
Abstract
Raloxifene hydrochloride (RAL), one of second generation of selective estrogen receptor modulators (SERMs), is usually used in preventing osteoporosis and breast cancer. The present study evaluated whether Raloxifene might sensitize multidrug resistant (MDR) breast cancers to chemotherapies, especially in estrogen receptor negative (ER−) breast cancer. The results showed that RAL could significantly sensitize ER- MDR breast tumors to paclitaxel both in vitro and in vivo. Combination of Raloxifene could significantly enhance paclitaxel-induced cell apoptosis, G2-M arrest as well as inhibition of cell proliferation in MDR tumors. Further studies showed that the combined treatment did not alter P-glycoprotein expression but increased P-gp ATPase activity. These results suggested that raloxifene might be a valuable chemosensitizer agent for breast cancer therapy.
Abbreviations
| RAL | = | Raloxifene hydrochloride |
| SERM | = | selective estrogen receptor modulators |
| TAM | = | tamoxifen |
| MDR | = | multidrug resistant |
| ER | = | estrogen receptor |
| P-gp | = | P-glycoprotein |
| Ki-67 | = | Ki-67 protein |
Introduction
Breast cancer is the second most prominent cause of mortality in women. Its incidence has been rapidly increased in recent years.Citation1 Chemotherapy is considered to be a promising therapy in the systematic treatment of breast cancer.Citation2 However, drug resistance has become a major barrier to overcome breast cancer. Multidrug resistance (MDR), is primarily responsible for ineffectiveness of drugs in clinical therapy.Citation2,3 Under long-term exposure, breast cancer cells gradually develop resistance to conventional drugs such as paclitaxel, vinblastine and vinorelbine,Citation4-6 and MDR has become a more and more severe problem.Citation7 Thus, the attention has been focused on the study how to enhance the response of tumors to chemotherapy with emphasis on reversing MDR and increasing the sensitivity of tumor cells to chemical drugs.Citation2,8
Selective estrogen receptor modulators (SERMs) have shown to be promising agents for reversing MDR.Citation8,9 In general, SERMs are known to function through regulation of estrogen receptors (ERs), acting as agonists or antagonists depending on the tissue type.Citation10,11 Specifically, most SERMs posses estrogen antagonist activity and are commonly used for the treatment of ER-positive breast cancer, which takes 65 percent of breast cancer.Citation5,10,11 Recent studies demonstrated that SERMs might act as a chemosensitizer when co-administered with other drugs either in ER-positive or ER-negative cancer cell lines,Citation6,9,12 which provide a potential strategy for clinicians to treat the patients with drug-resistant breast tumors. The mechanisms of the reversal effects of SERMS are not fully understood.Citation8,12,13 At present, the studies mainly focused on tamoxifen (TAM) and its derivatives.Citation4,8,9,12 TAM, a widely used agent in the hormonal therapy of breast cancer, is also an antagonist of P-glycoprotein (P-gp), a cell surface protein which confers drug resistance to cells.Citation10,13 Liu et al (2010) reported that high-dose TAM could reverse the MDR of human multidrug resistant cholangio carcinoma cells, due to competitive inhibition of over-expressed P-gp, although others might not completely agree.Citation12,13
In recent years, raloxifene (RAL) has been used as a second generation SERM and mainly used to treat metastatic breast cancer.Citation10,11 When compared with TAM, RAL was as effective as TAM in the prevention and treatment of breast cancer.Citation14 However, clinical trials demonstrated that TAM may significantly increase the risk of endometrial carcinoma, stroke, and venous thrombosis,Citation15-18 whereas the adverse effects of RAL are significantly decreased.Citation14,19 These findings suggest that RAL might be a reasonable agent for reversing MDR in patients. However, whether RAL could reverse MDR in breast cancer cells, especially in ER-negative cells, has not been reported. In present study, we focused on evaluating the ability of RAL to reverse MDR and attempted to explore its underlying mechanisms.
Results
Raloxifene sensitizes Bats-72 and Bads-200 cell lines to paclitaxel
The morphologic changes of the cells were observed after 24 hours of treatment as described in methods. Exposure to raloxifene alone had little effect on the morphology of 3 cell lines, indicating that raloxifene was non-toxic to the cells. Treatment to paclitaxel alone induced cell death in paclitaxel-sensitive cell line BCap37, and floated cells were displayed in the plates (). Growth of paclitaxel-treated BCap37 cells was also slowed down compared to the untreated BCap37 cells. Bats-72 and Bads-200 had little changes after paclitaxel treatment alone for 24 hours, showing their resistance to paclitaxel. However, paclitaxel with raloxifene-pretreatment caused massive cells to death in both Bats-72 and Bads-200 compared to the cells with paclitaxel-treated alone, indicating the lessened cytotoxity of paclitaxel were restored by raloxifene for these 2 cells ().
Figure 1. Raloxifene sensitizes Bats-72 and Bads-200 cells to paclitaxel. (A) Cell morphology after distinct treatments. Bcap37, Bats-72 and Bads-200 cells were plated in 6-well plates at a density of 105 cells per well for 24h before exposed to paclitaxel (5nM, 200nM and 2000nM respectively) with or without raloxifene (10μM, 10μM and 20μM respectively) for another 24h. (B) MTT assays were performed to evaluate drug-induced cytotoxicity. BCap37, Bats-72 and Bads-200 cells were cultured in 96-well plates at 104 cells per well. Density of drugs was as same as described in , and processing time extended to 72h. *P < 0.05 vs. PTX.
