
ABSTRACT
Methyl-CpG binding domain protein 4 (MBD4) is a DNA glycosylase that can remove 5-fluorodeoxyuracil from DNA as well as repair T:G or U:G mismatches. MBD4 is a target for frameshift mutation with DNA mismatch repair (MMR) deficiency, creating a truncated MBD4 protein (TruMBD4) that lacks its glycosylase domain. Here we show that TruMBD4 plays an important role for enhancing 5-fluorouracil (5FU) sensitivity in MMR-deficient colorectal cancer cells. We found biochemically that TruMBD4 binds to 5FU incorporated into DNA with higher affinity than MBD4. TruMBD4 reduced the 5FU affinity of the MMR recognition complexes that determined 5FU sensitivity by previous reports, suggesting other mechanisms might be operative to trigger cytotoxicity. To analyze overall 5FU sensitivity with TruMBD4, we established TruMBD4 overexpression in hMLH1-proficient or -deficient colorectal cancer cells followed by treatment with 5FU. 5FU-treated TruMBD4 cells demonstrated diminished growth characteristics compared to controls, independently of hMLH1 status. Flow cytometry revealed more 5FU-treated TruMBD4 cells in S phase than controls. We conclude that patients with MMR-deficient cancers, which show characteristic resistance to 5FU therapy, may be increased for 5FU sensitivity via secondary frameshift mutation of the base excision repair gene MBD4.
Abbreviations
| 5FU | = | 5-fluorouracil |
| CRC | = | colorectal cancer |
| MMR | = | DNA mismatch repair |
| PCR | = | polymerase chain reaction |
| MTS | = | 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt |
| MBD4 | = | Methyl-CpG binding domain protein 4 |
| BER | = | base excision repair |
| MSI | = | microsatellite instability |
| I/D | = | insertion/deletion |
| Smug1 | = | monofunctional uracil-DNA glycosylase 1 |
| UNG | = | uracil-DNA glycosylase |
Introduction
5-Fluorouracil (5FU) is the principal chemotherapeutic agent used to treat patients with advanced colorectal cancer. 5FU-based chemotherapy improves survival in patients with stage III colon cancer and stage II and III rectal cancer.Citation1-4 Although 5FU-based chemotherapy is the gold standard for advanced staged colorectal cancer patients, individual patient tumor response is low, but does have an impact on survival.Citation3-5 Retrospective and prospective studies of patients with colorectal cancer (CRC) indicate that those patients with intact DNA mismatch repair (MMR) within their tumors have improved survival with 5FU treatment, whereas patients whose tumors lost DNA MMR with subsequent microsatellite instability (MSI) do not have improved survival.Citation6-9
The DNA MMR system plays an important role in maintaining DNA fidelity after DNA synthesis for cell replication. DNA MMR has 2 recognition complexes for DNA alterations. hMutSα, a heterodimer of the DNA MMR proteins hMSH2 and hMSH6, recognizes base-base mispairs and insertion/deletion (I/D) loops less than 2 nucleotides,Citation10,11 whereas I/D loops more than 2 nucleotides are recognized by hMutSβ, an hMSH2-hMSH3 heterodimer.Citation11-17 hMutSα not only recognizes nucleotide mispairs but can also recognize altered nucleotides that are intercalated or formed by chemotherapy, such as intercrosslinks induced by cisplatin and the adduct O6-methylguanine.Citation11,18 We and others have demonstrated that 5FU incorporated into DNA is also recognized by hMutSα as well as hMutSβ,Citation19-22 and recognition of 5FU by these DNA MMR complexes directly correlates with subsequent cytotoxicity.Citation23 On the other hand, temporary hMLH1 downregulation within an acidic tumor microenvironment does not increase 5FU resistance because certain base excision repair (BER) proteins contribute to 5FU cytotoxicity in that acidic environment.Citation24 Some groups have reported that thymine DNA glycosylase (TDG) or methyl-CpG binding domain protein 4 (MBD4) downregulation induces 5FU cytotoxicity, and single-strand-selective monofunctional uracil-DNA glycosylase 1 (Smug1) overexpression enhances 5FU resistance, with uracil-DNA glycosylase (UNG) not involved in 5FU cytotoxicity despite its ability to recognize 5FU within DNA.Citation25-28 These previous findings indicate that dysfunction of BER molecules as well as DNA MMR complexes may modify cellular 5FU sensitivity.
MBD4 is a methyl-CpG-binding DNA glycosylase involved in the repair of mismatches arising from deamination of methyl-C in mammalian cells,Citation29 and has been shown in vitro to excise 5FU from DNA as well as mismatched thymine bases from oligonucleotide templates.Citation30,31 The MBD4 coding sequence contains an A10 repeat at codons 310–313 that can be subject to shortening frameshift mutation [A10 to A9] in MSI cancers, producing a truncated MBD4 protein (TruMBD4) lacking its glycosylase domain.Citation32-34 Experimentally, TruMBD4 is known to inhibit the glycosylase activities of normal MBD4 via a dominant-negative effect, leading to a hypermutable state further impairing DNA repair (i.e. possessing both MMR and BER defects).Citation35 Although Abdel-Rahman et al. reported that TruMBD4 overexpression alters sensitivity for cisplatin or etoposide,Citation36 it is not known if 5FU sensitivity is modified with TruMBD4. 5FU is highly important as the key therapy for patients with advanced CRC. Here, we examined the binding ability of 5FU in DNA by TruMBD4 and show how TruMBD4 induces 5FU sensitivity independently of the DNA MMR protein hMLH1, one of the potential binding partners of MBD4.
