
ABSTRACT
VX680 is a potent and selective inhibitor that targets the Aurora kinase family. The p38 mitogen-activated protein kinase (MAPK) regulates a large number of cellular pathways and plays an important role in the regulation of cell survival and apoptosis. This study aimed to evaluate the effect of VX680 on cervical cancer cells and investigate whether the effects on apoptosis are enhanced by the ablation of p38 MAPK activation. The results suggested that VX680 inhibited the proliferation of cervical cancer cells by causing G2/M phase arrest and endoreduplication and that the apoptotic effect was attenuated by the activation of p38 MAPK. However, the addition of BIRB796, which is an important p38 MAPK inhibitor, effectively eliminated the expression of p-p38 and hence significantly enhanced the cell death induced by VX680 in vitro. Further study demonstrated that BIRB796 cooperated with VX680 to suppress cervical cancer cell growth in a mouse xenograft model. Taken together, our results demonstrated that VX680 induced cell cycle arrest and endoreduplication in human cervical cancer cells. Combined treatment with VX680 and BIRB796 synergistically inhibited tumor growth both in vitro and in vivo. Dual blockade of Aurora kinases and p38 MAPK is therefore a promising strategy for cervical cancer treatment.
Introduction
Cervical cancer is the second most common cancer and has a high mortality rate among women in developing countries.Citation1,2 The novel therapy strategy of concurrent chemoradiotherapy (CCRT) effectively improves the prognoses of patients, especially among patients with advanced stage (IB2 or greater) cervical cancer.Citation3 Neoadjuvant chemotherapy drugs, such as cisplatin and paclitaxel, were designed to halt cell proliferation and destroy tumor cells mainly by interfering with DNA replication, affecting chromosome alignment and/or segregation and causing DNA damage, which leads to cell cycle arrest and apoptosis.Citation4
The Aurora kinase family participates in the regulation of the cell cycle in an ordered process. The over-expressions of Aurora A and B kinases have been observed in many types of human cancer, including cervical cancer, ovarian cancer, breast cancer, colorectal cancer, and gastric cancer.Citation5-8 Aurora A kinase controls centrosome maturation, mitotic spindle formation, chromosomes segregation and the process of cytokinesis in mammalian cells.Citation9-11 Aurora A kinase affects DNA repair and thus controls the radio- and chemosensitivities of cancer cells.Citation12 The inhibition of Aurora A kinase causes DNA damage and renders cancer cells more sensitive to radiation.Citation13-17 Cancer cells responds to DNA damage by initiating the DNA-damage response, which induces cell cycle delay, more prolonged growth arrests and apoptosis of the lethally damaged cells. Gamma-H2AX is involved in recruitment of DNA damage repair factors to sites of double-strand breaks (DSB). In androgen-resistant prostate cancer cells, inhibition of Aurora A leads to increased γ-H2AX and renders cells more sensitive to radiation.Citation18 Aurora B kinase is a chromosomal passenger protein that interacts with inner centromere protein (INCENP), survivin and borealin to form the chromosomal passenger complex.Citation19 Aurora B kinase coordinates with BUB1, BUB3, MAD2, MAD1 and BUBR1 to form the spindle assembly checkpoint (SAC) complex and hence regulates the G2/M transition during cell cycle progression.Citation19 Aurora B kinase phosphorylates p53 at S183, T211, and S215 to accelerate the degradation of p53 through the polyubiquitination-proteasome pathway, thus functionally suppressing the expression of p53 target genes involved in cell cycle inhibition and apoptosis.Citation7 Inhibition of Aurora B kinase contributes to the aneuploidy, loss of mitochondrial membrane potential, activation of caspase-9 followed by activation of caspase-3.Citation20,21 In a preclinical study, the inhibition of Aurora B kinase via AZD1152 was found to inhibit the growth of colon, lung and hematologic tumor xenografts in immunodeficient mice.Citation22
VX680 inhibits the activities of both the Aurora A and B kinases.Citation23 VX680 has been reported to inhibit the growths of cancer cells of various origins by causing cell cycle arrest and apoptosis. In a phase I/II study, VX680 was proven to be active in patients with refractory hematologic malignancies.Citation24 In another report, VX680 was found to inhibit the growth of hepatic carcinomas in combination with cisplatin.Citation25 However, the benefit of VX680 alone in the treatment of solid tumors in clinical trials has been somewhat limited due to its dose-limiting toxicities and 3 of 5 clinical trials have been halted for this reason.Citation26 Therefore, additional studies are required to identify a potential therapeutic partner for VX680 to enhance its inhibitory effect on tumor cells.
The p38 mitogen-activated protein kinase (MAPK) is a stress-activated protein with a primary function of extracellular signal transduction.Citation27-29 Once activated, p38 proteins translocate from the cytosol to the nucleus where they phosphorylate the serine/threonine residues of their many substrates. The relationship between p38 MAPK and cancer is complex. The constitutive activation of the p38 MAPK pathway has been observed in malignant tumors, and the down-regulation of p38 MAPK elicits anti-tumor effects.Citation30-32 On the other hand, p38 MAPK inhibits the tumor initiation induced by oncogenes that produce reactive oxygen species (ROS).Citation33 Additionally, p38 MAPK is pivotal in the apoptosis induced by chemotherapy drugs.Citation34-36 In recent studies, the activation of p38 MAPK has been suggested to be induced by Aurora B kinase inhibition.Citation37,38 However, the role of p38 MAPK in VX680-induced effects remains unclear.
In the current study, we examined the effects of VX680 on cervical cancer cells and observed p38 MAPK activation following VX680 treatment. We further used the agent BIRB796, which is a novel p38 MAPK inhibitor, to examine whether the ablation of p38 MAPK activation could facilitate the apoptosis induced by VX680. The results revealed that combined treatment with VX680 and BIRB796 synergistically inhibited cervical cancer growth both in vitro and in vivo. These data suggest that the combined inhibition of Aurora kinases and p38 MAPK may represent a remarkable strategy for pharmacological interventions in cervical cancer.
