
ABSTRACT
This review embarks upon a cell death journey from the discovery of apoptosis and necrosis through to the coalescence of these: necroptosis. The mechanisms of 2 emerging necrotic cell death pathways, pyroptosis and ferroptosis, will be explored before delving into apoptotic and necroptotic signaling cascades, highlighting the complex interplay between molecular players. The involvement of the ripoptosome, interferon signaling and DNA damage in necroptosis will be discussed briefly. The major focus is on necroptosis initiation by tumor necrosis factor-α (TNFα) and its cognate receptor TNFR1, caspase-independent RIP1/RIP3/MLKL necrosome activation and cell death propagation by damage-associated molecular pattern (DAMP) release. Finally, the implications of a complex cell death signaling network will be revealed in the context of cancer biology and therapy. The clinical contribution of the discovery of necroptosis as an unequivocally new way of dying is monumental and could drastically alter cancer therapy strategies in the future.
Introduction
Eukaryotic cell death is an evolutionarily conserved mechanism with implications in organismal development, physiological homeostasis and disease.Citation1,2 The distinction between life and death at the cellular level as well as the trajectory toward expiration itself is far hazier than one would imagine. The Nomenclature Committee on Cell Death (NCCD) proposed a shift from a purely morphological diagnosis of cell death toward molecular-based identification in 2012.Citation3 An updated narrative on cell death classification was published in 2015 with the potential to improve communication and understanding of cell death and expose avenues which call for deeper exploration.Citation4 As scientists continue to reveal key molecular players and contextualise these in terms of specific types of cell death and their morphologies, more of the currently hazy picture of networks that function within and between dying cells will be exposed, verified and even altered. The major focus of this review is on apoptosis and its contemporary counterpart, necroptosis, and how these forms of cell death contribute to cancer biology with a brief inquest into other emerging forms of cell death, crucial for our understanding of the entire cell death signaling network.
As the cell death journey toward the discovery of necroptosis begins, a basic understanding of the foundation that has elevated the eyes of research to a viewpoint from which we are now able to observe the workings of key necroptotic players is necessary. For centuries it has been known that cells age, senesce and perish over varying time frames and for a range of purposes. The programmable nature of cell death was first alluded to in a study describing sillkmoth development some 50 y ago.Citation5 Before the revelation of any particular molecular mechanisms involved, the morphological nature of dying cells as viewed under a microscope was identifiable and thus cell death was described as either active or passive in nature. It was not until 1972 that the term apoptosis was coined and documented by Kerr and colleagues.Citation1 ‘Apoptosis’ is a direct translation from Greek for the ‘falling off’ of petals from a flower, or leaves from a tree, and this is extrapolated to the way in which cells ‘drop off’ from the life source within a being without disrupting the integrity of the whole. The physical appearance of cell death by accidental necrosis was well-documented even before the term ‘apoptosis’ was coined. ‘Necrosis’ translates from Greek simply as a descriptor of ‘death’. This stems from a tangible cell death morphology as opposed to apoptosis which is more elusive in its quiescent, ‘falling off’ nature.
Microscopy blinkered research efforts toward purely apoptotic molecular signaling and from the possibility of existence of other non-accidental cell death mechanisms. On a morphological basis of cell death, necrosis and apoptosis are polar extremes of each other. Necrotic plasma membrane permeabilisation (PMP) and explosive release of damage-associated molecular patterns (DAMPs), including a range of pro-inflammatory cytokines, is far removed from the quiescent and localized implosion incurred by an apoptotic cell.Citation6 It is remarkable therefore that the routes traveled to these ends are initiated by interwoven and mutual signaling cascades. As a consequence, from within 2 phenotypically diverse cell death events, a hybrid was born and designated necroptosis, a revelation which has greatly impacted on multiple fields of scientific research, especially oncology and disease. This has exposed a number of additional unique cell death modalities all of which contribute to the greater understanding of the cell death phenomenon.
