.1004594107
- Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, Mendonca RV, Sweeney MD, Scott KC, Grothaus PG, et al. Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem 2007; 2:58-61; PMID:17154430; http://dx.doi.org/10.1002/cmdc.200600221
- Eda H, Santo L, Cirstea DD, Yee AJ, Scullen TA, Nemani N, Mishima Y, Waterman PR, Arastu-Kapur S, Evans E, et al. A novel Bruton's tyrosine kinase inhibitor CC-292 in combination with the proteasome inhibitor carfilzomib impacts the bone microenvironment in a multiple myeloma model with resultant antimyeloma activity. Leukemia 2014; 28:1892-901; PMID:24518207; http://dx.doi.org/10.1038/leu.2014.69
- Di Paolo JA, Huang T, Balazs M, Barbosa J, Barck KH, Bravo BJ, Carano RA, Darrow J, Davies DR, DeForge LE, et al. Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat Chem Biol 2011; 7:41-50; PMID:21113169; http://dx.doi.org/10.1038/nchembio.481
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37:1-13; PMID:19033363; http://dx.doi.org/10.1093/nar/gkn923
- Baumann J, Sevinsky C, Conklin DS. Lipid biology of breast cancer. Biochimica Biophys Acta 2013; 1831:1509-17; PMID:23562840; http://dx.doi.org/10.1016/j.bbalip.2013.03.011
- Hochachka PW, Rupert JL, Goldenberg L, Gleave M, Kozlowski P. Going malignant: the hypoxia-cancer connection in the prostate. Bioessays 2002; 24:749-57; PMID:12210536; http://dx.doi.org/10.1002/bies.10131
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 2012; 22:1813-31; PMID:22955991; http://dx.doi.org/10.1101/gr.136184.111
- Chang BY, Huang MM, Francesco M, Chen J, Sokolove J, Magadala P, Robinson WH, Buggy JJ. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res Ther 2011; 13:R115; PMID:21752263; http://dx.doi.org/10.1186/ar3400
- Evans EK, Tester R, Aslanian S, Karp R, Sheets M, Labenski MT, Witowski SR, Lounsbury H, Chaturvedi P, Mazdiyasni H, et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther 2013; 346:219-28; PMID:23709115; http://dx.doi.org/10.1124/jpet.113.203489
- Kowalski PJ, Rubin MA, Kleer CG. E-cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res 2003; 5:R217-22; PMID:14580257; http://dx.doi.org/10.1186/bcr651
- Paredes J, Albergaria A, Oliveira JT, Jeronimo C, Milanezi F, Schmitt FC. P-cadherin overexpression is an indicator of clinical outcome in invasive breast carcinomas and is associated with CDH3 promoter hypomethylation. Clin Cancer Res 2005; 11:5869-77; PMID:16115928; http://dx.doi.org/10.1158/1078-0432.CCR-05-0059
- Chu K, Cheng CJ, Ye X, Lee YC, Zurita AJ, Chen DT, Yu-Lee LY, Zhang S, Yeh ET, Hu MC, et al. Cadherin-11 promotes the metastasis of prostate cancer cells to bone. Mol Cancer Res 2008; 6:1259-67; PMID:18708358; http://dx.doi.org/10.1158/1541-7786.MCR-08-0077
- Ortolano S, Hwang IY, Han SB, Kehrl JH. Roles for phosphoinositide 3-kinases, Bruton's tyrosine kinase, and Jun kinases in B lymphocyte chemotaxis and homing. Eur J Immunol 2006; 36:1285-95; PMID:16619289; http://dx.doi.org/10.1002/eji.200535799
- Tsui KH, Chang YL, Feng TH, Chang PL, Juang HH. Glycoprotein transmembrane nmb: an androgen-downregulated gene attenuates cell invasion and tumorigenesis in prostate carcinoma cells. Prostate 2012; 72:1431-42; PMID:22290289; http://dx.doi.org/10.1002/pros.22494
- Schaefer L, Evan L, Whiteford J, Dyer D, Rowan D. Cell signalling in inflammation. Int J Exp Pathol 2011; 92:A6-A7
- Currie E, Schulze A, Zechner R, Walther TC, Farese RV, Jr. Cellular fatty acid metabolism and cancer. Cell Metab 2013; 18:153-61; PMID:23791484; http://dx.doi.org/10.1016/j.cmet.2013.05.017
- Silva JM, Li MZ, Chang K, Ge W, Golding MC, Rickles RJ, Siolas D, Hu G, Paddison PJ, Schlabach MR, et al. Second-generation shRNA libraries covering the mouse and human genomes. Nat Genet 2005; 37:1281-8; PMID:16200065
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met 1995; 57:289-300
- Lin DY. An efficient Monte Carlo approach to assessing statistical significance in genomic studies. Bioinformatics 2005; 21:781-7; PMID:15454414; http://dx.doi.org/10.1093/bioinformatics/bti053