
ABSTRACT
Invasion and metastasis via induction of matrix metalloproteinases are the main causes of death in melanoma cancer. In this study, we investigated the inhibitory effects of heat killed saprophytic bacterium Mycobacterium indicus pranii (Mw) on B16F10 melanoma cell invasion. Mw reported to be an immunomodulator has antitumor activity however, its effect on cancer cell invasion has not been studied. Highly invasive B16F10 melanoma was found sensitive to Mw which downregulated MMP-9 expression. Mw treatment inhibited nuclear factor-κB (NF-κB) and activator protein-1 (AP-1) transcriptional activity and respective DNA binding to MMP-9 promoter. Moreover, Mw also overcame the promoting effects of PMA on B16F10 cell invasion. Mw decreased PMA-induced transcriptional activation of NF-κB and AP-1 by inhibiting phosphorylation of AKT and ERK-1/2. Furthermore, Mw strongly suppressed PMA-induced membrane localization of protein kinase C α (PKCα) since PKCα inhibition caused a marked decrease in PMA-induced MMP-9 secretion as well as AKT/ERK-1/2 activation. These results suggest that Mw may be a promising anti-invasive agent as it blocks tumor growth and inhibits B16F10 cell invasion by reducing MMP-9 activation through inhibition of PKCα/ AKT/ ERK-1/2 phosphorylation and NF-κB/AP-1 activation.
Abbreviations
| Mw | = | Mycobacterium w / Mycobacterium indicus pranii |
| MMP | = | Matrix metalloproteinase |
| PKCα | = | Protein Kinase Cα |
| PKCδ | = | Protein Kinase Cδ |
| NF-κB | = | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| AP-1 | = | Activator protein 1 |
| PMA | = | Phorbol 12-myristate 13-acetate |
| PI3K | = | phosphatidylinositol 3-kinase |
| ERK-1/2 | = | extracellular signal-regulated kinase. |
Introduction
Metastatic melanoma tumor frequently diagnosed in both men and women is characterized by abnormal proliferation of melanocytes and increasing rapidly in all industrialized countries.Citation1 Invasion and metastasis are the main reasons for the high mortality rates associated with melanoma cancer. Because of local invasion and metastasis, neither radiation therapy nor chemotherapy substantially increases the length or quality of life for patients with advanced melanoma cancer.Citation2,3 Therefore, controlling the invasion and metastasis along with development of effective anti-invasive agents as a therapeutic strategy would be very probable to improve the treatment.
Invasion and metastasis consist of several interdependent processes including uncontrolled growth of cancer cells, migration or invasion to distant sites of surrounding tissues as well as adhesion or colonization of other organs and tissues.Citation4 Although a number of proteinases are involved, activation of matrix metalloproteinases (MMPs) in tumor invasion and metastasis has frequently been reported.Citation5-8 MMPs belongs to zinc-dependent protease family and divided into 4 subclasses based on the substrates comprising collagenase, gelatinase, stromelysin and membrane-associated MMPs.Citation9,10 Tumor-secreted MMPs result in organ failure and patient mortality by destroying extracellular matrix components in the surrounding tissues of the tumor and facilitate the spread to distant organs. Among various MMPs identified, MMP-2 (gelatinase-A) and MMP-9 (gelatinase-B) are the most important mediators of tumor migration and invasion.Citation11 The extent of MMP-2 and MMP-9 expression in tumors are highly correlated with the metastatic potential.Citation12-14 Generally, MMP-2 is constitutive and overexpressed in highly metastatic tumors, whereas MMP-9 can be stimulated by the inflammatory cytokines, tumor necrosis factor-α, epidermal growth factor or phorbol ester (PMA), through activation of different intracellular signaling pathways.Citation12,15-17 Induction of MMP-9 is particularly important for the invasiveness of skin cancers, including melanoma cancer, thereby blockade of MMP-9 suppresses melanoma cell invasion into other organs.Citation18-20 Mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K), and protein kinase C (PKC) signaling pathways are the predominant cascades participating in MMP-9 expression.Citation21-23 Furthermore, stimulators, such as cytokines or PMA, control the expression of MMP-9 by modulating the activation of transcription factors such as nuclear factor-κB (NF-κB) and activator protein-1 (AP-1) through MAPKs and PI-3K/AKT pathways.Citation22,24 NF-κB and AP-1 are well-known transcription factors which regulate the expression of genes involved in metastasis, tumorigenesis, and inflammation.Citation25,26 Several studies have indicated that inhibition of MMP-9 expression or enzyme activity can be used as early targets for preventing cancer metastasis.Citation27,28 Therefore, agents possessing the ability to suppress the expression of MMP-9 deserve development for anti-melanoma cancer invasion and metastasis.
Recent studies to investigate new anti-invasive agents have demonstrated that heat killed Mycobacterium indicus pranii (Mw) with chemopreventive potential can inhibit cell invasiveness by modifying MMP-9 expression in murine macrophages in vitro.Citation29 Mw is a saprophytic bacterium which shares antigen with Mycobacterium leprae and Mycobacterium tuberculosis.Citation30-35 Mw can be used as a general immunomodulator which alone or together with standard multidrug treatment, proved effective against various cancer and infectious diseases.Citation30-35 Despite these observations, the mechanism by which Mw mediates anti-invasive responses is unknown.
In this study, we investigated the molecular mechanisms by which heat killed Mw inhibits MMP-9 expression and subsequently the invasiveness of B16F10 melanoma cancer. Mw significantly suppressed MMP-9 gene expression through blocking the activation of NF-κB and AP-1 transcription factors via PKCα-mediated PI3K/AKT and ERK-1/2 signaling, therefore reducing invasion and metastasis of B16F10 cells.
Results
Mw affects proliferation and invasion of melanoma cancer cell
We first examined the cytotoxicity of Mw via MTT assay on melanoma cancer cell lines and control melanocytes. Mw (dose; 106 and 107 cells/ml) had moderate cytotoxicity respectively on B16F10 and B16F1 compared to control melanocytes. Among the melanoma cancer cell lines, highly invasive B16F10 was found sensitive to Mw than B16F1 cells (). Mw treatment inhibited the growth and cell proliferation of melanoma cells in a time and dose-dependent manner observed by Trypan-Blue exclusion and [3H]-Thymidine incorporation assay (). Mw also suppressed the clonogenic activity of these 2 cell lines (). Thus, Mw (106 cells/ml) inhibited the anchorage-dependent (cell proliferation) and anchorage-independent (colony formation) growth of highly and poorly invasive melanoma cancer cells, with the highly invasive B16F10 being more sensitive. Considering the sensitivity of B16F10 to Mw treatment, we determined the invasive behavior of B16F10 cells. As shown, Mw (106 cells/ml) markedly suppressed the invasion of B16F10 cells (). To explore the effect on migration, B16F10 cells were treated with Mw, and cell migration was determined. Mw (106 cells/ml) significantly decreased B16F10 cell migration in a dose dependent manner (). Finally, we evaluated the effect of Mw on cell adhesion. Mw (106 cells/ml) treatment also inhibited the adhesion of B16F10 cells onto the matrigel in a concentration-dependent manner compared with the untreated control (). Henceforth, result suggested that Mw (106 cells/ml) exhibited anti-invasive behavior toward metastatic B16F10 melanoma at non-cytotoxic concentrations.
Figure 1. Mw suppresses proliferation and invasion of B16F10 cells. (A) MTT assay of Mw (dose; 0, 104 −108 cells/ml) for 24hr and 48hr on melanocyte, B16F10 and B16F1 cells were analyzed. The experiment was repeated thrice and expressed as mean ± SD.π P< 0.05,ππ P< 0.01; *P< 0.05, **P< 0.01 versus untreated for 24hr and 48hr. (B) Effects of Mw on cell viability were assayed by Trypan blue exclusion assay for 24hr and 48hr. The experiment was repeated thrice and expressed as mean ± SD.ππ P < 0.01; **P < 0.01 vs. untreated for 24hr and 48hr. (C) Antiproliferative effect of Mw for 24hr and 48hr were measured by [3 H]–Thymidine incorporation. Triplicate results were expressed as mean ± SD.ππ P < 0.01; **P < 0.01 versus untreated cells for 24hr and 48hr. (D) Clonogenicity of B16F10 and B16F1 cells treated with Mw was assessed by soft agar colony assay. Results were expressed as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (E) Invasion assay was carried out in 12-well transwell after Mw treatment for 2hr. The randomly chosen fields were photographed (20X), and the number of cells migrated to the lower surface was calculated. Data are mean ± SD of 3 independent experiments. *P < 0.05, **P < 0.001 vs untreated. (F) Confluent cells were treated with Mw and scratched. After 24hr, the number of cells migrated into the scratched area was photographed (20X) and calculated. Data are mean ± SD of 3 independent experiments. *P< 0.05, and **P< 0.001 vs untreated. (G) Cell adhesion was carried out in a 12-well plate coated with matrigel and treated with Mw for 2hr. Attached cells were photographed (20X) and calculated. Data are mean ± SD of 3 independent experiments.*P< 0.05, **P< 0.001 vs untreated.