Classical to emerging cell death processes
Apoptosis – a classic
Apoptosis functions as a regulatory mechanism for the natural removal of unwanted cells such as in gut epithelial turnover,Citation7 T cell homeostasis,Citation8 irrevocable DNA damage Citation9 or upon infection with a pathogen.Citation10 Classical apoptotic signaling culminates in the formation of apoptotic bodies containing putatively intact organelles, which can be engulfed by phagocytes in a contained and non-destructive manner.Citation1 This signaling pathway is suppressed by the players of a pro-survival cascade Citation11,12 which is leveraged by tumorigenic cells to enable cellular immortalisation. Some of the key features of apoptosis will be revised and should be referred to throughout the section.
Figure 1. A schematic overview of the intrinsic and extrinsic arms of apoptotic signaling. The extrinsic portion can be activated by ligands binding their cell surface death receptors such as FASL binding FAS, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) binding death receptor 5 (DR5) or tumor necrosis factor-α (TNFα) binding TNF receptor 1 (TNFR1). FAS-associated death domain protein (FADD) is recruited to the FASL-FAS or TRAIL-DR5 receptor complex as well as to the cytosolic complex IIa for activation of Caspase-8 in the context of FLIP inhibition and loss of cellular inhibitors of apoptosis (cIAPs). TNFR1–associated death domain protein (TRADD), receptor-interacting protein kinase 1 (RIP1) and TNFR2-associated factor-2 (TRAF2) are recruited to TNFα-bound TNFR1 forming the pro-survival complex I as a prerequisite to apoptotic signaling via this death receptor. K-63 and M-1 linked poly-ubiquitination of RIP 1 by cIAPs and LUBAC respectively, antagonised by the second mitochondria-derived activator of caspases (SMAC) and cylandromatosis (CYLD), stabilises complex I formation. This allows binding of the NF-κB essential modifier (NEMO) and transforming growth factor β-activated protein kinase (TAK) binding protein (TAB) to associate with RIP1 via the ubiquitin chains thus activating NF-κB and cell survival signaling. Pro-survival genes such as opa1, c-iaps, traf2, a20 and c-flip are transcribed in the nucleus. Intrinsic signaling requires removal of inhibition by BCL2 proteins as well as the activation of BAX and BAK and truncation of BID to tBID by active Caspase-8 for mitochondrial outer membrane permeabilisation (MOMP). This releases anti-apoptotic molecules such as SMAC and pro-apoptotic molecules, such as Cytochrome c. The apoptosome can then form by activated Caspase-9, apoptotic protease activating factor 1 (APAF1) and Cytochrome c and the 2 signaling arms culminate in executioner Caspase-3 and −7 activation for apoptosis. DNA damage results in autocrine TNFα signaling by activation of the E3 ubiquitin ligase EDD, association with RIP1 and c-Jun N-terminal kinase (JNK) pathway signaling to transcription of TNFα.
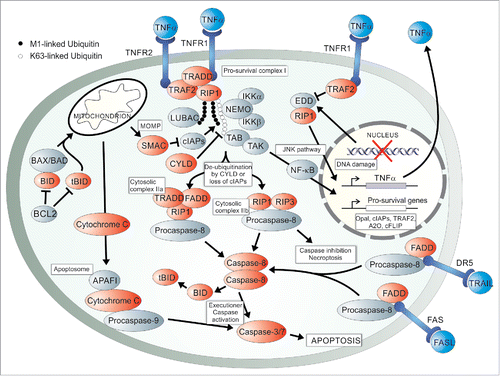
Execution of apoptotic cell death requires signal induction by extrinsic as well as intrinsic stimuli. The extrinsic pathway is activated by various death receptor-ligand interactions, to name a few, FAS ligand (FASL) binding FAS, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) binding death receptor 5 (DR5) and tumor necrosis factor-α (TNFα) binding TNF receptor 1 (TNFR1).Citation13,14 FAS activation results in the formation of a death-inducing signaling complex (DISC),Citation15 consisting of FASL-bound FAS or TRAIL-bound DR5, the intracellular FAS-associated death domain protein (FADD), cellular FLICE-like inhibitory protein (cFLIP) and Procaspase-8.Citation16 TNFα-bound TNFR1induces the formation of an independent complex, the pro-survival complex I, which will be the main focus in this review. The integral role of TNFα signaling in apoptosis will be described in the section on necroptosis initiating signals as this is a major point of overlap between these 2 modes of cell death and merits direct comparison.