Results
Truncated MBD4 proteins accumulate in the nucleus of colorectal cancer cells
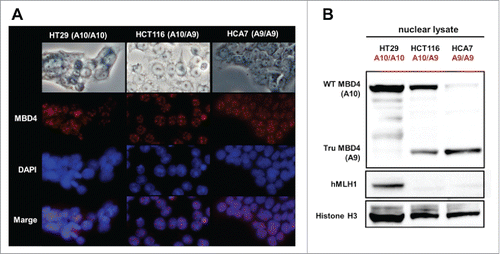
We utilized HT29 (MMR-proficient, MBD4A10/A10), HCT116 (hMLH1−/-, MBD4A10/A9), and HCA7 (hMLH1−/-, MBD4A9/A9) cells, and confirmed nuclear localization and expression of both wild type MBD4 (expressed in HT29 and HCT116, but not in HCA7) and TruMBD4 (expressed in HCT116 and HCA7, but not in HT29) as Bader et al. previously reported.Citation35 Indirect immunofluorescent analysis using polyclonal anti-MBD4 antibody and Alexa Fluor 594-conjugated secondary antibody showed that both wild type and TruMBD4 localized to nucleus forming foci (), indicating that TruMBD4 as well as wild type MBD4 can interact with genomic DNA. Western blotting using nuclear lysate with the polyclonal anti-MBD4 antibody showed that HT29 has only wild type MBD4 protein, HCT116 has both wild type and TruMBD4, and HCA7 has only TruMBD4 protein ().Citation36,37
Figure 1. Both normal MBD4 protein and TruMBD4 localize to the nucleus in colorectal cancer cells. (A) Upper row: light microscopy of human colon cancer cell lines HT29 (MMR-proficient, MBD4A10/A10), HCT116 (hMLH1−/-, MBD4A10/A9) and HCA7 (hMLH1−/-, MBD4A9/A9); Second row: Indirect immunofluorescence microscopy utilizing anti-MBD4 antibody with Alexa Fluor 594-conjugated secondary antibody (staining red); Third row: DAPI staining of nuclei; Fourth row: merge of second and third row images. (B) Western blot of colorectal cancer cell nuclear lysates for MBD4 and TruMBD4 expression. Signals were detected by an LAS-4000 luminescent image analyzer (GE Healthcare Bio-Sciences) utilizing a chemiluminescent solution.
Truncated MBD4 has a higher affinity for 5FU within DNA and reduces the 5FU affinity of DNA MMR proteins
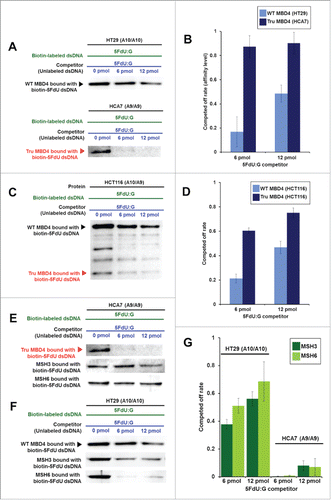
To examine relative binding of TruMBD4 for 5FU within DNA, we performed DNA pull down and “compete off” assays (). TruMBD4 extracted from homozygous MBD4 frameshift mutated HCA7 cells () as well as MBD4 extracted from homozygous wild type MBD4 HT29 cells () bound oligomers containing 5FdU:G, and was competed off by a 5FdU:G but not by a complementary paired oligomer (complementary oligomer not shown), indicating higher affinity for 5FU within DNA over normal cDNA. When we calculated the reduction (competed off) rate determining the relative affinity level of MBD4/TruMBD4 protein for 5FU within DNA, the affinity level of TruMBD4 for 5FU was substantially higher than that of MBD4 (). Similarly, with heterozygous MBD4 mutated HCT116 cells that express both MBD4 and TruMBD4, the 5FU binding affinity of TruMBD4 was higher for 5FU than that for MBD4 (). Overall, these results indicate that TruMBD4 binds to 5FU incorporated into DNA with higher affinity than normal MBD4 protein. We then examined how MBD4/TruMBD4 affects the binding of the DNA MMR recognition complexes. The affinity level of hMSH3 (a component of hMutSβ) and hMSH6 (a component of hMutSα) were substantially lower in TruMBD4-expressed cells compared to normal MBD4-expressed cells (), suggesting that TruMBD4 alters access to 5FU within DNA by the DNA MMR complexes. The hMSH6 component affinity was higher for 5FU as compared to the hMSH3 in wild type MBD4 as we have previously reported.Citation20,23,24 Our data showing high affinity of TruMBD4 for 5FU coupled with inhibition of the MMR protein complexes for access and binding to 5FU within DNA suggest that TruMBD4 may alter cellular 5FU sensitivity.
Figure 2. Schematic diagram for our DNA pull down assay.
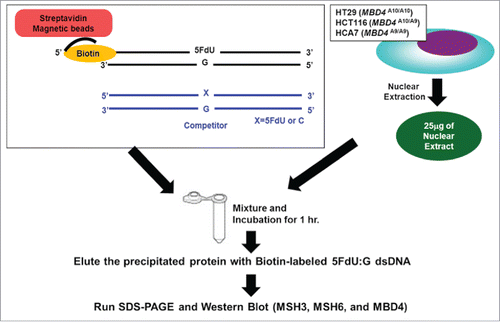
Figure 3. TruMBD4 binds to 5FU incorporated into DNA with higher affinity than normal MBD4 protein, and reduces 5FU affinity of DNA mismatch repair proteins in DNA pull down assays. (A) (Upper panel) Nuclear lysates from HT29 cells containing protein derived from wild type MBD4. As amounts of 5FdU:G competitor is increased relative to bound biotin-labeled 5FdU:G, the amount of MBD4 protein precipitated by biotin-labeled 5FdU:G is partially reduced, indicating some MBD4 protein was bound (or stolen) by the 5FdU:G competitor. This was not the case with a control complementary competitor, indicating MBD4 specifically recognizes 5FU within DNA (not shown). (Lower panel) Nuclear lysates from HT29 containing protein derived from frameshifted mutant MBD4 (TruMBD4). Here, as amounts of 5FdU:G competitor increased relative to biotin-labeled 5FdU:G, the amount of TruMBD4 precipitated by biotin-labeled 5FdU:G DNA is markedly reduced, suggesting a relative higher affinity for 5FdU:G as compared to normal MBD4 protein. (B) Bar graphs representing the reduction (competed off) rate by the protein for the 5FdU:G competitor, equating to the relative affinity level of the protein for 5FU within DNA. The affinity level of TruMBD4 for 5FdU:G is markedly higher than that of normal MBD4 protein. (C) Nuclear lysates from HCT116 cells containing both normal MBD4 protein and TruMBD4. As amounts of 5FdU:G competitor is increased, both normal MBD4 protein and TruMBD4 are competed off, but at apparently different rates. (D) Bar graph representing the competed off rate for both normal MBD4 protein and TruMBD4 from HCT116 cells. The relative affinity level of TruMBD4 was higher than normal MBD4 protein for the 5FdU:G competitor. (E, F) Nuclear lysates from HCA7 cells (E) and HT29 cells (F), demonstrating the relative pull down and competition off binding by MBD4/TruMBD4, MSH3 (key component of the hMutSβ MMR recognition complex) and MSH6 (key component of the hMutSα MMR recognition complex) for 5FdU:G. Note the relative difficulty for “compete off” bound reduction for MSH3 and MSH6 by the 5FdU:G competitor when TruMBD4 is present, compared to the “compete off” reduction for MSH3 and MSH6 when normal MBD4 protein is present. (F) Bar graph representing the competed off rate for MSH3 and MSH6 for the 5FdU:G competitor in the presence of normal MBD4 protein or TruMBD4. With normal MBD4 protein, MSH6 shows higher affinity for 5FU within DNA (as expected). However the binding affinity rates of both MMR proteins are markedly lower in TruMBD4-expressed cells than that of normal MBD4-expressed cells.