![Figure 1. Mw suppresses proliferation and invasion of B16F10 cells. (A) MTT assay of Mw (dose; 0, 104 −108 cells/ml) for 24hr and 48hr on melanocyte, B16F10 and B16F1 cells were analyzed. The experiment was repeated thrice and expressed as mean ± SD.π P< 0.05,ππ P< 0.01; *P< 0.05, **P< 0.01 versus untreated for 24hr and 48hr. (B) Effects of Mw on cell viability were assayed by Trypan blue exclusion assay for 24hr and 48hr. The experiment was repeated thrice and expressed as mean ± SD.ππ P < 0.01; **P < 0.01 vs. untreated for 24hr and 48hr. (C) Antiproliferative effect of Mw for 24hr and 48hr were measured by [3 H]–Thymidine incorporation. Triplicate results were expressed as mean ± SD.ππ P < 0.01; **P < 0.01 versus untreated cells for 24hr and 48hr. (D) Clonogenicity of B16F10 and B16F1 cells treated with Mw was assessed by soft agar colony assay. Results were expressed as mean ± SD. *P < 0.05, **P < 0.001 vs untreated. (E) Invasion assay was carried out in 12-well transwell after Mw treatment for 2hr. The randomly chosen fields were photographed (20X), and the number of cells migrated to the lower surface was calculated. Data are mean ± SD of 3 independent experiments. *P < 0.05, **P < 0.001 vs untreated. (F) Confluent cells were treated with Mw and scratched. After 24hr, the number of cells migrated into the scratched area was photographed (20X) and calculated. Data are mean ± SD of 3 independent experiments. *P< 0.05, and **P< 0.001 vs untreated. (G) Cell adhesion was carried out in a 12-well plate coated with matrigel and treated with Mw for 2hr. Attached cells were photographed (20X) and calculated. Data are mean ± SD of 3 independent experiments.*P< 0.05, **P< 0.001 vs untreated.](/cms/asset/8c587ddb-78c0-47ca-b40b-9e41f0373bdd/kcbt_a_1078024_f0001_oc.gif)
Mw suppresses B16F10 cell invasion by inhibiting MMP-9 through NF-κB and AP-1
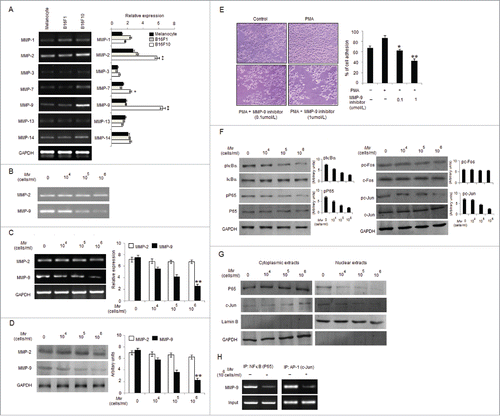
Cancer invasiveness and metastasis are associated with increased expression of MMPs.Citation36,37 Among various MMPs examined, mRNA levels of MMP-2 and MMP-9 were found high in B16F10 compared to B16F1 and control melanocytes (). Therefore, we examined whether the anti-invasive effect of Mw can be mediated by suppressing MMP-2 and MMP-9 activities. Gelatin zymography performed, using the conditioned medium (CM) from the Mw treated cells showed minimal MMP-9 activity, suggesting that Mw inhibited the invasiveness of B16F10 cells by reducing MMP-9 activity (). In order to determine whether the inhibitory effect of Mw on MMP-9 secretion was resulted from a downregulated level of MMP-9 mRNA expression, real-time PCR was conducted (). Consistently western blot analysis also revealed downregulated MMP-9 expression via Mw (). Thus, Mw regulated MMP-9 expression at the transcriptional level and inhibited the invasion of B16F10 cells. MMP-2 expression was found unaffected. Furthermore, to understand the interrelationship between invasion and MMP-9 in B16F10, we performed matrigel invasion assay with MMP-9 inhibitor. Result showed that MMP-9 inhibitor blocked invasion in a dose-dependent manner, suggesting the involvement of MMP-9 in B16F10 invasion (). Since, NF-κB and AP-1 are critical transcription factors in the regulation of MMP-9 expression.Citation23,38 Thus, to understand the inhibitory mechanisms of Mw on MMP-9 transcriptional regulation we checked the expression level of pIκBα, pP65, pc-Fos and pc-Jun. As shown, Mw inhibited the phosphorylation of IκBα, P65 and c-Jun only in B16F10 cells and prevented the translocation of P65 and c-Jun from cytoplasm to nucleus (). This indicated that IκBα degradation, P65 translocation and c-Jun activation blocked by Mw inhibits the NF-κB and AP-1 dependent B16F10 cell invasion. Mw exhibited no significant effect on c-Fos expression. ChIP analysis further confirmed the inhibitory effect of Mw on the DNA binding of transcription factors to MMP-9 promoter. Mw (106 cells/ml) inhibited the DNA binding of NF-κB (P65) and AP-1 (c-Jun) to MMP-9 promoter (). Hence, these result indicated that Mw inhibited the NF-κB and AP-1 dependent invasive behavior of B16F10 cells.
Figure 2. Mw suppresses transcription factors mediated B16F10 cell invasion. (A) 2×106 melanocytes, B16F1 and B16F10 cells were collected for mRNA extraction and real time PCR analyses of MMP-1, MMP-2, MMP-3, MMP-7, MMP-9, MMP-13 and MMP-14. Relative expression of target gene with respect to GAPDH was presented as mean ± SD, *P< 0.05, **P< 0.01 vs melanocytes. (B) 2×106 B16F10 cells were treated with Mw for 2hr and the activity of MMP-2 and MMP-9 was assessed by gelatin zymography. Data are representative of 3 independent experiments. (C) 2×106 B16F10 cells were treated with Mw and MMP-2 and MMP-9 gene expression was detected by real time PCR. Relative expression of target gene with respect to GAPDH were performed thrice and expressed as mean ± SD. **P< 0.01 vs. untreated cells. (D) Western blot was performed in similarly treated cell to detect MMP-2 and MMP-9 using specific antibodies with anti-GAPDH as reference. (E) B16F10 cells were incubated in Matrigel-coated transwell with or without PMA and MMP-9 inhibitor for 24hr. Attached cells were photographed (20X) and calculated. Data are mean ± SD of 3 independent experiments.*P< 0.05, **P< 0.001 vs untreated. (F) 2×106 B16F10 cells treated with Mw were subjected to western blot to detect pIκBα, IκBα, pP65, P65, pc-Fos, c-Fos, pc-Jun, c-Jun using specific antibodies. (G) Cytoplasmic and nuclear fractions of B16F10 cells were subjected to protein gel blot to study nuclear translocation of P65 and c-Jun with GAPDH and LaminB as reference. (H) 2×106 B16F10 cells treated with Mw (106 cells/ml) were immunoprecipitated using NF-κB (IP: NF-κBP65) and AP-1(IP: c-Jun) specific antibody. Semi quantitative RT-PCR was performed to amplify the putative NF-κB and AP-1 binding sites at the MMP-9 promoter. Data are from one representative experiment performed at least thrice.
