
Abstract
Anaplastic lymphoma kinase (ALK) is correlated with oncogenesis in different types of cancers, such as anaplastic large cell lymphoma, lung cancer, neuroblastoma, and even breast cancer, by abnormal fusion of ALK or non-fusion ALK activation. ALK is a receptor tyrosine kinase, with a single transmembrane domain, that plays an important role in development. Upon ligand binding to the extracellular domain, the receptor undergoes dimerization and subsequent autophosphorylation of the intracellular kinase domain. In recent years, ALK inhibitors have been developed for cancer treatment. These inhibitors target ALK activity and show effectiveness in ALK-positive non-small cell lung cancer. However, acquired treatment resistance makes the future of this therapy unclear; new strategies are underway to overcome the limitations of current ALK inhibitors.
Abbreviations
| ALCL | = | anaplastic large cell lymphoma |
| ALK | = | anaplastic lymphoma kinase |
| CNS | = | central nervous system |
| DLBCL | = | diffuse large B-cell lymphoma |
| EGFR | = | epidermal growth factor receptor |
| EML4 | = | echinoderm microtubule associated protein like 4 |
| IMT | = | inflammatory myofibroblastic tumor |
| JAK | = | Janus kinase |
| LTK | = | leukocyte tyrosine kinase |
| NHL | = | non-Hodgkin lymphoma |
| NPM | = | nucleophosmin |
| NSCLC | = | non-small cell lung cancer |
| STAT | = | signal transducer and activator of transcription |
| TKI | = | tyrosine kinase inhibitor |
| TPM | = | tropomyosin |
Structure and Function of Anaplastic Lymphoma Kinase
Tyrosine kinases are involved in the pathogenesis of most cancers. Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase belonging to the insulin receptor superfamily, and shares a high degree of homology with leukocyte tyrosine kinase (LTK).Citation1 ALK was initially identified as the product of a gene rearrangement in anaplastic large cell lymphoma (ALCL).Citation2,3 ALK was subsequently found to be rearranged, mutated, or amplified in a series of tumors including lymphoma, neuroblastoma, and non-small cell lung cancer (NSCLC).Citation4 There is strong preclinical evidence that ALK may be a driving force of oncogenesis in these cases.
ALK Structure
The human ALK gene is located on the 2p23 chromosomal segment, from base pair 29,192,773 to 29,921,610. The ALK gene is composed of 26 exons. The 6226-base pair cDNA encodes a 1620 amino acid (aa), 177,442 Da polypeptide; its post-translational modifications generate mature ALK of approximately 200–220 kDa.Citation5,6 ALK is highly conserved across species, and is transiently expressed in specific regions of the central and peripheral nervous systems. The single ALK locus encodes a classical receptor tyrosine kinase that comprises an extracellular ligand-binding domain (1030 aa), a transmembrane domain (28 aa), and an intracellular tyrosine kinase domain (561 aa) ().Citation7
Figure 1. The ALK receptor kinase: its domains, fusion break point, mutations, and resistance mutations. Reproduced with modification from Wellstein A, Toretsky JA. Hunting ALK to feed targeted cancer therapy. Nat Med. 2011; 17: 290–1. PMID: 21383740; doi: 10.1038/nm0311–290. © 2011 Nature America, Inc. By permission.

The extracellular domain of human ALK comprises 2 MAM (meprin, A5 protein, and receptor protein tyrosine phosphatase µ) domains separated by a low density lipoprotein class A domain. A glycine-rich domain precedes the transmembrane-spanning domain. The protein tyrosine kinase domain lies in the cytoplasmic portion. The ALK kinase domain shares the 3-tyrosine motif YxxxYY, with the other kinases of the same family. The three tyrosine residues (Tyr1287, Tyr1282 and Tyr1283) are located in the activation loop and represent the major autophosphorylation sites; the sequential phosphorylation of the tyrosine triplet regulates kinase activity.Citation8,9 ALK becomes activated only upon ligand-induced homo-dimerization, and inactivated through de-phosphorylation by receptor protein tyrosine phosphatase beta and zeta complex (PTPRB/PTPRZ1) when there is no stimulation by a ligand.Citation10
ALK Function
Immunohistochemical analysis of adult human tissues reveals that ALK expression is sparsely scattered in neural cells, endothelial cells and pericytes in the brain.Citation6 Morris et al.Citation2 report that ALK mRNA is expressed in the adult human brain, small intestine, testis, prostate, and colon; but not in normal human lymphoid cells, spleen, thymus, ovary, heart, placenta, lung, liver, skeletal muscle, kidney, or pancreas.
Mammalian ALK is thought to play a role in the development and function of the nervous system based upon the expression of its mRNA throughout the nervous system during mouse embryogenesis.Citation11,12 ALK mRNA and protein levels seem to diminish in all tissues after birth — they reach minimum levels at 3 weeks of age, but are maintained at low levels in adult animals.Citation5,7 ALK knockout mice have provided further clues to possible physiological roles of the receptor in the nervous system. These mice develop normally and have a full life span. They do not display any anatomical abnormalities, but intriguingly exhibit better performance relative to wild-type littermates in experimental models of clinical depression, such as behavioral despair tests.Citation11 Thus, at present, the normal function of ALK remains an open question.
ALK Signaling
Information regarding ALK signaling mostly comes from studies of ALK fusion proteins such as NPM-ALK and EML4-ALK, and has been complemented in recent years by studies of full-length ALK mutants. Different fusion partners affect ALK homo-dimerization, as well as ALK signaling potential.
In terms of general signaling output, ALK activates multiple pathways. These include phospholipase Cγ, Janus kinase (JAK)-signal transducer and activator of transcription (STAT), PI3K-AKT, mTOR, sonic hedgehog, JUNB, CRKL-C3G (also known as RAPGEF1)-RAP1 GTPase and MAPK signaling cascades, which affect cell growth, transformation and anti-apoptotic signaling ().Citation13 It has been reported that in this scenario, a number of additional genes are transactivated by NPM-ALK activity. Some of these genes, such as JUNB, YBX1, BCL2A1, HIF1A, have been validated by siRNA analyses.Citation14–17 MYCN is identified in neuroblastoma as a transcriptional target of activated full-length ALK.Citation18 There are reports of ALK signaling involving microRNAs, with miR-135b, miR-29a and miR-16 being downstream of NPM-ALK, and miR-96-mediated regulation of ALK expression.Citation19-22
Figure 2. The ALK receptor kinase signaling pathways.
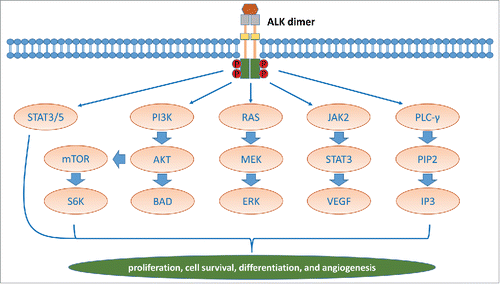
Two proteins, midkine and pleiotrophin, have been reported to be activating ligands for mammalian ALK.Citation23 They are also able to activate other receptors, including receptor protein tyrosine phosphatase-β (RPTPβ) and RPTPζ, N-syndecan, low-density lipoprotein receptor-related protein, and integrins.Citation13 Pleiotrophin can also function via RPTPβ and RPTPζ phosphatases to activate ALK signaling.Citation23 However, they are not confirmed to stimulate mammalian ALK under conditions where monoclonal antibodies directed against the ALK extracellular domain are able to activate its signaling.Citation24 Thus, the identity of physiological ligands for ALK is uncertain.Citation25,26
ALK Fusion Proteins in Cancers
A gene fusion may occur when a translocation joins 2 otherwise separated genes; gene fusion is relatively common in cancers. Reciprocal translocations are usually an exchange of materials between nonhomologous chromosomes, and incidence in human newborns is estimated from 1 in 500 to 1 in 625. Such translocations are usually harmless and may be found through prenatal diagnosis. Most balanced translocation carriers are healthy and do not have any symptoms, but about 6% have a spectrum of symptoms that may include autism, intellectual disability, or congenital anomalies. A gene disrupted or disregulated at the breakpoint of the translocation carrier is likely the cause of these symptoms. Several forms of cancer, mainly leukemia, are caused by acquired translocations. Translocations have also been described in solid malignancies such as Ewing's sarcoma.Citation27
As an oncogene, ALK is transiently expressed in specific regions. When ALK fusion occurs, the formation of dimers by the amino-terminal protein of the ALK fusion proteins will result in the activation of the ALK protein kinase domain that plays a key role in the tumorigenic process. The partner protein, which is the C-terminal protein, controls the fusion's behavior, such as expression level and activation; its effect may be to constitutively activate the tyrosine kinase. Therefore, these cells uncontrolledly proliferate, survive, differentiate, and migrate, consequently leading to cancer.Citation28
ALCL and NMP-ALK
Anaplastic large cell lymphoma (ALCL) is a rare type of non-Hodgkin lymphoma (NHL), but one of the more common subtypes of T-cell lymphoma. ALCL comprises about 3% of all NHLs but an estimated 10 to 30% of all NHL's in children. ALCL can clinically present in 2 main ways: cutaneous ALCL involving the skin usually grows slowly; systemic ALCL tends to grow in lymph nodes and other organs and often spreads quickly. Systemic ALCL is sub-divided into ALK-positive and ALK-negative entities. ALK-positive lymphomas define a distinct class of NHL. More recently, another category of ALCL has been increasingly reported in the literature and is associated with the presence of breast implants.Citation29,30 ALK-positive cancers are more common in young patients and usually respond well to chemotherapy. ALK-negative cancers are more common in people over 60. This type may need more aggressive treatment because of increased recurrence rates. Accordingly, systemic ALK-positive ALCLs have 5-year survivals of 70–80%, while systemic ALK-negative ALCLs have estimated 5-year survivals of 15–45%. Thus, not only has the presence of ALK mutations been used to aid physicians in chemotherapeutic regimens, ALK mutations also are prognostic markers.