Results
VX680-induced growth inhibition in cervical cancer cells
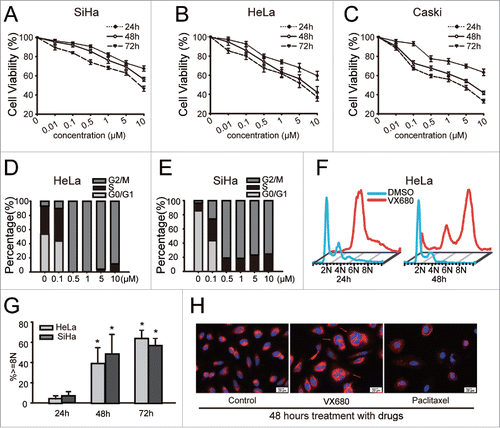
To study the effects of VX680 on cervical cancer cells, we treated SiHa, HeLa and Caski cells in 96-well plates with various concentrations of VX680 (0.1–10 μM) for as long as 72 h. VX680 was found to inhibit the growth of cervical cancer cells in a time- and dose-dependent manner (). Compared with the SiHa cells, the HeLa and Caski cells were much more sensitive to treatment with VX680. Seventy-two-hour treatments with 0.5 μM VX680 resulted in growth inhibition rates of 33.6% and 40.5% in the HeLa and Caski cells, respectively, whereas a growth inhibition rate of only 25.8% was observed in the SiHa cells. To explore the mechanisms of growth inhibition induced by VX680 in the cervical cancer cell lines, we analyzed the cell cycle profiles using flow cytometry and found that VX680 prominently induced G2/M arrest in HeLa and SiHa cells (). Additionally, endoreduplication induced by VX680 was also detected in the HeLa and SiHa cells after the longer treatment. The percentages of cells with 8N DNA content accumulations increased from ∼10% to above 50% when the time of treatment was extended from 24 h to 72 h (). As illustrated in , the nuclei changed into forms with larger volumes and a more irregular shapes following VX680 treatment compared with the control and paclitaxel groups.
Figure 1. VX680-induced growth inhibition and cell cycle arrest in cervical cancer cell lines. (A, B, C) VX680 induced cell growth inhibition in dose- and time- dependent manners in cervical cancer cell lines. Cell viability was measured using the Cell Count Kit-8 (CCK8) assay following VX680 treatment. The results are presented as the percentages of viable cells in the VX680 treatment group compared with the DMSO treatment group. The graphs illustrate the means ± the SDs of triplicate results. (D, E) Cell cycle distributions of the HeLa (D) and SiHa (E) cells after 24-h exposures to various concentrations of VX680 (0.1–10 μM). (F) A time-course study of the effects of VX680 on cell cycle progression. Polyploidy was induced after a 48-h treatment with VX680. (G) Quantification of the polyploidy induced by VX680. The cells were treated with 500 nM for 24, 48 or 72 h, and the DNA contents were measured by flow cytometry. (H) VX680 treatment caused abnormal cytokinesis. HeLa cells were treated with DMSO, VX680 (500 nM), or paclitaxel (1 μM) for 48 h and were then stained with Hoechst-33342 and the membrane probe Dil to visualize the nuclei (blue) and membranes (orange red), respectively. Abnormal cytokinesis was identified by the multinuclearity (red arrows). Scale bar, 20 μm.
Tumor cells escaped from apoptosis induced by VX680 via the activation of the p38 MAPK signal pathway
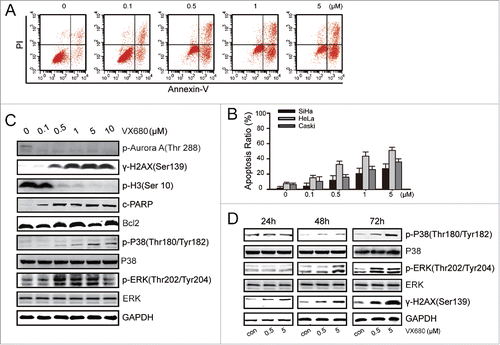
In a pre-clinical study, MLN8237 treatment markedly slowed the growth of >75% of patient-derived metastatic melanoma tumors, but the apoptotic effect of MLN8237 was not obvious.Citation39 We next tested the apoptotic effect of VX680 via flow cytometry with an Annexin V-FITC/PI kit. The fraction of Annexin V + was defined as apoptotic cells. The apoptosis rates were 32% and 18% with the treatment of 0.5 μM VX680 in the HeLa cells and SiHa cells, respectively (). To improve the treatment efficiency, we sought to identify a potential therapeutic partner for this Aurora kinase inhibitor. We treated HeLa cells with various concentrations of VX680 for 48 h and then performed western blot analyses to explore the potential molecular targets (). We found that the expression of p-Aurora A kinase was dramatically inhibited following exposure to 0.1 μM VX680 for 48 h. Moreover, the expression of γ-H2AX, which is an important marker of DNA damage, was enhanced after VX680 treatment. Additionally, the phosphorylation of histone H3 (at Ser10), which is a substrate of Aurora B kinase, was severely suppressed. These results indicated that VX680 effectively inhibited both the Aurora A and B kinases. In the HeLa cells, the expression of cleaved PARP increased after treatment, whereas the mitochondrial apoptosis associated protein Bcl2 was not affected. Interestingly, the expressions of p-p38 and p-ERK were up-regulated after VX680 treatment while the total proteins of p38 and ERK were not affected. The same changes were detected after VX680 treatments for different periods of time (). Based on all of these results, we assumed that the increased expression of p38 MAPK might be involved in the survival signal pathway by which cancer cells resist VX680 treatment.
Figure 2. p38-MAPK activation led to the evasion of apoptosis induced by VX680. (A) HeLa cells were treated with various concentrations of VX680 for 72 h, and apoptosis was then analyzed by flow cytometry. Annexin V + was identified as apoptosis. (B) Results of Annexin V assays for apoptosis detection. The graphs illustrate the means ± the SDs of triplicate results. (C) HeLa cells were treated with various concentrations of VX680 for 48 h. The expressions of related proteins were determined by immunoblotting. (D) HeLa cells were treated with various concentrations of VX680 and cultured for 24, 48, or 72 h. The expressions of p38-related proteins were determined by immunoblotting. GAPDH served as a loading control.