Mw overcome the promoting effects of PMA on B16F10 cell invasion
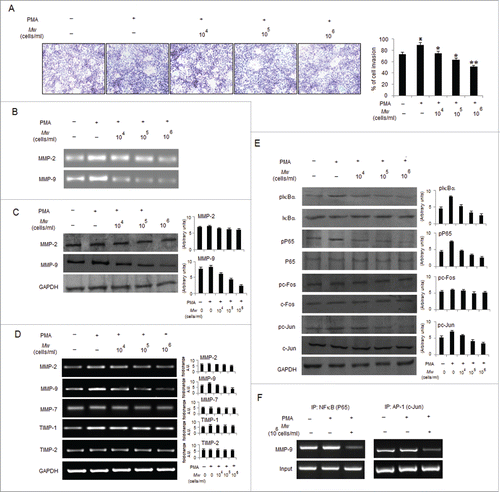
PMA has been shown to enable cancer cell invasion via inducing NF-κB and/or AP-1 signaling pathways to mediate MMP-9 expression.Citation23,38 Accordingly, we determined whether Mw can abrogate the promoting effects of PMA on B16F10 cells. As shown, Mw abrogated PMA-induced invasion by downregulating MMP-9 gelatinolytic activity and protein expression in a dose-dependent manner, indicating that Mw inhibits MMP-9 enzyme activity by reducing the protein level of MMP-9 (). Similar to prior observations, Mw had little effect on MMP-2 protein expression. Furthermore, we performed RT-PCR to determine Mw mediated regulation of MMPs at the mRNA level. Mw significantly inhibited PMA-induced MMP-9 mRNA expression in a dose-dependent manner but had no effect on the mRNA level of MMP-2. Since the activity of MMP-9 is tightly regulated by endogenous inhibitors, tissue inhibitors of metalloproteinases (TIMP), we examined the expression levels of TIMP-1 and TIMP-2 by RT-PCR. As shown TIMP-1, but not TIMP-2, was slightly stimulated by PMA. However, Mw had no effect on the mRNA levels of both TIMP-1 and TIMP-2. Furthermore, we examined the expression level of MMP-7 in Mw-treated B16F10 cells and showed that MMP-7 remained essentially unchanged (). These results indicate that Mw selectively suppressed MMP-9 expression both at the protein and mRNA levels in a dose-dependent manner. Interestingly, we examined the effects of Mw on NF-κB and AP-1 signal cascades, which play a major role in MMP-9 transcription. Our results showed that pIκBα and pP65 were upregulated by PMA treatment at 80nM. Mw treatment markedly inhibited the PMA-induced upregulation of pIκBα and pP65 respectively. We further investigated the effect of Mw on the PMA-induced upregulation of pc-Fos and pc-Jun expression. Results showed that Mw inhibited PMA-induced upregulation of c-Jun only, thus affecting the AP-1 signal cascades (). In addition, ChIP analysis also suggested Mw mediated significant inhibition on PMA-induced NF-κB (P65) and AP-1(c-Jun) binding to MMP-9 promoter respectively (). Taken together, these results indicated that Mw can overcome the facilitative effects of PMA on B16F10 cells by blocking NF-κB and AP-1 mediated MMP-9 expression and cell invasion.
Figure 3. Mw overcomes the facilitative effects of PMA on B16F10 cell invasion. (A) B16F10 cells preincubated with Mw for 2hr and the cell suspension containing Mw and PMA (80nM) was seeded onto the upper well of 12-well microchemotaxis chambers. The randomly chosen fields were photographed (20X). The number of cells migrated to the lower surface was calculated. Data are mean ± SD of 3 independent experiments.πP < 0.05 versus untreated, *P< 0.05, **P< 0.001 vs. PMA alone. (B) 2×106 B16F10 cells were pretreated with Mw for 2hr and then stimulated with 80nM PMA for 24hr. The activity of MMP-2 and MMP-9 was assessed by gelatin zymography. (C) Western blot was performed in similarly treated cell to detect MMP-2 and MMP-9 using specific antibodies with anti-GAPDH as reference. (D) 2×106 B16F10 cells pretreated with Mw for 2hr and stimulated with 80nM PMA for 24hr. Gene expression of MMP-2, MMP-9, MMP-7, TIMP-1 and TIMP-2 was detected by RT- PCR analysis with GAPDH as control. (E) The protein expression of pIκBα, IκBα, pP65, P65, pc-Fos, c-Fos, pc-Jun, c-Jun and anti-GAPDH was assessed by western blot in B16F10 cells pretreated with Mw for 2hr and stimulated with 80nM PMA for 24hr using specific antibodies. (F) B16F10 cells treated similarly were immunoprecipitated using NF-κB (IP: NF-κB) and AP1 (IP: c-Jun) specific antibody. Semi quantitative RT-PCR amplified the putative NF-κB and AP1 binding sites at the MMP-9 promoter. Data are from one representative experiment performed at least thrice.
Mw regulates PKC isotype mediated MMP-9 expression in B16F10 melanoma
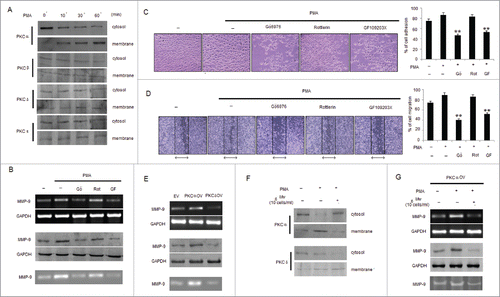
Differential PKC activation correlates with B16F10 cell metastasis, proliferation and apoptosis.Citation39 PMA has been reported as a PKC activator, and activation of the PKC isoforms including α, β, δ and ϵ by PMA has been identified.Citation40,41 However, contributions of PKC isoforms in PMA-induced invasion and migration of B16F10 cells are still unclear. To determine whether PMA causes the activation of PKC isotypes in B16F10 cells, we analyzed the levels of PKC isotypes in cytosol and membrane fractions. PMA stimulated the translocation of PKCα from the cytosol to the membrane only after 10min of stimulation compared to PKCβ, PKCδ and PKCϵ isotypes expressed in B16F10 cells (). Furthermore, to determine the involvement of PKCα isotype in PMA-induced MMP-9 activation and cell invasion, B16F10 cells were exposed to PMA together with PKC inhibitors including Gö6976, Rottlerin and GF109203X. As shown, incubation of B16F10 cells with Gö6976 and GF109203X, but not Rottlerin, inhibited PMA-induced MMP-9 expression and activity (). Data from the cell invasion and migration assay revealed that the addition of GF109203X and Gö6976 compared to Rottlerin significantly inhibited the PMA-induced invasion and migration of B16F10 cells (). Since, PKCα and PKCδ exhibited reciprocal effect on each otherCitation39 so melanoma cells transfected with PKCαOV and PKCδOV had also shown differential effect in the expression and activity of MMP-9 where PKCαOV cell had augmented MMP- 9 expression compared to PKCδOV cells (). To study the effect of Mw on PKC isotypes, translocation of PKCα by PMA from the cytosol to membrane was studied which was found blocked by Mw addition, however no blocking effect was detected on PKCδ translocation (). Furthermore, treatment with Mw had shown decreased MMP-9 expression and activity in PMA treated PKCαOV transfected B16F10 melanoma cells (). Thus suggesting that Mw inhibited PKCα activation involved in PMA-induced MMP-9 activation and migration.
