
ABSTRACT
Specific and efficient delivery of siRNA into intended tumor cells remains as a challenge, even though RNAi has been exploited as a new strategy for prostate cancer therapy. This work aims to address both specificity and efficiency of SURVIVIN-siRNA delivery by constructing a therapeutic complex using combinatorial strategies. A fusion protein STD was first expressed to serve as a backbone, consisting of streptavidin, a cell-penetrating peptide called Trans-Activator of Transcription (TAT) and a double-stranded RNA binding domain. A biotinylated Prostate Specific Membrane Antigen (PSMA) specific aptamer A10 and SURVIVIN-siRNA were then linked to STD protein to form the therapeutic complex. This complex could specifically targeted PSMA+ tumor cells. Compared to lipofectamine and A10-siRNA chimera, it demonstrated higher efficiency in delivering siRNA into target cells by 19.2% and 59.9%, and increased apoptosis by 16.8% and 26.1% respectively. Upon systemic administration, this complex also showed significant efficacy in suppressing tumor growth in athymic mice (p <0.001). We conclude that this therapeutic complex could specifically and efficiently deliver SURVIVIN-siRNA to target cells and suppressed tumor growth in vivo, which indicates its potential to be used as a new strategy in prostate cancer therapy
Abbreviations
| BPH | = | Benign Prostate Hyperplasia |
| BSA | = | Bull Serum Albumin |
| CPPs | = | Cell Penetrating Peptides |
| CLSM | = | Confocal Laser Scanning Microscopy |
| DRBD | = | Double Strand RNA Binding Domain |
| EMSA | = | Electrophoretic Mobility Shift Assay |
| MFI | = | Mean Fluorescence Intensity |
| PCa | = | Prostate Cancer |
| PSMA | = | Prostate Specific Membrane Antigen |
| SA | = | Streptavidin |
| STD | = | STREPTAVIDIN-TAT-DRBD fusion protein |
| sur-siRNA | = | SURVIVIN siRNA |
| TAT | = | Trans-Activator of Transcription |
Introduction
Currently, selective knockdown of abnormal genes has prompted notable activity in developing RNAi-based drugs for prostate cancer (PCa) therapy. Anti-apoptotic protein SURVIVIN (GenBank accession no. AAC51660.1) is highly expressed in most human cancer cells, but has very low expression in normal differentiated cells.Citation1-3 In PCa it has also been confirmed that the expression of SURVIVIN is augmented compared to benign prostate hyperplasia (BPH) and positively correlated with tumor aggressiveness.Citation4 Thus, SURVIVIN gene (GenBank accession numberU75285) is considered as a therapeutic target for PCa.Citation5 Our previous work has demonstrated that knocking down the expression of SURVIVIN gene by lipid-based delivery of siRNAs-plasmid could significantly enhance apoptosis in PCa cell line PC-3 and increase its sensitivity to the anti-cancer drug cisplatin in vitro.Citation6 However, for clinical application, a robust method of delivery is required to ensure that the therapeutic siRNAs could specifically reach target cells or tissues and avoid generalized cytotoxicity to normal cells. Furthermore, the siRNAs need to be delivered into cancer cells effectively to knock down the expression of target mRNAs.Citation7
Due to their instability, vulnerability and immunogenicity, the delivery of non-encapsulated siRNAs needs to be facilitated by transfection vehicles.Citation8 Over recent years, while progress has been made in delivery agents to enhance specificity and efficacy, yet most of the delivery strategies focus on only one of these 2 aspects. Cationic lipids,Citation9 ligands, nanoparticles,Citation10 polymers Citation11 and viral vectors provide protection of siRNAs and allow for easy uptake by cells, but offer little in targeting specific cells. Aptamers, antibodies are useful for cell-type-specific delivery of siRNAs, but not able to enhance intracellular uptake. It has become apparent that a combinatorial approach is highly desirable to enhance both of specificity and efficiency of siRNA delivery, especially for systemic administration in vivo.
Studies have showed that cell penetrating peptides (CPPs) can transport a variety of macromolecules, e.g. peptides, proteins, nucleic acids into cells rapidly via micropinocytosis.Citation12-14 One of the best known CPPs is HIV1 Trans-Activator of Transcription (TAT).Citation15 For siRNAs delivery, cationic CPPs and anionic siRNAs will aggregate due to charge neutralization, which may affect biological functions of siRNA. Eguchi A et al.Citation16-18 solved this problem by construction of a fusion protein of CPPs with double strand RNA binding domain (DRBD) to mask siRNA's charges, which can efficiently deliver siRNA into primary cells. For specific delivery of siRNA, there is an increasing interest in aptamers. These small RNA/DNA molecules can form secondary and tertiary structures capable of specifically binding proteins or other cellular targets. The first aptamer based therapeutic was FDA approved in 2004 for the treatment of age-related macular degeneration and several other aptamers for cancer therapy such as NOX-A12, AS1411 are currently being evaluated in clinical trials. Citation19,20 Aptamer A10 is known for its high affinity (KD ∼10−9M) for Prostate Specific Membrane Antigen (PSMA) Citation21 and has been used widely in development of PCa therapy. Currently, A10 has been mainly employed to delivery therapeutic siRNA in the form of aptamer-siRNA chimeras and showed to promote regression of PSMA+ tumors through intratumor injection.Citation22-25 In this work our aim is, using a combinatorial strategy, to construct a novel PCa therapeutic complex, consist of aptamer A10, TAT-DRBD and siRNAs, and to assess its specific and efficient delivery as well as the effect of PCa suppression.