Inhibition of p38 by BIRB796 sensitized the cervical cancer cells to VX680-induced cytotoxicity
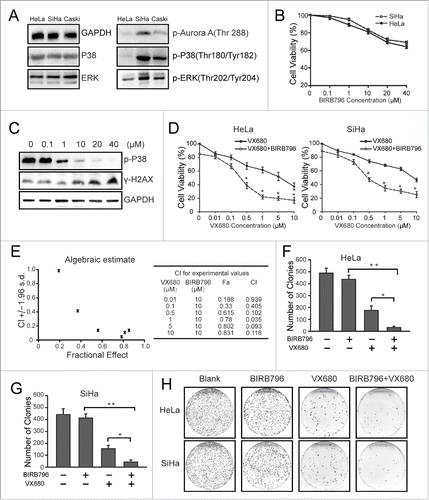
To verify our hypothesis, we first examined the baseline expressions of p-p38 and p-ERK in the 3 cell lines. As illustrated in , the SiHa cells showed the highest expression level of p-p38 and p-ERK. No obvious differences were observed in the expressions of total p38 and ERK in these cell lines. Next, we explored the effects of BIRB796 on tumor cells proliferation. CCK8 results showed moderate inhibition of proliferation following BIRB796 treatment for 72 h (). As shown in , BIRB796 effectively inhibited the phosphorylation of p38 MAPK. A sublethal dose (10 μM BIRB796) that induced less than 20% cell death was chosen for the combination treatment. The results of a CCK8 assay indicated that the additional treatment with 10 μM BIRB796 reduced the viability of the HeLa cells by 40% compared with 0.5 μM VX680 alone (). Similar results were observed in the SiHa cells (). The combination index of 0.5 μM VX680 and 10 μM BIRB796 was 0.102 (implies synergistic effect) calculated with the Calcusyn software (). To study the long-term effects of the combination of BIRB796 and VX680 on cervical cancer cells, we performed colony formation assays with SiHa and HeLa cells (). The group of cells exposed to the combined treatment exhibited reduced colony formation compared with the single agent group. These findings suggested that BIRB796 significantly enhanced the growth inhibition effects of VX680 via the inhibition of p38 MAPK.
Figure 3. The inhibition of p38 with BIRB796 sensitized the cervical cancer cells to VX680 cytotoxicity. (A) The baseline expressions of p38 and ERK in HeLa, SiHa and Caski cells. (B) SiHa and HeLa cells were treated with various concentrations of BIRB796 for 72 h and then subjected to CCK-8 assays for cell viability analyses. The results are presented as percentages of viable cells in the BIRB796 treatment group compared with the DMSO treatment group. (C) The expression of p-P38 was tested via protein gel blot after the cells were treated with BIRB796. (D) HeLa and SiHa cells were treated with VX680 alone or in combination with 10 μM BIRB796 for 72 h and then subjected to CCK-8 assays for cell viability analyses. The combined treatment with the 2 agents resulted in reduced cell viability compared with the VX680 alone treatment (* P < 0.05). (E) Chou-talalay analyses for CCK8 results of the HeLa cells () using Calcusyn software. (F, G) The numbers of colonies formed in (H) were counted. The histogram data indicate the means ± the SDs of triplicate results. * P < 0.05 for the combined treatment group versus the VX680 group, ** P < 0.01 for the combined treatment group vs. the BIRB796 group. (H) Representative images from the colony formation assay. The black-stained dots in the culture dishes represent surviving colonies.
BIRB796 enhanced the apoptosis induced by VX680 in cervical cancer cells
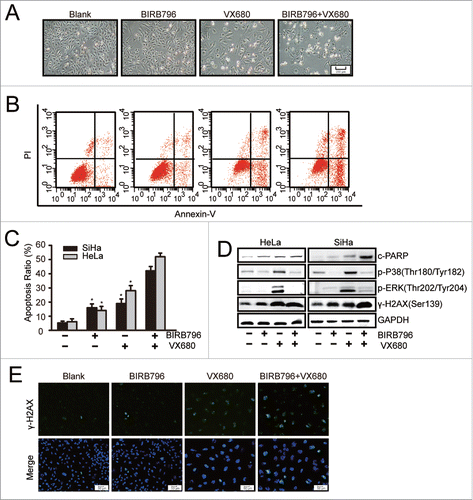
To further study the effects of combination therapy, we examined the apoptotic effect of VX680 in combination with BIRB796. As illustrated in , following exposure to 10 μM BIRB796 for 48 hours, no obvious inhibition of proliferation was observed in the HeLa cells. Regarding the VX680 group, only moderate apoptosis was induced after treatment. However, much more apparent apoptosis was observed following combined treatment with BIRB796 and VX680 for the same period of time. This finding is consistent with the results of the CCK8 assay illustrated in . Additionally, the synergistic apoptosis-inducing effect of the combination of BIRB796 and VX680 was confirmed via flow cytometry results (). In the SiHa cells, apoptosis ratio of the BIRB796 group was 16%. A 2.1 ± 0.12-fold increase (P < 0.05) in apoptosis induced by the combined treatment (42%) compared with the VX680 (19%) alone was observed. Similar results were observed in the HeLa cells. The protein gel blot results confirmed that BIRB796 enhanced the expressions of cleaved PARP and γ-H2AX than were induced by VX680 through the inhibition of p38 MAPK activation (). These results implied that the DNA damage responses were involved in apoptosis induced by combination treatment. The same results were observed based on immunofluorescence staining for γ-H2AX expression in the HeLa cells following treatment with the drugs (). The combined treatment group was associated with more γ-H2AX-positive cells than the single-agent treatment group. These findings indicated that inhibition of p-p38 by BIRB796 enhanced VX680-induced DNA damage and cell death in cervical cancer cells.
Figure 4. BIRB796 enhanced the apoptosis effect induced by VX680 in vitro. (A-C) HeLa cells were plated in 6-well plates and treated with DMSO as a control, 10 μM BIRB796, 500 nM VX680, or both drugs for 48 h. (A,B) Morphologic changes were captured using a Nikon inverted microscope (magnification: 40X), and the apoptotic effects were analyzed by flow cytometry using an Annexin V-FITC/PI kit. (C) The apoptosis results are presented as the means ± the SDs of 3 independent experiments. * P < 0.05 for the BRB796 group versus the combined group or the VX680 group vs. the combine group. (D) After combined treatment with BIRB796 and VX680, the expressions of cleaved PARP and γ-H2AX as well as components of the p38 signal pathway were tested via western blot. GAPDH served as a loading control. Representative images of 3 independent experiments are shown. (E) HeLa cells were treated as in A and then stained with anti-γ-H2AX (green) and Hoechst 33342 (chromosomes; blue). The fluorescence images were captured using a Leica microscope (magnification: 100X).