Truncated MBD4 overexpression enhances 5FU cytotoxicity through S phase arrest independently of hMLH1 status
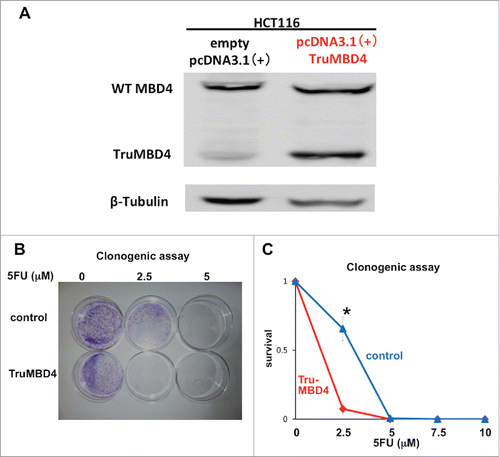
To analyze 5FU sensitivity under TruMBD4-expressed conditions, and to verify our data using natively-expressed TruMBD4 extracts, we constructed a TruMBD4-expression plasmid by inserting the frameshift mutated MBD4 coding sequence into pcDNA3 and amplified it utilizing competent E. Coli DH5α. We then stably-transfected constructs into HT29 (hMLH1-proficient and wild type MBD4) cells and selected TruMBD4-expressed cell clones. TruMBD4 overexpression was confirmed by Western blotting ().
Figure 4. TruMBD4 enhances 5FU cytotoxicity in hMLH1-proficient cells. (A) Establishment of stable, TruMBD4-expressed HT29 cell clones as shown by Western blot (right lane). Cells were transfected with a pcDNA3 plasmid (Invitrogen) encoding TruMBD4, and selected by G418. HCT116 lysates served as a positive control since it expresses both normal MBD4 protein and TruMBD4 (left lane). HT29 cells transfected with an empty pcDNA3 plasmid served as negative control (middle lane). β-actin served as a loading control. (B) MTS assay. Cells were seeded at a density of 5000 cells per well into 96-well plates in culture medium treated with 5 μM, 10 μM;, 15 μM;, 20 μM of 5FU. After 5 d of growth, the number of viable cells was counted via the assay. (C) Clonogenic assay. Cells were plated in growth medium supplemented by 10% FBS and containing various concentrations of 5-FU (0, 5, and 10 µM). After 10 d of growth, the culture plates were washed, fixed with methanol, and stained with 3% Giemsa. From both MTS and clonogenic assays, TruMBD4 enhances 5FU-induced cytotoxicity.
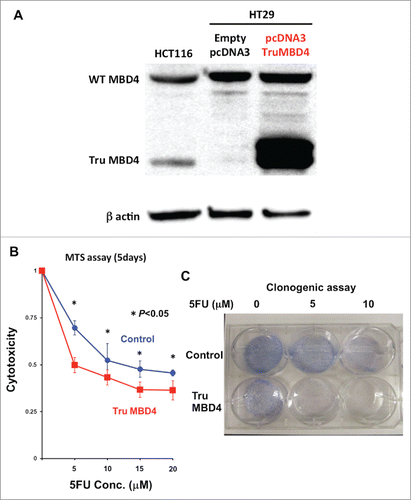
Comparing TruMBD4 overexpressed cells with control plasmid cells, we demonstrate reduced cellular proliferation with 5FU-treatment for TruMBD4 cells by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) (P< 0 .05; ) and clonogenic assays (P< 0 .01; ), confirming enhanced 5FU sensitivity with TruMBD4. Fluorescent-activated cell sorting (FACS) revealed that 5 μM 5FU-treated TruMBD4 cells slowed its cell cycle accumulating cells in S phase (18.9% of cells pre-5FU treatment vs 55.0% of cells post-5FU treatment; ) as compared to control cells (16.2% pre-treatment vs 24.3% post-5FU treatment). Because TruMBD4 lacks its hMLH1 binding domain in addition to its glycosylase domain as was reported previously,Citation38 we suspected that hMLH1 would not mediate the TruMBD4-directed enhanced cytotoxicity. To confirm this hypothesis, we additionally overexpressed TruMBD4 in heterozygous MBD4 mutated HCT116 (hMLH1-deficient) cells, increasing the ratio of TruMBD4 expression relative to normal MBD4 protein (). We demonstrate via clonogenic assay that forced TruMBD4 expression induces 5FU sensitivity in the absence of hMLH1 protein (), similar to cells proficient for hMLH1 (). These results suggest that TruMBD4 expression enhances 5FU cytotoxicity independently of hMLH1 expression.