The cytoplasmic complex I consists of TNFR1, TNFR1–associated death domain protein (TRADD), serine/threonine poly-ubiquitinated receptor interacting protein kinase 1 (RIP1) and cell death inhibitory TNFR2-associated factor 2 (TRAF2) bound to TNFR2.Citation7,12,17 Upon formation, one of 2 events can occur – cell death or cell survival. Under pro-survival cellular conditions, complex I activates the c-Jun N-terminal kinase (JNK) pathway and the IκB kinase (IKK) complex for transcription of the nuclear factor-κB (NF-κB) and pro-survival genes.Citation17 Contrary to survival signaling, death receptor activation culminating in apoptosis is dependent on caspase activity.Citation13 Complex I formation induced by TNFα-bound TNFR1 therefore has the paradoxical effect of stimulating cell death or survival while FAS activation is a more rapid and committing cell death event.Citation11,12
Initially it was believed that FADD and Procaspase-8 are recruited to the death domain of TNFR1 upon activation, as is the case with FAS signaling, but it was subsequently discovered that TRADD is recruited to the receptor and FADD plays an essential downstream role.Citation12,18 At this point, a molecule with pleiotropic functions is necessary for signaling transduction toward either cell survival or its functional antithesis, cell death. RIP1 volunteers itself as one of these crucial molecules, dependant on the cellular context. TRAF2 plays a pro-survival role within complex I as it recruits cellular inhibitors of apoptosis (cIAPs) to perform E3 ubiquitin ligase activity on RIP1.Citation19,20 RIP1 is then able to activate transcription factor NF-κB Citation11,21,22 which initiates transcription of pro-survival genes such as opa1,Citation23 ciaps, traf2, a20 and cflips.Citation13,24 TRAF2 also facilitates the addition of K48-linked ubiquitin to Caspase-8 thus inducing its proteasomal degradation and preventing apoptosis.Citation22 Upon loss of complex I-associated inhibitors, such as cIAPs and cFLIP, the cytosolic apoptotic complex IIa is allowed to form Citation12 and caspase activation must occur by relieving the inhibitory effect of cFLIPL.Citation13 cFLIPL forms an active heterodimer with Capsase-8 but reduces its canonical downstream cleavage potential. Removal of the inhibitor allows for auto-cleavage of Procaspase-8, formation of functional Caspase-8 homodimers and activation of the apoptotic executioner Caspases-3 and −7.Citation25,26 This point of the extrinsic apoptotic signaling arm converges with the classically termed ‘intrinsic’ pathway.
The internal wave of apoptotic signaling results in the formation of cytoplasmic inclusions known as apoptosomes which give an apoptotic cell its characteristic morphology.Citation27 These are the death effector complexes activated by mitochondrial outer membrane permeabilisation (MOMP), believed to be carried out by pro-apoptotic proteins such as BAD, BAX, BAK, BID and BIM, to name a few. Recently it has been shown that BAD is most likely dispensable and thus was omitted from . Citation28 BID is cleaved to a truncated form (tBID) by active Caspase-8 homodimers resulting in activation and dimerization of BAK/BAX and consequently MOMP.Citation29 Cytochrome c is released from the mitcochondria and binds to intracellular molecules, such as APAF-1, for apoptosome activation.Citation27 SMAC is another molecule released during MOMP and functions to enhance the apoptosis signaling pathway through cIAP inhibition.Citation30 SMAC-mimetic drugs are employed to activate apoptosis through cIAP degradation and consequent removal of a level of cell death signaling inhibition.Citation31,32 The intrinsic initiator Caspase-9 has recently been shown to bind the apoptosome in an allosteric manner Citation33 causing activation and downstream cleavage of executioner Caspase-7 or −3.Citation27 Classical apoptosis ensues resulting in the quiescent and non-inflammatory demise of the afflicted cell. It is of interest that the destructive potential of MOMP has been leveraged in a clinical setting for improved acute myeloid leukemia chemotherapy by priming mitochondria and causing increased tumor cell sensitivity to apoptotic death.Citation34 Manipulation of key apoptotic molecular players is a promising strategy for the treatment of disease both through its activation or inhibition, depending on the pathology and desired outcome.