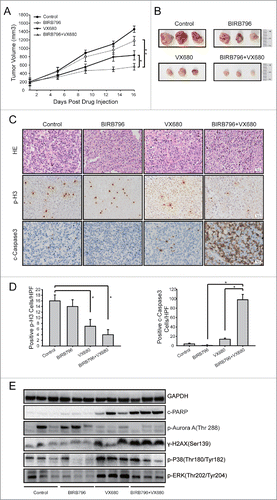
BIRB796 enhanced the apoptosis induced by VX680 in a xenograft model
The HeLa cell xenograft model was used to probe whether BIRB796 could enhance the tumor growth inhibition induced by VX680 in vivo. HeLa cells resuspended in 100 μl PBS were subcutaneously injected into the right axillary areas of nude mice. When the tumors reached volumes of 150–200 mm3, the mice were divided randomly into 4 groups (n = 6 per group) and treated either with vehicle, 25 mg/kg of VX680, 10 mg/kg of BIRB796, or a combination of VX680 and BIRB796. The mice were sacrificed 3 d after the final administration of VX680. As illustrated in , VX680 effectively inhibited tumor growth in vivo, and BIRB796 obviously enhanced this effect. In the VX680 treatment group, the mean tumor volumes were reduced by 43% (P < 0.05) compared with the control group. The combination of BIRB796 and VX680 treatment reduced tumor volume by 62% (P < 0.01) compared with the control group. HE staining indicated swelling of the nuclei and nuclear fragmentation in the combined treatment group (). The immunohistochemistry (IHC) results revealed an obvious reduction in H3 phosphorylation after VX680 treatment, indicating the inhibition of Aurora B kinase activity in vivo. Increased cleaved-caspase-3 expression in the tumor tissues of the combined treatment group compared with the control was observed (). Western blot results from the samples from the tumor tissues indicated that p-p38 and p-ERK were effectively inhibited by BIRB796 in vivo. Compared with the VX680 alone group, the expressions of DNA damage marker γ-H2AX and cleaved PARP (an indicator of enhanced apoptosis) were upregulated in the VX680 and BIRB796 combination treatment group ().
Figure 5. BIRB796 enhanced the anti-tumor effects of VX680 in vivo. Nude mice bearing HeLa tumor xenografts were administered vehicle,BIRB796, VX680, or BIRB796 combined with VX680 as described in the Materials and Methods. (A) Growth curves of the tumor xenografts in the nude mice. The data points represent the averages ± the SDs for each group. Tumor growth was significantly inhibited in the combined BIRB796 and VX680 treatment group (**P < 0.01, the combined group versus the BIRB796 group; *P < 0.05, the combined group vs. the VX680 group). (B) Photograph of tumor xenografts isolated from the nude mice. (C) Representative images of H&E staining (the upper panel) and immunohistochemical staining for p-H3 (the middle panel) and cleaved-caspase-3 in the tumor tissues from each group. Magnification: 200X (D) Left panels: p-H3-positive cells were counted under high magnification in 10 casual fields and the mean was calculated. *P < 0.05 the control group versus the VX680 group or the control group vs. the combined group. Right panels: cleaved-caspase-3-positive cells were calculated in the same manner. *P < 0.05 the BIRB796 group versus the combined group or the VX680 group vs. the combined group (E) Western blot analyses of expressions of related proteins in the samples obtained from the xenograft tumors. GAPDH was used as a loading control.
Discussion
An important hallmark of malignant tumors is uncontrolled rapid growth caused by genetic abnormities. Cancer cells benefit from compromised cell cycle check points and disrupted cell cycle kinetics and thus obtain proliferative and survival advantages compared with normal cells.Citation40,41 Chemotherapy drugs targeting the mitotic phase of cell cycle have achieved great success in improving the prognoses of cancer patients. The Aurora kinases belong to the serine/threonine family and regulate many steps in the mitotic phase, including the formation of the mitotic spindle, chromosome organization and alignment, and the exit from mitosis.Citation42-45 A large body of literature has proven that the overexpression of Aurora kinases causes the override of mitotic checkpoints in human cancers.Citation12,46-49 Given the Aurora kinases' pivotal roles in the mitotic process during cell cycle, their over-expressions in malignancies and their cross-talk with tumor suppressors and oncogenic signaling pathways, small molecules targeting the Aurora kinases have attracted intense attention in the field of tumor therapy. Citation19,50
VX680 is a potent and selective inhibitor that targets both the Aurora A and B kinases and has demonstrated significant potential as an anti-cancer agent. Regarding cervical cancer cells, VX680 induced cell cycle arrest and inhibited cell proliferation in time- and dose-dependent manners (). VX680 exhibited strong inhibition activity against Aurora A kinase at the concentration of 0.1 μM. With the treatment of VX680 at the higher concentration of 0.5 μM, the activities of Aurora A and B kinases could be blocked at the same time (). Thus inhibition of both the Aurora A and B kinases could be contributing to the effects of cell cycle arrest and DNA content accumulation () caused by VX680 in cervical cancer cells. In a recent study, dual inhibition of Aurora A and B kinases showed impressive anti-tumor activity against Triple-Negative breast cancer by causing tetraploid accumulation followed by apoptosis, senescence or surviving octaploid (8N) cells.Citation51 8N cells caused by KW-2450 did not undergo apoptosis or senescence. Elimination of the surviving octaploid (8N) cells enhanced apoptosis induced by KW-2450.Citation51 To improve apoptotic effects and avoid drug resistance, combined treatment is needed for VX680. In previous studies, combinations of VX680 with other drugs, such as paclitaxel, cis-platinum, TSA and nutlin-3, have been proven to produce synergistic effects in malignant tumors.Citation25,52-54
In the present study, we sought to identify a potential therapeutic partner for VX680 with a targeted and complimentary anti-cancer mechanism. We found that p-p38 and p-ERK were up-regulated after VX680 treatment, whereas the expression of Bcl-2 was not affected. In a previous study, Sun et al have found downregulation of p-ERK after overexpression of Aurora A kinase in the SiHa cells. They have also found a negative correlation between AURKA and p-ERK in cervical cancer tissues from clinical patients.Citation52 The up-regulation of p-ERK might be induced by Aurora A kinase inhibition via VX680. A study suggested that phosphorylation of p38 MAPK was induced by ROS following by ZM447439 (a selective inhibitor of Aurora B kinase) treatment, while no change in expression of γ-H2AX could be detected.Citation20 Inhibition of Aurora A induced DNA damage and increased the accumulation of γ-H2AX in cancer cells.Citation18 In this study, treatment of VX680 induced apoptosis and activated H2AX in a dose dependent manner. This implied that DNA damage and γ-H2AX accumulation caused by Aurora A inhibition might be involved in the apoptotic effect of VX680. Gamma-H2AX is usually used as a marker of DNA damage response. It is also a target of p-p38 in the ROS/p-p38 MAPK/γ-H2AX signal pathway. This signal pathway is usually activated by chemotherapy drugs and leads to apoptosis.