Figure 5. TruMBD4 induces S phase cell cycle arrest upon 5FU treatment. (A) Using fluorescence-activated cell sorting (FACS) analysis, empty plasmid transfected HT29 cells expressing normal MBD4 protein showed a modest increase in S phase cells after 5FU treatment (16.2% pre-treatment, 24.3% post 5FU treatment). (B) TruMBD4-expressing HT29 cells, in response to 5FU treatment, demonstrated marked increase in S phase cells (18.9% pre-5FU treatment, 55.0% post 5FU treatment).
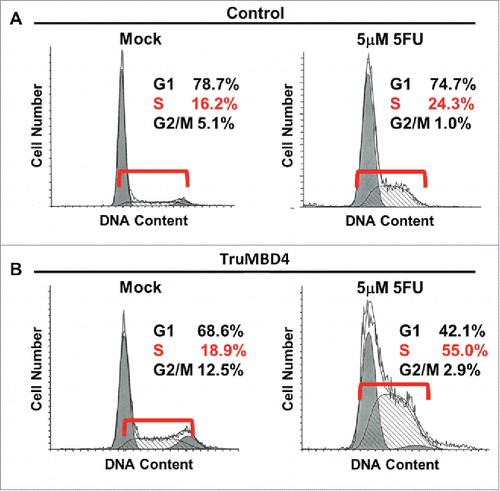
Figure 6. TruMBD4 enhances 5FU cytotoxicity independent of hMLH1 status. (A) Establishment of stable, TruMBD4-expressed HCT116 cell clones as shown by Western blot (right lane). Cells were transfected with a pcDNA3 plasmid (Invitrogen) encoding TruMBD4, and selected by G418. HCT116 cells transfected with an empty pcDNA3 plasmid served as negative control (left lane). β-actin served as a loading control. Note the marked increase in TruMBD4 expression relative to normal MBD4 protein expression. (B, C) Clonogenic assay. Cells were plated in growth medium supplemented by 10% FBS and containing various concentrations of 5-FU (0, 5, and 10 µM). After 10 d of growth, the culture plates were washed, fixed with methanol, and stained with 3% Giemsa (B). Previously viable clonal colonies of at least 50 cells were counted. The relative surviving fraction for each cell line was expressed as a ratio of the plating efficiency in treated cultures to that observed in the controls (C). The enhanced TruMBD4-overexpressed HCT116 cells increased 5FU cytotoxicity over control cells (*P<0.05 at 2.5 µM 5FU).
Discussion
DNA MMR deficiency induces a hypermutable state for the CRC cellCitation39,40 that drives frameshift mutation of target genes such as TGFBR2 and ACVR2, among others.Citation41-45 MBD4 is one frameshift mutation target gene that provides a very unique but complex character for CRC, in that: (a) MBD4 excises 5FU from DNA as well as mismatched thymine bases from oligomer templates,Citation30,31 (b) frameshift mutation of MBD4 by A10 to A9 deletion (codons 310–313) produce TruMBD4 lacking its glycosylase domain in MSI cancers,Citation32-34 and (c) wild type MBD4 interacts with hMLH1 at its glycosylase domain.Citation38 We previously reported that MBD4, as part of BER, may salvage 5FU cytotoxicity in DNA MMR-deficient CRC in an acidic tumor microenvironment, but little was known how TruMBD4 affects 5FU cytotoxicity.Citation24 Our study demonstrates that: (a) TruMBD4 has higher affinity for 5FU that is incorporated into DNA than normal MBD4 protein, (b) TruMBD4 reduces the binding affinity of DNA MMR complexes to 5FU within DNA, and (c) TruMBD4 overexpression enhances 5FU cytotoxicity independently of hMLH1. Our study demonstrates an enhancement of cytotoxicity from 5FU, the key drug used for patients with advanced colorectal cancers, by the TruMBD4 protein that arises from frameshift mutation as consequence of deficient DNA MMR. Our finding may have implications for patients with MSI CRCs.
We demonstrated the nuclear localization of TruMBD4 by immunofluorescence microscopy and Western blotting of nuclear extracts. We originally hypothesized that expression of TruMBD4 would affect the 5FU-directed DNA damage response because: (i) TruMBD4 protein possesses its methyl CpG binding domain but lacks its glycosylase domain (rendering MBD4 incapable of removing 5FU from DNA), and (ii) TruMBD4 protein lacks its predicted nuclear export signal (codons 506- 511; LGLYDL) that is homologous to the murine MBD4 protein, suggesting that TruMBD4 would accumulate in nucleus.Citation46
The effects of TruMBD4 appear to be different than wild type MBD4 deficiency (or deletion), although both conditions obviously lack MBD4s glycosylase function. In this study, we show that TruMBD4 binds 5FU in DNA with higher affinity than normal MBD4 protein. Prior demonstration regarding 5FU resistance was solely under MBD4-deficient conditions, not truncated protein conditions. Sansom et al. demonstrated resistance to 5FU treatment in intestinal crypts of Mbd4−/− mice compared to wild type mice.Citation28 Cortellino et al. also showed Mbd4−/− mouse embryonic fibroblasts are resistant to 5FU.Citation47 In terms of damage response triggered by platinum chemotherapeutic agents such as cisplatin, TruMBD4 enhances cisplatin cytotoxicity but Mbd4−/− mouse embryonic fibroblasts are resistant.Citation36,47 In these experiments, Mbd4-null mice were used to examine the Mbd4-deficient condition, but it is impossible to observe the TruMbd4-expressed condition because the mouse Mbd4 locus does not contain a coding poly A microsatellite equivalent to human MBD4.Citation28,48,49 In humans, frameshift mutation of MBD4s polyadenine tract has been identified in 20–43% of MSI CRCs and other MSI cancersCitation32-34 and little is clinically reported about TruMBD4 effects within CRC. Given this background, our results of higher affinity by TruMBD4 to 5FU within DNA, coupled with inhibition of DNA MMR binding of 5FU due to the presence of TruMBD4, suggests that TruMBD4 plays an important role for 5FU cytotoxicity in CRC.