Necrosis – a misunderstood classic
The second classical cell death modality, necrosis, is categorised by its putatively accidental occurrence. Morphologically, it involves the ‘explosive’ release of cellular contents, swelling of mitochondria and disruption of the cytoplasmic membrane,Citation35 all features unlike those found with typical apoptotic cell death. Necrosis was up until the turn of the 21st century believed to occur uncontrollably as a result of physical or chemical harm to the cell. This was such a firm belief to the extent that very little to no effort was made to understand how this occurs at a biochemical level.Citation36 It was during elucidative studies on apoptosis that the inkling of a programmable necrotic pathway was sparked.Citation37 Upon inhibition of apoptosis in mouse thymocyte cell culture with the pan-caspase inhibitor zVAD.fmk, Hirsch and colleagues observed a switch to necrosis instead of the expected death resistance.Citation37 While apoptosis has been shown to be highly efficient, non-destructive and pathophysiologically relevant, a crucial reason for the existence of non-apoptotic means of cell death could be to compensate for the inhibition of apoptosis by the ensuing cellular context, caused by pathogens, cancer biology or otherwise, thus preventing this type of cell death from occurring. Hirsch and colleagues thus fortuitously led the inquisition into programmable forms of necrosis. It seems that the line between accidental and programmed necrosis is growing hazier as a greater number of non-apoptotic, programmable forms of necrotic cell death emerge.
Emerging forms of programmed cell death
The previously black-and-white fallacy that cell death occurs either by apoptosis or accidental necrosis has been warped by the discovery of not one, but many programmable non-apoptotic forms of cell death. Ultimately, the various forms of cell death are more complex than a mono-categorical classification system and should be considered in isolation from, as well as within, the greater network that exists. Two of the many fundamentally independent modes of programmed necrotic cell death, the highly inflammatory ‘pyroptosis’ and the iron-dependent ‘ferroptosis’, will be explored briefly before delving into necroptosis as a new way of dying.
Death by fire, pyroptosis
One example of an important emerging form of programmed necrosis is pyroptosis, a strongly inflammatory response to certain pathogenic infections, such as Shigella flexneri.Citation38 Pyroptosis, like apoptosis and unlike necrosis, requires caspase activation and thus was initially classified as apoptosis but since has been fitted to a unique cell death model. As ‘pyro’ and ‘ptosis’ translate from Greek to mean ‘fire’ and ‘to fall’, the name pyroptosis was coined to evoke a sense of ‘falling fire’ attributed to the pro-inflammatory cytokines, namely IL-1β and IL-18, which burst forth from the dying cell.Citation38,39 Pyroptosis engages the ‘inflammasome’ Citation40 for cell death execution, a complex also activated during an innate immune response hence the highly inflammatory cell death characteristic of pyroptosis.Citation41 Signal induction activates Caspase-1, −4 and −5 in humans for cleavage of pro-IL-1β and –IL-18 as well as for cellular ‘explosion’, unlike the result of caspase activation in apoptosis which is the defining difference between these cell death modalities.Citation38 Gasdermin D was identified in 2015 as a key mediator of pyroptosis induced through cleavage by both non-canonical and canonical inflammasomes containing Caspases-4 and −5 or Caspase-1 respectively.Citation42,43 This was discovered through a forward genetics approach Citation43 and confirmed by CRISPR/Cas9 gene knock out and short interfering RNA (siRNA) mediated knock down of Gasdermin D.Citation42 The lack of Gasdermin D functioning in apoptosis and necroptosis was confirmed. The full complement of cell-death related functions as well as downstream mechanisms of action of Gasdermin D remain to be elucidated.