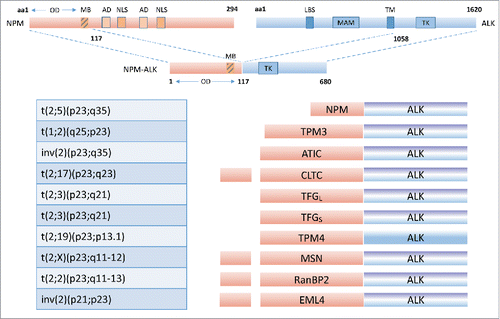
ALCL is frequently associated with the t(2;5)(p23;q35) translocation.Citation31 It creates a fusion gene, most commonly fusing the ALK gene (2p23) and the NPM gene (5q35) occurring in approximately 70–80% of all ALK-positive ALCL patients. The frequency of the NPM-ALK rearrangement among 1,084 reported ALCL patients is 43%; for children 86/103 (83%), but for adults only 106/339 (31%).Citation32 Several t(2;5) translocation breakpoint variants have been described. Some of the breakpoints have been cloned, and the fusion proteins have been identified. NPM (also known as NMP1, nucleolar phosphoprotein B23, numatrin) encodes a 294 aa, 32,575 Da multifunctional protein which is involved in the transport of pre-ribosomal particles and ribosome biogenesis, regulation of cell division, DNA repair, transcription and genomic stability.Citation33 The NPM protein includes a nucleolar localization signal and a dimerization domain, favoring the generation of large homo-complexesCitation34 and hetero-complexes of NPM and ALK activation.Citation35,36 The NPM-ALK chimeric protein is constitutively expressed from the NPM promoter, leading to the ectopic overexpression of the ALK catalytic domain. NPM-ALK fusion proteins homodimerize and the kinase becomes constitutively active. The constitutively active fusion proteins are responsible for 5–10% of NHL.
Many other rearrangements involving the ALK gene have recently been shown to be associated with ALCL, including ALO17-ALK, TFG-ALK, MSN-ALK, TPM3-ALK, TPM4-ALK, ATIC-ALK, MYH9-ALK, CLTC-ALK (), among which the ATIC-ALK rearrangement resulting from the inv(2)(p23q35) translocation is probably the most recurrent.Citation37
Figure 3. The ALK receptor kinase: fusion proteins. Reproduced with modification from Ardini E, Magnaghi P, Orsini P, Galvani A, Menichincheri M. Anaplastic Lymphoma Kinase: role in specific tumors, and development of small molecule inhibitors for cancer therapy. Cancer Lett 2010; 299: 81–94. PMID: 20934803; doi: 10.1016/j.canlet.2010.09.001. © 2010 Elsevier Ireland Ltd. By permission.
There is evidence of an immune response to the ALK protein. Autologous antibodies against NPM-ALK can be detected in ALK-positive patients, but not in control subjects; this might be a reason for ALK-positive ALCL having a relatively good prognosis.Citation38
NPM-ALK downstream signaling includes phospholipase C-γ, phosphatidylinositol 3-kinase, the Ras/ERK1/2 module, and the JAK/STAT pathway. In vitro studies revealed the functional relationship between ALK and CD30.Citation39 In ALK-positive ALCL, CD30 transcription is upregulated by ALK via the ERK1/2 pathway and by phosphorylated STAT3. The effect of CD30 engagement in ALCL cells is the activation of NF-κB pathways, which results in apoptosis and p21-mediated cell-cycle arrest. It follows that anti-CD30 antibodies are of great interest in ALK-positive ALCL treatment, nowadays with routine use in select patients in clinics across the world.Citation40
IMT and TPM3-ALK
Inflammatory myofibroblastic tumors (IMT) possess a pseudosarcomatous inflammatory appearance and can occur in nearly every soft tissue and viscera of the body. IMT most commonly arise in the lung, abdomen, pelvis, and retroperitoneum. However, IMT also arise in other sites, including but not limited to soft tissue, central nervous system (CNS), and bone. It occurs primarily in children and young adults, though it can occur at any age.
Approximately 50% of IMT display clonal rearrangements of chromosome 2's short arm (where ALK is located) fused to TPM3 or to TPM4.Citation41,42 TPM3 is located on chromosome 1 and encodes a non-muscle tropomyosin, of which the N-terminal residues are fused to ALK. TPM4 is a homolog of TPM3 located on the short arm of chromosome 19. Both TPM3-ALK and TPM4-ALK proteins cause constitutive autophosphorylation and activation of ALK as a result of homodimerization through the TPM coiled domain.Citation43
The TPM3-ALK downstream signaling involves STAT3. Many other fusion proteins are found in IMT, including CLTC-ALK, ATIC-ALK, SEC31A-ALK, RANBP2-ALK, PPFIBP1-ALK, and CARS-ALK. IMT were the first solid tumor to be associated with ALK translocation. The presence of ALK translocations is associated with a better prognosis in both ALCL and IMT, which can be exploited for diagnostic and therapeutic purposes.
NSCLC and EML4-ALK
Lung cancer is the leading cause of cancer-related deaths worldwide (about 1.8 million new cases per year and 1.4 million deaths per year) with a 5-year survival rate of about 5% (www.cancer.gov). Small-cell lung cancer accounts for 10–15% of all lung cancers, and non-small cell lung cancer (NSCLC) accounts for 85–90% of lung cancers. In 2007, Prof. Mano's group reported that the chromosomal rearrangement involving the ALK and EML4 (echinoderm microtubule-associated protein like 4) genes showed potent transforming activity in NSCLC.Citation4 About 5% of NSCLCs harbor an EML4-ALK fusion protein; the rearrangement is frequently observed in relatively younger patients, non- or light smokers, and those with adenocarcinoma histology without other genetic disorders, such as mutations of the epidermal growth factor receptor (EGFR) gene.Citation44,45 All 13 fusion variants of EML4-ALK contain exons 20–29 of ALK, which encode the entire intracellular segment of ALK, and 8 different EML4 exons (2, 6, 13, 14, 15, 17, 18, and 20). Other ALK fusion proteins are also found in NSCLC, including KIF5B-ALK, TFG-ALK, KLC1-ALK, PTPN3-ALK, and STRN-ALK. Downstream signaling of this group of fusion proteins includes Ras/ERK1/2, Akt, and JAK/STAT. Importantly, inhibitors of ALK markedly inhibit the growth of BA/F3 cells that express EML4-ALK;Citation46 thus, ALK rearrangements ware thought of as potential therapeutic targets. Although the proportion of NSCLCs with EML4-ALK fusion proteins is low (5%), the absolute number remains high due to the relatively high incidence of NSCLC. Thus, the total number of cases of NSCLC amenable to treatment with ALK inhibitors is greater than that for all other known ALK-related cancers combined. The EML4-ALK fusion protein was discovered in NSCLCs in 2007, and a clinically effective inhibitor (crizotinib) was approved by the U.S. FDA in 2011. These are now increasingly being recognized as first-line therapy for ALK-positive NSCLC instead of conventional chemotherapy regimens, along with increased use of routine ALK testing in all NSCLC (adenocarcinoma) patients at some centers.