It has been reported that MBD4 interacts with hMLH1, a component of the DNA MMR complex hMutLα[38]. Although TruMBD4 lacks the predicted hMLH1-binding domain (codons 406–580),Citation38 we needed to confirm if TruMBD4 affected 5FU cytotoxicity independently of the hMLH1 status because it has not been directly shown that TruMBD4 physically interacts with hMLH1. We observed that TruMBD4 induces cytotoxicity via MTS and clonogenic assays. This was first shown using hMLH1-proficient HT29 cells. We further analyzed the effects of TruMBD4 on 5FU cytotoxicity using hMLH1-deficient cells with the understanding that there are 2 scenarios that can drive MBD4 frameshift mutations with DNA MMR deficiency. One is the hMLH1-deficient condition (i.e., hMLH1 silencing by hypermethylation of its promoter) seen in almost all sporadic MSI CRCs,Citation39,50 and the other is hMLH1-proficiency with deficiency of a different MMR protein (e.g. hMSH2 or hMSH6 mutation) as seen with Lynch syndrome cancers.Citation51 By showing similar 5FU sensitivity in TruMBD4-overexpressed hMLH1-deficient as well as in hMLH1-proficient cells, we demonstrate that 5FU sensitivity with TruMBD4 expression is independent of hMLH1.
Collura et al. clinically investigated beneficial molecular markers among stage II/III colorectal cancer patients who received 5FU-based chemotherapy, and identified that HSP110 T17 mutation predicted excellent progression free survival, and notably, the next best progression free survival was seen with MBD4 mutation.Citation52 Additional studies will need to examine the context of MBD4 mutation and its modification of a clinical response to 5FU-based chemotherapy for patients with stage IV CRC as well as in the setting of adjuvant chemotherapy for stage II/III CRC patients.
In conclusion, TruMBD4 enhances 5FU cytotoxicity in MSI CRC cells independently of hMLH1. Our finding may have implications in the approach to 5FU-based chemotherapy for patients with MSI CRCs.
Materials and methods
Cell lines and cultures
The human colon cancer cell lines HT29 (MMR-proficient, MBD4A10/A10) and HCT116 (hMLH1−/−, MBD4A10/A9) were obtained from American Type Culture Collection (Rockville, MD, USA), and HCA7 (hMLH1−/−, MBD4A9/A9) was kindly provided by Minoru Koi, Ph.D. (Baylor University Medical Center). Cells were maintained in growth medium containing 10% fetal bovine serum (FBS).
Extraction of nuclear proteins and western blotting
We utilized 106 cells that were washed with cold PBS, and proteins were extracted from nuclei by using a Nuclear Extraction Kit (Cayman Chemical, MI, USA) following the manufacturer's instructions. The nuclear extract was mixed with 4×protein sample buffer (4×NuPAGE®LDS Sample Buffer [Life Technologies, NY, USA], 3% 2-mercaptoethanol) and heated for 10 min at 98°C. The proteins were then separated by electrophoresis on 4–12% NuPAGE®Bis-Tris Mini Gels (Life Technologies, CA USA), and transferred to Protoran™ Nitrocellulose membranes (GE Healthcare Bio-Sciences, CA, USA) in a transfer apparatus (Life Technologies). The membranes were blocked with 5% skim milk and 0.1% Tween in Tris-buffered saline (TBS).
DNA pull down assay
We utilized 106 cells that were washed with cold PBS, and proteins were extracted from nuclei by using a Nuclear Extraction Kit (Cayman Chemical, MI, USA) following the manufacturer's instructions. For immobilization, Biotin-labeled dsDNA probes (12.5 pmol) was mixed with 20 μg Dynabeads® M-280 Streptavidin (Invitrogen) in washing buffer (5 mM Tris-HCl [pH7.5], 0.5 mM EDTA, and 1M NaCl) and incubated for 40 min at room temperature utilizing a rotator. After the washing procedure using Magnetic Rack (Magna GrIP™ Rack supplied by Millipore, CA, USA), immobilized Biotin-labeled dsDNA probes with or without unlabeled DNA probe (competitor) were added in incubation buffer (50 mM Tris-HCl [pH7.2], 1 mM EDTA, 5% Glycerol, 0.01% NP-40, and 1mM DTT) followed by mixture with 25 μg of nuclear lysate, and incubated using a rotator for 1 hr at room temperature. After washing, precipitated proteins were mixed with 30 μL 1×protein sample buffer and boiled for 3 min, and the supernatant was collected for Western blowing.
Transfection of frameshift mutant MBD4
For isolation of stable truncated MBD4 overexpressed clone, HT29 or HCT116 cells were transfected with a pcDNA3(for HT29) or pcDNA 3.1(+) (for HCT116) plasmid vector (Invitrogen) that encodes truncated MBD4 by using Nucleofecter Kit R (for HT29) or V (for HCT116) (Lonza, Germany), and selection was done using both 400 μg/ml of G418. After selection, colonies were pooled and cultured for following analysis. The stable truncated MBD4 expression was confirmed by western blotting.
Cell growth assay
For the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) assay, cells were seeded at a density of 5000 cells per well into 96-well plates in culture medium treated with 5 μM, 10 μM, 15 μM, 20 μM of 5FU, respectively. After 5days, the number of viable cells was counted by using a CellTiter 96® AQueous One Solution Cell Proliferation Assay Kit (Promega) according to the manufacturer's instructions. The kit detects mitochondrial nicotinamide adenine dinucleotide dehydrogenase activity in live cells by measuring reduction of the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS). Measurement of the absorbance of the formazan was carried out in 96 well microplates read at 490nm. For clonogenic assays, cells were plated on 60 mm (for HT29) or 100mm (for HCT116) Tissue Culture Dish (Becton Dickinson Labware, NJ) in Iscove's modified Dulbecco's medium supplemented with 10% FBS and containing various concentrations of 5-FU (0, 5, and 10 µM), then incubated at 37°C and 5% CO2. After 10 d (for HT29) or 14days (for HCT116) of growth, the culture plates were washed with PBS, fixed with methanol for 15 minutes, and then rewashed with PBS. The colonies were stained with 3% Giemsa (Sigma, St Louis, MO) for 15 minutes and rinsed with water.