Tumor cell growth might be regulated by the coordination of cell proliferation and apoptosis signals. P38 MAPK plays dual roles in the regulation of tumor cell growth; i.e., p38 can mediate survival and cell death signals at the same time. P38 MAPK has been proven to be protective with respect to the initiation of cancer.Citation33 However, cancer cells with high tumorigenicity can bypass the apoptotic effects induced by p38 activation following ROS generation. The inability of malignant cells to respond to p38 MAPK signaling with a shift toward apoptosis could possibly be due to alterations in the regulators of apoptosis that help in cell survival.Citation55 The ultimate effect of p38 MAPK activation depends on the type of stimulus in a cell-type-specific manner.Citation56-58
To further explore the role of p38 activation in response to VX680 treatment, we treated cells with BIRB796. BIRB796 is a novel p38 MAPK inhibitor that inhibits all p38 MAPK isoforms in vitro and vivo.Citation59 It also shows weak inhibitory activity to c-Raf, IKK2 and ERK2, et al.Citation59 BIRB796 reverses ATP-binding-cassette family (ABCB1)-mediated multidrug resistance in malignant cells.Citation60 The addition of BIRB796 eliminated p38 and ERK activation induced by VX680. Specifically, apoptosis was apparently enhanced in cervical cancer cells accompanied with the upregulation of γ-H2AX after combined treatment. Based on our results, we assumed that the activation of p38 MAPK might be involved in the survival-signal pathway by which cancer cells resist VX680 treatment. The significance of p38 activation following VX680 treatment differs from that in the ROS/p-p38 MAPK/γ-H2AX signal pathway. And further study is still needed to explore the modulator of p38 activation.
In conclusion, our studies demonstrated that BIRB796 synergized with VX680, which resulted in the inhibition of the growth of cervical cancer cells and the activation of death pathways. These findings may aid the design of novel targeted therapeutic regimens to achieve effective cancer treatments and help to overcome Aurora kinase inhibitor resistance that is likely to develop during the course of long-term therapy.
Materials and methods
Cell lines and culture
HeLa, Caski and SiHa cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in DMEM (Gibco) containing 10% fetal bovine serum (Gibco), 100 units/ml penicillin, and 100 mg/ml streptomycin. Cells were maintained at 37°C in a humidified atmosphere with 5% CO2.
CCK8 assay for cell growth inhibition
Cells (4 × 103 in 200 μl culture media per well) were seeded in 96-well plates and maintained for 24 h. Then culture medium was changed for fresh medium containing drugs. Cells were maintained at 37°C in a humidified atmosphere with 5% CO2 for another 24, 48 or 72 h. The cell counting kit-8 (CCK-8) (Dojindo, Mashikimachi, Japan) assay was performed following the manufacturer's instructions. OD values of each sample were measured at 450 nm and 620 nm (as the reference wavelength) by automatic enzyme-linked immunosorbent assay readers.
Cell cycle and apoptosis analysis
For cell cycle analyses, cells were harvested and fixed in ice-cold 70% ethanol overnight at −20°C. Samples were then washed with phosphate-buffered saline (PBS) and stained with buffer containing propidiumiodide (PtdIns: 50 μg/mL) and RNaseA (50 μg/mL) for 30 minutes in the dark at room temperature. The PI fluorescence of individual nuclei was measured with flow cytometers (BD FACS Calibur, Becton Dickinson, USA). The cell cycle profiles were analyzed with Cell Quest Pro V 3.2.1 software. For apoptosis detection, cells were assayed with an Annexin V-FITC/PtdIns apoptosis kit (MultiSciences Biotech Co., Ltd. Nanjing, China) and analyzed by flow cytometry.
Colony formation assay
Cells were tripsinized and planted in a 6-cm dish at a density of 1000 cells per dish. After 24 h, cultured supernatants were changed for fresh completed culture medium containing 0.5μM VX680, 10 μM BIRB796, or both. Culture media were then changed every 3 d. After a treatment of 14 d with drugs, colonies were fixed with 4% paraformaldehyde, stained with 0.5% crystal violet and counted.
Immuofluorescence staining
Cells were implanted on glass chamber slides and allowed for 16–24 h attachment. After treatment of VX680 for 48 h, they were fixed with 4% paraformaldehyde and permeablized with 0.2% Triton X-100. After being blocked with 5% BSA at room temperature for 15 minutes, the cells were incubated with primary antibody at 4°C overnight. Next day, the cells were washed by PBS and incubated with FITC-conjugated secondary antibody at 37°C for 1 h. Cells were then washed and incubated with Hoechst 33342 at a dilution of 1:2000 (Beyontime Biotechnilogy, Nantong, China) for nucleus staining. Images were captured with a fluorescent microscope (Nikon ECLIPSE Ti-S, Japan).
Western blot analysis
Cells were harvested and lysed on ice in RIPA (Beyotime, Nantong) buffer containing protease inhibitor cocktail. For xenograft models, the tissues near the surface of tumor were chosen for western blot analyses. Tumor tissues were preserved at −80°C and thawed on ice. Then they were grinded in glass homogenizer in RIPA buffer. After ultrasound pyrolysis processing, samples were centrifuged at 12000 rpm for 20 minutes. The supernatants were kept for next step. Cell lysates (50 μg) were resolved on an SDS polyacrylamide gel by electrophoresis and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, MA). After being blocked in 5% non-fat milk in TBS buffer for half an hour, the membranes were incubated with primary antibody overnight at 4°C. On the next day, they were incubated with the appropriate secondary antibodies for 1 h at room temperature and washed twice before immunoblotting analysis was performed with the Enhanced Chemiluminescence (ECL) Western Blotting Detection reagents (Pierce, USA).
Chemicals and reagents
VX680 and BIRB796 were purchased from Selleck Chemicals. A 10 mM stock solution of VX680, the same as BIRB796, was dissolved in dimethyl sulfoxide (DMSO) (Sigma–Aldrich, St. Louis, MO, USA), and stored at −80°C, and diluted in fresh medium before use. Antibodies specific for phosphor-Aurora A kinase (Thr 288) (#2914), γ-H2AX (Ser139) (#9718), phospho-p38 MAPK (Thr180/Tyr182) (#4511), phospho-ERK (Thr202/Tyr204) (#4370), cleaved-caspase-3 (Asp175) (#9661) and cleaved poly (ADP-ribose) polymerase (PARP1; 89 KD) (#5625) were purchased from Cell Signaling Technology. The primary antibody against p-H3 (Ser10) was purchased from abcam (ab5176). ERK1/2 (mab1576) was purchased from R&D. GAPDH (10494-1-AP), MAPK14 (14064-1-AP) and Bcl-2 (12789-1-AP) were purchased from Proteintech.