Figure 4. Mw inhibits PMA-induced PKCα mediated B16F10 cell invasion. (A) B16F10 cells were treated with 80nM PMA, and PKC isotypes were analyzed in cytosol and membrane fractions by protein gel blotting. (B) B16F10 cells were pretreated for 1hr with Gö6976 (2uM), Rottlerin (0.5uM) or GF109203X (2uM), followed by 80nM PMA stimulation for 24hr. The MMP-9 mRNA (top), protein (middle) expression and activity (bottom) was analyzed by semi-quantitative RT-PCR, western blot and gelatin zymography respectively. Data are from one of 3 representative experiments. (C) Invasion assay was carried out in cells stimulated with 80nM PMA for 24hr after pretreatment with PKC inhibitors for 1hr. The randomly chosen fields were photographed (20X), and the number of cells migrated to the lower surface was calculated. Data are mean ± SD of 3 independent experiments. **P < 0.001 vs PMA alone treatment. (D) Confluent cells were scratched and treated with 80nM PMA for 24hr after pretreatment with PKC inhibitors for 1hr. The number of cells migrated into the scratched area was photographed (20X) and calculated. Data are shown as the mean ± SD of 3 independent experiments. **P< 0.001 vs PMA alone treatment. (E) B16F10 cells transfected with empty vector (EV), overexpressed full-length mouse PKCα (PKCαOV) or PKCδ (PKCδOV) isotypes as mentioned in materials and methods were subjected to semi-quantitative RT-PCR, protein gel blot and gelatin zymography for MMP-9 expression and activity respectively. Data are from one of 3 representative experiments. (F) B16F10 cells were pretreated with Mw for 2hr and then stimulated with 80nM PMA for 24hr. Western blot analysis was performed to detect the PKCα and PKCδ level in cytosol and membrane fractions. (G) B16F10 cells, transfected with overexpressed full-length mouse PKCα (PKCαOV) isotype were treated with Mw for 2hr followed by 80nM PMA stimulation for 24hr. The MMP-9 mRNA (top), protein (middle) expression and activity (bottom) were analyzed by semi-quantitative RT-PCR, western blot and gelatin zymography respectively. Data are from one of 3 representative experiments.
Mw suppresses ERK-1/2 and AKT mediated activation of transcription factors downstream of PKCα
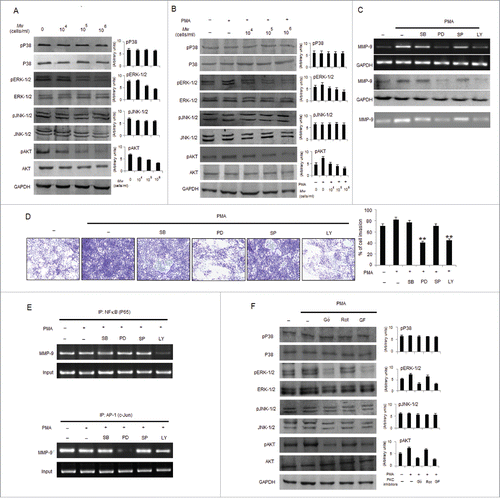
NF-κB and AP-1 controls MMP-9 expression and are regulated by MAPKs such as PI3K/AKT, ERK-1/2, JNK-1/2, and P38 kinase. Hence, induction of MAPK or PI3K signaling pathways are involved in the expression of MMP-9; however, studies regarding the mechanism of Mw mediated downregulation of MMP-9 through suppression of MAPK or PI3K have not been carried out. As examined, Mw in a dose dependent manner inhibited the phosphorylation of ERK-1/2 and AKT, but not p38 MAPK or JNK-1/2 in B16F10 cells. PMA mediated induction of ERK-1/2 and AKT phosphorylation were also blocked by Mw pretreatment but expressions of p38 MAPK or JNK-1/2 remain unchanged (). Pharmacological inhibitors of MAPK and AKT were used to identify the molecular mechanisms by which PMA induces MMP-9 expression. As observed, treatment of cells with PD95059 and LY294002 abrogated PMA induced MMP-9 expression and activity as well as B16F10 cell invasion, whereas treatment with SB203550 or SP600125 did not (). Furthermore, ChIP analysis investigated that PMA induced DNA binding of NF-κB (P65) and AP-1 (c-Jun) to MMP-9 promoter were found to be inhibited by the LY294002 and PD95059 respectively (). This indicates that PMA stimulates cell invasion and MMP-9 secretion by modulating AKT-mediated NF-κB activation and ERK-1/2-mediated AP-1 activation. Furthermore, phosphorylation of ERK-1/2 and AKT was blocked by treatment with GF109203X and Gö6976, but not with Rottlerin thereby suggesting that Mw mediated suppression of ERK-1/2 and AKT is located downstream of PKCα and consequently inhibits MMP-9 expression in B16F10 cells (Fig. 5F).
Figure 5. Mw inhibits PMA-induced transcriptional activation of MMP-9 and phosphorylation of ERK1/2 and AKT in B16F10 cells. (A) 2×106 B16F10 cells were treated with Mw and the expression of phospho/total MAPKs and AKT were determined by protein gel blotting. (B) 2×106 B16F10 cells preincubated with Mw for 2hr followed by 80nM PMA for 24hr were subjected to western blot using antibodies specific to total/phospho MAPKs and AKT. Data are from one of 3 representative experiments with similar results. (C) Cells were treated with specific inhibitors of MAPKs or AKT for 1hr and then stimulated with PMA for 24hr. The MMP-9 mRNA (top), protein expression (middle) and activity (bottom) were analyzed by semi-quantitative RT-PCR, protein gel blot and gelatin zymography respectively. (D) Similarly cells were preincubated with specific inhibitors of MAPKs or AKT for 1hr and then stimulated with PMA for 24hr was seeded onto the upper chamber wells. After incubation for 24hr at 37°C, the filter was fixed and stained. The randomly chosen fields were photographed (20X). Data are shown as the mean ± SD of 3 independent experiments. **P< 0.001 vs PMA alone treatment. (E) B16F10 cells treated with specific inhibitors of MAPKs or AKT for 1hr and then stimulated with PMA for 24hr were subjected to immunoprecipitation using NF-κB (IP: NF-κBP65) and AP-1 (IP: c-Jun) specific antibody. Semi quantitative RT-PCR amplify the putative NF-κB and AP1 binding sites at the MMP-9 promoter. Data are from one representative experiment performed at least thrice. (F) Cells were stimulated with 80nM PMA for 24hr after pretreatment with PKC inhibitors for 1hr, and the levels of total/phospho MAPKs and AKT were determined by western blotting. Data are from one of 3 representative experiments.
Effect of Mw on melanoma tumor growth in vivo
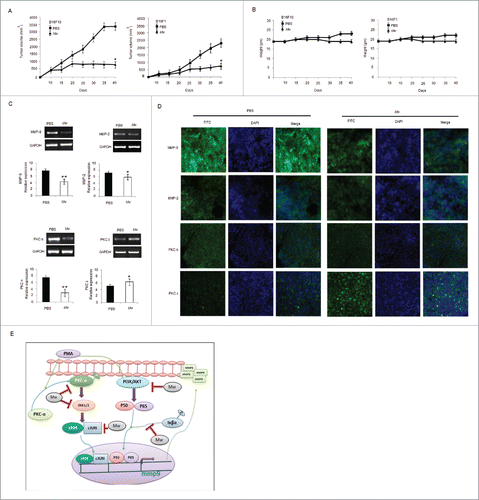
To evaluate the effect of Mw in vivo, B16F10 and B16F1 murine models were generated. Tumor-bearing mice were treated with PBS or Mw intradermally at a dose (107 bacilli/ mouse) twice within 15 d interval starting from day 3 of tumor cell implantation. Animals were sacrificed when the tumors reached a palpable size. As shown, Mw significantly inhibited B16F10 and B16F1 tumor growth (). Moreover, Mw treatment did not reduce the body weight of mice, suggesting that Mw had no toxicity effect (). The tumor samples of mice bearing B16F10 cells were isolated, cells were harvested, and experiments were conducted to determine whether Mw perturbed MMP-9 expression in vivo. Interestingly, samples from mice treated with Mw showed minimal MMP-9 and PKCα expression with respect to MMP-2 and PKCδ compared with PBS treated mice (). Furthermore, immunohistochemical analysis also indicated that the expression of MMP-9 and PKCα in the tumors was noticeably higher in PBS treatment and inhibited by Mw treatment (). Hence, these results demonstrated that the administration of Mw had a great potential in melanoma cancer therapy.
Figure 6. In vivo therapeutic efficacy of Mw on melanoma cancer models. (A) 3×105 B16F10 cells were injected in the right flank of C57BL/6 mice on day 0. Mw (107 bacilli/mouse) was given intradermally (i.d.) at the tumor site twice within 15 d interval starting from day 3 of tumor implantation. Tumor growth and animal body weight was measured twice per week. Data are mean ± SD values obtained from 5-mice/group. *P< 0.05 vs PBS treated mouse. (B) Mw treatment did not affect animal body weight. (C) MMP-9, MMP-2, PKCα, PKCδ expression in B16F10 tumor treated with Mw was detected by real time PCR analysis. Data are mean ± SD obtained from 5-mice/group. *P< 0.05, **P< 0.01 vs PBS treated mouse. (D) Expression of MMP-9, MMP-2, PKCα, and PKCδ in B16F10 tumor treated with Mw was detected by tumor immunofluorescence staining. (E) Signaling pathway where Mw inhibits B16F10 melanoma cancer cell invasion and migration.