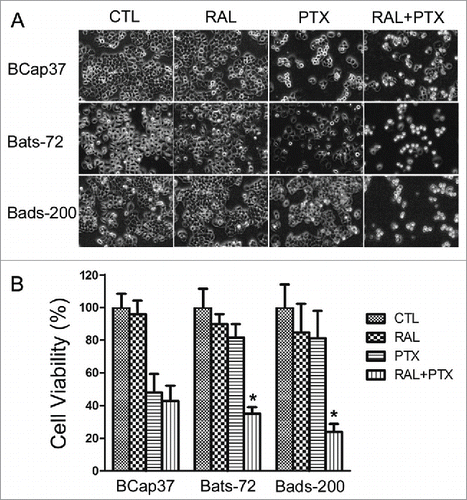
As MTT results shown in , cell viability of all the 3 cell lines was above 80% under the condition of raloxifene treatment alone. BCap37 cells were sensitive to paclitaxel, with a sharp decrease of 52.04% in cell viability. And Bats-72 and Bads-200 cells retained survival rate of 81.63% and 81.29%, respectively, under the treatment of paclitaxel alone, which verified our previous results.Citation20 However, raloxifene significantly sensitized Bats-72 and Bads-200 to paclitaxel. With the pre-treatment of raloxifene, the survivals of Bats-72 and Bads-200 declined 46.55% and 57.37% compared to paclitaxel-treatment alone. And the corresponding decrease of cell viability was only 5.13% for BCap37 cells.
Raloxifene reverses resistance to paclitaxel-induced mitotic arrest in MDR cell lines
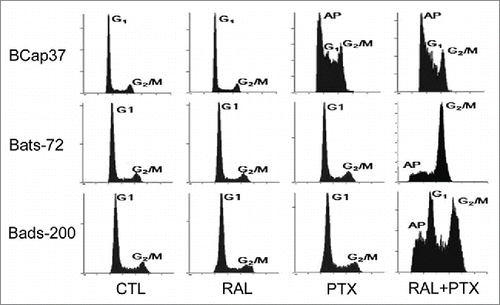
Flow cytometric analysis of propidium iodide staining was performed to analyze the cell cycle distribution after drug-treatments. As shown in , raloxifene had little effect upon cell cycle distribution for all the 3 cell lines. BCap37 was significantly induced of apoptosis peak and G2/M arrest by paclitaxel, without much change by adding raloxifene. As expected in MDR cells, paclitaxel alone had slight impact on Bats-72 and Bads-200 cells, which showed resistance to paclitaxel-induced cell arrest. But the 2 drug-resistant cells showed different changes from BCap37 cells by co-incubation of paclitaxel and raloxifene. Both of Bats-72 and Bads-200 cells shown with distinct apoptotic peak and G2/M phase arrest, though the G1 phase was not very obvious for Bats-72 cells.
Figure 2. The reversal effect of raloxifene via enhanced paclitaxel-induced mitotic arrest. Flow cytometric analysis of cell cycle distribution was performed after BCap37, Bats-72 and Bads-200 cells were treated for 48 h.
Raloxifene reverses resistance to paclitaxel-induced apoptosis in MDR cell lines
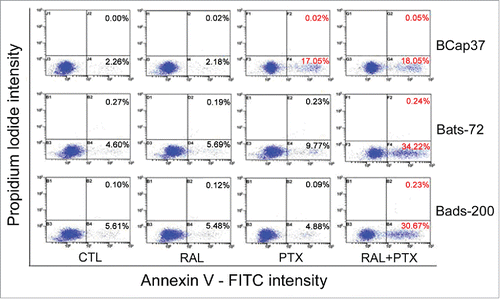
We also used flow cytometric analysis to determine whether raloxifene affected the paclitaxel-induced apoptosis for MDR cells treated with paclitaxel for 48 h. As shown in , raloxifene alone had no pronounced apoptosis effects on BCap37, Bats-72 and Bads-200 cell lines. However, following the pre-treatment of raloxifene and paclitaxel for 48 hours, 34.22% of Bats-72 cells had undergone early apoptosis, which was 24.45% more than the early apoptosis percentage by paclitaxel-treatment alone. Similarly, the percentage of early apoptosis for Bads-200 cells rose from 4.88% to 30.67% after pre-treatment of raloxifene. While for BCap37 cells, the cell undergoing early apoptosis was only slightly increase from 17.05% to 18.05% (p < 0.01).
Figure 3. The reversal effect of raloxifene via enhanced paclitaxel-induced cell apoptosis. Early apoptosis was quantified by combined staining with Annexin V and propidium iodide and flow cytometric analysis after treated for 48h (the percentages in right bottom stand for early apoptotic cells, and the percentages in top right present late apoptotic cells).