The quintessence of carcinogenesis is the collapse of cell death signaling such that immortality prevails. Colon cancer cell proliferation can be inhibited by Gasdermin D, now known to be a gastrointestinal cell tumor suppressor, by activation of pyroptosis.Citation44 Additionally in this cancer model, the liver X receptor β, a cytoplasmically located transcription factor, is also able to function in a non-transcriptional manner to induce cell death by pyroptosis through Caspase-1 activation.Citation45 These have the potential for development into novel treatment targets for colon cancer, one of the most prevalent and deadly global cancers. In the context of most cancers, an inflammatory cell death response would promote tumourigenesis by induction of angiogenesis and the potential for malignancy and thus pyroptosis becomes unfavourable. However, a surprising recent finding emerged that pyroptosis can proceed in a non-inflammatory manner in a malignant mesothelioma cell model thus favoring this cell death pathway for treatment of this cancer, usually promoted through inflammation. Caspase-1 activation but no IL-1β and IL-18 activation results in pyroptotic death when cells are treated with curcumin, a natural anticarcinogenic agent found in turmeric.Citation46 These 2 cancer models and one type of cell death alone are able to expose the intricate complexities within the system. The reflexive strategy may be to avoid pyroptosis induction in certain tumor models yet it is now evident that, upon deeper investigation, an alternative angle may exist to achieve the desired clinical outcome.
Death by iron, ferroptosis
Another important emerging, non-apoptotic cell death modality is ferroptosis, named for its iron-dependent mode of killing carried out by the excessive cellular production of reactive oxygen species (ROS).Citation47 The fate of such death is necrotic-like but results from a unique activation pathway to other programmable forms of necrosis and is morphologically characterized by condensation and outer membrane rupture of mitochondria.Citation48 Identification of ferroptosis occurred by the discovery of a small molecule activator called erastin from a screen of over 24 000 compounds.Citation49 Intracellular iron, its transporter transferrin, lipid-ROS Citation47 and functional lysosomes Citation50 are all essential for execution of ferroptosis. The role of iron and ROS in cell death are discussed in a recent review.Citation51
Artemisinin and its derivatives, drugs typically used for the treatment of malaria, have been shown to induce ferroptosis and kill pancreatic cancer cells Citation52 as well as those expressing high levels of transferrin-receptor when the drug is tagged to transferrin as these cells have a higher level of, and dependence on, intracellular iron.Citation53,54 A small molecule called sulfasalazine (SAS), commonly used to treat ulcerative colitis and rheumatoid arthritis, is an inducer of ferroptosis Citation47 and thus SAS and artemisinin derivatives are attractive avenues for chemotherapy development owing to the high current level of knowledge around these drugs as well as their availability. Ferroptosis has additional clinical relevance in that it has the propensity for selective lethality in RAS-mutant cancer cells when activated but prevents excitotoxic death of damaged neuronal cells when inhibited.Citation47 It is clear therefore that understanding whether ferroptosis activation or inhibition is the cause of an associated disease is imperative and could have a monumental impact on treatment strategies.
Pyroptosis and ferroptosis are only 2 of many newly described forms of cell death that are fundamentally unlike both classical apoptosis and necrosis. These and others have the potential to contribute immensely to our understanding of cell death in the context of cancer biology, disease pathogenesis and the development of appropriate treatment strategies.
Necroptosis – a revolutionary chimaera of apoptosis and necrosis
The novel chimera born from apoptosis and necrosis, aptly named necroptosis, occurs by a programmable mechanism with a characteristic necrotic cell death phenotype.Citation36 The term necroptosis has been used on occasion interchangeably with programmed or regulated necrosis Citation55-57 but this is too general a description, especially considering alternative programmed necrotic cell death modalities which are now known to exist such as pyroptosis and ferroptosis. Necroptosis is thus a unique cell death pathway which functions in the greater network of cell death signaling and has provided a pivotal novel outlook on death.
In 1972, Kerr and colleagues Citation1 coined the term ‘apoptosis’ and proposed this to be the only form of programmed cell death. A mere 3 y later, Carswell and colleagues Citation58 named tumor necrosis factor (TNF) and the inquest into a form of cell death which was necrotic yet programmable and undeniably non-apoptotic was sparked. TNF was found to be a necrosis-inducing endotoxin for neoplastic cells, hence the designation, but was later shown to induce necroptosis as well as apoptosis. The molecular switches involved in this deathly decision will be discussed further.