Discussion
The current study was designed to investigate the anti-invasive potential of heat killed Mw and to explore the molecular mechanisms underlying its activity. Mw is a nonpathogenic, saprophytic cultivable mycobacterium which played a potent immunomodulatory role against leprosy, tuberculosis, leishmaniasis and cancer.Citation30-35 Several studies have documented the anti-cancer activity of Mw.Citation31,32 Mw can modulate the effector responses via interacting with TLRs on the cell surface since TLRs play an important role in the recognition of various microbial molecules.Citation33,34 However, the role of Mw in suppressing invasion and migration are limited and its effect against MMP-9 expression in B16F10 melanoma remains unknown. Migration and invasion are the important prerequisites of cancer cell progression and metastasis.Citation4,5 It is well established that tumor cell migration and invasion depend on gelatinase activity.Citation8,9 MMP-9 and 2 are the important enzymes in tumor metastasis, resulting from ECM degradation.Citation11-14 MMPs are highly expressed in melanoma cancer, and a direct relationship between cancer progression and MMPs expression has been well established.Citation16,17 Recent study has shown statistically significant different expression patterns of MMP-9 between well-differentiated and poorly differentiated tissue samples which play key roles during the development of cancer.Citation42 The current study demonstrated that heat killed Mw significantly inhibited B16F10 melanoma cancer cell proliferation, growth, invasion, adhesion and colony formation by modulating the activity and expression of MMP-9, suggesting its potential in controlling melanoma cancer progression. MMPs activity is also regulated by tissue-specific inhibitors (TIMPs) which bind MMPs and directly affect the level of MMP activity. Because TIMP-1 is a major inhibitor of MMP-9, TIMP-1 and TIMP-2 are differentially regulated in vivo as well as in cultured cells.Citation43,44 The mRNA levels of these 2 proteins ruled out the effects of Mw on TIMP-1 and TIMP-2. Therefore, the inhibitory effect of Mw on MMP-9 activity was mainly due to the transcriptional regulation and protein expression of MMP-9. The MMP-9 promoter region contains NF-κB and AP-1 binding sites. NF-κB and AP-1 are ubiquitous eukaryotic transcription factors and can be induced by multiple stimuli.Citation22,24 NF-κB, a heterodimer is sequestered in the cytoplasm due to its association with the inhibitory protein IκBα. Stimulation by inflammatory cytokines or tumor promoters leads to the dissociation of IκBα from NF-κB through the ubiquitin/proteasome dependent pathway following its phosphorylation. The released NF-κB translocates into the nucleus and binds to the promoter region of MMP-9, leading to gene expression.Citation45 On the other hand, AP-1 is a nuclear transcription factor comprising homodimers and heterodimers of members of the Fos and Jun families.Citation46 NF-κB and/or AP-1 dependent MMP-9 expression is regulated by mitogen-activated protein kinases (MAPKs) and by the phosphatidylinositol 3-kinase (PI3K)/AKT pathway, depending on the type of cell and stimuli.Citation47 Therefore, these upstream molecules that regulates MMP-9 expression or enzymatic activity can be used as a target for treating melanoma cancer metastasis. Our results demonstrated that Mw exerted anti MMP-9 effect by blocking IκBα, P65 and c-Jun phosphorylation but has no effect on c-Fos. Furthermore Mw inhibited the respective DNA binding of NF-κB and AP-1 to MMP-9 promoter.
Next we evaluated the inhibitory effect of Mw on PMA induced invasion and migration in B16F10 cells. PMA is a well-known inflammatory stimulus and tumor promoter that activates almost all PKC isozymes by direct binding.Citation38,48 PMA increases the invasiveness of cancer cells by activating MMP-9 via transcription factors, PKCs, MAPKs or PI3K/AKT pathways.Citation23,49 Mw treatment in a dose-dependent manner impaired MMP-9 activity and gene expression. Moreover, Mw inhibited PMA induced activation of NF-κB and AP-1 levels. However, the distinct mechanisms regulating PMA induced MMP-9 expression in B16F10 melanoma are not defined clearly. To gain a comprehensive understanding of the PMA-induced signaling cascade underlying MMP-9 expression in B16F10 cells, we assessed the effects of specific inhibitors of MAPKs and PI3K on PMA-induced invasion. Our data show that PMA-induced invasion was significantly inhibited by treatment with an ERK-1/2 inhibitor (PD98059) or PI3K/AKT inhibitor (LY294002), than with p38MAPK inhibitor (SB203580) or JNK inhibitor (SP600125). PMA-induced MMP-9 expression and DNA binding of NF-κB was also inhibited by the PI3K/AKT inhibitor, whereas DNA binding of AP-1 was abolished only by an ERK-1/2 inhibitor. Thus results indicate that cell invasion and MMP-9 expression is mainly regulated by NF-κB activation via PI3K/AKT and by AP-1 activation via ERK-1/2. Moreover, Mw suppressed PMA-induced phosphorylation of ERK-1/2 and AKT, key pathways in PMA-induced cell invasion via MMP-9 expression. These results demonstrate that Mw reduces MMP-9 expression by blocking NF-κB activation via PI3K/AKT as well as AP-1 activation via ERK-1/2 signaling. PMA also activates classical (α, βI, βII and γ) and novel (δ, ϵ, θ and η) PKCs by binding to the C1 domain of these isoforms. Activation of PKC by PMA involves PKC translocation, thereby causing proliferation, differentiation, malignant transformation and cell death in cancer cells.Citation39,40,41 Hence, PKC isoforms are promising targets for the prevention and treatment of melanoma cancer. In this study, PMA stimulation resulted in PKCα translocation from cytosol to membrane however, minimal translocation of PKCδ and other isotypes was observed. Treatment with a specific PKCα inhibitor Gö6976 and a broad PKC inhibitor GF109203X caused marked inhibition in PMA-induced ERK-1/2 and AKT activation, MMP-9 expression and cell invasion but not with PKCδ inhibitor Rottlerin. Mw treatment abridged membrane localization of PKCα induced by PMA. Furthermore, B16F10 cells transfected with PKCαOV and PKCδOV exhibit differential effect in MMP-9 expression where PKCαOV cell augmented MMP-9 expression compared to PKCδOV cells and Mw treatment decreased PKCαOV mediated MMP-9 expression and activity. This suggests that PKCα activation is involved in MMP-9 activation and migration, which was inhibited by Mw treatment. Intradermal administration of Mw in murine model led to a substantial inhibition of tumor growth. MMP-9 and PKCα expression was also inhibited in the tumor tissues of mice administered with Mw. These results indicate that Mw can suppress the melanoma cancer growth and MMP-9 expression.
Collectively, Mw inhibit the invasion and migration of B16F10 cells. The anti-invasive effects of Mw in B16F10 cells is through inhibiting the PI3K/AKT/NF-κB axis and PKCα/ERK/AP-1 cascades, with consequent suppression of MMP-9 expression. Intradermal administration of Mw in the tumor site suppressed tumor growth and invasiveness in mice implanted with B16F10 cells. Thus, Mw may be a useful anti-invasive agent in therapeutic strategies against metastatic melanoma tumor.
Materials Methods
Reagents and chemicals
Mw was a kind gift from Dr. B. M. Khamer (Cadila Pharmaceuticals Limited, Gujarat, India). Dulbecco's modified Eagle's medium (DMEM) (AT007) were from Himedia. Penicillin-streptomycin (P4333), collagenase (S5138), SB203580 (S8307), PD98059 (P215), LY294002 (L9908), SP600125 (S5567), Gö6976 (G1171), GF109203X (G2911), Rottlerin (R5648), PMA (P1585) were from Sigma. Fetal calf serum (16140071), Trypsin-EDTA (25200–056) were from Gibco BRL. MMP-9 inhibitor (444249) were obtained from Calbiochem. MMP-2 (sc-10736), MMP-9 (sc-6841), PKCα (sc-208), pPKCα (sc-12356), PKCβII (sc-210), pPKCβII (sc-11760), PKCδ (sc-213), pPKCδ (sc-11770), PKCϵ (sc-214), pPKCϵ (sc-12355), GAPDH (sc-25778), P38 (sc-7972), pP38 (sc-7973), AKT (sc-1618), pAKT (sc-135650) ERK-1/2 (sc-135900), pERK-1/2 (sc-7383), JNK-1/2 (sc-7345), pJNK-1/2 (sc-81502), NFκBP65 (sc-372), NFκBpP65 (sc-33020), pIκBα (sc-52943), IκBα (sc- 371), c-Jun (sc-1694), pc-Jun (sc-16312) antibodies were obtained from Santa Cruz Biotechnology and c-Fos(5348), pc-Fos (2250) antibodies were from cell signaling.