DLBCL and CLTC-ALK
Diffuse large B-cell lymphoma (DLBCL) is a malignancy of B cells, a type of leukocyte that is responsible for antibody production. It is the most common type of non-Hodgkin lymphoma diagnosed in adults and accounts for 30–40% of newly diagnosed lymphomas in the United States, equating to 21,000–28,000 cases per year (www.cancer.gov). The most frequent chromosomal rearrangement of ALK in DLBCL is the t(2;17)(p23;q23) translocation, which generates CLTC-ALK. A small portion of DLBCLs, less than 0.5–1%, possess the NPM-ALK fusion protein or other fusion proteins such as SQSTM1-ALK and SEC31A-ALK. This subset of B-cell lymphoma is associated with poor prognosis. However, ALK-targeted therapy is likely to benefit ALK-positive DLBCL patients.Citation47
So far, 22 different genes have been described as being translocated with ALK () and, adding to the complexity, within the different ALK fusion there are examples of several breakpoint variants, as illustrated by the EML4-ALK translocations observed in NSCLC, by which multiple EML4 exon breakpoints fuse in-frame with exon 20 of ALK. Differences have been reported between a number of ALK fusion proteins with regard to proliferation rate, colony formation, invasion, and tumorigenicity owing to activation of various signaling pathways. The variance of ALK fusion proteins should relate to the pathogenesis of cancer.
Despite the variety of ALK fusion partners, some general features can be noted. The initiation of transcription of ALK fusion proteins is driven by the regulatory regions of the partner gene. The subcellular localization of the fusion protein is determined by the partner protein, which means that ALK activity can occur in the nucleus and/or in the cytoplasm. The dimerization of ALK fusions occurs through the ALK partner protein and involves trans-autophosphorylation, and thus activation of the ALK kinase domain.
NPM-ALK or ATIC transcripts are detected in cells of healthy individuals with relatively high frequency.Citation48,49 Assuming reliability of RT-PCR detection, we can conclude that ALK translocations might be relatively common, necessary but not sufficient to induce transformation of cancer. Other molecular event(s) might be required for normal cells to be fully transformed or bypass oncogene-induced senescence.
Full Length ALK-positive Cancers (non-fusion ALK)
ALK amplification and overexpression
Amplification of the ALK locus and overexpression of ALK protein has been reported in many different types of cancer cell lines and human tumor samples,Citation38,50 including melanoma, NSCLC, neuroblastoma, glioblastoma, rhabdomyosarcoma, ovarian cancer, breast cancer, astrocytoma, Ewing's sarcoma, and retinoblastoma.
ALK expression is increased about 250-fold in nearly 100% of basal cell carcinoma.Citation45 ALK is overexpressed in over 50% of neuroblastomas, and accounts for 12.4% of tumors carrying activating ALK point mutations that are also common in familial. Approximately 10% of primary neuroblastomas and cell lines display ALK gene amplification.
van Gaal et al.Citation51 report that ALK protein is highly expressed in alveolar rhabdomyosarcoma and less so in the embryonal subtype (81% vs 32%). Elevated ALK gene copy number occurs in both the alveolar (88%) and embryonal subtypes (52%). Increased ALK expression is found in all metastatic tumors but in only one third of the tumors without metastasis. ALK expression is correlated with a poor prognosis as is the alveolar histology. These investigations suggest that ALK may play a role in rhabdomyosarcoma biology therefore may provide a potential therapeutic target for these tumors.
In neuroblastoma cell lines, overexpression activates ALK, which is shown to form a stable complex with hyperphosphorylated ShcC, a SHC adaptor molecule, and to modify the responsiveness of the MAPK pathway to growth factors. The signaling transduction by amplified ALK in neuroblastoma cell involves Akt, ERK1/2, and STAT3. About 20% of neuroblastoma patients possess a high level of N-Myc amplification at chromosome 2p24, which is associated with a poorer prognosis.
ALK point mutations
ALK mutation is found in 7% of sporadic neuroblastomas and 50% of familial neuroblastomas. Gain-of-function ALK mutations are observed in a substantial number of cases of familial neuroblastoma, as well as in sporadic cases, with 2 hotspot mutations occurring in the kinase domain: F1174 (mutated to L, S, I, C or V) and R1275 (mutated to Q or L and resulting in a loss of the basic side chain).Citation52 These hotspot mutations are observed in around 85% of cases with mutant ALK.
Most of the ALK mutations described are located within the kinase domain, and several – K1062M, F1174L and R1275Q – have been shown to behave in an oncogenic manner in mice and cell culture models.Citation50,53,54
ALK point mutations have been found mainly in neuroblastoma, as well as in NSCLC and ATC (anaplastic thyroid cancer). NSCLC and IMT gateway mutations and other resistance mutations are notable. Secondary mutations in the context of ALK fusions have been described in NSCLC, IMT and ATC.
About 10% of patient samples and cell lines derived from ATC exhibit mutations in the ALK gene.Citation55–57 The incidence of new cases of thyroid cancer in the US is about 62,980 and the annual number of deaths is about 1,890 (www.cancer.gov), of which about 6% may be associated with an ALK missense mutation. There is considerable room for improvement in the treatment of ATC, and targeting tumors with ALK mutations represents an important step in producing better clinical outcomes.
So far the accumulated data from such analyses suggest that the different cancer-associated ALK mutations fall into 3 classes. The first class is gain-of-function ligand-independent mutations (such as F1174I, F1174S and F1174L), and the second class is ligand-dependent mutations that are not constitutively active and require activation with agonist antibodies (such as D1091N, T1151M and A1234T).Citation58 The importance of these mutations, and whether they represent passenger or driver mutations in neuroblastoma, is currently unclear. The third class of mutation is kinase dead, which so far only includes the I1250T mutation that has been found in 2 patients.Citation59
ALK Diagnostic Methods and Target Cancer Therapy
ALK diagnosis
A number of methods are readily available for ALK diagnosis. Break-apart FISH is the currently recommended methodology for the identification of ALK translocations in NSCLC.Citation60 In 2011, the FDA approved the Vysis ALK Break Apart FISH Probe Kit (Abbott Molecular) for molecular diagnostic testing.Citation61 The FISH detects all rearrangements regardless of the fusion partner, and is accurate and reliable. However, the FISH test is at the DNA level, so a positive result may not be entirely related to fusion, and thus is not useful for mutations and overexpression.
Immunohistochemistry (IHC) readily identifies ALK at the expression level. However, ALK protein levels in ALK-rearranged NSCLC are comparatively low, and hardly detectable by IHC. If higher sensitivity ALK antibodies become available, IHC may find more extensive clinical applications.Citation6,62
RT-PCR is a precise, sensitive, and reproducible technique that can detect ALK fusion transcripts. The amplicons can be sequenced to identify the specific fusion variants.Citation63–65 The RT-PCR method is targeted to one partner gene per run; it is limited to the primer sequence located on the partner gene and ALK gene. Quantitative RT-PCR can detect mRNA amount differences, which are appropriate to test overexpression of fusion and non-fusion ALK.
Next generation sequencing may be used for NSCLC patients with a high likelihood of harboring driver mutations not detected by other methods.Citation66 There is some research on next generation sequencing for rearrangement in cancer; by using pair-end sequencing,Citation67 a panel of all possible partner genes can be tested together with the ALK gene. RNA-Seq is the most powerful method; it can test not only the expression level, but also translocation and its variances.Citation68
Lin et al.Citation69 used exon array profiling (Affymetrix Human Exon 1.0 Arrays) to detect ALK rearrangements in breast, colorectal, and NSCLCs. Potential gene fusion candidates showed discordant 5' and 3' ALK transcript expression. RNA arrays are useful methods that can measure relative amounts of ALK, but may not be able to tell positives by translocation, amplification, or even simple overexpression.
ALK targeted cancer therapy
ALK tyrosine kinase receptors are therapeutic targets in NSCLC and other malignancies with ALK fusion. The clinical strategy is to inhibit ALK kinase activity using inhibitors such as crizotinib and alectinib, given that ALK is not widely expressed in adult tissue. Therefore, few toxic effects might be expected from treatment aimed at blocking ALK function.
Immune-based therapeutic strategies can potentially be employed for ALK-positive ALCL. Vaccination against lymphoma depends on an ALK-specific antigen. It has been demonstrated that many patients with NPM-ALK-positive lymphomas have remarkable levels of circulating antibodies directed against the ALK portion of NPM-ALK, suggesting that ALK fusions are quite immunogenic.Citation38 Thus, several strategies for anti-ALK immune-based treatments of chemotherapy-resistant ALCL can be imagined.