The necroptotic signaling pathway
Necroptosis forms part of a complex network of cross-talk interactions with a number of critical molecular switches. Important studies have been conducted contributing to the paradigm shift in our understanding of programmed necrosis.Citation55,59-61 The 2 major necroptotic death effector complexes, the necrosome and ripoptosome, are induced by TNFR1 and toll like receptor 3 (TLR3) signaling respectively.Citation62,63 Caspase inactivity or absence must prevail for necroptosis to ensue, characterized by cellular rounding, an increase in cytosolic calcium ions and ROS, depletion of ATP and intracellular acidification and ultimately cellular swelling and release of DAMPs following plasma membrane rupture.Citation64,65 The most recent NCCD guidelines advocate the use of 3 terms for the description of specific phases of cell death.Citation4 These include ‘initiation’ for reversible and non-committing signaling, ‘execution’ for the irreversible progression toward death and ‘propagation’ for the consequent waves of inflammation or release of molecular patterns. These terms will dictate the upcoming narrative around the necroptotic molecular players and recent scientific research findings, depicted in .
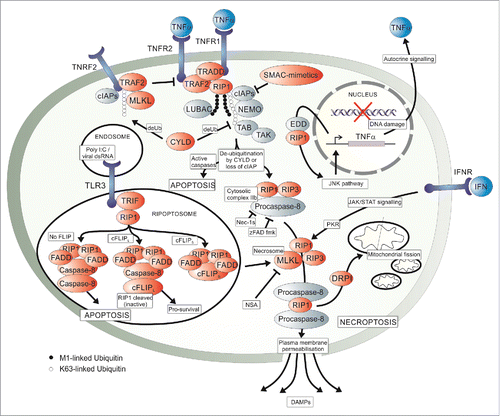
Figure 2. A schematic overview of the molecular pathways involved in necroptosis. Signaling can occur via 4 mentioned modes, namely TNFα binding TNFR1 in a paracrine as well as autocrine manner, interferon (IFN) binding IFN receptors (IFNR) and endosomal viral double stranded RNA (dsRNA) binding toll-like receptor 3 (TLR3). TNFα-induced signaling results in the formation of the pro-survival complex I which signals to apoptosis, necroptosis or, as seen in , to cell survival. An NF-κB-independent JNK signaling pathway via EDD activation following DNA damage and association with RIP1 results in autocrine TNFα production, also seen in . TRAF2 stabilization of complex I is inhibited by the interaction between the mixed lineage kinase domain-like protein (MLKL), TRAF2 and TNFR2 as promoted by cIAP-induced TRAF2 ubiquitination. Removal of caspase inhibition and ubiquitination of RIP1, either by loss of cellular inhibitors of apoptosis (cIAPs) or deubiquitination by cylindromatosis (CYLD), allows for the formation of the cytosolic complex IIa and IIb which may result in apoptosis or, upon inhibition of caspases, necroptosis via the formation of the necrosome. The necrosome forms by the interaction between RIP1, RIP3 and MLKL which then results in the interaction between RIP3 and the long and short isoforms of the phosphoglycerate mutase family member 5 protein (PGAM5) to activate dynamin-related protein (DRP1) or induce plasma membrane permeabilisation (PMP). DRP1 causes mitochondrial fission and PMP results in the release of damage-associated molecular patterns (DAMPs). The ripoptosome can be recruited to the necrosome by an unknown mechanism to result in necroptotic cell death. Within the ripotosome, cellular FLIP isoforms either promote or inhibit cell death. Dimerised RIP1 interacts with FADD to either activate Caspase-8 or, if cFLIPL dimerises with Caspase-8 a non-canonical catalytic complex is formed both resulting in inactivation of RIP1/3. One outcome is apoptosis and the other pro-survival signaling. Another pro-survival cascade involves NF-κB-dependent JNK and IKK signaling, as seen in . The pathway can be artificially activated by SMAC-mimetics, as well as zFAD.fmk through the inhibition of apoptosis or by activation of ripoptosome formation with Poly(I:C). Necroptosis is inhibited experimentally by inhibition of RIP1 kinase activity with Nec-1s and at MLKL by Necrosulfonamide (NSA).