Cell culture
B16F10 and B16F1 cell lines were obtained from ATCC and maintained in DMEM supplemented with 10% FBS at 37°C with 5% CO2. Primary melanocytes were prepared and maintained as described elsewhere.Citation50
Cytotoxic assay and cell viability
5×103 melanoma and melanocyte cells were treated with Mw for 24hr or 48hr. The medium was replaced with fresh DMEM (without Phenol Red) containing 1 mg/ml MTT and incubated at 37°C for 3hr, the untransformed MTT was removed and 50μl of 0.04 M HCl isopropanolic solution was added to each well. After 15min, absorbance was measured on an automatic plate reader (Polarstar optima, BMG Labtech, Victoria) at a reference wavelength of 690 nm and test wavelength of 650 nm. Cell viability was also estimated by trypan blue dye exclusion assay.
Soft-agar colony formation assay
5×104 melanoma cells treated with Mw were suspended in 1ml of DMEM containing 0.3% low-melting-point agarose (Amresco, USA) and 10% FBS, plated on a bottom layer containing 0.6% agarose and 10% FBS. After 2 weeks, plates were stained with 0.2% gentian violet and the colonies were counted under light microscope (20X) (Leica Microsystem, Germany).
Invasion assay
5×104 melanoma cells preincubated with Mw for 2hr was seeded in a 12-well transwell chamber (Corning Costar, Cambridge, MA).The lower chamber was filled with medium containing 20% FBS. After overnight incubation at 37°C, the filter was fixed and stained with 2% ethanol containing 0.2% crystal violet (15min). The stained cells were enumerated under light microscope (20X) (Leica Microsystem, Germany). For quantification, the invaded stained cells were extracted with 33% acetic acid and the absorbance was determined at 570 nm.
Wound healing assay
4×105 melanoma cells were seeded in a 12-well plate and incubated. The confluent cells were scratched with a pipet tip, washed with PBS, and then treated with Mw. After 24hr of incubation, the cells were fixed and stained with 2% ethanol containing 0.2% crystal violet powder (15min), and randomly chosen fields were photographed (20X) (Leica Microsystem, Germany). The number of cells migrated into the scratched area was calculated.
Adhesion assay
5×104 melanoma cells preincubated with Mw for 2hr at 37°C were seeded in a 12-well plate coated with matrigel. Unattached cells were removed by washing with PBS. Attached cells were fixed in 4% paraformaldehyde (15min), stained with 0.02% crystal violet solution (10min), and randomly chosen fields were photographed (20X) (Leica Microsystem, Germany). To quantify the number of attached cells, crystal violet was dissolved with 70% ethanol and O.D. was measured at 570 nm.
Cell proliferation
5×104 melanoma cells were treated with Mw for 24hr and 48hr. Each well was then pulsed with 1 μCi [3 H]–Thymidine (specific activity 20 Ci/mmol). [3 H]-Thymidine incorporation was determined on liquid scintillation counter (Tri-Carb 2800TR; Perkin Elmer, Waltham, USA).Citation39
Gelatin zymography
1×105 B16F10 cells were incubated in serum-free medium including 80nM PMA for 24hr, following pretreatment with Mw for 2hr, or specific inhibitors of MAPKs, PI3K and PKC isotypes for 1hr, respectively. The conditioned medium was collected and protein concentration was determined. Equal amounts of protein (30μg) were separated on a 10% SDS-PAGE gel containing 0.1% gelatin. After electrophoresis, the gels were washed in renaturing buffer, equilibrated in developing buffer and finally incubated in fresh developing buffer at 37°C for 24hr. The gelatinolytic activity of MMPs was visualized by staining the gels with 0.5% Coomassie blue R-250 in 45% methanol, 10% acetic acid and destained with 45% methanol, 10% acetic acid until clear bands appeared.
RNA Isolation and PCR
Total RNA was extracted from cell via TRI reagent.Citation51 For cDNA synthesis, 1μg of total RNA was reverse-transcribed as per manufacturer's protocol and amplified with 0.5 unit Taq DNA polymerase (Primers in ). PCR products were run on 1.5% agarose gel, stained with EtBr and visualized under UV-light. Quantitative real-time PCR was performed using SYBR-Green mix and the ABI-7500 RT-PCR system (Applied Biosystems, Waltham, USA) according to the manufacturer's instruction.Citation52
Table 1 List of primers
Immunoblotting
Cell lysates were prepared as described elsewhere.Citation53 Equal amounts of protein (40μg) were subjected to 10% SDS-PAGE and were subsequently transferred to a nitrocellulose membrane. The membrane was blocked overnight with 3% bovine serum albumin in Tris-saline buffer (pH, 7.5), and immunoblotting was performed keeping anti-GAPDH antibodies as control.Citation54
Nuclear and cytoplasmic fractionation
Nuclear and cytoplasmic extracts were prepared as described.Citation33 The purity of nuclear and cytoplasmic extracts was assessed by protein gel blot with anti-laminB and anti-GAPDH antibodies as control respectively.
Chromatin Immunoprecipitation (ChIP)
B16F10 cells treated with Mw for 24hr were processed for ChIP assay using a ChIP assay kit (Millipore) as per manufacturer's protocol.Citation55 Briefly, immunoprecipitation was performed with NF-κB (P65), AP-1 (c-Jun) specific antibody. The NF-κB and AP-1 binding site of MMP-9 promoter was detected by PCR (Primers in ).
PKCα and PKCδ cloning
The gene products were cloned in TA vector (Promega, USA) and subsequently sub-cloned in pd2EGFP-N1 vector using the restriction enzymes. The clones were ligated and sequenced as mentioned elsewhere.Citation39
In vivo experiments
Female C57BL6 mice, 6–8 weeks old, were purchased from National Center for Laboratory Animal Sciences, Hyderabad, India. The mice were injected subcutaneously with B16F1 or B16F10 cells (3×105 cells) in 100µl PBS into the right flank of each mouse. Mice were divided into 2 groups (n = 5), the control group received vehicle (PBS), while treatment group received Mw (total dose: 107 cells of Mw/100ul PBS/mouse) intradermally (i.d.) after 3 d of tumor implantation. The intradermal injection was performed twice in adjacent site of tumor (50µl per injection). The injection was repeated within 15 d interval. The tumor volume were estimated using the following formula: 4π/3X (width/2)2X (length/2). Mice were euthanized after day 35 and excised surgically.
Immunofluorescence staining
Surgically excised tumors were cryosectioned to 7mm thick sections. The frozen sections were thawed and fixed with 4% paraformaldehyde for 30min. After blocking with 3% BSA/0.2% Triton X-100 in PBS for 1hr, sections were incubated with respective antibody at 4°C overnight. For visualization of cell nucleus, DAPI was used. Sections were observed by confocal laser scanning microscope (Leica Microsystems, USA).
Statistical analysis
In vitro assays were done in triplicate and a minimum of 5 mice were used per group for in vivo experiments. Data, including densitometry analysis, shown as means ± SD, are from an experiment performed at least 3 times. Student's t-test or one-way ANOVA was used to assess the significance of differences between the mean values for control and experimental groups. A difference with P < 0.05 was considered significant, and a difference with P < 0.001 was considered highly significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank The Council of Scientific and Industrial Research (CSIR), Government of India, New Delhi, for providing fellowship to Kuntal Halder.
Funding
This work was supported by The Council of Scientific and Industrial Research (CSIR), Government of India, New Delhi.