RNAi technology is currently one of the most promising classes of nucleic acid molecules for the next generation of pharmaceutical drugs. RNA interference molecules have some advantages as cancer therapeutics, including proven efficacy on both wild-type and mutated transcripts and an extremely high sequence-specificity. If exogenous small interfering RNA (siRNA) is to be used therapeutically, the most significant hurdle to be overcome is the specific, effective, nontoxic delivery of siRNA to its intracellular site of action.Citation70
Crizotinib
Crizotinib (XALKORI®, Pfizer), is a small molecule TKI that inhibits the activity of the ALK fusion proteins, MET and ROS1,Citation47,71 by competitive binding within the ATP-binding pocket of target kinases.Citation72 It was approved in an accelerated manner by the U.S. FDA for treatment of late stage NSCLC on August 26, 2011.Citation73 Crizotinib was initially investigated as a c-MET inhibitor.Citation74 Early results of an initial phase I trial with 82 patients with ALK-positive lung cancer showed an overall response rate of 57%, a disease control rate at 8 weeks of 87% and progression free survival (PFS) at 6 months of 72%.Citation75 These data were encouraging especially in light of known similar data for conventional chemotherapy for NSCLC. Consistent with initial data, phase 2 studies of crizotinib demonstrated a response rate of 59.8% and a median PFS of 8.1 months.Citation76 In phase 3 trials comparing crizotinib with chemotherapy in previously treated patients, crizotinib was associated with significantly longer median PFS (7.7 vs 3.0 months) and over triple the response rate (65% vs 20%)Citation77 with similar results reported for previously untreated patients.Citation78 Thus, ALK-positive NSCLCs are highly responsive to crizotinib treatment, which may also be extrapolated to treating palliative cases.Citation79,80 There have been studies of crizotinib on other cancers: IMT,Citation81,82 ALCL,Citation82,83 neuroblastoma with R1275Q and F1174L mutations,Citation84 thyroid carcinoma, and leukemia.Citation79
The toxicity of crizotinib treatment was compared with that of chemotherapy. Patients receiving crizotinib remained on therapy 2 to 3 times as long as those receiving chemotherapy. 100% of crizotinib-treated patients experienced adverse reactions, 17% discontinued crizotinib due to adverse reactions, 16% required dose reduction, and 39% required temporary discontinuation of crizotinib. Among chemotherapy patients, 98% experienced adverse reactions, 14% discontinued chemotherapy due to adverse reactions, 15% required dose reduction, and 16% required temporary discontinuation of chemotherapy. The most common adverse drug reactions for crizotinib were vision disorders, nausea, diarrhea, vomiting, constipation, edema, elevated transaminases, and fatigue. The incidence of vision disorders (including diplopia, photopia, photophobia, blurred vision, visual brightness) was high (60%) with crizotinib treatment. Severe or life-threatening adverse drug reactions of crizotinib included interstitial lung disease or pneumonitis, drug-induced liver injury, and QT-interval prolongation.Citation73 Clinical thinking supported the use of crizotinib over cytotoxic chemotherapy despite these risks, mostly because of the increased efficacy and improved quality of life. However, its side effect profile led pharmaceutical industries to later explore other small molecule inhibitors, discussed shortly.
PROFILE 1014 was the second positive global phase 3 study that evaluated crizotinib against chemotherapy, a standard of care for patients with advanced NSCLC. The PROFILE 1014 study was a randomized study of 343 patients with previously untreated, advanced, nonsquamous ALK-positive NSCLC in which the patients were randomized to receive crizotinib or pemetrexed plus either cisplatin or carboplatin (PPC).Citation85 The primary endpoint for the study was progression-free survival (PFS), with secondary endpoints of overall response rate (ORR), overall survival (OS), and safety. The study reported that the median PFS was 10.9 months in the crizotinib group compared with 7.0 months in the PPC group (P < .0001). ORR was 74% in the crizotinib group versus 45% in the PPC group (P < .0001). Thus, in patients with previously untreated ALK-positive NSCLC, crizotinib treatment was superior to pemetrexed-platinum chemotherapy with respect to PFS, objective response rate, reduction of lung-cancer symptoms, and improvement in quality of life.Citation78
Crizotinib is active against ALK-positive lung cancer; it is now the standard of care for ALK-positive NSCLC patients as recognized by NCCN and ASCO guidelines. However, acquired resistance invariably develops over time. Current mechanisms of resistance seem to center on upregulation of bypass signaling pathways, de novo mutations in the target kinase, and overexpression of the primary oncogene.Citation86 Secondary mutations in the ALK kinase domain were detected in 20 out of 69 patients who were reported to have crizotinib resistance (29%). Most commonly, L1196M (the “gatekeeper” mutation) and C1156Y in the ALK kinase domain were the first-reported secondary mutations conferring resistance to crizotinib.Citation87 L1196M interferes with the binding of crizotinib, allowing continued ALK activation in the presence of crizotinib.
Copy number gain, or amplification of ALK fusion genes, is another way of overcoming ALK inhibition. The non-dominant mechanisms of resistance to crizotinib include mutations of other oncogenes, such as EGFR and KRAS genes, amplification of the KIT gene, increased autophosphorylation of EGFR and transformation to sarcomatoid carcinoma. Other oncogenes also bypass the ALK requirement for tumor cells.Citation46,88-93
Further research on mechanisms of acquired resistance as well as accordingly finding alternative strategies is of paramount importance for future clinical efficacy of ALK inhibitors. A recent report has demonstrated that the presence of new ALK mutations can correlate with clinical relapse in patients, which is clinically meaningful because not only can new mutations imply a causative clinical effect of relapse (although further studies are needed to prove causation), but also highlights the importance of being able to detect potential mutations and accordingly change treatment strategies and give clinicians an overall view of prognosis prior to clinical relapse.Citation94
Another significant limitation of crizotinib appears to be poor activity in the CNS. The high rate of CNS relapse in patients treated with crizotinib is likely due to poor blood-brain barrier penetration of crizotinib.Citation95,96
The second-generation ALK inhibitors
The toxicity, resistance and limitations of crizotinib led to investigation of the second-generation ALK inhibitors. A number of second-generation ALK inhibitors are currently in or have completed clinical trials. These inhibitors, such as ceritinib, alectinib and AP26133, are able to inhibit secondary 'resistance' mutations found in patients treated with crizotinib. Most of these inhibitors have lower IC50 and exhibit some degree of blocking activity against crizotinib resistance.
The preclinical data of ceritinib (Zykadia™, formerly called LDK378, Novartis) show an overall response rate of 56% (95%CI 45–67), which is similar to the data with crizotinib. The U.S. FDA approved its use against ALK-positive NSCLC following treatment with crizotinib. In ALK-positive cell line models, ceritinib is able to effectively inhibit ALK harboring the crizotinib-resistant mutations L1196M, G1269A, and I1171T, and S1206Y; however it is ineffective in inhibiting ALK containing the G1202R and F1174C mutations. Ceritinib is also highly active in patients with ALK-positive NSCLC, both in the crizotinib-native and crizotinib-treated settings. The overall response rate was 58% in 114 patients with NSCLC who received ceritinib. The response rate for 80 patients who had previously been treated with crizotinib was 56%, while the response rate among patients who were crizotinib-native was 62%. However, like those treated with crizotinib, patients treated with ceritinib invariably relapsed because of the emergence of resistance. The mechanisms of resistance might be similar to that of crizotinib, i.e., point mutations and ALK bypass activation.Citation97-99
Alectinib (Chugai/Roche, CH/RO5424802) is a potent, relatively selective and orally administered inhibitor of ALK with ten-fold greater potency than crizotinib, and can effectively inhibit ALK harboring the gatekeeper mutation (L1196M). It was also shown to effectively inhibit cyclin G associated kinase (GAK) and leukocyte receptor tyrosine kinase. Preliminary data suggested that alectinib achieved an overall response rate of 55.4% in NSCLC patients, also similar to crizotinib or ceritinib.Citation100 In addition to activity against ALK, it has activity against the kinases LTK and GAK, but it is not active against INSR, IGF-1R, MET, or ROS1. Preclinical studies have demonstrated that alectinib is active against the crizotinib-resistant ALK mutations L1196M, C1156Y, and F1174L. Neuroblastoma ALK mutation at F1174L might be potentially treated by alectinib as well. The overall response rate for 46 patients who had 300 mg twice daily was 93.5%. Alectinib showed superior toxicity profiles when compared to older drugs such as critotinib. The most common side effects of alectinib at this dose included predominantly grade 1 or 2 dysgeusia, elevated AST, increased bilirubin, decreased neutrophil count, increased creatinine, increased creatine phosphokinase, myalgias, gastrointestinal symptoms, and rash. The only grade 3 adverse event that occurred at this dose was decreased neutrophil count, and no grade 4 toxicities were observed. Similar to its fellow aforementioned drugs, resistance still was an issue. A possible mechanism of resistance to alectinib is via amplification of the MET gene, which can be overcome with crizotinib therapy.Citation101–104
ASP3026 (Astellas Pharma) decreased the viability, proliferation, and colony formation, and also induced apoptotic cell death of NPM-ALK ALCL cells. ASP3026 significantly reduced the proliferation of 293T cells transfected with NPM-ALK mutants that were resistant to crizotinib and down-regulated tyrosine phosphorylation of these mutants. ASP3026 abrogated systemic NPM-ALK ALCL growth in mice. This agent also displayed activity against the neuroblastoma-activating ALK mutation F1174L and R1275Q and against the crizotinib-resistance gatekeeper mutation L1196M. There are 2 clinical trials on ASP3026, one completed on solid tumor, one ongoing on solid tumor and B-cell lymphoma (www.clinicaltrials.gov).Citation105,106
AP26113 (Ariad, recently renamed Brigatinib) is an orally-active TKI that inhibits native and rearranged EML4-ALK fusion proteins and the T790 (but not native) EGFR protein.Citation107–109 In vitro studies revealed that AP26113 inhibited the native and F1174C, L1196M, S1206R, E1210K, F1245C and G1269S EML-ALK fusions. AP26113 is undergoing phase 1/2 human clinical trials for the treatment of NSCLC, DLBCL and IMT (www.clinicaltrials.gov).