Materials and methods
1. General materials
Unless otherwise noted, all primers and restriction enzymes were purchased from Takara Biotechnology (catalog number Xho I 1635, Sma I 1629). All primary antibodies were purchased from Abcam: anti-HIV tat antibody, (N3), ab63957; anti-human PKR DRBD motif1 antibody, (YE350), ab32052; anti-PSMA antibody, (YPSMA-1) ab19071; anti-SURVIVIN antibody, (EP2880Y), ab76424; anti-β-actin antibody, ACTN05 (C4) (ab3280). The secondary antibody was purchased from Odyssey (Goat anti-Mouse antibody, IRDye 680). Lipofectamine2000 was purchased from Life Technology (11668027). Plasmid pET-44b+ vector was purchased from Novagen (71123-3). T4 DNA ligase was purchased from New England BioLabs (M0202S). His GraviTrap Kit for protein purification was purchased from GE Healthcare (11-0036-90 AA).
All cell lines were purchased from the Type Culture Collection of Shanghai, the Chinese Academy of Sciences. The main PCa cell line LNCaP was tested by detection of PSMA in the cellular supernatant. All culture medium was purchased from Gibco BRL/Life Technologies. Tumor cells were maintained at 37°C and 5% CO2 in RPMI 1640 supplemented with 10% fetal bovine serum.
2. Complex construction
2.1. Complex design
The designed complex is composed of 3 parts: PSMA specific aptamer A10, a STREPTAVIDIN-TAT-DRBD fusion protein (STD) as a backbone, and a functional anti-cancer agent SURVIVIN siRNA (sur-siRNA). To link these 3 parts together, sur-siRNA binds with DRBD, and biotinylated A10 with STREPTAVIDIN (SA). Sequence information details of these complex components were showed in .
Table 1. Sequence information of each component of the complex.
2.2. Expression of STD fusion protein
The DNA fragments of SA and 3TAT-DRBD were synthesized by Sangon Biotech respectively. The recombinant fragment of SA-3TAT-DRBD was obtained by PCR and inserted between Sma I and Xho I of pET-44b, downstream of a soluble Nus·Tag and a thrombin cleavage site, and upstream of a His·Tag. Recombinant plasmid pET44b-STD was transformed to E. coli strain Rosetta-gami and the bacteria were cultured 6 hours in Luria Broth at 25°C with 0.1 mM IPTG. The E. coli pellet was disrupted by sonication. The STD fusion protein was purified from the soluble lysate using Ni-NTA affinity chromatography. The purified product was analyzed by SDS-PAGE and Western Blotting using LI-COR Odyssey infrared fluorescence imaging system. The eluted fraction was dialysed to remove imidazole. After freeze-drying, purified STD was reconstituted into 1 mg/mL and stored at −70°C.
2.3. Complex assembling assay
siRNA and A10 were labeled with Cy7 when synthesized by RiboBio Co. Ltd. and stored at 20 μM. DRBD binds to a dsRNA backbone on 90° surface quadrants of the dsRNA helix, resulting in 4 DRBDs encompassing a single molecule siRNA, and each monomeric SA binds to one biotinylated A10.Citation25 Theoretically these will form a complex at a ratio of 4:4:1 for A10, STD and sur-siRNA. To construct the A10-STD-(sur-siRNA) complex 10 μL 5 μM sur-siRNA in water was mixed with 10 μL 20–50 μM STD in PBS with 10% glycerol on ice for 30 min, then mixed with 10 μL 20 μM biotinylated A10 in PBS (pH 7.4) for another 30 min.Citation16
To confirm the binding between STD and sur-siRNA or A10, 10 μL sur-siRNA (diluted to 1/32 μM) and 10 μL A10 (diluted to 1/4 μM) were incubated with STD fusion protein in PBS with 10% glycerol on ice for 2 hours respectively. STD fusion protein (20 μg) was 2-folds diluted up to 1/128. Then 5 x Electrophoretic Mobility Shift Assay (EMSA)/Gel-Shift binding buffer (Beyotime Biotechnology, GS002) was added to the reaction before A10 or siRNA were shifted in 2% agarose gel. Bull Serum Albumin (BSA) without DRBD was used as negative control. The result was analyzed using LI-COR Odyssey infrared fluorescence imaging system.
3. Specific targeting assay
3.1. In vitro Specificity assay
To determine whether A10 could mediate specific cell-surface binding, one PSMA+ prostate tumor cell line (LNCaP) and 3 PSMA− tumor cell lines (one derived from prostate tumor PC-3 and 2 from non-prostate tumor cells Hela and HepG2) were selected to incubate with the complex. For all cell culture based assays, complex containing 200 pmol sur-siRNA was used per 5 × 105 LNCaP cells with final siRNA concentrations between 100 to 400 nM. To confirm the conjugation was mediated between A10 and PSMA, a parallel group was set with anti-PSMA antibody to compete with the complex.
Tumor cells in culture were harvested and fixed. Cell pellets were washed with PBS and resuspended with 1 × Binding Buffer (1×BB, BD, 51-66121E). Each cell type was divided into 3 groups: Group 1: cell culture medium only; Group 2: with the complex (200 pmol sur-siRNA / 5 × 105 cells); and Group 3: with the complex in the presence of anti-PSMA antibody (0.1 μg / 1 × 106 cells) at 25°C for 30 min. Tumor cells were washed and >5,000 cell events were acquired and analyzed (FL4-H channel) on a BD FACS flow cytometer (FACS Calibur) based on mean fluorescence intensity (MFI). Raw data was analyzed by FCS Express software.