Fluorescence-activated cell sorting (FACS) analysis
For FACS analysis, cells were washed in phosphate-buffered saline (PBS), incubated in PBS for 10 min, and then trypsinized and fixed in 80% ethanol. The cells were washed again, resusupended in 50 µg/ml propidium iodide and analyzed by using a Beckman Coulter Epics XL (Beckman Coulter, CA, USA).
Statistical analysis
Comparisons were made using Student's t-test: P values less than 0.05 were taken as statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
MI and JMC conceived the study. SS and MI performed Western blotting and cell growth assays. MI and STR performed DNA pull down assays. SS, MI and HY constructed truncated MBD4 expression plasmid. SK, KS HM and JMC interpreted data. MI and JMC drafted and critically revised the manuscript. All authors read and approved the final manuscript.
Funding
Supported by the United States Public Health Service (DK067287, CA162147 and CA206010 to JMC) and Japan Society for the Promotion of Science (JSPS) (KAKENHI Grant Number 25860530 to MI).
References
- Moertel CG, Fleming TR, Macdonald JS, Haller DG, Laurie JA, Tangen CM, Ungerleider JS, Emerson WA, Tormey DC, Glick JH, et al. Fluorouracil plus levamisole as effective adjuvant therapy after resection of stage III colon carcinoma: a final report. Ann Intern Med 1995; 122(5):321-6; PMID:7847642; http://dx.doi.org/10.7326/0003-4819-122-5-199503010-00001
- Moertel CG, Fleming TR, Macdonald JS, Haller DG, Laurie JA, Goodman PJ, Ungerleider JS, Emerson WA, Tormey DC, Glick JH, et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N Engl J Med 1990; 322(6):352-8; PMID:2300087; http://dx.doi.org/10.1056/NEJM199002083220602
- Boland CR, Sinicrope FA, Brenner DE, Carethers JM. Colorectal cancer prevention and treatment. Gastroenterology 2000; 118(2 Suppl 1):S115-28; PMID:10868902; http://dx.doi.org/10.1016/S0016-5085(00)70010-2
- Carethers JM. Systemic treatment of advanced colorectal cancer – tailoring therapy to the tumor. Ther Adv Gastroenterol 2008; 1:33-42; PMID:21180512; http://dx.doi.org/10.1177/1756283X08093607
- Meta-analysis Group In Cancer. Efficacy of intravenous continuous infusion of fluorouracil compared with bolus administration in advanced colorectal cancer. J Clin Oncol. 1998; 16(1):301-8; PMID:9440757
- Carethers JM, Smith EJ, Behling CA, Nguyen L, Tajima A, Doctolero RT, Cabrera BL, Goel A, Arnold CA, Miyai K, et al. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology 2004; 126(2):394-401; PMID:14762775; http://dx.doi.org/10.1053/j.gastro.2003.12.023
- Jover R, Zapater P, Castells A, Llor X, Andreu M, Cubiella J, Balaguer F, Sempere L, Xicola RM, Bujanda L, et al. The efficacy of adjuvant chemotherapy with 5-fluorouracil in colorectal cancer depends on the mismatch repair status. Eur J Cancer 2009; 45(3):365-73; PMID:18722765; http://dx.doi.org/10.1016/j.ejca.2008.07.016
- Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003; 349(3):247-57; PMID:12867608; http://dx.doi.org/10.1056/NEJMoa022289
- Hamaya Y, Guarinos C, Tseng-Rogenski SS, Iwaizumi M, Das R, Jover R, Castells A, Llor X, Andreu M, Carethers JM. Efficacy of 5-fluorouracil adjuvant therapy for patients with EMAST-positive stage II/III colorectal cancers. PLoS One 2015; 10:e0127591; PMID:25996601; http://dx.doi.org/10.1371/journal.pone.0127591
- Marsischky GT, Kolodner RD. Biochemical characterization of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 complex and mispaired bases in DNA. J Biol Chem 1999; 274(38):26668-82; PMID:10480869; http://dx.doi.org/10.1074/jbc.274.38.26668
- Carethers JM, Koi M, Tseng-Rogenski S. EMAST is a form of microsatellite instability that is initiated by inflammation and modulates colorectal cancer progression. Genes 2015; 6:185-205; PMID:25836926; http://dx.doi.org/10.3390/genes6020185
- Acharya S, Wilson T, Gradia S, Kane MF, Guerrette S, Marsischky GT, Kolodner R, Fishel R. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc Natl Acad Sci USA 1996; 93(24):13629-34; PMID:8942985; http://dx.doi.org/10.1073/pnas.93.24.13629
- Blackwell LJ, Bjornson KP, Modrich P. DNA-dependent activation of the hMutSalpha ATPase. J Biol Chem 1998; 273(48):32049-54; PMID:9822679; http://dx.doi.org/10.1074/jbc.273.48.32049
- de Wind N, Dekker M, Claij N, Jansen L, van Klink Y, Radman M, Riggins G, van der Valk M, van't Wout K, te Riele H. HNPCC-like cancer predisposition in mice through simultaneous loss of Msh3 and Msh6 mismatch-repair protein functions. Nat Genet 1999; 23(3):359-62; PMID:10545954; http://dx.doi.org/10.1038/15544
- Genschel J, Littman SJ, Drummond JT, Modrich P. Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J Biol Chem 1998; 273(31):19895-901; PMID:9677427; http://dx.doi.org/10.1074/jbc.273.31.19895