PF-06463922, the dual ALK and ROS1 inhibitor developed by Pfizer, was designed to penetrate the blood–brain barrier and showed a 30–40% brain drug exposure vs. plasma concentrations in rats. Zou et al.Citation110,111 presented that PF-06463922 is the only inhibitor with low IC50 against the G1202R mutation.Citation112
Hsp90 (heat shock protein 90) inhibitors induce fusion protein misfolding and subsequent degradation by the proteasome system.Citation113 Fusion proteins formed as the result of chromosomal rearrangements are thought to be particularly dependent on Hsp90 for protein folding, transport, and stability.Citation114 At least 3 Hsp90 inhibitors have been tested in patients with ALK rearrangement: retaspimycin hydrochloride (IPI-504), genetespid (STA-9090), and AUY922. Preclinical studies have shown that crizotinib-resistant ALK-positive cell lines are highly sensitive to the Hsp90 inhibitor 17-AAG (analog of geldanamycin with reduced toxicity).Citation46,115,116
Some ongoing clinical trials address combinatorial treatments for ALK positive tumors, including the use of Hsp90 inhibitors together with either of the ALK TKIs crizotinib or LDK378 for NSCLC. Further large-scale data is thus eagerly anticipated in efforts to optimize biological therapies for potentially more robust responses and patient outcomes.
More new inhibitors have been developed: the inhibitor of ALK ROS1 TRK-A TRK-B TRK-C known as RXDX-101 (Nerviano);Citation93,117 X-376 and X-396 (Xcovery), the selective ALK inhibitors CEP-28122 and CEP-37440 (Teva); the ALK inhibitor TSR-011 (Tesaro). Pfizer has announced that in 2015 it will start trials that combine immunotherapy drugs (PDL-1 and PD-1) with its 2 ALK inhibitor drugs, likely also aiming to optimize combinatorial therapy to assess synergism and synchronous activity that has been proven for other cancer therapies. Pfizer is also planning a potential pivotal study of PF-06463922 starting in 2015, as reported by the website http://alkinhibitors.com. lists the information of several ALK inhibitors that are on clinical trials. It will be important to characterize potential susceptibilities to resistance and other pitfalls in these new agents, in hopes that they offer novel answers to clinical roadblocks of the past.
Table 1. List of ALK inhibitor drugs currently in clinical trials
Conclusions
Next-generation ALK inhibitors may overcome resistance to mutations or copy number gain, but they are unlikely to overcome resistance to up-regulation of other pathways, such as EGFR or Kit. However, the combination strategy represents a potential therapy. Though there has not been much progress on the RNAi therapy and antibody therapy by now, one may still expect that these methods have the opportunity to become treatment options in the future.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2010; 141:1117-34; PMID:20602996; http://dx.doi.org/10.1016/j.cell.2010.06.011
- Morris SW, Kirstein MN, Valentine MB, Dittmer K, Shapiro DN, Look AT, Saltman DL. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science 1995; 267:316-7; PMID:7824924
- Shiota M, Fujimoto J, Semba T, Satoh H, Yamamoto T, Mori S. Hyperphosphorylation of a novel 80 kDa protein-tyrosine kinase similar to Ltk in a human Ki-1 lymphoma cell line, AMS3. Oncogene 1994; 9:1567-74; PMID:8183550
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007; 448:561-6; PMID:17625570
- Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, Witte DP. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin's lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 1997; 14:2175-88; PMID:9174053
- Pulford K, Lamant L, Morris SW, Butler LH, Wood KM, Stroud D, Delsol G, Mason DY. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)–ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood 1997; 89:1394-404; PMID:9028963
- Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997; 14:439-49; PMID:9053841
- Lee CC, Jia Y, Li N, Sun X, Ng K, Ambing E, Gao MY, Hua S, Chen C, Kim S, et al. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem J 2010; 430:425-37; PMID:20632993; http://dx.doi.org/10.1042/BJ20100609
- Bossi RT, Saccardo MB, Ardini E, Menichincheri M, Rusconi L, Magnaghi P, Orsini P, Avanzi N, Borgia AL, Nesi M, et al. Crystal structures of anaplastic lymphoma kinase in complex with ATP competitive inhibitors. Biochemistry 2010; 49:6813-25; PMID:20695522; http://dx.doi.org/10.1021/bi1005514
- Perez-Pinera P, Zhang W, Chang Y, Vega JA, Deuel TF. Anaplastic lymphoma kinase is activated through the pleiotrophin/receptor protein-tyrosine phosphatase beta/zeta signaling pathway: an alternative mechanism of receptor tyrosine kinase activation. J Biol Chem 2007; 282:28683-90; PMID:17681947
- Bilsland JG, Wheeldon A, Mead A, Znamenskiy P, Almond S, Waters KA, Thakur M, Beaumont V, Bonnert TP, Heavens R, et al. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacology 2008; 33:685-700; PMID:17487225
- Vernersson E, Khoo NK, Henriksson ML, Roos G, Palmer RH, Hallberg B. Characterization of the expression of the ALK receptor tyrosine kinase in mice. Gene Expr Patterns 2006; 6:448-61; PMID:16458083
- Palmer RH, Vernersson E, Grabbe C, Hallberg B. Anaplastic lymphoma kinase: signalling in development and disease. Biochem J 2009; 420:345-61; PMID:19459784; http://dx.doi.org/10.1042/BJ20090387
- Piva R, Pellegrino E, Mattioli M, Agnelli L, Lombardi L, Boccalatte F, Costa G, Ruggeri BA, Cheng M, Chiarle R, et al. Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J Clin Invest 2006; 116:3171-82; PMID:17111047
- Marzec M, Liu X, Wong W, Yang Y, Pasha T, Kantekure K, Zhang P, Woetmann A, Cheng M, Odum N, et al. Oncogenic kinase NPM/ALK induces expression of HIF1α mRNA. Oncogene 2011; 30:1372-8; PMID:21102525; http://dx.doi.org/10.1038/onc.2010.505
- Pearson JD, Lee JK, Bacani JT, Lai R, Ingham RJ. NPM-ALK and the JunB transcription factor regulate the expression of cytotoxic molecules in ALK-positive, anaplastic large cell lymphoma. Int J Clin Exp Pathol 2011; 4:124-33; PMID:21326808
- Staber PB, Vesely P, Haq N, Ott RG, Funato K, Bambach I, Fuchs C, Schauer S, Linkesch W, Hrzenjak A, et al. The oncoprotein NPM–ALK of anaplastic large-cell lymphoma induces JUNB transcription via ERK1/2 and JunB translation via mTOR signaling. Blood 2007; 110:3374-83; PMID:17690253
- Schönherr C, Ruuth K, Kamaraj S, Wang CL, Yang HL, Combaret V, Djos A, Martinsson T, Christensen JG, Palmer RH, et al. Anaplastic lymphoma kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene 2012; 31:5193-200; PMID:22286764; http://dx.doi.org/10.1038/onc.2012.12
- Dejean E, Renalier MH, Foisseau M, Agirre X, Joseph N, de Paiva GR, Al Saati T, Soulier J, Desjobert C, Lamant L, et al. Hypoxia-microRNA 16 downregulation induces VEGF expression in anaplastic lymphoma kinase (ALK)-positive anaplastic large-cell lymphomas. Leukemia 2011; 25:1882-90; PMID:21778999; http://dx.doi.org/10.1038/leu.2011.168
- Desjobert C, Renalier MH, Bergalet J, Dejean E, Joseph N, Kruczynski A, Soulier J, Espinos E, Meggetto F, Cavaillé J, et al. MiR29a downregulation in ALK-positive anaplastic large-cell lymphomas contributes to apoptosis blockade through MCL1 overexpression. Blood 2011; 117:6627-37; PMID:21471522; http://dx.doi.org/10.1182/blood-2010-09-301994
- Matsuyama H, Suzuki HI, Nishimori H, Noguchi M, Yao T, Komatsu N, Mano H, Sugimoto K, Miyazono K. miR135b mediates NPM-ALK-driven oncogenicity and renders IL17 producing immunophenotype to anaplastic large cell lymphoma. Blood 2011; 118:6881-92; PMID:22042699; http://dx.doi.org/10.1182/blood-2011-05-354654
- Vishwamitra D, Li Y, Wilson D, Manshouri R, Curry CV, Shi B, Tang XM, Sheehan AM, Wistuba II, Shi P, et al. MicroRNA 96 is a post-transcriptional suppressor of anaplastic lymphoma kinase expression. Am J Pathol 2012; 180:1772-80; PMID:22414602; http://dx.doi.org/10.1016/j.ajpath.2012.01.008
- Stoica GE, Kuo A, Powers C, Bowden ET, Sale EB, Riegel AT, Wellstein A. Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. J Biol Chem 2002; 277:35990-8; PMID:12122009
- Motegi A, Fujimoto J, Kotani M, Sakuraba H, Yamamoto T. ALK receptor tyrosine kinase promotes cell growth and neurite outgrowth. J Cell Sci 2004; 117:3319-29; PMID:15226403
- Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer 2013; 13:685-700; PMID:24060861; http://dx.doi.org/10.1038/nrc3580
- Roskoski R Jr. Anaplastic lymphoma kinase (ALK): Structure, oncologyic activation, and pharmacological inhibition. Pharmacol Res 2013; 68:68-94; PMID:23201355; http://dx.doi.org/10.1016/j.phrs.2012.11.007
- Hartwell, Leland H. Genetics: From Genes to Genomes. New York: McGraw-Hill; 2011. p. 443.