3.2. In vivo specificity assay on LNCaP xenografts in nude mice
Male athymic BALB/c nude mice, 4 to 6 week old, weight 18 to 20 g, were purchased from the Animal Experimental Center of Slaccas (Shanghai, China), and were maintained in a laminar airflow cabinet under pathogen-free conditions. All animal protocols were approved by the Animal Welfare and Ethics Committee of the Fourth Military Medical University. 200 μL cell suspension (1×107 cells/mL, in the logarithmic phase) with 50% matrigel (BD, 356234) were inoculated in the hip subcutaneously. About 28 d later the volume of the tumor exceeded 0.1 cm3.
To confirm A10 could also function in vivo, athymic mice bearing PSMA+ (LNCaP) and PSMA− (PC-3 and Hela) xenografts received the complex (containing 400 pmol Cy7 labeled sur-siRNA) through caudal vein injection. For LNCaP xenografts, free sur-siRNA and lipofectamine + sur-siRNA and A10-(sur-siRNA) chimera (22) were also used as control. Twenty-four hours later images of Cy7-fluorescence distribution were captured using a caliper IVIS Lumina II system.
4. siRNA intracellular delivery efficiency assay
4.1. Localization of complex
To determine whether the complex could efficiently deliver siRNA into cytoplasm, Confocal Laser Scanning Microscopy (CLSM) was used to observe the distribution of sur-siRNA labeled with FAM and indicated by green fluorescence. To evaluate efficiency, the complex was compared with lipofectamine and A10-siRNA chimera.
Five × 105 LNCaP cells of each group were seeded in Class Bottom Cell Culture Dishes (NEST Biotechnology, 801002) in serum-free medium. After attached to the wall, the cell monolayer was transfected with 200 pmol FAM-labeled sur-siRNA in 5 groups: Group 1: mock control; Group 2: incubated with free sur-siRNA (negative control); Group 3: transfected by lipofectamine 2000 (lipofectamine + sur-siRNA); Group 4: transfected with A10-(sur-siRNA) chimera; Group 5: transfected with A10-STD-(sur-siRNA) complex. Six hours later the cultural medium was removed and LNCaP cells were washed to remove extracellular siRNA, fixed with 4% formaldehyde and stained with DAPI. Images were captured on an Olympus FV10i confocal microscope at 240-folds magnification.
4.2. siRNA transfection efficiency assays
To further confirm the intracellular siRNA-transfection efficiency of the complex, LNCaP cells were seeded in 6-well plates and treated similarly as in 4.1. Eight hours later, >5,000 cells from each group were collected and analyzed using flow cytometry (FL1-H channel) to test the MFI, detailed as in 3.1.
5. Tumor suppression efficacy
5.1. SURVIVIN gene scilencing assay
Gene silencing effect was assessed at siRNA and protein level respectively. LNCaP cells grouped as in 4.1 were seeded in 6-well plates and were transfected with 200 pmol sur-siRNA at 70% confluency. Real-time quantitative PCR was performed using iScript One-Step RT-PCR Kit with SYBR Green (Takara) to quantify mRNA 48 hours later. β-actin was used as internal control. Primers for human SURVIVIN gene were: forward 5′-TCGCTCTCTGCTCCTCCTGTTC-3′; reverse 5′-CGCCCAATACGACCAAATCC-3′; Primers for reference β-actin gene were: forward 5′-GACAAGTACGGCCTTGGGTA-3′; reverse 5′-GTGCCGTCACGCTCTATGTA-3′; Relative amount of SURVIVIN mRNA was normalized to β-actin mRNA applying 2−△△CT algorithm. Three independent experiments were performed in triplicate and Mean±SEM values were calculated. Specificity was verified by melting curve analysis and agarose gel electrophoresis.
For western blotting, 72 hours after the transfection LNCaP cells from each group were collected and 50 μg of total protein from the supernatants of lysate were used to detect the expression of SURVIVIN protein in LI-COR Odyssey infrared fluorescence imaging system.
5.2. Cell apoptosis assay
To determine the apoptosis-inducing effect of SURVIVIN silence, LNCaP cells grouped as in 4.1 were transfected with 200 pmol sur-siRNA at 70% confluency. 72 hours later Annexin V-FITC/PI method (BD, 556547) using flow cytometry (FITC, FL1-H channel; PtdIns, FL2-H channel) was adopted to measure apoptosis of LNCaP cells. A parallel cisplatin-treated group (cisplatin 5 µg/ml, Dalian Mei Lun Biology Technology, 15663-27-1) was included to test if the apoptosis-promoting effect of the complex was enhanced in the presence of cisplatin.
5.3. Tumor growth curves
To assess the tumor suppression effect of the complex in vivo, athymic nude mice bearing LNCaP xenografts were prepared as mentioned in 3.2. After tumor volume reached 0.1 cm3, 40 athymic mice were randomly divided into 5 groups. 400 pmol sur-siRNA was injected through caudal vein every other day for a total of 7 times, delivered by different strategies as in 4.1. Day 0 marked the first day of injection. Athymic nude mice injected with PBS were included as mock control. Tumors were measured every 3 d with calipers and the tumor volume was calculated using the formula: VT = a×b2×π/6 (a, the longest dimension; b, the shortest dimension). The growth curves are plotted as mean tumor volume ± SEM. The mice were killed 3 d after the last treatment (day 21), and the tumors were excised and weighed. Statistical analysis was conducted using a one-way ANOVA in Prism (Version 5.0, GraphPad). A p ≤ 0.05 was considered to indicate a significant difference. In addition to a one-way ANOVA, 2-tailed unpaired t-tests were conducted between groups, and the p value was corrected with Bonfferoni Correction.