- Habraken Y, Sung P, Prakash L, Prakash S. Binding of insertion/deletion DNA mismatches by the heterodimer of yeast mismatch repair proteins MSH2 and MSH3. Curr Biol 1996; 6(9):1185-7; PMID:8805366; http://dx.doi.org/10.1016/S0960-9822(02)70686-6
- Umar A, Risinger JI, Glaab WE, Tindall KR, Barrett JC, Kunkel TA. Functional overlap in mismatch repair by human MSH3 and MSH6. Genetics 1998; 148(4):1637-46; PMID:9560383
- Duckett DR, Drummond JT, Murchie AI, Reardon JT, Sancar A, Lilley DM, Modrich P. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc Natl Acad Sci USA 1996; 93(13):6443-7; PMID:8692834; http://dx.doi.org/10.1073/pnas.93.13.6443
- Tajima A, Hess MT, Cabrera BL, Kolodner RD, Carethers JM. The mismatch repair complex hMutS alpha recognizes 5-fluorouracil-modified DNA: implications for chemosensitivity and resistance. Gastroenterology 2004; 127(6):1678-84; PMID:15578504; http://dx.doi.org/10.1053/j.gastro.2004.10.001
- Tajima A, Iwaizumi M, Tseng-Rogenski S, Cabrera BL, Carethers JM. Both hMutSalpha and hMutSbeta DNA mismatch repair complexes participate in 5-Fluorouracil cytotoxicity. PLoS One 2011; 6(12):e28117; PMID:22164234; http://dx.doi.org/10.1371/journal.pone.0028117
- Fischer F, Baerenfaller K, Jiricny J. 5-Fluorouracil is efficiently removed from DNA by the base excision and mismatch repair systems. Gastroenterology 2007; 133(6):1858-68; PMID:18054558; http://dx.doi.org/10.1053/j.gastro.2007.09.003
- Chung H, Chaudhry J, Lopez, CG, Carethers JM. Cyclin E and histone H3 are regulated by 5-fluorouracil in a DNA mismatch repair-dependent manner. Cancer Biol Ther 2010; 10:1147-56; PMID:20930505; http://dx.doi.org/10.4161/cbt.10.11.13447
- Iwaizumi M, Tseng-Rogenski S, Carethers JM. DNA mismatch repair proficiency executing 5-fluorouracil cytotoxicity in colorectal cancer cells. Cancer Biol Ther 2011; 12(8):756-64; PMID:21814034; http://dx.doi.org/10.4161/cbt.12.8.17169
- Iwaizumi M, Tseng-Rogenski S, Carethers JM. Acidic tumor microenvironment downregulates hMLH1 but does not diminish 5-fluorouracil chemosensitivity. Mutat Res 2013; 747-8: 19-27; PMID:23643670; http://dx.doi.org/10.1016/j.mrfmmm.2013.04.006
- Andersen S, Heine T, Sneve R, Konig I, Krokan HE, Epe B, Nilsen H. Incorporation of dUMP into DNA is a major source of spontaneous DNA damage, while excision of uracil is not required for cytotoxicity of fluoropyrimidines in mouse embryonic fibroblasts. Carcinogenesis 2005; 26(3):547-55; PMID:15564287; http://dx.doi.org/10.1093/carcin/bgh347
- An Q, Robins P, Lindahl T, Barnes DE. 5-Fluorouracil incorporated into DNA is excised by the Smug1 DNA glycosylase to reduce drug cytotoxicity. Cancer Res 2007; 67(3):940-5; PMID:17283124; http://dx.doi.org/10.1158/0008-5472.CAN-06-2960
- Kunz C, Focke F, Saito Y, Schuermann D, Lettieri T, Selfridge J, Schär P. Base excision by thymine DNA glycosylase mediates DNA-directed cytotoxicity of 5-fluorouracil. PLoS Biol 2009; 7(4):e91; PMID:19402749; http://dx.doi.org/10.1371/journal.pbio.1000091
- Sansom OJ, Zabkiewicz J, Bishop SM, Guy J, Bird A, Clarke AR. MBD4 deficiency reduces the apoptotic response to DNA-damaging agents in the murine small intestine. Oncogene 2003; 22(46):7130-6; PMID:14562041; http://dx.doi.org/10.1038/sj.onc.1206850
- Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature 1999; 401(6750):301-4; PMID:10499592; http://dx.doi.org/10.1038/45843
- Petronzelli F, Riccio A, Markham GD, Seeholzer SH, Genuardi M, Karbowski M, Yeung AT, Matsumoto Y, Bellacosa A. Investigation of the substrate spectrum of the human mismatch-specific DNA N-glycosylase MED1 (MBD4): fundamental role of the catalytic domain. J Cell Physiol 2000; 185(3):473-80; PMID:11056019; http://dx.doi.org/10.1002/1097-4652(200012)185:3%3c473::AID-JCP19%3e3.0.CO;2-
- Petronzelli F, Riccio A, Markham GD, Seeholzer SH, Stoerker J, Genuardi M, Yeung AT, Matsumoto Y, Bellacosa A. Biphasic kinetics of the human DNA repair protein MED1 (MBD4), a mismatch-specific DNA N-glycosylase. J Biol Chem 2000; 275(42):32422-9; PMID:10930409; http://dx.doi.org/10.1074/jbc.M004535200
- Bader S, Walker M, Hendrich B, Bird A, Bird C, Hooper M, Wyllie A. Somatic frameshift mutations in the MBD4 gene of sporadic colon cancers with mismatch repair deficiency. Oncogene 1999; 18(56):8044-7; PMID:10637515; http://dx.doi.org/10.1038/sj.onc.1203229
- Riccio A, Aaltonen LA, Godwin AK, Loukola A, Percesepe A, Salovaara R, Masciullo V, Genuardi M, Paravatou-Petsotas M, Bassi DE, et al. The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nat Genet 1999; 23(3):266-8; PMID:10545939; http://dx.doi.org/10.1038/15443
- Yamada T, Koyama T, Ohwada S, Tago K, Sakamoto I, Yoshimura S, Hamada K, Takeyoshi I, Morishita Y. Frameshift mutations in the MBD4/MED1 gene in primary gastric cancer with high-frequency microsatellite instability. Cancer Lett 2002; 181(1):115-20; PMID:12430186; http://dx.doi.org/10.1016/S0304-3835(02)00043-5