Raloxifene sensitizes MDR cells to paclitaxel through up-regulation of CyclinB1, phosphorylation of CDC25 and Bcl-2
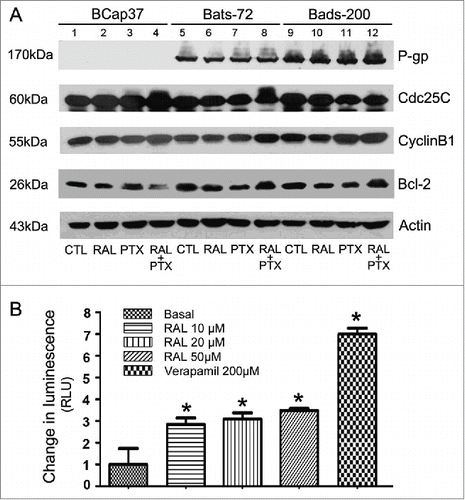
In the BCap37 cells, phosphorylation of Cdc25C emerged in the groups treated paclitaxel (, lanes 3, 4). Similarly, paclitaxel with the combination of raloxifene notably caused phosphorylation of Cdc25C in Bats-72 cells (, lanes 8), while phosphorylation of Cdc25C in Bads-200 cells was not distinct. Paclitaxel with the pre-treatment of raloxifene led upregulation of CyclinB1 in all the 3 cell lines (, lanes 4, 8, 12), which coincided with paclitaxel-induced G2/M arrest of the cells. Moreover, a down-regulation/phosphorylation of bcl-2 was also seen in BCap37 cells treated with paclitaxel (, lanes 3, 4). Meanwhile, the phosphorylation of Bcl-2 was also noticed by co-incubation of paclitaxel and raloxifene in either Bats-72 or Bads-200 cells.(, lanes 8, 12).
Figure 4. Raloxifene affect the activities of P-gp, Cdc25C, Cyclin B 1 and Bcl-2 proteins. (A) Western blotting analyses for the P-gp, Cdc25C, Cyclin B 1 and bcl−2 proteins. Cells were treated with different concentrations of raloxifene or paclitaxel or their various combinations as described. Equal amounts (40μg/lane) of cellular protein analyzed by immunoblotting with anti-P-gp, Cdc25C, Cyclin B 1 and bcl−2 antibodies. β-Actin protein was blotted as a control. (B) Stimulation of P-gp ATPase activity by raloxifene. Untreated (NT), 100µM Na3VO4-, 10-50µM raloxifene and 200µM Verapamil-treated P-gp reactions were performed according to the protocol. The decrease in luminescence of NT samples compared to samples plus Na3VO4 (ΔRLUbasal) represents basal P-gp ATPase activity, which was transformed as 1. The change in luminescence of Verapamil-treated samples (ΔRLUTC) represents Verapamil-stimulated P-gp ATPase activity. The change in luminescence of raloxifene-treated samples (ΔRLURAL) represents raloxifene-stimulated P-gp ATPase activity. ΔRLUTC and ΔRLURAL were transformed as ratios of ΔRLUbasal to illustrate the stimulation of P-gp ATPase activity by raloxifene. *P < 0.05 versus basal.
Raloxifene stimulates P-gp ATPase activity, but does not affect P-gp expression
BCap37 cells did not express P-glycoprotein, while both of Bats-72 and Bads-200 cell lines expressed P-glycoprotein in varying protein levels, which confirmed our previous reports.Citation20 However, Western blotting revealed no distinct changes by different treatments in any cell lines, showing that raloxifene did not affect P-glycoprotein expression ().
A variety of P-gp inhibitors and substrates can stimulate ATPase activity. By comparing the basal activity of P-gp with the P-gp activity in Raloxifene-treated cells, Raloxifene can be ranked as stimulator, inhibitor or having no effect on P-gp ATPase activities. Verapamil was used as positive control for its activation of P-gp ATPase, while verapamil is also referred to as a P-gp inhibitor because as a substrate for transport, it inhibits P-gp activity with other substrates in a competitive mode. By comparing with basal and verapamil-stimulated P-gp ATPase activities, we observed that raloxifene had the similar function with verapamil which increased the ATPase activity in recombinant human P-gp membrane protein (). These results suggest that raloxifene could affect P-gp ATPase activity by interacting with P-gp directly.
Raloxifene sensitizes resistance of MDR cell lines to paclitaxel in vivo
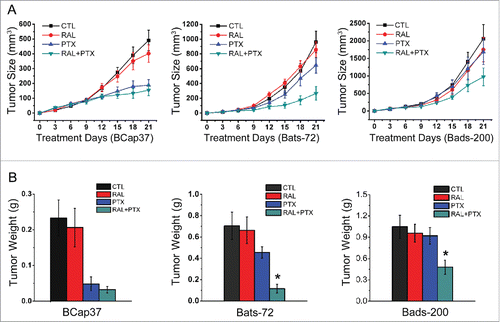
Animal studies were performed to determine whether the effects of raloxifene also occurred in xenograft models. The results showed that raloxifene alone had little suppressive effects on the growth of tumors generated from 3 cell lines. The tumor size of Bcap37 cells was well-controlled by treated with paclitaxel with or without raloxifene, with inhibition rate without essential distinction (62% and 68%, respectively). Consistent with in vitro experiments, tumor masses from 2 MDR cell lines possessed significant resistance to paclitaxel in vivo. However, compared with paclitaxel alone, co-treated with raloxifene significantly strengthened toxicity of paclitaxel to Bats-72 and Bads-200 tumor masses, with inhibitory rate from 33% to 73% and 18% to 53%, respectively (). Two MDR cell lines had distinct properties of proliferative ability and drug-sensitivity in vivo. Bats72 cell line was more sensitive to paclitaxel than Bads-200 cell line, and showed better response to raloxifene. This conclusion was also confirmed by the tumor weight ().
Figure 5. Raloxifene sensitizes resistance of MDR cell lines to paclitaxel in vivo. Nude mice bearing Bcap37, Bats-72 and Bads-200 tumors were treated with raloxifene, paclitaxel and their combination in indicated doses. Tumor size (A) and tumor weight (B) were measured and calculated as described in materials and methods. Data are presented as mean ± standard error based on 12 mice for each group in 2 independent experiments. *P < 0.05 vs. PTX.