Antitumor study of human tumor xenografts in nude mice
Nude mice aged 4–5 weeks were purchased from Huafukang (Beijing, China). Animals were maintained and treated pursuant to the Hubei Province animal experiment regulations. 4 × 106 HeLa cells resuspended in 100 μl PBS were subcutaneously injected to the right axillary areas of nude mice. After tumors reached 150–200 mm3, the mice were divided into 4 groups. Each group (n = 6) received either PBS, VX680, BIRB796, or combined of the 2 agents for treatment. For VX680 treatment, drugs (25 mg/kg) were administered intraperitoneally (i.p.) twice a day for 14 d.Citation11 BIRB796 (10 mg/kg) was administrated orally the next day of VX680 treatment for the first time, and then it was applied every 3 d.Citation61 The tumor sizes were estimated with a caliper. A standard formula (width2 ×length × 0.52) was used for calculating the volumes of tumors. The curves of tumor growth were drawn according to tumor volume and time of drugs administration. Mice were sacrificed and executed to death 3 d after the final treatment of VX680. Tumors were surgically removed and tumor tissues were fixed in formalin, embedded in paraffin, sectioned and stained with hematoxylin and eosin (HE) for histological analysis. For immunohistochemical inspection, tissue sections were stained with phosphorylated histone H3 and cleaved-caspase-3 according to the manufactures' protocol. Sections observation and images collection were performed using a Zeiss KS400 image analysis system (Jena, Germany). Ten randomized fields of each section were selected for quantitative analysis.
Statistical analysis
All major dependent variables in this study were continuous measurements, and thus they were expressed as means ± standard deviation (SD). Statistical analyses were performed with SPSS software (version 9.1.3, SAS Institute Inc., Cary, NC, USA). Student's t-tests were used to compare groups. *P < 0.05 was considered statistically significant, **P < 0.01.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This study is supported by a grant from National Natural Science Foundation of China (no. 81302264).
References
- Osman M. The role of neoadjuvant chemotherapy in the management of locally advanced cervix cancer: a systematic review. Oncol Rev 2014; 8:250; PMID:25992238; http://dx.doi.org/10.4081/oncol.2014.250
- Denny L. Cervical cancer: prevention and treatment. Discov Med 2012; 14:125-31; PMID:22935209
- Kokka F, Bryant A, Brockbank E, Powell M, Oram D. Hysterectomy with radiotherapy or chemotherapy or both for women with locally advanced cervical cancer. Cochrane Database Syst Rev 2015; 4:CD010260; PMID:25847525
- Taylor SS, Scott MI, Holland AJ. The spindle checkpoint: a quality control mechanism which ensures accurate chromosome segregation. Chromosome Res 2004; 12:599-616; PMID:15289666; http://dx.doi.org/10.1023/B:CHRO.0000036610.78380.51
- Lassus H, Staff S, Leminen A, Isola J, Butzow R. Aurora-A overexpression and aneuploidy predict poor outcome in serous ovarian carcinoma. Gynecol Oncol 2011; 120:11-7; PMID:20937525; http://dx.doi.org/10.1016/j.ygyno.2010.09.003
- Wang J, Yang S, Zhang H, Song Y, Zhang X, Qian H, Han X, Shi Y. Aurora-A as an independent molecular prognostic marker in gastric cancer. Oncol Rep 2011; 26:23-32; PMID:21479365; http://dx.doi.org/10.3892/or.2011.1250
- Gully CP, Velazquez-Torres G, Shin JH, Fuentes-Mattei E, Wang E, Carlock C, Chen J, Rothenberg D, Adams HP, Choi HH, et al. Aurora B kinase phosphorylates and instigates degradation of p53. Proc Natl Acad Sci U S A 2012; 109:E1513-22; PMID:22611192; http://dx.doi.org/10.1073/pnas.1110287109
- Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J 1998; 17:3052-65; PMID:9606188; http://dx.doi.org/10.1093/emboj/17.11.3052
- Marumoto T, Honda S, Hara T, Nitta M, Hirota T, Kohmura E, Saya H. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem 2003; 278:51786-95; PMID:14523000; http://dx.doi.org/10.1074/jbc.M306275200
- Zhou S-F, Li J-P, Yang Y-X, Liu Q-L, Pan S, He Z, Zhang X, Yang T, Chen X-W, Wang D, et al. The investigational Aurora kinase A inhibitor alisertib (MLN8237) induces cell cycle G2/M arrest, apoptosis, and autophagy via p38 MAPK and Akt/mTOR signaling pathways in human breast cancer cells. Drug Des Devel Ther 2015:1627; PMID:25834401; http://dx.doi.org/10.2147/DDDT.S75378
- Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med 2004; 10:262-7; PMID:14981513; http://dx.doi.org/10.1038/nm1003
- Katayama H, Wang J, Treekitkarnmongkol W, Kawai H, Sasai K, Zhang H, Wang H, Adams HP, Jiang S, Chakraborty SN, et al. Aurora kinase-A inactivates DNA damage-induced apoptosis and spindle assembly checkpoint response functions of p73. Cancer Cell 2012; 21:196-211; PMID:22340593; http://dx.doi.org/10.1016/j.ccr.2011.12.025
- Oh ET, Byun MS, Lee H, Park MT, Jue DM, Lee CW, Lim BU, Park HJ. Aurora-a contributes to radioresistance by increasing NF-kappaB DNA binding. Radiat Res 2010; 174:265-73; PMID:20726719; http://dx.doi.org/10.1667/RR2017.1
- Wu CC, Yu CT, Chang GC, Lai JM, Hsu SL. Aurora-A promotes gefitinib resistance via a NF-kappaB signaling pathway in p53 knockdown lung cancer cells. Biochem Biophys Res Commun 2011; 405:168-72; PMID:21216229; http://dx.doi.org/10.1016/j.bbrc.2011.01.001
- Opyrchal M, Salisbury JL, Zhang S, McCubrey J, Hawse J, Goetz MP, Lomberk GA, Haddad T, Degnim A, Lange C, et al. Aurora-A mitotic kinase induces endocrine resistance through down-regulation of ERalpha expression in initially ERalpha+ breast cancer cells. PloS One 2014; 9:e96995; PMID:24816249; http://dx.doi.org/10.1371/journal.pone.0096995