Results
1. Complex construction
1.1. Identification of STD fusion protein
A protein was purified using Ni-NTA affinity chromatography column from the supernatant containing soluble lysate of the transformed E. coli. As expected, this protein is approximately 70 KDa as shown on SDS-PAGE. It was identified by the specific anti-TAT antibody or anti-DRBD antibody using protein gel blotting (), indicating that this is the constructed STD fusion protein.
Figure 1. Expression of STD fusion protein and construction of the complex (a) Expression and identification of STD fusion protein. SDS–PAGE stained with Coomassie brilliant blue (left graph). Lane M: standard molecular weight markers of protein; Lane 1: ultrasound supernatant of E. coli Rosetta-gami transformed with pET-44b-STD; Lane 2: purified STD protein corresponding to Lane 1. Western Blotting analysis of purified STD protein with anti-TAT or anti-DRBD antibody (right graph). (b) EMSA analysis of interaction between siRNA or A10 and STD fusion protein. Ten μL siRNA (1/32 μM) and 10 μL A10 (1/4 μM) pre-incubated with serial 2-fold diluted STD protein. Untreated siRNA or A10 was used as mock control and Bovine Serum Albumin (BSA) as negative control. (c) Schematic diagram of assembled A10-STD-(sur-siRNA) complex.
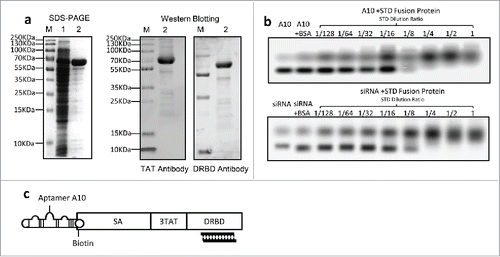
1.2. siRNA and A10 bind with STD fusion protein
Compared with free A10 and sur-siRNA, agarose gel electrophoresis showed that their migration was blocked in the presence of STD protein, and the blocking effect was STD protein concentration-dependent. As a negative control, the migration was not blocked by BSA. These results indicate that biotinylated A10 bound to SA and sur-siRNA bound to DRBD of the STD fusion protein respectively. The complete blocking of A10 and siRNA by STD was achieved at a dilution of 1/8 and 1/4 respectively (). The structure of assembled A10-STD-(sur-siRNA) complex was showed in a schematic diagram ().
2. A10 mediates specific targeting in vitro or in vivo
Unstained cells of each cell lines were used to define background signal for flow cytometry in vitro. Markers were set to cover Cy7 positive cells, allowing for a maximum coverage of unstained cells of 2%. It showed that almost all LNCaP cells (99.7%) transfected with the complex were fluorescent and the fluorescent percentage dramatically decreased to 24.3% in the presence of anti-PSMA antibody. All the other 3 PSMA− cell lines transfected with the complex did not show increased fluorescence, similar to the unstained cells. These results indicate that the therapeutic complex could only bind to PSMA+ PCa cells (LNCaP), but not PSMA− cells (PC-3, Hela and HepG2), and the specific binding could be blocked by anti-PSMA antibody competition ().
Figure 2. A10 mediates specific targeting to LNCaP cells or xenografts. (a) Flow Cytometry analysis of the specificity of A10 to PSMA+(LNCaP) and PSMA-(PC-3, Hela, HepG2) cell lines. Unstained cells are shown in purple. Percentage of fluorescent cells of each cell line was showed above the marker. The group transfected with Cy7-labeled A10-STD-(sur-siRNA) complex was showed in green line. A parallel group transfected with the complex and anti-PSMA antibody was showed in red line. (b) Caliper IVIS Lumina II system analysis of the specificity of A10 to PSMA+ xenografts in vivo. The red arrow indicates the xenografts grown in the hip subcutaneously. Aggregation of Cy7 fluorescence in the xenografts was marked with yellow arrows.
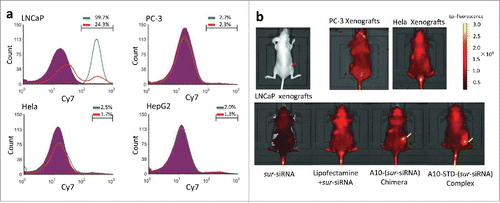
In athymic mice, Cy7 fluorescence specifically aggregated in LNCaP xenografts, but not in PSMA− xenografts PC-3 or Hela. In LNCaP xenografts the fluorescence signal only aggregated in the 2 groups containing A10, which showed that it was aptamer A10 in the complex that mediated sur-siRNA targeting to LNCaP xenografts ().
3. TAT mediates high efficient delivery of siRNA into LNCaP cells
The sur-siRNA was labeled with green fluorescence FAM. The fluorescence in the cytoplasm of LNCaP cells captured by CLSM indicates that sur-siRNA has been successfully delivered into LNCaP cells. In comparison, the fluorescence signal was similar to the mock control in the free siRNA group, which demonstrates the free siRNAs could not enter cells directly without any delivery reagents. The relative high fluorescence intensity in the therapeutic complex group demonstrates that it could efficiently delivery siRNA into LNCaP cells. Compared with the therapeutic complex, the A10-(sur-siRNA) chimera group showed relative low fluorescence signal in the cytoplasm, but instead aggregation of fluorescence was seen on the cell membrane. These indicate the chimera has poor delivery efficiency for siRNA into cytoplasm ().