Combination of raloxifene with paclitaxel reduced proliferative effects of MDR cell lines in vivo
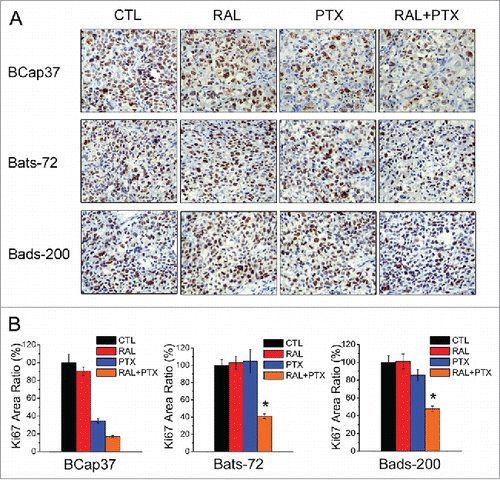
Paraffin-embedded tissue sections of tumors derived from 3 cell lines with different treatment of drugs were stained with proliferation marker Ki-67. As shown in , no significant difference on the percentage of Ki-67-positive areas was observed between the groups treated with PBS, raloxifene with or without paclitaxel in Bats-72 and Bads-200 tumors. So did the control and raloxifene groups in Bcap37 tumors. These data suggested that raloxifene itself had little impact on the tumor cell proliferation in these 3 xenograft models. However, 7 cycles of co-administration of paclitaxel with raloxifene significantly reduced the percentages of Ki-67-positive areas to 41% in Bats-72 tumors, and 48% in Bads-200 tumors, respectively (both P < 0.05 versus PTX, Figure 6B). These results indicate that raloxifene could reinforce paclitaxel-induced tumor growth inhibition in vivo.
Figure 6. Raloxifene with paclitaxel reduces proliferation of MDR cell lines in vivo. (A) Ki-67 immunohistochemistry staining of Bcap37, Bats-72, Bads-200 tumors after various treatments. (B) Quantitative analysis of Ki-67 positive area percentage. Data are presented as mean ± standard error based on 5 randomly selected microscopic fields for each group. *P < 0.05 versus PTX.
Discussions
Quite a few clinical trials have suggested that tamoxifen, a typical SERM, could reduce the risk of ER positive breast cancer.Citation17,18,21,22 Further studies revealed that raloxifene was as effective as tamoxifen in reducing the risk of invasive breast cancer. Particularly, it has a lower risk of thromboembolic events and cataracts, suggesting its bright prospect in breast cancer prevention.Citation14,23,24 Although tamoxifen and fulvestrant (ICI 182,780), the pure estrogen receptor antagonists, have been demonstrated their sensitizing effects on chemotherapeutics in breast cancer cells expressing ER-α, very few studies have investigated whether raloxifene could sensitize multidrug resistant breast cancer cells to anti-microtubule agents,Citation5,13 especially ER negative breast tumor which usually results in poor prognosis.
BCcap37 was originally quite sensitive to antimicrotubule agent induced apoptosis and G2/M arrest.Citation20 However, through repeated selection and screening with paclitaxel, Bads-200 and Bats-72 were born with multidrug resistance, high expression of P-gp and no expression of ER. In the current study, we found that co-treatment with raloxifene could significantly reverse the resistance to paclitaxel in ER− MDR breast cancer cell lines Bads-200 and Bats-72 by enhancing paclitaxel induced mitotic arrest and apoptosis in vitro and in vivo.
Several experiments have suggested that paclitaxel might induce apoptosis independently of a prior G2/M-phase arrest.Citation25,26 The 2 drug-resistant cells were induced analogous effects by co-incubation of paclitaxel and raloxifene, meanwhile Bads-200 cells also presented G1 phase arrest. CyclinB1 is periodically expressed in the cell cycle and accumulates in the G2/M phase,Citation6,27 and causes dose-dependent mitotic arrest phenotypes.Citation28 Our results indicated that paclitaxel with the pre-treatment of raloxifene caused increased levels of CyclinB1 in all the 3 cell lines, which coincided with paclitaxel-induced G2-M arrest. In addition, cell division cycle 25 (CDC25) phosphatases regulate key transitions between cell cycle phases during normal cell division, and they are key targets of the checkpoint machinery in the event of DNA damage.Citation29,30 It was demonstrated that phosphorylation of Cdc25C was required for the activation of cdc2-cyclin B and entry into M-phase.Citation31 In our experiments, paclitaxel with raloxifene induced Cdc25C phosphorylation in bats-72 cells, which also emerged in BCap37 cells when treated with paclitaxel. Consistent with the in vivo test, compared to the group treated with raloxifene or paclitaxel alone, the Bats-72 and Bads-200 tumors exposed to combination of raloxifene and paclitaxel exhibited much less Ki-67 positive cells and smaller tumor volumes.
Additionally, previous studies have demonstrated that Bcl-2 protects cancer cells from apoptosis induced by a variety of anticancer agents and Bcl-2 phosphorylation/downregulation play important roles in paclitaxel-induced apoptosis.Citation32-35 Bcl-2 phosphorylation is supposed to be a specific hallmark of paclitaxel cytotoxicity and a significant step from microtubule damage to apoptosis.Citation34,36 Cells are more susceptible to a death signal during the G2/M when Bcl-2 phosphorylation occurs, indicating that Bcl-2 phosphorylation may lower the threshold for apoptosis in the G2/M phase.Citation37 Through our experiments, western blot revealed that pre-treatment of raloxifene with paclitaxel caused phosphorylation of Bcl-2 in both Bats-72 and Bads-200 cells (down-regulation of Bcl-2 was also seen in BCap37 cells), coincided with the apoptosis assay results determined by flow cytometry. These combined treatments also disturbed cell-cycle progression in the G2/M stage in parallel with phosphorylation of Bcl-2. Our results suggest that raloxifene may sensitize Bats-72 and Bads-200 cells to paclitaxel through Bcl-2 phosphorylation and reactivation of apoptotic signal pathways.