- Xu J, Yue CF, Zhou WH, Qian YM, Zhang Y, Wang SW, Liu AW, Liu Q. Aurora-A contributes to cisplatin resistance and lymphatic metastasis in non-small cell lung cancer and predicts poor prognosis. J Transl Med 2014; 12:200; PMID:25082261; http://dx.doi.org/10.1186/1479-5876-12-200
- Wang Y, Sun H, Wang Z, Liu M, Qi Z, Meng J, Sun J, Yang G. Aurora-A: a potential DNA repair modulator. Tumor Biol 2013; 35:2831-6; PMID:24277377; http://dx.doi.org/10.1007/s13277-013-1393-8
- Moretti L, Niermann K, Schleicher S, Giacalone NJ, Varki V, Kim KW, Kopsombut P, Jung DK, Lu B. MLN8054, a small molecule inhibitor of aurora kinase a, sensitizes androgen-resistant prostate cancer to radiation. Int J Radiat Oncol Biol Phys 2011; 80:1189-97; PMID:21514073; http://dx.doi.org/10.1016/j.ijrobp.2011.01.060
- Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer 2010; 10:825-41; PMID:21102634; http://dx.doi.org/10.1038/nrc2964
- Kumari G, Ulrich T, Krause M, Finkernagel F, Gaubatz S. Induction of p21CIP1 protein and cell cycle arrest after inhibition of Aurora B kinase is attributed to aneuploidy and reactive oxygen species. J Biol Chem 2014; 289:16072-84; PMID:24782314; http://dx.doi.org/10.1074/jbc.M114.555060
- Mori N, Ishikawa C, Senba M, Kimura M, Okano Y. Effects of AZD1152, a selective Aurora B kinase inhibitor, on Burkitt's and Hodgkin's lymphomas. Biochem Pharmacol 2011; 81:1106-15; PMID:21371446; http://dx.doi.org/10.1016/j.bcp.2011.02.010
- Wilkinson RW, Odedra R, Heaton SP, Wedge SR, Keen NJ, Crafter C, Foster JR, Brady MC, Bigley A, Brown E, et al. AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin Cancer Res 2007; 13:3682-8; PMID:17575233; http://dx.doi.org/10.1158/1078-0432.CCR-06-2979
- Tyler RK, Shpiro N, Marquez R, Eyers PA. VX-680 inhibits Aurora A and Aurora B kinase activity in human cells. Cell Cycle 2007; 6:2846-54; PMID:18032922; http://dx.doi.org/10.4161/cc.6.22.4940
- Giles FJ, Swords RT, Nagler A, Hochhaus A, Ottmann OG, Rizzieri DA, Talpaz M, Clark J, Watson P, Xiao A, et al. MK-0457, an Aurora kinase and BCR-ABL inhibitor, is active in patients with BCR-ABL T315I leukemia. Leukemia 2013; 27:113-7; PMID:22772060; http://dx.doi.org/10.1038/leu.2012.186
- Yao R, Zheng J, Zheng W, Gong Y, Liu W, Xing R. VX680 suppresses the growth of HepG2 cells and enhances the chemosensitivity to cisplatin. Oncol Lett 2014; 7:121-4; PMID:24348832; http://dx.doi.org/10.3892/ol.2013.1648
- Cheung CH, Sarvagalla S, Lee JY, Huang YC, Coumar MS. Aurora kinase inhibitor patents and agents in clinical testing: an update (2011 – 2013). Expert opin Ther Pat 2014; 24:1021-38; PMID:24965505; http://dx.doi.org/10.1517/13543776.2014.931374
- Liu H, He J, Yang J. Tumor cell p38 MAPK: A trigger of cancer bone osteolysis. Cancer Cell Microenviron 2015; 2; PMID:26029733
- Kim HG, Shi C, Bode AM, Dong Z. p38alpha MAPK is required for arsenic-induced cell transformation. Mol carcinog 2015; 55:910-7 PMID:25969347; http://dx.doi.org/10.1002/mc.22331
- Plotnikov A, Zehorai E, Procaccia S, Seger R. The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim Biophys Acta 2011; 1813:1619-33; PMID:21167873; http://dx.doi.org/10.1016/j.bbamcr.2010.12.012
- Furukawa T. Impacts of activation of the mitogen-activated protein kinase pathway in pancreatic cancer. Front Oncol 2015; 5:23; PMID:25699241; http://dx.doi.org/10.3389/fonc.2015.00023
- Fu Y, O'Connor LM, Shepherd TG, Nachtigal MW. The p38 MAPK inhibitor, PD169316, inhibits transforming growth factor beta-induced Smad signaling in human ovarian cancer cells. Biochem Biophys Res Commun 2003; 310:391-7; PMID:14521923; http://dx.doi.org/10.1016/j.bbrc.2003.09.021
- Tsuchiya T, Tsuno NH, Asakage M, Yamada J, Yoneyama S, Okaji Y, Sasaki S, Kitayama J, Osada T, Takahashi K, et al. Apoptosis induction by p38 MAPK inhibitor in human colon cancer cells. Hepato-gastroenterology 2008; 55:930-5; PMID:18705300
- Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell 2007; 11:191-205; PMID:17292829; http://dx.doi.org/10.1016/j.ccr.2006.12.013
- Ravindran J, Gupta N, Agrawal M, Bala Bhaskar AS, Lakshmana Rao PV. Modulation of ROS/MAPK signaling pathways by okadaic acid leads to cell death via, mitochondrial mediated caspase-dependent mechanism. Apoptosis 2011; 16:145-61; PMID:21082355; http://dx.doi.org/10.1007/s10495-010-0554-0
- Liu WH, Cheng YC, Chang LS. ROS-mediated p38alpha MAPK activation and ERK inactivation responsible for upregulation of Fas and FasL and autocrine Fas-mediated cell death in Taiwan cobra phospholipase A(2)-treated U937 cells. J Cell Physiol 2009; 219:642-51; PMID:19180563; http://dx.doi.org/10.1002/jcp.21713
- Bu HQ, Cai K, Shen F, Bao XD, Xu Y, Yu F, Pan HQ, Chen CH, Du ZJ, Cui JH. Induction of apoptosis by capsaicin in hepatocellular cancer cell line SMMC-7721 is mediated through ROS generation and activation of JNK and p38 MAPK pathways. Neoplasma 2015; 62:582-91; PMID:25997958; http://dx.doi.org/10.4149/neo_2015_070
- Kumari G, Ulrich T, Krause M, Finkernagel F, Gaubatz S. Induction of p21CIP1 Protein and Cell Cycle Arrest after Inhibition of Aurora B Kinase Is Attributed to Aneuploidy and Reactive Oxygen Species. J Biol Chem 2014; 289:16072-84; PMID:24782314; http://dx.doi.org/10.1074/jbc.M114.555060
- Kumari G, Ulrich T, Gaubatz S. A role for p38 in transcriptional elongation of p21 (CIP1) in response to Aurora B inhibition. Cell Cycle 2013; 12:2051-60; PMID:23759594; http://dx.doi.org/10.4161/cc.25100