Figure 3. TAT mediates efficient delivery of siRNA into LNCaP cells. (a) CLSM analysis of the location of siRNA. FAM labeled sur-siRNA was shown in green fluorescence. Phase contrast channel (gray) and the nuclei stained with DAPI (blue) was used to show the position of LNCaP cells. Channels were merged to co-locate the position of sur-siRNA in LNCaP cells. Scale bar = 20 μm. (b) Flow Cytometry analysis of transfection efficiency of sur-siRNA into LNCaP cells corresponding to (a). Unstained cells are shown in purple. The complex group was showed with black lines. Free sur-siRNA (red line) was set as negative control, while the lipofectamine (blue line) and chimera (green line) groups were set as positive control. Marker 1 was used to cover FAM positive cells by defining unstained cells as negative, allowing for a maximum coverage of unstained cells of 2%. The percentage of fluorescence positive cells covered by Marker 1 was showed on the right of (b).
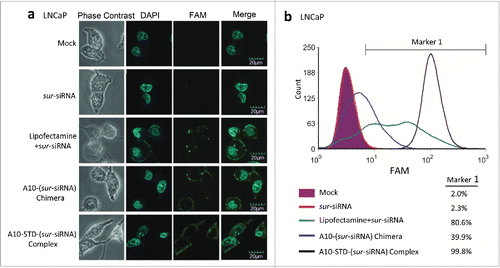
To confirm delivery efficiency, fluorescent cells percentage was further analyzed using flow cytometry. Compared with lipofectamine group (80.6%) and chimera group (39.9%), the complex group demonstrates better siRNA delivery efficiency with higher fluorescent cells percentage (99.8%). It also showed higher cells fluorescence intensity with right-shift fluorescence peak channel ().
4. Tumor suppression efficacy
4.1. The therapeutic complex silences SURVIVIN gene expression and promotes apoptosis of LNCaP cells
Comparing with the mock control, the down-regulation effect of SURVIVIN gene was showed by reduced expression of SURVIVIN mRNA and protein (). The complex group demonstrated a stronger silencing effect than lipofectamine and chimera group (P<0.01).
Figure 4. Down regulation of SURVIVIN gene and its effect on apoptosis of LNCaP cells (a) Expression analysis of SURVIVIN gene. Real-time quantitative PCR (left graph) was used to detect the expression of SURVIVIN mRNA. Relative amount of SURVIVIN mRNA was normalized to β-actin mRNA applying 2−△△CT algorithm. Relative expression of untreated LNCaP cells was normalized as 100%. Data represent Mean +/− SEM of at least 3 separate experiments. Comparison was between the complex group with other groups. ***: P < 0.0001; **: P < 0.001; *: P < 0.01. Western blotting (right graph) was used to detect the expression of SURVIVIN protein corresponding to mRNA. (b) Flow cytometry analysis of apoptosis induced by down-regulation of SURVIVIN gene using Annexin V assay. The histogram showed the sur-siRNA transfected group with blank bars, while the parallel group with sur-siRNA and cisplatin was showed with slash bars. The percentage of total apoptosis (early and late) of LNCaP cells was showed above the bars.
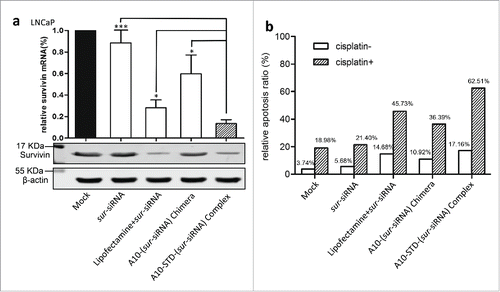
In vitro, therapeutic effect on SURVIVIN expression was showed by increased apoptosis ratio of LNCaP cells. The therapeutic complex promoted apoptosis of LNCaP cells more effectively than the other 2 groups lipofectamine and chimera, especially in the presence of cisplatin, by 16.8% and 26.5% respectively ().
4.2. Four.2 The therapeutic complex restricts LNCaP tumor growth in vivo
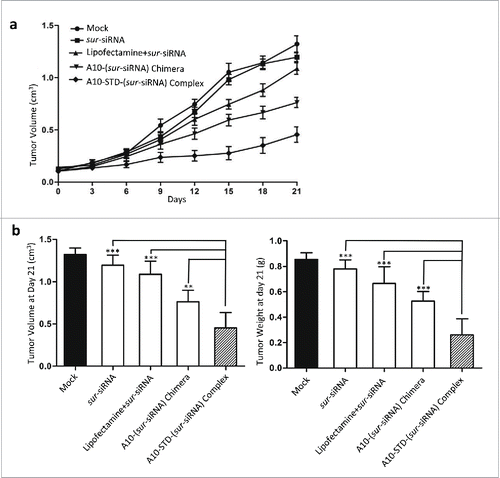
Upon systemic administration of this therapeutic complex in vivo, growth of LNCaP xenografts in athymic mice was suppressed throughout the experiment period. By day 21 the mean tumor volume increased only 3.8 folds, much lower than the untreated tumors (12.4 folds) (). Accidental death happened in 2 mice in the complex group on day 13 or day 15. At the last day (day21), the therapeutic complex showed better tumor growth suppression effect both in tumor volume and tumor weight (), compared with lipofectamine and chimera groups (P<0.001), demonstrating superior therapeutic efficacy. Tumor growth was also significantly suppressed in the A10-(sur-siRNA) chimera group compared to the lipofectamine group (P < 0.05).