Above results agreed with previous reports on MDR reversion by SERM, such as Tamoxifen, Toremifene and Droloxifene.Citation8,9,38-40 Our results also suggested that raloxifene could reverse MDR independent of antagonizing ER mediated signal pathway. P-glycoprotein is overexpressed in many multidrug resistant cells as an energy-dependent membrane transporter, including Bats-72 and Bads-200 cells. P-glycoprotein could extrude excessive chemotherapeutic drugs from cells and prevent the cytotoxic effects of the drugs.Citation3 Raloxifene might act as a chemosensitizer when co-administered with paclitaxel and we suspected that the mechanism of reversal modulation of multidrug resistance by raloxifene was related to its down regulation of over-expression of P-glycoprotein. In previous studies, tamoxifen inhibited P-glycoprotein overexpression or decreased P-glycoprotein mRNA levels and therefore reversed the multidrug resistance of cancer cells.Citation9 Another selective estrogen receptor modulator, Doxoloxifene, could directly inhibited P-glycoprotein pump-efflux functions.Citation40 Our protein gel blotting showed that pre-treatment of raloxifene at 10 μM or 20 μM for 48 hours did not inhibit the expression of P-glycoprotein but stimulated P-gp ATPase activity with a tendency similar to verapamil. These results support the hypothesis that raloxifene reverses MDR in tumor cells by uncoupling P-gp ATPase activity from the drug substrate efflux activity of P-glycoprotein as a substrate competitive inhibitor. Therefore, our studies suggested that raloxifene might not only be used as ER antagonist for endocrine therapy in ER+ breast cancers, but also be valuable as a chemosensitizer to reverse P-gp mediated MDR breast cancers, particularly for ER- breast cancers in clinic.
In summary, through a series of assays, we found that the combination of raloxifene, a selective ER modulator, could dramatically reverse the drug resistance to paclitaxel in Bats-72 and Bads-200 cells, and even produced synergistic effects. Further assays showed that the reversal of MDR by raloxifene was mediated through selective and potent inhibition of P-glycoprotein function. Other potential mechanisms might also be involved. Particularly, raloxifene hydrochloride has little or even no cytotoxicity cell lines or animal models used in the experiments, it might be valuable as a potential safe chemosensitizer agent for cancer therapy.
Materials and Methods
Cell lines and mice
The multidrug resistant cell lines Bats-72 and Bads-200 were respectively screened from ER- human breast cancer cell line BCap37 by time-stepwise and dose-stepwise increment exposure of paclitaxel as described previously.Citation20 BCap37 and Bats-72 cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin,Citation5,20 whereas Bads-200 cells were cultured with additional 200 nM paclitaxel. Female aged 5 weeks of athymic nude mice were purchased from Shanghai SLAC animal facility. All animal cares and experiments were conducted by the Guidelines of Zhejiang University Animal Care Committee.
Drugs and treatments
Raloxifene hydrochloride were purchased from Sigma and dissolved in DMSO. Paclitaxel (Taxol® injection) was purchased from Mead Johnson Co, (Princeton. NJ). Drugs were diluted with culture media to obtain the desired concentrations prior to usage. Cells were cultured into 96-well or 6-well plates in drug-free medium for 24 h. Then the cells were treated with distinct concentrations of paclitaxel (5 nM, 200 nM and 2000 nM for BCap37, Bats-72 and Bads-200 cells, respectively) with or without 3 h pre-treatment of raloxifene (10 μM for BCap37 and Bats-72 cells, 20 μM for Bads-200 cells).
3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide assay
Drug-induced cytotoxicity was determined by MTT assay as described previously.Citation41 Briefly, cells were seeded into 96-well plates at 104 cells per well. After incubation overnight, the cells were treated with drugs designated as described above for 72 h. Four hours before the end of treatments, MTT solution was added. Finally, the medium was removed and 200 μL of DMSO was added to dissolve the formazan crystals. The absorbance in individual wells at 570 nm was measured by a microplate reader.
Cell morphology in vitro
The cytotoxicity of paclitaxel, raloxifene, and their combination was further evaluated by cell morphology. Cells were plated on 6-well plates (35 mm) with 105 cells/well. The tumor cells were incubated in drug-free medium for about 24 h before they were exposed to different concentrations of paclitaxel with or without pre-treatment of raloxifene as described above. After treatment for 24 h, cells on the plates were examined and photographed using bright-field microscope.
Cell cycle analysis
Flow cytometric analysis was used to determine the cell cycle distribution and induction of apoptosis. Cells were plated on 6-well plates (35 mm) with approximately 105 cells /well. After 24 h, the medium was replaced with fresh medium containing paclitaxel alone or in combination with raloxifene as mentioned. At the end of treatments for 48 h, both detached and attached cells were harvested and washed twice with PBS. Cell sample preparation, including propidium iodide staining, was performed as described previously.Citation42 Cell cycle distribution and DNA content were determined using a Coulter Epics V instrument (Beckman Coulter, Inc., Fullerton, CA).