References
- Erdei E, Torres SM. A new understanding in the epidemiology of melanoma. Expert Rev Anticancer Ther 2010; 11:1811-23; http://dx.doi.org/10.1586/era.10.170
- Madhunapantula SV, Robertson GP. Chemoprevention of melanoma. Adv Pharmacol 2012; 65:361-98; PMID:22959032; http://dx.doi.org/10.1016/B978-0-12-397927-8.00012-9
- Inamdar GS, Madhunapantula SV, Robertson GP. Targeting the MAPK pathway in melanoma: why some approaches succeed and other fail. Biochem Pharmacol 2010; 5:624-37; http://dx.doi.org/10.1016/j.bcp.2010.04.029
- Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 2009; 4:274-84; http://dx.doi.org/10.1038/nrc2622
- Salo T, Mäkelä M, Kylmäniemi M, Autio-Harmainen H, Larjava H. Expression of matrix metalloproteinase-2 and −9 during early human wound healing. Lab Invest 1994; 2:176-82.
- Deryugina EI, Bourdon MA, Reisfeld RA, Strongin A. Remodeling of collagen matrix by human tumor cells requires activation and cell surface association of matrix metalloproteinase-2. Cancer Res 1998; 16:3743-50.
- Deryugina EI, Luo GX, Reisfeld RA, Bourdon MA, Strongin A. Tumor cell invasion through matrigel is regulated by activated matrix metalloproteinase-2. Anticancer Res 1997; 17:3201-10; PMID:9413149
- Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2002; 3:161-74; http://dx.doi.org/10.1038/nrc745
- Gong Y, Chippada-Venkata UD, Oh WK. Roles of matrix metalloproteinases and their natural inhibitors in prostate cancer progression. Cancers (Basel) 2014; 3:1298-327; http://dx.doi.org/10.3390/cancers6031298
- Yan C, Boyd DD. Regulation of matrix metalloproteinase gene expression. J Cell Physiol 2007; 1:19-26; http://dx.doi.org/10.1002/jcp.20948
- Sun J. Matrix metalloproteinases and tissue inhibitor of metalloproteinases are essential for the inflammatory response in cancer cells. J Signal Transduct 2010; 2010:985132; PMID:21152266; http://dx.doi.org/10.1155/2010/985132
- Duffy MJ, Maguire TM, Hill A, McDermott E, O'Higgins N. Metalloproteinases: role in breast carcinogenesis, invasion and metastasis. Breast Cancer Res 2000; 4:252-7; http://dx.doi.org/10.1186/bcr65
- Hanemaaijer R, Verheijen JH, Maguire TM, Visser H, Toet K, McDermott E, O'Higgins N, Duffy MJ. Increased gelatinase-A and gelatinase-B activities in malignant vs. benign breast tumors. Int J Cancer 2000; 2:204-7; http://dx.doi.org/10.1002/(SICI)1097-0215(20000415)86:2%3c204::AID-IJC9%3e3.0.CO;2-6
- Chambers AF, Matrisian LM. Changing views of the role of matrix metalloproteinases in metastasis. J Natl Cancer Inst 1997; 17:1260-70; http://dx.doi.org/10.1093/jnci/89.17.1260
- Davies KJ. The Complex Interaction of Matrix Metalloproteinases in the Migration of Cancer Cells through Breast Tissue Stroma. Int J Breast Cancer 2014; 2014:839094; PMID:24800085
- Määttä M, Soini Y, Liakka A, Autio-Harmainen H. Differential expression of matrix metalloproteinase (MMP)-2, MMP-9, and membrane type 1-MMP in hepatocellular and pancreatic adenocarcinoma: implications for tumor progression and clinical prognosis. Clin Cancer Res 2000; 7:2726-2734.
- Said AH, Raufman JP, Xie G. The role of matrix metalloproteinases in colorectal cancer. Cancers (Basel) 2014; 6:366-375; PMID:24518611; http://dx.doi.org/10.3390/cancers6010366
- Vaday GG, Schor H, Rahat, MA, Lahat N, Lider O. Transforming growth factor-β suppresses tumor necrosis factor α-induced matrix metalloproteinase-9 expression in monocytes. J Leukoc Biol 2001; 4:613-621.
- Aalinkeel R, Nair MP, Sufrin G, Mahajan SD, Chadha KC, Chawda RP, Schwartz SA. Gene expression of angiogenic factors correlates with metastatic potential of prostate cancer cells. Cancer Res 2004; 15:5311-21; http://dx.doi.org/10.1158/0008-5472.CAN-2506-2
- Pellikainen JM, Ropponen KM, Kataja VV, Kellokoski JK, Eskelinen MJ, Kosma VM. Expression of matrix metalloproteinase (MMP)-2 and MMP-9 in breast cancer with a special reference to activator protein-2, HER2, and prognosis. Clin Cancer Res 2004; 22:7621-8; http://dx.doi.org/10.1158/1078-0432.CCR-04-1061
- Kim D, Kim S, Koh H, Yoon SO, Chung AS, Cho KS, Chung J. Akt/PKB promotes cancer cell invasion via increased motility and metalloproteinase production. FASEB J 2001; 15:1953-62; PMID:11532975; http://dx.doi.org/10.1096/fj.01-0198com
- Sato T, Koike L, Miyata Y, Hirata M, Mimaki Y, Sashida Y, Yano M, Ito A. Inhibition of activator protein-1 binding activity and phosphatidylinositol 3-kinase pathway by nobiletin, a polymethoxy flavonoid, results in augmentation of tissue inhibitor of metalloproteinases-1 production and suppression of production of matrix metalloproteinases-1 and −9 in human fibrosarcoma HT-1080 cells. Cancer Res 2002; 62:1025-9; PMID:11861377
- Shin Y, Yoon SH, Choe EY, Cho SH, Woo CH, Rho JY, Kim JH. PMA-induced upregulation of MMP-9 is regulated by a PKCalpha-NF-kappaB cascade in human lung epithelial cells. Exp Mol Med 2007; 39:97-105; PMID:17334233; http://dx.doi.org/10.1038/emm.2007.11
- Eberhardt W, Huwiler A, Beck KF, Walpen S, Pfeilschifter J. Amplification of IL- 1 β-induced matrix metalloproteinase-9 expression by superoxide in rat glomerular mesangial cells is mediated by increased activities of NF-kappa B and activating protein-1 and involves activation of the mitogen-activated protein kinase pathways. J Immunol 2000; 165:5788-97; PMID:11067938; http://dx.doi.org/10.4049/jimmunol.165.10.5788
- Garg A, Aggarwal BB. Nuclear transcription factor-kB as a target for cancer drug development. Leukemia 2002; 16:1053-68; PMID:12040437; http://dx.doi.org/10.1038/sj.leu.2402482
- Pastore S, Mascia F, Mariotti F, Dattilo C, Mariani V, Girolomoni G. ERK1/2 regulates epidermal chemokine expression and skin inflammation. J Immunol 2005; 174:5047-56; PMID:15814736; http://dx.doi.org/10.4049/jimmunol.174.8.5047
- Johnson LL, Dyer R, Hupe DJ. Matrix metalloproteinases. Curr Opin Chem Biol 1998; 2:466-71; PMID:9736919; http://dx.doi.org/10.1016/S1367-5931(98)80122-1
- Liabakk NB, Talbot I, Smith RA, Wilkinson K, Balkwill F. Matrix metalloprotease 2 (MMP-2) and matrix metalloprotease 9 (MMP-9) type IV collagenases in colorectal cancer. Cancer Res 1996; 56:190-6; PMID:8548762
- Pandey RK, Dahiya Y, Sodhi A. Mycobacterium indicus pranii downregulates MMP-9 and iNOS through COX-2 dependent and TNF-α independent pathway in mouse peritoneal macrophages in vitro. Microbes Infect 2012; 4:348-56; http://dx.doi.org/10.1016/j.micinf.2011.11.004
- Gupta A, Geetha N, Mani J, Upadhyay P, Katoch VM, Natrajan M, Gupta UD, Bhaskar S. Immunogenicity and protective efficacy of “Mycobacterium w” against Mycobacterium tuberculosis in mice immunized with live versus heat-killed M.w by the aerosol or parenteral route. Infect Immun 2009; 1:223-31; http://dx.doi.org/10.1128/IAI.00526-08
- Rakshit S, Ponnusamy M, Papanna S, Saha B, Ahmed A, Nandi D. Immunotherapeutic efficacy of Mycobacterium indicus pranii in eliciting anti-tumor T cell responses: critical roles of IFNγ. Int J Cancer 2012; 4:865-75; http://dx.doi.org/10.1002/ijc.26099
- Ahmad F, Man J, Kumar P, Haridas S, Upadhyay P, Bhaskar S. Activation of anti-tumor immune response and reduction of regulatory T cells with Mycobacterium indicus pranii (MIP) therapy in tumor bearing mice. PLoS One 2011; 9:e25424; http://dx.doi.org/10.1371/journal.pone.0025424
- Adhikari A, Majumder S, Banerjee S, Gupta G, Bhattacharya P, Majumdar SB, Saha B, Majumdar S. Mycobacterium indicus pranii (Mw)-mediated protection against visceral leishmaniasis: involvement of TLR4 signalling. J Antimicrob Chemother 2012; 12:2892-2902; http://dx.doi.org/10.1093/jac/dks315
- Kumar P, Tyagi R, Das G, Bhaskar S. Mycobacterium indicus pranii and Mycobacterium bovis BCG lead to differential macrophage activation in Toll-like receptor-dependent manner. Immunology 2014; 143:258-68; PMID:24766519; http://dx.doi.org/10.1111/imm.12306
- Adhikari A, Gupta G, Majumder S, Banerjee S, Bhattacharjee S, Bhattacharya P, Kumari S, Haldar S, Majumdar SB, Saha B, Majumdar S. Mycobacterium indicus pranii (Mw) re-establishes host protective immune response in Leishmania donovani infected macrophages: critical role of IL-12. PLoS One 2012; 7:e40265; PMID:22792256; http://dx.doi.org/10.1371/journal.pone.0040265
- Purcell WT, Rudek MA, Hidalgo M. Development of matrix metalloproteinase inhibitors in cancer therapy. Hematol Oncol Clin North Am 2002; 5:1189-227; http://dx.doi.org/10.1016/S0889-8588(02)00044-8
- Hua J, Muschel RJ. Inhibition of matrix metalloproteinase 9 expression by a ribozyme blocks metastasis in a rat sarcoma model system. Cancer Res 1996; 22:5279-84.