Figure 5. Tumor growth suppression effect of the complex. (a) Tumor Growth Curve analysis of the suppression effect of the therapeutic complex. Tumor growth curve was drawn by tumor volume of mice in each group at 8 time points (every 3 day after the first injection). Data represent Mean +/− SEM of at least 3 separate measurements. The diagram of each group was showed above the curves. (b) On day 21 mice were killed and LNCaP tumors were incised. Mean ± SEM of tumor volume (left graph) and weight (right graph) were measured and showed in histogram. Comparison was between the complex group and negative control group (free sur-siRNA) or the other 2 positive control groups (lipofectamine and chimera). ***: P < 0.0001; **: P < 0.001; *: P < 0.01. The complex group n = 6, other groups n = 8.
Discussion
Ever since the discovery of siRNA and realization of its potential of as a new therapeutic strategy for many diseases, e.g., various malignancies, studies have been mainly carried out along 2 lines. One is focused on understanding of the mechanisms of siRNA in order to improve its design as a potent therapeutic agent for particular disease and the other is to address how to deliver well-designed drug candidate siRNA into target cells or tissues specifically as well as efficiently, and this is particularly pertinent in cancer therapy. In most studies up to now, efforts have been made separately to improve either specific targeting or efficiency of intracellular delivery, reviewed by Lares MR.Citation26
Antibody-directed delivery was first experimented to guide anti-cancer drugs and used for specific delivery of siRNA as well.Citation27,28 In recent years, nucleic acid aptamers have showed new promise as specific ligands for diagnostic or therapeutic use.Citation29 Aptamers have the advantages of being highly specific, relatively small in size, and easily synthesized, which could be regarded as a chemical equivalent of antibodies.Citation30,31 In this study, we adopted a truncated aptamer A10-3.2 with 39 nucleotides for its simplicity of chemical synthesis and enhanced specificity. It is about 55% of the full A10 in size. Citation32 Both A10-(sur-siRNA) chimera and A10-STD-(sur-siRNA) complex showed specificity to target PSMA+ LNCaP cells or xenografts, and better tumor suppression effect than the free sur-siRNA group in athymic mice. These confirm the functionality of the truncated A10 in this therapeutic complex. TAT was introduced to improve cell uptake of siRNA, and the therapeutic complex showed higher delivery efficiency by nearly 60% than A10-(sur-siRNA) chimera, and better tumor growth suppression effect (P < 0.001) in athymic mice, which indicated successful efficiency-improvement strategy of the complex.
To improve delivery efficiency, consideration should include how to overcome endosomal entrapment and make therapeutic siRNA access to the cytoplasm when designing siRNA delivery strategies. Despite various approaches have been explored, such as osmotic methods, lipid conjugation and virus vectors, most of them are limited for research and perform the best on adherent tumor cells. CPPs have demonstrated strong transfection ability by delivery siRNA into some acknowledged hard-transfected primary cells like T cells, HUVECs and hESCs Citation16 and even the blood-brain barrier, which indicates its potential use in vivo.Citation33,34 In this study, we used 3TAT-DRBD to bind sur-siRNA and delivered it to the cytoplasm. DRBD is a highly conserved α−β−β−β−α domain in numerous proteins, responsible for almost all protein-RNA duplex interactions with a high affinity (KD˜10−9M). Citation35-37 Compared with commonly used transfection reagent lipofectamine 2000, the therapeutic complex demonstrated similar delivery efficiency and SURVIVIN downregulation effect in vitro. However, without specific targeting strategy, the lipofectamine group showed significantly poorer tumor growth suppression effect than the 2 A10-containing groups in athymic mice, even though it showed higher delivery efficiency than the chimera group in vitro. These support the importance of specific delivery of therapeutic siRNA for systematic administration.
To achieve improvement of both specificity (A10) and efficiency (TAT-DRBD) in the same complex, STREPTAVIDIN, which was expressed together with TAT-DRBD, was used to bind biotinylated A10 for their ultrahigh affinity (KD∼10−15M).Citation38 A truncated but still functional natural core SA (126 amino acids) Citation39 was employed with a D128A mutation to avoid tetramer and produce monomeric SA.Citation40 The tight interaction between A10 or sur-siRNA and STD protein allows for a steady and easy construction of the therapeutic complex by a simple 2-steps incubation, exempted from the high demand of complicated formation techniques required for nanoparticles or polymer carriers of siRNA.
Even though some strategies have been adopted to decrease the molecular weight of STD protein, such as use of truncated A10 an SA, the STD protein (approximately 70 KDa) is considered still big enough to possess significant immunogenicity. To reduce the size of this therapeutic complex further, work needs to be done by getting rid of Nus·Tag through thrombin cleavage or changing to better expression system. Further characterization of the therapeutic complex, such as pharmacokinetics, biodistribution and side effects also needs to be carried out in future.
Currently, the complex is designed to target PSMA+ PCa relying on the specificity of A10. Because the aptamer A10 and siRNA bind with STD protein in a sequence-independence fashion in this complex, this strategy could have broader applications for other cancers treatment, depending on different specificity of aptamers targeting particular biomarkers and more therapeutic siRNAs.
In summary, in this proof-of-concept study, we achieved the construction of a therapeutic complex to improve both specificity and efficiency of siRNA delivery using a combinatorial approach. This therapeutic complex specifically and efficiently delivered SURVIVIN siRNA to target cells, promoted apoptosis and suppressed tumor growth in vivo. This approach could be used as a new strategy in prostate cancer therapy and shows a potential for broader applications.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by National Natural Science Foundation of China (NSFC) grant 81172446 to XK Hao, 30973463 to JY Liu.