Apoptotic analysis
Annexin V/propidium iodide (PI) apoptosis detection kit (Beyotime, Haimen, China) was used to detect cell apoptosis according to the manufacturer's instructions.Citation20 Briefly, cells were harvested and washed twice with PBS after treatment for 48 h. Then cells were suspended with 400 μL of Annexin V binding buffer. 5 μL of fluorescein isothiocyanate (FITC) Annexin V and 10 μL of PtdIns were added and the cells were incubated in the mixture for 15 min at 4°C in the dark. Finally, the percentage of apoptotic cells was determined by flow cytometry.
Western blot analysis
After various treatments for 24 h, cells were harvested and washed twice with PBS. Equal amounts (40 μg/lane) of proteins were fractionated on 10 to 12% SDS–PAGE gels and transferred to polyvinylidene difluoride membranes. The membranes were incubated with anti-Bcl-2 (DAKO), anti-MDR1, anti-Cdc25C and anti_CyclinB1 (Santa Cruz, CA) primary antibodies, respectively. After washing with PBS containing 0.1% (v/v) Tween 20, the membranes were incubated with peroxidase-conjugated secondary antibodies (Santa Cruz, CA) followed by enhanced chemiluminescence staining using the ECL system (Amersham Biosciences, UK). β-actin (Santa Cruz, CA) was used for normalization of protein loading.
P-glycoprotein ATPase activity assay
The P-gp-Glo™ Assay Systems (Promega) provide the necessary reagents for performing luminescent P-glycoprotein ATPase assays.Citation43 The P-gp-Glo™ Assay relies on the ATP-dependence of the light-generating reaction of firefly luciferase, which is a valuable screening tool for determining if a drug interacts with P-gp. This assay system consists of human P-gp/MDR1 membrane and P-gp/MDR1-negative control membrane fractions, buffers, solutions, and relevant reagents. The effect of raloxifene (10, 20 and 50 μM) on the ATPase activity of P-glycoprotein was measured according to the manufacturer's protocol.
Animal studies
Tumor cells (4 × 106 cells in 0.2 ml PBS) were injected via subcutaneous route in the left flank of Nude mice (female, 6-weeks old). When tumors reached a mean diameter of 0.3–0.4 cm, each type of xenografts were randomly divided into 4 groups and treated with (i) vehicle; (ii) raloxifene (60 mg/kg, p.o.); (iii) paclitaxel (20 mg/kg, i.v.); (iiii) raloxifene (60 mg/kg, p.o.) and paclitaxel (20 mg/kg, i.v.). The paclitaxel treatment was repeated every 3 days for total of 8 cycles. In each cycle, raloxifene was administrated at the same day with paclitaxel and the day before paclitaxel. Two perpendicular diameters (width and length) of the tumors were collected every 3 days. The tumor volume was calculated according to the following formula: volume (mm3) = π(L × W2)/6.Citation44 Data were representative of 2 separate experiments.
Histology assays
Tumor tissues were collected at the indicated time, fixed in 10% neutral formalin, embedded in paraffin, and stained with mAb to Ki-67 followed by examination and photography under microscopy. Image-Pro Plus was used to figure up the brown areas which were stained by mAb to Ki-67 of 5 randomly non-necrotic microscopic fields (400 ×) for each group.
Statistical analysis
Data are presented as mean ± standard error of 3 independent experiments. Data were evaluated by variance analysis of single factor (ANOVA) and Tukey–Kramer post-hoc comparison to determine the statistical difference between various experimental and control groups. Differences were considered statistically significant at a level of *P < 0.05.
Disclosure of Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported in part by Grants NNSF–81071880 and NNSF-81372462.
References
- Lind P, Igerc I, Beyer T, Reinprecht P, Hausegger K. Advantages and limitations of FDG PET in the follow-up of breast cancer. Eur J Nucl Med Mol Imaging 2004; 31 Suppl 1:S125-34; PMID:15085295.
- Li J, Xu LZ, He KL, Guo WJ, Zheng YH, Xia P, Chen Y. Reversal effects of nomegestrol acetate on multidrug resistance in adriamycin-resistant MCF7 breast cancer cell line. Breast Cancer Res 2001; 3:253-63; PMID:11434876.; http://dx.doi.org/10.1186/bcr303.
- Baird RD, Kaye SB. Drug resistance reversal - are we getting closer? European J Cancer 2003; 39:2450-61; http://dx.doi.org/10.1016/S0959-8049(03)00619-1.
- Jeansonne DP, Koh GY, Zhang F, Kirk-Ballard H, Wolff L, Liu D, Eilertsen K, Liu Z. Paclitaxel-induced apoptosis is blocked by camptothecin in human breast and pancreatic cancer cells. Oncol Rep 2011; 25:1473-80; PMID:21331447.
- Sui M, Jiang D, Hinsch C, Fan W. Fulvestrant (ICI 182,780) sensitizes breast cancer cells expressing estrogen receptor α to vinblastine and vinorelbine. Breast Cancer Res Treat 2010; 121:335-45; PMID:19626437.; http://dx.doi.org/10.1007/s10549-009-0472-4.
- Sui M, Huang Y, Park BH, Davidson NE, Fan W. Estrogen receptor α mediates breast cancer cell resistance to paclitaxel through inhibition of apoptotic cell death. Cancer Res 2007; 67:5337-44; PMID:17545614.; http://dx.doi.org/10.1158/0008-5472.CAN-06-4582.