- Duyster J, Bai RY, Morris SW. Translocation involving anaplastic lymphoma kinase (ALK). Oncogene 2001; 20:5623-37; PMID:11607814
- Ye X, Shokrollahi K, Rozen WM, Conyers R, Wright P, Kenner L, Turner SD, Whitaker IS. Anaplastic large cell lymphoma (ALCL) and breast implants: breaking down the evidence. Mutat Res Rev Mutat Res 2014; 762:23-32; PMID:25475421; http://dx.doi.org/10.1016/j.mrrev.2014.08.002
- Gidengil CA, Predmore Z, Mattke S, van Busum K, Kim B. Breast implant-associated anaplastic large cell lymphoma: a systematic review. Plast Reconstr Surg 2015; 135:713-20; PMID:25490539; http://dx.doi.org/10.1097/PRS.0000000000001037
- Bitter MA, Franklin WA, Larson RA, McKeithan TW, Rubin CM, Le Beau MM, Stephens JK, Vardiman JW. Morphology in Ki-1(CD30)-positive non-Hodgkin's lymphoma is correlated with clinical features and the presence of a unique chromosomal abnormality, t(2;5)(p23;q35). Am J Surg Pathol 1990; 14:305-16; PMID:2157342
- Drexler HG, Gignac SM, von Wasielewski R, Werner M, Dirks WG. Pathobiology of NPM-ALK and variant fusion gene in anaplastic large cell lymphoma and other lymphomas. Leukemia 2000; 14:1533-59; PMID:10994999
- Grisendi S, Mecucci C, Falini B, Pandolfi PP. Nucleophosmin and cancer. Nat Rev Cancer 2006; 6:493-505; PMID:16794633
- Chan PK, Chan FY. Nucleophosmin/B23 (NPM) oligomer is a major and stable entity in HeLa cells. Biochim Biophys Acta 1995; 1262:37-42; PMID:7772597
- Liu QR, Chan PK. Formation of nucleophosmin/B23 oligomers requires both the amino- and the carboxyl-terminal domains of the protein. Eur J Biochem 1991; 200:715-21; PMID:1915343
- Bischof D, Pulford K, Mason DY, Morris SW. Role of the nucleophosmin (NPM) portion of the non-Hodgkin's lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein in oncogenesis. Mol Cell Biol 1997; 17:2312-25; PMID:9121481
- Boi M, Zucca E, Inghirami G, Bertoni F. Advances in understanding the pathogenesis of systemic anaplastic large cell lymphomas. Br J Haematol 2015; 168:771-83; PMID:25559471; http://dx.doi.org/10.1111/bjh.13265
- Pulford K, Falini B, Banham AH, Codrington D, Roberton H, Hatton C, Mason DY. Immune response to the ALK oncogenic tyrosine kinase in patients with anaplastic large-cell lymphoma. Blood 2000; 96:1605-7; PMID:10942417
- Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997; 14:439-49; PMID:9053841
- Eyre TA, Khan D, Hall GW, Collins GP. Anaplastic lymphoma kinase‐positive anaplastic large cell lymphoma: current and future perspectives in adult and paediatric disease. Eur J Haematol 2014; 93:455-68; PMID:24766435; http://dx.doi.org/10.1111/ejh.12360
- Coffin CM, Patel A, Perkins S, Elenitoba-Johnson KS, Perlman E, Griffin CA. ALK1 and p80 expression and chromosomal rearrangements involving 2p23 in inflammatory myofibroblastic tumor. Mod Pathol 2001; 14:569-76; PMID:11406658
- Griffin CA, Hawkins AL, Dvorak C, Henkle C, Ellingham T, Perlman EJ. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res 1999; 59:2776-80; PMID:10383129
- Chiarle R, Martinengo C, Mastini C, Ambrogio C, D'Escamard V, Forni G, Inghirami G. The anaplastic lymphoma kinase is an effective oncoantigen for lymphoma vaccination. Nat Med 2008; 14:676-80; PMID:18469826; http://dx.doi.org/10.1038/nm1769
- Toyokawa G, Seto T. Anaplastic Lymphoma Kinase rearrangement in lung cancer: Its biological and clinical significance. Respir Investig 2014; 52:330-8; PMID:25453376; http://dx.doi.org/10.1016/j.resinv.2014.06.005
- Shackelford RE, Vora M, Mayhall K, Cotelingam J. ALK-rearrangements and testing methods in non-small cell lung cancer: a review. Genes Cancer 2014; 5:1-14; PMID:24955213
- Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, Shakespeare WC, Iafrate AJ, Engelman JA, Shaw AT. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A 2011; 108:7535-40; PMID:21502504; http://dx.doi.org/10.1073/pnas.1019559108
- Webb TR, Slavish J, George RE, Look AT, Xue L, Jiang Q, Cui X, Rentrop WB, Morris SW. Anaplastic lymphoma kinase: role in cancer pathogenesis and small-molecule inhibitor development for therapy. Expert Rev Anticancer Ther 2009; 9:331-56; PMID:19275511; http://dx.doi.org/10.1586/14737140.9.3.331
- Trümper L, Pfreundschuh M, Bonin FV, Daus H. Detection of the t(2;5)-associated NPM/ALK fusion cDNA in peripheral blood cells of healthy individuals. Br J Haematol 1998; 103:1138-44; PMID:9886332
- Maes B, Vanhentenrijk V, Wlodarska I, Cools J, Peeters B, Marynen P, de Wolf-Peeters C. The NPM-ALK and the ATIC-ALK fusion genes can be detected in non-neoplastic cells. Am J Pathol 2001; 158:2185-93; PMID:11395396
- Lamant L, Pulford K, Bischof D, Morris SW, Mason DY, Delsol G, Mariamé B. Expression of the ALK tyrosine kinase gene in neuroblastoma. Am J Pathol 2000; 156:1711-21; PMID:10793082
- van Gaal JC, Flucke UE, Roeffen MH, de Bont ES, Sleijfer S, Mavinkurve-Groothuis AM, Suurmeijer AJ, van der Graaf WT, Versleijen-Jonkers YM. Anaplastic lymphoma kinase aberrations in rhabdomyosarcoma: clinical and prognostic implications. J Clin Oncol 2012; 30:308-15; PMID:22184391; http://dx.doi.org/10.1200/JCO.2011.37.8588
- Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, et al. Identification of ALK as a major familial neuroblastoma tredisposition gene. Nature 2008; 455:930-5; PMID:18724359; http://dx.doi.org/10.1038/nature07261
- Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008; 455:971-4; PMID:18923524; http://dx.doi.org/10.1038/nature07399
- Janoueix-Lerosey I, Lequin D, Brugières L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008; 455:967-70; PMID:18923523; http://dx.doi.org/10.1038/nature07398
- Nagaiah G, Hossain A, Mooney CJ, Parmentier J, Remick SC. Anaplastic thyroid cancer: a review of epidemiology, pathogenesis, and treatment. J Oncol 2011; 2011:542358; PMID:21772843; http://dx.doi.org/10.1155/2011/542358
- Neff RL, Farrar WB, Kloos RT, Burman KD. Anaplastic thyroid cancer. Endocrinol Metab Clin North Am 2008; 37:525-38, xi; PMID:18502341; http://dx.doi.org/10.1016/j.ecl.2008.02.003
- Murugan AK, Xing M. Anaplastic thyroid cancers harbor novel oncogenic mutations of the ALK gene. Cancer Res 2011; 71:4403-11; PMID:21596819; http://dx.doi.org/10.1158/0008-5472.CAN-10-4041
- George RE, Sanda T, Hanna M, Fröhling S, Luther W 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008; 455:975-8; PMID:18923525; http://dx.doi.org/10.1038/nature07397
- Schönherr C, Ruuth K, Yamazaki Y, Eriksson T, Christensen J, Palmer RH, Hallberg B. Activating ALK mutations found in neuroblastoma are inhibited by Crizotinib and NVP-TAE684. Biochem J 2011; 440:405-13; PMID:21838707; http://dx.doi.org/10.1042/BJ20101796
- Lira ME, Kim TM, Huang D, Deng S, Koh Y, Jang B, Go H, Lee SH, Chung DH, Kim WH, et al. Multiplexed gene expression and fusion transcript analysis to detect ALK fusions in lung cancer. J Mol Diagn 2013; 15:51-61; PMID:23246132; http://dx.doi.org/10.1016/j.jmoldx.2012.08.006
- U.S. Food and Drug Administration. FDA approves Xalkori with companion diagnostic for a type of late-stage lung cancer [Internet]. 2011 Aug 26 [cited 2015 Oct 30]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm269856.htm.