References
- Dong H, Ji X, Wang Y, Meng L, Chen D, Feng W. Survivin expression and serum levels in pancreatic cancer. World J Surg Oncol 2015; 13:189; PMID:26016480; http://dx.doi.org/10.1186/s12957-015-0605-7
- Vanderstraeten A, Everaert T, Van Bree R, Verbist G, Luyten C, Amant F, Tuyaerts S. In Vitro Validation of Survivin as Target Tumor-associated Antigen for Immunotherapy in Uterine Cancer. J Immunother 2015; 38:239-49; PMID:26049547; http://dx.doi.org/10.1097/CJI.0000000000000085
- Pluta P, Jeziorski A, Cebula-Obrzut AP, Wierzbowska A, Piekarski J, Smolewski P. Expression of IAP family proteins and its clinical importance in breast cancer patients. Neoplasma 2015; 62:666-73; PMID:25997966; http://dx.doi.org/10.4149/neo_2015_080
- Danilewicz M, Stasikowska-Kanicka O, Wągrowska-Danilewicz M. Augmented immune expression of survivin correlates with parameters of aggressiveness in prostate cancer. Pol J Pathol 2015; 66:44-8; PMID:26017879; http://dx.doi.org/10.5114/pjp.2015.51152
- Xiao M, Wang J, Lin Z, Lu Y, Li Z, White SW, Miller DD, Li W. Design, Synthesis and Structure-Activity Relationship Studies of Novel Survivin Inhibitors with Potent Anti-Proliferative Properties. PLoS One 2015; 10:e0129807; PMID:26070194; http://dx.doi.org/10.1371/journal.pone.0129807
- Shen J, Liu J, Long Y, Miao Y, Su M, Zhang Q, Han H, Hao X. Knockdown of Survivin expression by siRNAs enhances chemosensitivity of prostate cancer cells and attenuates its tumorigenicity. Acta Biochim Biophys Sin (Shanghai) 2009; 41:223-30; PMID:19280061; http://dx.doi.org/10.1093/abbs/gmp005
- Scomparin A, Polyak D, Krivitsky A, Satchi-Fainaro R. Achieving successful delivery of oligonucleotides - From physico-chemical characterization to in vivo evaluation. Biotechnol Adv 2015; 33:1294-309; PMID:25916823; http://dx.doi.org/10.1016/j.biotechadv.2015.04.008
- Juliano RL, Alahari S, Yoo H, Kole R, Cho M. Antisense pharmacodynamics: critical issues in the transport and delivery of antisense oligonucleotides, Pharm Res 16:494-502; PMID:10227702; http://dx.doi.org/10.1023/A:1011958726518
- Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev 2013; 65:36-48; PMID:23036225; http://dx.doi.org/10.1016/j.addr.2012.09.037
- Baetke SC, Lammers T, Kiessling F. Applications of nanoparticles for diagnosis and therapy of cancer. Br J Radiol 2015; 88:20150207; PMID:25969868; http://dx.doi.org/10.1259/bjr.20150207
- Kopeček J. Polymer-drug conjugates: origins, progress to date and future directions. Adv Drug Deliv Rev 2013; 65:49-59; PMID:23123294; http://dx.doi.org/10.1016/j.addr.2012.10.014
- Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med 2004; 10:310-5; PMID:14770178; http://dx.doi.org/10.1038/nm996
- Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. Arginine-rich Peptides: An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem 2001; 276:5836-5840; PMID:11084031; http://dx.doi.org/10.1074/jbc.M007540200
- Ruczynski J, Wierzbicki PM, Kogut-Wierzbicka M, Mucha P, Siedlecka-Kroplewska K, Rekowski P. Cell-penetrating peptides as a promising tool for delivery of various molecules into the cells. Folia Histochem Cytobiol 2014; 52:257-69; PMID:25530464; http://dx.doi.org/10.5603/FHC.a2014.0034
- Vivès E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem 1997; 272:16010-7; PMID:Can't; http://dx.doi.org/10.1074/jbc.272.25.16010
- Eguchi A, Meade BR, Chang YC, Fredrickson CT, Willert K, Puri N, Dowdy SF. Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein. Nat Biotechnol 2009; 27:567-71; PMID:19448630; http://dx.doi.org/10.1038/nbt.1541
- Eguchi A, Dowdy SF. Efficient siRNA delivery by novel PTD-DRBD fusion proteins. Cell Cycle 2010; 9:424-5; PMID:20090414; http://dx.doi.org/10.4161/cc.9.3.10693
- Palm-Apergi C, Eguchi A, Dowdy SF. PTD-DRBD siRNA delivery. Methods Mol Biol 2011; 683:339-47; PMID:21053141; http://dx.doi.org/10.1007/978-1-60761-919-2_24
- Ni X, Castanares M, Mukherjee A, Lupold SE. Nucleic acid aptamers: clinical applications and promising new horizons Curr Med Chem. 2011; 18:4206-14; PMID:21838685; http://dx.doi.org/10.2174/092986711797189600
- Kanwar JR, Roy K, Maremanda NG, Subramanian K, Veedu RN, Bawa R, Kanwar RK. Nucleic Acid-Based Aptamers: Applications, Development And Clinical Trials. Curr Med Chem 2015; 22:2539-57; PMID:25723512; http://dx.doi.org/10.2174/0929867322666150227144909