- Kerb R, Hoffmeyer S, Brinkmann U. ABC drug transporters: hereditary polymorphisms and pharmacological impact in MDR1, MRP1 and MRP2. Pharmacogenomics 2001; 2:51-64; PMID:11258197.; http://dx.doi.org/10.1517/14622416.2.1.51.
- Sugimoto Y, Tsukahara S, Imai Y, Ueda K, Tsuruo T. Reversal of breast cancer resistance protein-mediated drug resistance by estrogen antagonists and agonists. Mol Cancer Ther 2003; 2:105-12; PMID:12533678.
- Liu ZH, Ma YL, He YP, Zhang P, Zhou YK, Qin H. Tamoxifen reverses the multi-drug-resistance of an established human cholangiocarcinoma cell line in combined chemotherapeutics. Mol Biol Rep 2011; 38:1769-75; PMID:20835928.; http://dx.doi.org/10.1007/s11033-010-0291-z.
- Pickar JH, MacNeil T, Ohleth K. SERMs: progress and future perspectives. Maturitas 2010; 67:129-38; PMID:20580502.; http://dx.doi.org/10.1016/j.maturitas.2010.05.009.
- Shanle EK, Xu W. Selectively targeting estrogen receptors for cancer treatment. Adv Drug Deliv Rev 2010; 62:1265-76; PMID:20708050.; http://dx.doi.org/10.1016/j.addr.2010.08.001.
- Kiss Z, Crilly KS. Tamoxifen inhibits uptake and metabolism of ethanolamine and choline in multidrug-resistant, but not in drug-sensitive, MCF-7 human breast carcinoma cells. FEBS Lett 1995; 360:165-8; PMID:7875322.; http://dx.doi.org/10.1016/0014-5793(95)00094-P.
- De Vincenzo R, Scambia G, Benedetti Panici P, Fattorossi A, Bonanno G, Ferlini C, Isola G, Pernisco S, Mancuso S. Modulatory effect of tamoxifen and ICI 182,780 on adriamycin resistance in MCF-7 human breast-cancer cells. Int J Cancer 1996; 68:340-8; PMID:8903476.; http://dx.doi.org/10.1002/(SICI)1097-0215(19961104)68:3%3c340::AID-IJC12%3e3.0.CO;2-C.
- Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, Bevers TB, Fehrenbacher L, Pajon ER, Jr, Wade JL 3rd, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA 2006; 295:2727-41; PMID:16754727.; http://dx.doi.org/10.1001/jama.295.23.joc60074.
- Swaby RF, Sharma CG, Jordan VC. SERMs for the treatment and prevention of breast cancer. Rev Endocr Metab Disord 2007; 8:229-39; PMID:17440819.; http://dx.doi.org/10.1007/s11154-007-9034-4.
- Veronesi U, Maisonneuve P, Rotmensz N, Bonanni B, Boyle P, Viale G, Costa A, Sacchini V, Travaglini R, D'Aiuto G, et al. Tamoxifen for the prevention of breast cancer: late results of the Italian Randomized Tamoxifen Prevention Trial among women with hysterectomy. J Natl Cancer Inst 2007; 99:727-37; PMID:17470740.; http://dx.doi.org/10.1093/jnci/djk154.
- Cuzick J, Powles T, Veronesi U, Forbes J, Edwards R, Ashley S, Boyle P. Overview of the main outcomes in breast-cancer prevention trials. Lancet 2003; 361:296-300; PMID:12559863.; http://dx.doi.org/10.1016/S0140-6736(03)12342-2.
- Fisher B, Costantino JP, Wickerham DL, Cecchini RS, Cronin WM, Robidoux A, Bevers TB, Kavanah MT, Atkins JN, Margolese RG, et al. Tamoxifen for the prevention of breast cancer: current status of the National Surgical Adjuvant Breast and Bowel Project P-1 study. J Natl Cancer Inst 2005; 97:1652-62; PMID:16288118.; http://dx.doi.org/10.1093/jnci/dji372.
- DeMichele A, Troxel AB, Berlin JA, Weber AL, Bunin GR, Turzo E, Schinnar R, Burgh D, Berlin M, Rubin SC, et al. Impact of raloxifene or tamoxifen use on endometrial cancer risk: a population-based case-control study. J Clin Oncol 2008; 26:4151-9; PMID:18757329.; http://dx.doi.org/10.1200/JCO.2007.14.0921.
- Jiang D, Sui M, Zhong W, Huang Y, Fan W. Different administration strategies with paclitaxel induce distinct phenotypes of multidrug resistance in breast cancer cells. Cancer Lett 2013; 335:404-11; PMID:23499896.; http://dx.doi.org/10.1016/j.canlet.2013.02.059.
- Vogel VG. Recent results from clinical trials using SERMs to reduce the risk of breast cancer. Ann N Y Acad Sci 2006; 1089:127-42; PMID:17261762.; http://dx.doi.org/10.1196/annals.1386.010.
- Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N, Atkins J, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 1998; 90:1371-88; PMID:9747868.; http://dx.doi.org/10.1093/jnci/90.18.1371.
- Jordan VC, Gapstur S, Morrow M. Selective estrogen receptor modulation and reduction in risk of breast cancer, osteoporosis, and coronary heart disease. J Natl Cancer Inst 2001; 93:1449-57; PMID:11584060.; http://dx.doi.org/10.1093/jnci/93.19.1449.
- Vogel VG. The NSABP Study of Tamoxifen and Raloxifene (STAR) trial. Expert Rev Anticancer Ther 2009; 9:51-60; PMID:19105706.; http://dx.doi.org/10.1586/14737140.9.1.51.