- Bader SA, Walker M, Harrison DJ. A human cancer-associated truncation of MBD4 causes dominant negative impairment of DNA repair in colon cancer cells. Br J Cancer 2007; 96(4):660-6; PMID:17285135; http://dx.doi.org/10.1038/sj.bjc.6603592
- Abdel-Rahman WM, Knuutila S, Peltomaki P, Harrison DJ, Bader SA. Truncation of MBD4 predisposes to reciprocal chromosomal translocations and alters the response to therapeutic agents in colon cancer cells. DNA Repair (Amst) 2008; 7(2):321-8; PMID:18162445; http://dx.doi.org/10.1016/j.dnarep.2007.11.009
- Meyers M, Wagner MW, Mazurek A, Schmutte C, Fishel R, Boothman DA. DNA mismatch repair-dependent response to fluoropyrimidine-generated damage. J Biol Chem 2005; 280(7):5516-26; PMID:15611052; http://dx.doi.org/10.1074/jbc.M412105200
- Bellacosa A, Cicchillitti L, Schepis F, Riccio A, Yeung AT, Matsumoto Y, Golemis EA, Genuardi M, Neri G. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc Natl Acad Sci USA 1999; 96(7):3969-74; PMID:10097147; http://dx.doi.org/10.1073/pnas.96.7.3969
- Carethers JM, Jung BH. Genetics and genetic biomarkers in sporadic colorectal cancer. Gastroenterology 2015; 149:1177-90; PMID:26216840; http://dx.doi.org/10.1053/j.gastro.2015.06.047
- Ashktorab H, Ahuja S, Kannan L, Llor X, Ellis N, Xicola RM, Adeyinka LO, Carethers JM, Brim H, Nouraie M. A meta-analysis of MSI frequency and race in colorectal cancer. Oncotarget 2016; PMID:2710810; http://dx.doi.org/10.18632/oncotarget.8945
- Chung H, Young DJ, Lopez C, Le T-AT, Lee JK, Ream-Robinson D, Huang SC, Carethers JM. Mutation rates of TGFBR2 and ACVR2 coding microsatellites in human cells with defective DNA mismatch repair. PLoS One 2008; 3:e3463; PMID:18941508; http://dx.doi.org/10.1371/journal.pone.0003463
- Chung H, Chaudhry J, Lai JF, Young DJ, Carethers JM. Flanking nucleotide specificity for DNA mismatch repair-deficient frameshifts within Activin Receptor 2 (ACVR2). Mutat Res 2012; 729:73-80; PMID:22001236; http://dx.doi.org/10.1016/j.mrfmmm.2011.09.009
- Chung H, Lopez CG, Holmstrom J, Young DJ, Lai JF, Ream-Robinson D, Carethers JM. Both microsatellite length and sequence context determine frameshift mutation rates in defective DNA mismatch repair. Hum Mol Genet 2010; 19:2638-4; PMID:20418486; http://dx.doi.org/10.1093/hmg/ddq151
- Chung H, Lopez CG, Young DJ, Lai JF, Holmstrom J, Ream-Robinson D, Cabrera BL, Carethers JM. Flanking sequence specificity determines coding microsatellite heteroduplex and mutation rates with defective DNA mismatch repair. Oncogene 2010; 29:2172-80; PMID:20140012; http://dx.doi.org/10.1038/onc.2009.508
- Jung B, Doctolero RT, Tajima A, Nguyen AK, Keku T, Sandler RS, Carethers JM. Loss of activin receptor type 2 protein expression in microsatellite unstable colon cancers. Gastroenterology 2004; 126:654-59; PMID:14988818; http://dx.doi.org/10.1053/j.gastro.2004.01.008
- Grigera F, Bellacosa A, Kenter AL. Complex relationship between mismatch repair proteins and MBD4 during immunoglobulin class switch recombination. PLoS One 2013; 8(10):e78370; PMID:24205214; http://dx.doi.org/10.1371/journal.pone.0078370
- Cortellino S, Turner D, Masciullo V, Schepis F, Albino D, Daniel R, Skalka AM, Meropol NJ, Alberti C, Larue L, et al. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proc Natl Acad Sci USA 2003; 100(25):15071-6; PMID:14614141; http://dx.doi.org/10.1073/pnas.2334585100
- Wong E, Yang K, Kuraguchi M, Werling U, Avdievich E, Fan K, Fazzari M, Jin B, Brown AM, Lipkin M, et al. Mbd4 inactivation increases Cright-arrowT transition mutations and promotes gastrointestinal tumor formation. Proc Natl Acad Sci USA 2002; 99(23):14937-42; PMID:12417741; http://dx.doi.org/10.1073/pnas.232579299
- Bader S, Walker M, Harrison D. Most microsatellite unstable sporadic colorectal carcinomas carry MBD4 mutations. Br J Cancer 2000; 83(12):1646-9; PMID:11104560; http://dx.doi.org/10.1054/bjoc.2000.1482
- Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology 2010; 138(6):2073-87. e3; PMID:20420947; http://dx.doi.org/10.1053/j.gastro.2009.12.064
- Carethers JM, Stoffel EM. Lynch syndrome and Lynch syndrome mimics: the growing complex landscape of hereditary colon cancer. World J Gastroenterology 2015; 21:9253-61; PMID:26309352; http://dx.doi.org/10.3748/wjg.v21.i31.9253
- Collura A, Lagrange A, Svrcek M, Marisa L, Buhard O, Guilloux A, Wanherdrick K, Dorard C, Taieb A, Saget A, et al. Patients with colorectal tumors with microsatellite instability and large deletions in HSP110 T17 have improved response to 5-fluorouracil-based chemotherapy. Gastroenterology 2014; 146(2):401-11. e1; PMID:24512910; http://dx.doi.org/10.1053/j.gastro.2013.10.054