- Lupold SE, Hicke BJ, Lin Y, Coffey DS. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res 2002; 62:4029-33; PMID:12124337; http://dx.doi.org/10.1158/0008-5472
- McNamara JO, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, Sullenger BA, Giangrande PH. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol 2006; 24:1005-15; PMID:16823371; http://dx.doi.org/10.1038/nbt1223
- Kim E, Jung Y, Choi H, Yang J, Suh JS, Huh YM, Kim K, Haam S. Prostate cancer cell death produced by the co-delivery of Bcl-xL shRNA and doxorubicin using an aptamer-conjugated polyplex. Biomaterials 2010; 31:4592-9; PMID:20206379; http://dx.doi.org/10.1016/j.biomaterials.2010.02.030
- Dassie JP, Liu XY, Thomas GS, Whitaker RM, Thiel KW, Stockdale KR, Meyerholz DK, McCaffrey AP, McNamara JO 2nd, Giangrande PH. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Stockdale KR, Nat Biotechnol 2009; 27:839-49; PMID:19701187; http://dx.doi.org/10.1038/nbt.1560
- Ryter JM, Schultz SC. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J 1998; 17:7505-13; PMID:9857205; http://dx.doi.org/10.1093/emboj/17.24.7505
- Lares MR, Rossi JJ, Ouellet DL. RNAi and small interfering RNAs in human disease therapeutic applications. Trends Biotechnol 2010; 28:570-9; PMID:20833440; http://dx.doi.org/10.1016/j.tibtech.2010.07.009
- Lu H, Wang D, Kazane S, Javahishvili T, Tian F, Song F, Sellers A, Barnett B, Schultz PG. Site-specific antibody-polymer conjugates for siRNA delivery. J Am Chem Soc 2013; 135:13885-91; PMID:23924037; http://dx.doi.org/10.1021/ja4059525
- Cuellar TL, Barnes D, Nelson C, Tanguay J, Yu SF, Wen X, Scales SJ, Gesch J, Davis D, Siebel CW, et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Res 2015; 43:1189-203; PMID:25550431; http://dx.doi.org/10.1093/nar/gku1362
- Kruspe S, Mittelberger F, Szameit K, Hahn U. Aptamers as drug delivery vehicles. ChemMedChem 2014; 9:1998-2011; PMID:25130604; http://dx.doi.org/10.1002/cmdc.201402163
- Chen A, Yang S. Replacing antibodies with aptamers in lateral flow immunoassay. Biosens Bioelectron 2015; 71:230-242; PMID:25912679; http://dx.doi.org/10.1016/j.bios.2015.04.041
- Toh SY, Citartan M, Gopinath SC, Tang TH. Aptamers as a replacement for antibodies in enzyme-linked immunosorbent assay. Biosens Bioelectron 2014; 64C:392-403; PMID:25278480; http://dx.doi.org/10.1016/j.bios
- Wu X, Ding B, Gao J, Wang H, Fan W, Wang X, Zhang W, Wang X, Ye L, Gao S, et al. Second-generation aptamer-conjugated PSMA-targeted delivery system for prostate cancer therapy. Int J Nanomedicine 2011; 6:1747-56; PMID:21980237; http://dx.doi.org/10.2147/IJN
- Wen X, Wang K, Zhao Z, Zhang Y, Sun T, Zhang F, Wu J, Fu Y, Du Y, Song Y, et al. Brain-targeted delivery of trans-activating transcriptor-conjugated magnetic PLGA/lipid nanoparticles. PLoS One 2014; 9:e106652; PMID:25187980; http://dx.doi.org/10.1371/journal.pone.0106652
- Niu F, Yao H, Liao K, Buch S. HIV Tat 101-mediated loss of pericytes at the blood-brain barrier involves PDGF-BB. Ther Targets Neurol Dis 2015; 2:e471; PMID:26023685; http://dx.doi.org/10.14800/ttnd.471
- Patel RC, Stanton P, McMillan NM, Williams BR, Sen GC. The interferon-inducible double-stranded RNA-activated protein kinase self-associates in vitro and in vivo. Proc Natl Acad Sci USA 1995; 92:8283-7; PMID:7545299; http://dx.doi.org/10.1073/pnas.92.18.8283
- Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J 1998; 17:5458-65; PMID:9736623; http://dx.doi.org/10.1093/emboj/17.18.5458
- Tian B, Bevilacqua PC, Diegelman-Parente A, Mathews MB. The double-stranded-RNA-binding motif: interference and much more. Nat Rev Mol Cell Biol 2004; 5:1013-23; PMID:15573138; http://dx.doi.org/10.1038/nrm1528
- Sano T, Cantor CR. Expression of a cloned streptavidin gene in Escherichia coli. Proc Natl Acad Sci USA 1990; 87:142-6; PMID:2404273; http://dx.doi.org/10.1073/pnas.87.1.142
- Sano T, Pandori MW, Chen X, Smith CL, Cantor CR. Recombinant core streptavidins. A minimum-sized core streptavidin has enhanced structural stability and higher accessibility to biotinylated macromolecules. J Biol Chem 1995; 270:28204-9; PMID:7499314; http://dx.doi.org/10.1074/jbc.270.36.21354
- Qureshi MH, Yeung JC, Wu SC, Wong SL. Development and characterization of a series of soluble tetrameric and monomeric streptavidin muteins with differential biotin binding affinities. J Biol Chem 2001; 276:46422-8; PMID:11584006; http://dx.doi.org/10.1074/jbc.M107398200