- Huang Y, Johnson KR, Norris JS, Fan W. Nuclear factor-kappaB/IkappaB signaling pathway may contribute to the mediation of paclitaxel-induced apoptosis in solid tumor cells. Cancer Res 2000; 60:4426-32; PMID:10969788.
- McDaid HM, Horwitz SB. Selective potentiation of paclitaxel (taxol)-induced cell death by mitogen-activated protein kinase kinase inhibition in human cancer cell lines. Mol Pharmacol 2001; 60:290-301; PMID:11455016.
- Sui M, Dziadyk JM, Zhu X, Fan W. Cell cycle-dependent antagonistic interactions between paclitaxel and gamma-radiation in combination therapy. Clin Cancer Res 2004; 10:4848-57; PMID:15269161.; http://dx.doi.org/10.1158/1078-0432.CCR-03-0707.
- Wolf F, Wandke C, Isenberg N, Geley S. Dose-dependent effects of stable cyclin B1 on progression through mitosis in human cells. EMBO J 2006; 25:2802-13; PMID:16724106.; http://dx.doi.org/10.1038/sj.emboj.7601163.
- Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer 2007; 7:495-507; PMID:17568790.; http://dx.doi.org/10.1038/nrc2169.
- Cazales M, Boutros R, Brezak MC, Chaumeron S, Prevost G, Ducommun B. Pharmacologic inhibition of CDC25 phosphatases impairs interphase microtubule dynamics and mitotic spindle assembly. Mol Cancer Ther 2007; 6:318-25; PMID:17237290.; http://dx.doi.org/10.1158/1535-7163.MCT-06-0299.
- Hoffmann I, Clarke PR, Marcote MJ, Karsenti E, Draetta G. Phosphorylation and activation of human cdc25-C by cdc2–cyclin B and its involvement in the self-amplification of MPF at mitosis. EMBO J 1993; 12:53-63; PMID:8428594.
- Dziadyk JM, Sui M, Zhu X, Fan W. Paclitaxel-induced apoptosis may occur without a prior G2/M-phase arrest. Anticancer Res 2004; 24:27-36; PMID:15015572.
- Salah-Eldin AE, Inoue S, Tsukamoto S, Aoi H, Tsuda M. An association of Bcl−2 phosphorylation and Bax localization with their functions after hyperthermia and paclitaxel treatment. Int J Cancer 2003; 103:53-60; PMID:12455053.; http://dx.doi.org/10.1002/ijc.10782.
- Haldar S, Jena N, Croce CM. Inactivation of Bcl−2 by phosphorylation. Proc Natl Acad Sci U S A 1995; 92:4507-11; PMID:7753834.; http://dx.doi.org/10.1073/pnas.92.10.4507.
- Srivastava RK, Srivastava AR, Korsmeyer SJ, Nesterova M, Cho-Chung YS, Longo DL. Involvement of microtubules in the regulation of Bcl2 phosphorylation and apoptosis through cyclic AMP-dependent protein kinase. Mol Cell Biol 1998; 18:3509-17; PMID:9584191.; http://dx.doi.org/10.1128/MCB.18.6.3509.
- Blagosklonny MV, Giannakakou P, El-Deiry WS, Kingston DG, Higgs PI, Neckers L, Fojo T. Raf-1/bcl−2 phosphorylation: a step from microtubule damage to cell death. Cancer Res 1997; 57:130-5; PMID:8988053.
- Yamamoto K, Ichijo H, Korsmeyer SJ. BCL−2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol 1999; 19:8469-78; PMID:10567572.
- Liu ZH, He YP, Qin H. The growth-inhibition effect of tamoxifen in the combination chemotherapeutics on the human cholangiocarcinoma cell line QBC939. Mol Biol Rep 2010; 37:2693-701; PMID:19757172.; http://dx.doi.org/10.1007/s11033-009-9801-2.
- Zhang Y, Wang H, Wei L, Li G, Yu J, Gao Y, Gao P, Zhang X, Wei F, Yin D, et al. Transcriptional modulation of BCRP gene to reverse multidrug resistance by toremifene in breast adenocarcinoma cells. Breast Cancer Res Treat 2010; 123:679-89; PMID:19967559.; http://dx.doi.org/10.1007/s10549-009-0660-2.
- Li J, Xu LZ, Yao JJ, Guo WJ, Xia P, Chen Y. Reversal effects of droloxifene on multidrug resistance in adriamycin-resistant K562 cell line. Acta Pharmacol Sin 2001; 22:1023-7; PMID:11749795.
- Scudiero DA, Shoemaker RH, Paull KD, Monks A, Tierney S, Nofziger TH, Currens MJ, Seniff D, Boyd MR. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res 1988; 48:4827-33; PMID:3409223.
- Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 1991; 139:271-9; PMID:1710634.; http://dx.doi.org/10.1016/0022-1759(91)90198-O.
- Duan Z, Choy E, Hornicek FJ. NSC23925, identified in a high-throughput cell-based screen, reverses multidrug resistance. PLoS One 2009; 4:e7415; PMID:19823672.; http://dx.doi.org/10.1371/journal.pone.0007415.
- Herr I, Ucur E, Herzer K, Okouoyo S, Ridder R, Krammer PH, von Knebel Doeberitz M, Debatin KM. Glucocorticoid cotreatment induces apoptosis resistance toward cancer therapy in carcinomas. Cancer Res 2003; 63:3112-20; PMID:12810637.