- Hwang YP, Yun HJ, Kim HG, Han EH, Lee GW, Jeong HG. Suppression of PMA-induced tumor cell invasion by dihydroartemisinin via inhibition of PKCalpha/Raf/MAPKs and NF-kappaB/AP-1-dependent mechanisms. Biochem Pharmacol 2010; 12:1714-26; http://dx.doi.org/10.1016/j.bcp.2010.02.003
- Halder K, Banerjee S, Bose A, Majumder S, Majumdar S. Overexpressed PKCδ downregulates the expression of PKCα in B16F10 melanoma: induction of apoptosis by PKCδ via ceramide generation. PLoS One 2014; 3:e91656; http://dx.doi.org/10.1371/journal.pone.0091656
- Tahara E, Kadara H, Lacroix L, Lotan D, Lotan R. Activation of protein kinase C by phorbol 12-myristate 13-acetate suppresses the growth of lung cancer cells through KLF6 induction. Cancer Biol Ther 2009; 9:801-7; http://dx.doi.org/10.4161/cbt.8.9.8186
- Shih SC, Mullen A, Abrams K, Mukhopadhyay D, Claffey KP. Role of protein kinase C isoforms in phorbol ester-induced vascular endothelial growth factor expression in human glioblastoma cells. J Biol Chem 1999; 22:15407-14; http://dx.doi.org/10.1074/jbc.274.22.15407
- Song G, Ouyang G, Mao Y, Ming Y, Bao S, Hu T. Osteopontin promotes gastric cancer metastasis by augmenting cell survival and invasion through Akt-mediated HIF-1α upregulation and MMP9 activation. J Cell Mol Med 2008; 13:1706-18; http://dx.doi.org/10.1111/j.1582-4934.2008.00540.x
- Stetler-Stevenson WG, Brown PD, Onisto M, Levy AT, Liotta LA. Tissue inhibitor of metalloproteinases-2 (TIMP-2) mRNA expression in tumor cell lines and human tumor tissues. J Biol Chem 1990; 265:13933-8; PMID:2380196
- Stetler-Stevenson WG, Brown PD, Onisto M, Levy AT, Liotta LA. Tissue inhibitor of metalloproteinases-2 (TIMP-2) mRNA expression in tumor cell lines and human tumor tissues. J Biol Chem 1990; 265:13933-8; PMID:2380196
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa] B activity. Annu Rev Immunol 2000; 18:621-63; PMID:10837071; http://dx.doi.org/10.1146/annurev.immunol.18.1.621
- Karin M, Liu Zg, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol 1997; 2:240-6; http://dx.doi.org/10.1016/S0955-0674(97)80068-3
- Woo JH, Lim JH, Kim YH, Suh SI, Min DS, Chang JS, Lee YH, Park JW, Kwon TK. Resveratrol inhibits phorbol myristate acetate-induced matrix metalloproteinase-9 expression by inhibiting JNK and PKC delta signal transduction.Oncogene 2004; 23:1845-53; PMID:14661062; http://dx.doi.org/10.1038/sj.onc.1207307
- Hsiang CY, Wu SL, Chen JC, Lo HY, Li CC, Chiang SY, Wu HC, Ho TY. Acetaldehyde induces matrix metalloproteinase-9 gene expression via nuclear factor-kappaB and activator protein 1 signaling pathways in human hepatocellular carcinoma cells: association with the invasive potential. Toxicol Lett 2007; 171:78-86; PMID:17543481; http://dx.doi.org/10.1016/j.toxlet.2007.04.009
- Park SK, Hwang YS, Park KK, Park HJ, Seo JY, Chung WY. Kalopanaxsaponin A inhibits PMA-induced invasion by reducing matrix metalloproteinase-9 via PI3K/Akt- and PKCdelta-mediated signaling in MCF-7 human breast cancer cells. Carcinogenesis 2009; 7:1225-33; http://dx.doi.org/10.1093/carcin/bgp111
- Guyonneau L, Murisier F, Rossier A, Moulin A, Beermann F. Melanocytes and pigmentation are affected in dopachrome tautomerase knockout mice. Mol Cell Biol 2004; 24:3396-403. Karin M, Liu Zg, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol 1997; 2:240-6; http://dx.doi.org/10.1128/MCB.24.8.3396-3403.2004
- Chomezynasky P, Sacchi N. A single step method of RNA isolation by guanidium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987; 162:156-9; PMID:2440339; http://dx.doi.org/10.1006/abio.1987.9999
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. NewYork: Cold Spring Harbor, Cold Spring Laboratory Press. 1989. Park SK, Hwang YS, Park KK, Park HJ, Seo JY, Chung WY. Kalopanaxsaponin A inhibits PMA-induced invasion by reducing matrix metalloproteinase-9 via PI3K/Akt- and PKCdelta-mediated signaling in MCF-7 human breast cancer cells. Carcinogenesis 2009; 7:1225-33.
- Majumdar S, Kane LH, Rossi MW, Volpp BD, Nauseef WM, Korchak HM. Protein kinase C isotypes and signal transduction in human neutrophils: selective substrate specificity of calcium dependent β-PKC and novel calcium independent n-PKC. Biochim Biophys Acta 1993; 1176:276-86; PMID:8471629; http://dx.doi.org/10.1016/0167-4889(93)90056-U
- Ghosh S, Bhattacharyya S, Sirkar M, Sa GS, Das T, Majumdar D, Roy S, Majumdar S. Leishmania donovani suppresses activator protein-1 and NF-kB in host macrophages via ceramide generation: involvement of extracellular signal regulated kinase. Infect Immun 2002; 70:6828-38; PMID:12438359; http://dx.doi.org/10.1128/IAI.70.12.6828-6838.2002
- Das S, Banerjee S, Majumder S, Paul Chowdhury B, Goswami A, Halder K, Chakraborty U, Pal NK, Majumdar S. Immune subversion by Mycobacterium tuberculosis through CCR5 mediated signaling: involvement of IL-10. PLoS One 2014; 4:e92477; http://dx.doi.org/10.1371/journal.pone.0092477