- Lamant L, Meggetto F, al Saati T, Brugières L, de Paillerets BB, Dastugue N, Bernheim A, Rubie H, Terrier-Lacombe MJ, Robert A, et al. High incidence of the t(2;5)(p23;q35) translocation in anaplastic large cell lymphoma and its lack of detection in Hodgkin's disease. Comparison of cytogenetic analysis, reverse transcriptase-polymerase chain reaction, and P-80 immunostaining. Blood 1996; 87:284-91; PMID:8547653
- Li Y, Pan Y, Wang R, Sun Y, Hu H, Shen X, Lu Y, Shen L, Zhu X, Chen H. ALK-rearranged lung cancer in Chinese: a comprehensive assessment of clinicopathology, IHC, FISH and RT-PCR. PLoS One 2013; 8:e69016; PMID:23922677; http://dx.doi.org/10.1371/journal.pone.0069016
- Wu YC, Chang IC, Wang CL, Chen TD, Chen YT, Liu HP, Chu Y, Chiu YT, Wu TH, Chou LH, et al. Comparison of IHC, FISH and RT-PCR methods for detection of ALK rearrangements in 312 non-small cell lung cancer patients in Taiwan. PLoS One 2013; 8:e70839; PMID:23951022; http://dx.doi.org/10.1371/journal.pone.0070839
- Zhang YG, Jin ML, Li L, Zhao HY, Zeng X, Jiang L, Wei P, Diao XL, Li X, Cao Q, et al. Evaluation of ALK rearrangement in Chinese non-small cell lung cancer using FISH, immunohistochemistry, and real-time quantitative RT- PCR on paraffin-embedded tissues. PLoS One 2013; 8:e64821; PMID:23741400; http://dx.doi.org/10.1371/journal.pone.0064821
- Peled N, Palmer G, Hirsch FR, Wynes MW, Ilouze M, Varella-Garcia M, Soussan-Gutman L, Otto GA, Stephens PJ, Ross JS, et al. Next-generation sequencing identifies and immunohistochemistry confirms a novel crizotinib-sensitive ALK rearrangement in a patient with metastatic non-small-cell lung cancer. J Thorac Oncol 2012; 7:e14-6; PMID:22895149; http://dx.doi.org/10.1097/JTO.0b013e3182614ab5
- Campbell PJ, Stephens PJ, Pleasance ED, O'Meara S, Li H, Santarius T, Stebbings LA, Leroy C, Edkins S, Hardy C, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet 2008; 40:722-9; PMID:18438408; http://dx.doi.org/10.1038/ng.128
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009; 10:57-63; PMID:19015660; http://dx.doi.org/10.1038/nrg2484
- Lin E, Li L, Guan Y, Soriano R, Rivers CS, Mohan S, Pandita A, Tang J, Modrusan Z. Exon array profiling detects EML4-ALK fusion in breast, colorectal, and non-small cell lung cancers. Mol Cancer Res 2009; 7:1466-76; PMID:19737969; http://dx.doi.org/10.1158/1541-7786.MCR-08-0522
- Di Paolo D, Brignole C, Pastorino F, Carosio R, Zorzoli A, Rossi M, Loi M, Pagnan G, Emionite L, Cilli M, et al. Neuroblastoma-targeted nanoparticles entrapping siRNA specifically knockdown ALK. Mol Ther 2011 19:1131-40; PMID:21487394; http://dx.doi.org/10.1038/mt.2011.54
- Ardini E, Magnaghi P, Orsini P, Galvani A, Menichincheri M. Anaplastic Lymphoma kinase: Role in specific tumours, and development of small molecule inhibitors for cancer therapy. Cancer lett 2010; 299:81-94; PMID:20934803; http://dx.doi.org/10.1016/j.canlet.2010.09.001
- Ohanian M, Cortes J, Kantarjian H, Jabbour E. Tyrosine kinase inhibitors in acute and chronic leukemias. Expert Opin Pharmacother 2012; 13:927-38; PMID:22519766; http://dx.doi.org/10.1517/14656566.2012.672974
- Kazandjian D, Blumenthal GM, Chen HY, He K, Patel M, Justice R, Keegan P, Pazdur R. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014; 19:e5-11; PMID:25170012; http://dx.doi.org/10.1634/theoncologist.2014-0241
- Cui JJ, Tran-Dubé M, Shen H, Nambu M, Kung PP, Pairish M, Jia L, Meng J, Funk L, Botrous I, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med Chem 2011; 54:6342-63; PMID:21812414; http://dx.doi.org/10.1021/jm2007613
- Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010; 363:1693-703; PMID:20979469; http://dx.doi.org/10.1056/NEJMoa1006448
- Kim D-W, Ahn M-J, Shi Y, De Pas TM, Yang P-C, Riely GJ, Crin∫ L, Evans TL, Liu X, Han J-Y, et al. Results of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC). J Clin Oncol 2012; 30:7533 (suppl; abstr 7533).
- Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, et al. Crizotinib vs. chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013; 368:2385-94; PMID:23724913; http://dx.doi.org/10.1056/NEJMoa1214886
- Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014; 371:2167-77; PMID:25470694; http://dx.doi.org/10.1056/NEJMoa1408440
- Awad MM, Shaw AT, ALK inhibitors in non-small cell lung cancer: crizotinib and beyond. Clin Adv Hematol Oncol 2014; 12:429-39; PMID:25322323
- Ou SH, Bazhenova L, Camidge DR, Solomon BJ, Herman J, Kain T, Bang YJ, Kwak EL, Shaw AT, Salgia R, et al. Rapid and dramatic radiographic and clinical response to an ALK inhibitor (crizotinib, PF02341006) in an ALK translocation-positive patient with non-small cell lung cancer. J Thorac Oncol 2010; 5:2044-6; PMID:21102269; http://dx.doi.org/10.1097/JTO.0b013e318200f9ff
- Butrynski JE, D'Adamo DR, Hornick JL, Dal Cin P, Antonescu CR, Jhanwar SC, Ladanyi M, Capelletti M, Rodig SJ, Ramaiya N, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med 2010; 363:1727-33; PMID:20979472; http://dx.doi.org/10.1056/NEJMoa1007056
- Mossé YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, Rolland D, Balis FM, Maris JM, Weigel BJ, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children's Oncology Group phase 1 consortium study. Lancet Oncol 2013; 14:472-80; PMID:23598171; http://dx.doi.org/10.1016/S1470-2045(13)70095-0
- Gambacorti-Passerini C, Messa C, Pogliani EM. Crizotinib in anaplastic large-cell lymphoma. N Engl J Med 2011; 364:775-6; PMID:21345110; http://dx.doi.org/10.1056/NEJMc1013224
- Bresler SC, Wood AC, Haglund EA, Courtright J, Belcastro LT, Plegaria JS, Cole K, Toporovskaya Y, Zhao H, Carpenter EL, et al. Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma. Sci Transl Med 2011; 3:108ra114; PMID:22072639; http://dx.doi.org/10.1126/scitranslmed.3002950
- Mok T, Kim DW, Wu YL, Solomon BJ, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, et al. First-line crizotinib versus pemetrexed-cisplatin or pemetrexed-carboplatin in patients with advanced ALK-positive non-squamous non-small cell lung cancer: results of a phase III study (PROFILE 1014). J Clin Oncol 2014; 32:5S (suppl; abstr 8002).