- Vilgelm AE, Pawlikowski JS, Liu Y, Hawkins OE, Davis TA, Smith J, Weller KP, Horton LW, McClain CM, Ayers GD, et al. Mdm2 and aurora kinase a inhibitors synergize to block melanoma growth by driving apoptosis and immune clearance of tumor cells. Cancer Res 2015; 75:181-93; PMID:25398437; http://dx.doi.org/10.1158/0008-5472.CAN-14-2405
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Kelly KR, Ecsedy J, Mahalingam D, Nawrocki ST, Padmanabhan S, Giles FJ, Carew JS. Targeting aurora kinases in cancer treatment. Curr Drug Targets 2011; 12:2067-78; PMID:21777198; http://dx.doi.org/10.2174/138945011798829410
- Silva VC, Cassimeris L. Stathmin and microtubules regulate mitotic entry in HeLa cells by controlling activation of both Aurora kinase A and Plk1. Mol Biol Cell 2013; 24:3819-31; PMID:24152729; http://dx.doi.org/10.1091/mbc.E13-02-0108
- Zhou N, Singh K, Mir MC, Parker Y, Lindner D, Dreicer R, Ecsedy JA, Zhang Z, Teh BT, Almasan A, et al. The investigational Aurora kinase A inhibitor MLN8237 induces defects in cell viability and cell-cycle progression in malignant bladder cancer cells in vitro and in vivo. Clin Cancer Res 2013; 19:1717-28; PMID:23403633; http://dx.doi.org/10.1158/1078-0432.CCR-12-2383
- Mannino M, Gomez-Roman N, Hochegger H, Chalmers AJ. Differential sensitivity of Glioma stem cells to Aurora kinase A inhibitors: implications for stem cell mitosis and centrosome dynamics. Stem Cell Res 2014; 13:135-43; PMID:24879067; http://dx.doi.org/10.1016/j.scr.2014.05.001
- Marxer M, Ma HT, Man WY, Poon RY. p53 deficiency enhances mitotic arrest and slippage induced by pharmacological inhibition of Aurora kinases. Oncogene 2014; 33:3550-60; PMID:23955083; http://dx.doi.org/10.1038/onc.2013.325
- Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, Gerlich DW. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 2009; 136:473-84; PMID:19203582; http://dx.doi.org/10.1016/j.cell.2008.12.020
- Yang G, Chang B, Yang F, Guo X, Cai KQ, Xiao XS, Wang H, Sen S, Hung MC, Mills GB, et al. Aurora kinase A promotes ovarian tumorigenesis through dysregulation of the cell cycle and suppression of BRCA2. Clin Cancer Res 2010; 16:3171-81; PMID:20423983; http://dx.doi.org/10.1158/1078-0432.CCR-09-3171
- Portella G, Passaro C, Chieffi P. Aurora B: a new prognostic marker and therapeutic target in cancer. Curr Med Chem 2011; 18:482-96; PMID:21143115; http://dx.doi.org/10.2174/092986711794480203
- Fu S, Hu W, Kavanagh JJ, Bast RC, Jr. Targeting Aurora kinases in ovarian cancer. Exp Opin Ther Targets 2006; 10:77-85; PMID:16441230; http://dx.doi.org/10.1517/14728222.10.1.77
- Green MR, Woolery JE, Mahadevan D. Update on Aurora Kinase Targeted Therapeutics in Oncology. Exp Opin Drug discov 2011; 6:291-307; PMID:21556291; http://dx.doi.org/10.1517/17460441.2011.555395
- Kai K, Kondo K, Wang X, Xie X, Pitner MK, Reyes ME, Torres-Adorno AM, Masuda H, Hortobagyi GN, Bartholomeusz C, et al. Antitumor Activity of KW-2450 against Triple-Negative Breast Cancer by Inhibiting Aurora A and B Kinases. Mol Cancer Ther 2015; 14:2687-99; PMID:26443806; http://dx.doi.org/10.1158/1535-7163.MCT-15-0096
- Sun JM, Yang LN, Xu H, Chang B, Wang HY, Yang G. Inhibition of Aurora A promotes chemosensitivity via inducing cell cycle arrest and apoptosis in cervical cancer cells. Am J Cancer Res 2015; 5:1133-45; PMID:26045992
- Cheok CF, Kua N, Kaldis P, Lane DP. Combination of nutlin-3 and VX-680 selectively targets p53 mutant cells with reversible effects on cells expressing wild-type p53. Cell Death Differ 2010; 17:1486-500; PMID:20203688; http://dx.doi.org/10.1038/cdd.2010.18
- Kretzner L, Scuto A, Dino PM, Kowolik CM, Wu J, Ventura P, Jove R, Forman SJ, Yen Y, Kirschbaum MH. Combining histone deacetylase inhibitor vorinostat with aurora kinase inhibitors enhances lymphoma cell killing with repression of c-Myc, hTERT, and microRNA levels. Cancer Res 2011; 71:3912-20; PMID:21502403; http://dx.doi.org/10.1158/0008-5472.CAN-10-2259
- Park JS, Carter S, Reardon DB, Schmidt-Ullrich R, Dent P, Fisher PB. Roles for basal and stimulated p21(Cip-1/WAF1/MDA6) expression and mitogen-activated protein kinase signaling in radiation-induced cell cycle checkpoint control in carcinoma cells. Mol Biol Cell 1999; 10:4231-46; PMID:10588655; http://dx.doi.org/10.1091/mbc.10.12.4231
- Grossi V, Peserico A, Tezil T, Simone C. p38alpha MAPK pathway: a key factor in colorectal cancer therapy and chemoresistance. World J Gastroenterol 2014; 20:9744-58; PMID:25110412; http://dx.doi.org/10.3748/wjg.v20.i29.9744
- Koul HK, Pal M, Koul S. Role of p38 MAP Kinase Signal Transduction in Solid Tumors. Genes Cancer 2013; 4:342-59; PMID:24349632; http://dx.doi.org/10.1177/1947601913507951
- Tormos AM, Talens-Visconti R, Nebreda AR, Sastre J. p38 MAPK: a dual role in hepatocyte proliferation through reactive oxygen species. Free Radical Res 2013; 47:905-16; PMID:23906070; http://dx.doi.org/10.3109/10715762.2013.821200
- Kuma Y, Sabio G, Bain J, Shpiro N, Marquez R, Cuenda A. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J Biol Chem 2005; 280:19472-9; PMID:15755732; http://dx.doi.org/10.1074/jbc.M414221200
- He D, Zhao XQ, Chen XG, Fang Y, Singh S, Talele TT, Qiu HJ, Liang YJ, Wang XK, Zhang GQ, et al. BIRB796, the inhibitor of p38 mitogen-activated protein kinase, enhances the efficacy of chemotherapeutic agents in ABCB1 overexpression cells. PloS One 2013; 8:e54181; PMID:23349819; http://dx.doi.org/10.1371/journal.pone.0054181
- Regan J, Breitfelder S, Cirillo P, Gilmore T, Graham AG, Hickey E, Klaus B, Madwed J, Moriak M, Moss N, et al. Pyrazole urea-based inhibitors of p38 MAP kinase: from lead compound to clinical candidate. J Med Chem 2002; 45:2994-3008; PMID:12086485; http://dx.doi.org/10.1021/jm020057r