- Gainor JF, Shaw AT. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J Clin Oncol 2013; 31:3987-96; PMID:24101047; http://dx.doi.org/10.1200/JCO.2012.45.2029
- Ai X, Shen S, Shen L, Lu S. An interaction map of small-molecule kinase inhibitors with anaplastic lymphoma kinase (ALK) mutants in ALK-positive non-small cell lung cancer. Biochimie 2015 112:111-20; PMID:25769414; http://dx.doi.org/10.1016/j.biochi.2015.03.003
- Yamaguchi N, Lucena-Araujo AR, Nakayama S, de Figueiredo-Pontes LL, Gonzalez DA, Yasuda H, Kobayashi S, Costa DB. Dual ALK and EGFR inhibition targets a mechanism of acquired resistance to the tyrosine kinase inhibitor crizotinib in ALK rearranged lung cancer. Lung Cancer 2014; 83:37-43; PMID:24199682; http://dx.doi.org/10.1016/j.lungcan.2013.09.019
- Gainor JF, Varghese AM, Ou SH, Kabraji S, Awad MM, Katayama R, Pawlak A, Mino-Kenudson M, Yeap BY, Riely GJ, et al. ALK rearrangements are mutually exclusive with mutation in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res 2013; 19:4273-81; PMID:23729361; http://dx.doi.org/10.1158/1078-0432.CCR-13-0318
- Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, Lathan C, Marcoux JP, Du J, Okuda K, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res 2011; 71:6051-60; PMID:21791641; http://dx.doi.org/10.1158/0008-5472.CAN-11-1340
- Jabbour E, Parikh SA, Kantarjian H, Cortes J. Chronic myeloid leukemia: mechanisms of resistance and treatment. Hematol Oncol Clin North Am 2011; 25:981-95, v; PMID:22054730; http://dx.doi.org/10.1016/j.hoc.2011.09.004
- Qi J, McTigue MA, Rogers A, Lifshits E, Christensen JG, Jänne PA, Engelman JA. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to MET inhibitors. Cancer Res 2011; 71:1081-91; PMID:21266357; http://dx.doi.org/10.1158/0008-5472.CAN-10-1623
- Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med 2012; 4:120ra17; PMID:22277784; http://dx.doi.org/10.1126/scitranslmed.3003316
- Schleiermacher G, Javanmardi N, Bernard V, Leroy Q, Cappo J, Rio Frio T, Pierron G, Lapouble E, Combaret V, Speleman F, et al. Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol 2014; 32:2727-34; PMID:25071110; http://dx.doi.org/10.1200/JCO.2013.54.0674
- Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, Yatabe Y, Takeuchi K, Hamada T, Haruta H, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 2010; 363:1734-9; PMID:20979473; http://dx.doi.org/10.1056/NEJMoa1007478
- Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, Riely GJ, Solomon B, Ou SH, Kim DW, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol 2012; 13:1011-9; PMID:22954507; http://dx.doi.org/10.1016/S1470-2045(12)70344-3
- Chen J, Jiang C, Wang S. LDK378: a promising anaplastic lymphoma kinase (ALK) inhibitor. J Med Chem 2013; 56:5673-4; PMID:23837797; http://dx.doi.org/10.1021/jm401005u
- Shaw AT, Engelman JA. Ceritinib in ALK- rearranged non-small-cell lung cancer. N Engl J Med 2014; 370:2537-9; PMID:24963575; http://dx.doi.org/10.1056/NEJMc1404894
- Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, Michellys PY, Awad MM, Yanagitani N, Kim S, et al. The ALK inhibitor LDK378 overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov 2014; 4:662-73; PMID:24675041; http://dx.doi.org/10.1158/2159-8290.CD-13-0846
- Gadgeel S, Ou SH, Chiappori AA, Riely G, Lee R, Garcia L, Sato J, Yokoyama S, Tanaka S, Gandhi L. A phase1 dose escalation study of a new ALK inhibitor, CH5424802/ RO5424802, in ALK+ non-small cell lung cancer (NSCLC) patients who have failed crizotinib (AF-002JG/NP28761, NCT01588028). J Thorac Oncol 2013; 8:S199 (suppl 2; abstr O16.06).
- Kinoshita K, Asoh K, Furuichi N, Ito T, Kawada H, Hara S, Ohwada J, Miyagi T, Kobayashi T, Takanashi K, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem 2012; 20:1271-80; PMID:22225917; http://dx.doi.org/10.1016/j.bmc.2011.12.021
- Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, Oikawa N, Tsukuda T, Ishii N, Aoki Y. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011; 19:679-90; PMID:21575866; http://dx.doi.org/10.1016/j.ccr.2011.04.004
- Seto T, Kiura K, Nishio M, Nakagawa K, Maemondo M, Inoue A, Hida T, Yamamoto N, Yoshioka H, Harada M, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1–2 study. Lancet Oncol 2013; 14:590-8; PMID:23639470; http://dx.doi.org/10.1016/S1470-2045(13)70142-6
- Nakagawa K, Kiura K, Nishio M, Seto T, Maemondo M, Inoue A, Hida T, Yamamoto N, Yoshioka H, Harada M, et al. A phase I/II study with a highly selective ALK inhibitor CH5424802 in ALK-positive non-small cell lung cancer (NSCLC) patients: updated safety and efficacy results from AF-001JP. J Clin Oncol 2013; 31:8033 (suppl; abstr 8033).
- Mori M, Ueno Y, Konagai S, Fushiki H, Shimada I, Kondoh Y, Saito R, Mori K, Shindou N, Soga T, et al. The selective anaplastic lymphoma receptor tyrosine kinase inhibitor ASP3026 induces tumor regression and prolongs survival in non-small cell lung cancer model mice. Mol Cancer Ther 2014; 13:329-40; PMID:24419060; http://dx.doi.org/10.1158/1535-7163.MCT-13-0395
- Patnaik A, LoRusso P, Ball HA, Bahceci E, Yuen G, Papadopoulos KP, Kittaneh M, Tolcher AW. Pharmacokinetics and safety of an oral ALK inhibitor, ASP3026, observed in a phase I dose escalation trial. J Clin Oncol 2013; 31:2602 (suppl; abstr 2602).
- Rivera VM, Anjum R, Wang F, Zhang S, Keats J, Ning Y, Wardwell SD, Moran L, Ye E, Chun DY, et al. Efficacy and pharmacodynamics analysis of AP26113, a potent and selective orally active inhibitor of anaplastic lymphoma kinase (ALK). Cancer Res 2010; 70:3623; http://dx.doi.org/10.1158/1538-7445.AM10-3623
- Rivera VM, Wang F, Anjum R, Zhang S, Squillace R, Keats J, Miller D, Ning Y, Wardwell SD, Moran L, et al. AP26113 is a dual ALK/EGFR inhibitor: characterization against EGFR T790M in cell and mouse models of NSCLC. Cancer Res 2012; 72:1794; http://dx.doi.org/10.1158/1538-7445.AM2012-1794
- Camidge DR, Bazhenova L, Salgia R, Weiss GJ, Langer CJ, Shaw AT, Narasimhan NI, Dorer DJ, Rivera VM, Zhang J, et al. First-in-human dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignancies: Updated results. J Clin Oncol 2013; 31:8031 (suppl; abstr 8031).
- Zou HY, Engstrom LR, Li Q, Lu MW, Tang RW, Wang H, Tsaparikos K, Timofeevski S, Lam J, Yamazaki S, et al. PF-06463922, a novel ROS1/ALK inhibitor, demonstrates sub-nanomolar potency against oncogenic ROS1 fusions and capable of blocking the resistant ROS1G2032R mutant in preclinical tumor models. Mol Cancer Ther 2013; 12:A277; http://dx.doi.org/10.1158/1535-7163.TARG-13-A277
- Zou HY, Li Q, Engstrom LD, West M, Appleman V, Wong KA, McTigue M, Deng YL, Liu W, Brooun A, et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc Natl Acad Sci U S A. 2015; 112:3493-8; PMID:25733882; http://dx.doi.org/10.1073/pnas.1420785112
- Alice T. Shaw. Current and Emerging Approaches to Acquired Resistance for ALK/ROS1. http://cancergrace.org/lung/2014/10/24/ar_forum_new_alk_ros1_approaches.
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer 2005; 5:761-72; PMID:16175177
- Pillai RN, Ramalingam SS. Heat shock protein 90 inhibitors in non-small-cell lung cancer. Curr Opin Oncol 2014; 26:159-64; PMID:24463348; http://dx.doi.org/10.1097/CCO.0000000000000047
- Sang J, Acquaviva J, Friedland JC, Smith DL, Sequeira M, Zhang C, Jiang Q, Xue L, Lovly CM, Jimenez JP, et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov 2013; 3:430-43; PMID:23533265; http://dx.doi.org/10.1158/2159-8290.CD-12-0440
- Normant E, Paez G, West KA, Lim AR, Slocum KL, Tunkey C, McDougall J, Wylie AA, Robison K, Caliri K, et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene 2011; 30:2581-6; PMID:21258415; http://dx.doi.org/10.1038/onc.2010.625
- De Braud FG, Pilla L, Niger M, Damian S, Bardazza B, Martinetti A, Pelosi G, Marrapese G, Palmeri L, Cerea G, et al. Phase 1 open label, dose escalation study of RXDX101, an oral pan-trk, ROS1,and ALK inhibitor, in patients with advanced solid tumors with relevant molecular alteration. J Clin Oncol 2014; 32:5S (suppl; abstr 2502).