Initiation
Ligand-receptor interactions are external stimuli for initiation of necroptosis, namely by TNFα-induced TNFR1, interferon (IFN)-induced IFN receptor (IFNR),Citation66 and FASL-induced FAS Citation67 signaling. IFNR activation, primarily by type-I IFN,Citation68 is believed to involve a Caspase- and FADD-independent, RIP3-dependent mode of cell death via the formation of the necrosome.Citation66 Following IFNR activation, JAK/STAT signaling and the activity of RNA-responsive protein kinase (PKR) upstream of RIP1/RIP3 necrosome formation is essential.Citation66 Downstream signaling is thought to occur in much the same manner as that following TNFα-TNFR1 induced necrosome formation. TNFα binding to TNFR1 causes recruitment of TRADD and RIP1 via their death domains for formation of the pro-survival complex I Citation7,12 and is stabilised by TNFα-bound TNFR2-TRAF2.Citation21 Internalisation of the TNFR1-TRADD-RIP1 complex is required for recruitment of Caspase-8 and FADD, necessary for apoptosis.Citation69 This is therefore a major cell death checkpoint as the absence of NF-κB activation and pro-survival signaling results in pro-apoptotic complexes IIa and IIb Citation12 or, alternatively, the pro-necroptotic complex known as the necrosome.
Internal initiating signals include viral dsRNA-induced TLR3 activation Citation14 and DNA damage,Citation17 among others. Viral dsRNA activates TLR3, an endosomally-located membrane receptor,Citation63 to recruit an adapter known as TRIF followed by RIP1 thus inducing the formation of the ripoptosome which in turn can engage the apoptosome or necrosome.Citation70 The ripoptosome consists of FADD, cFLIP and Caspase-8 and allows necroptosis to prevail if active cleavage of RIP1 by Caspase-8 is prevented by cFLIPL.Citation63 The TLR3-induced pathway therefore converges with the TNFR1-induced pathway at the necrosome. Whether there is inclusion of the necrosome into the ripoptosome or not is yet to be uncovered.Citation63 Alternatively, upon DNA damage, the tumor suppressor protein EDD is activated and triggers TNFα transcription via the JNK pathway.Citation17 This results in autocrine TNFα signaling and cell death if damage is extensive enough (). If the intracellular conditions allow, all initiating signals received by the cell as described above have the potential to be transduced toward cell death by necroptosis.
Intracellular conditions associated with initiation or suppression of necroptosis are set by a range of modulatory proteins or experimental molecules. Poly(I:C) is a synthetic dsRNA analog and used for initiation of the TLR3-mediated pathway.Citation63 cIAPs, already alluded to earlier (), function within complex I to prevent both apoptosis and necroptosis.Citation11,71 M1-linked ubiquitin chains, loaded by the linear ubiquitin chain assembly complex (LUBAC), and linear K-63 ubiquitin chains are added to RIP1 to stabilize the pro-survival complex I.Citation72 The hybrid ubiquitination of RIP1 allows for simultaneous binding of the pro-survival molecules, transforming growth factor β-activated protein kinase (TAK) and its binding protein (TAB) Citation73 and the NF-κB essential modifier (NEMO).Citation74,75 Both types of ubiquitin need to be removed, or prevented from forming, before death signaling can proceed.Citation76,77 To achieve this, SMAC or synthetic SMAC-mimetics, result in cIAP degradation and thus reduction in RIP1 ubiquitination, NF-κB is suppressed and cFLIPs are not produced allowing for Caspase-8 activation.Citation17,71 To prevent Caspase-8 activation, the long cFLIP isoform (cFLIPL) binds to Procaspase-8, inducing its autocleavage and forming a proteolytic complex containing active, but non-homodimerised Caspase-8.Citation25 This prevents the interaction of Caspase-8 with FADD, required for apoptosis.Citation78 While apoptosis is inhibited, non-canonical Caspase-8/cFLIPL dimer functioning results in the cleavage of RIP1/RIP3 for simultaneous inhibition of necroptosis.Citation25 Loss of cFLIPL or activation of cFLIPS within the ripoptosome induces caspase-dependent apoptosis or caspase-independent necroptosis. The involvement of cFLIPs in necrosome or complex II activation has not been fully clarified but it is thought to be essential.Citation63
A mechanism for necroptosis inhibition involves de-ubiquitination of RIP1 by cylindromatosis (CYLD).Citation77 Caspase-8 activity, besides resulting in cleavage of RIP1 and RIP3,Citation79 causes cleavage of CYLD and this is thought to enhance the inhibition of necroptosis by increasing survival signaling through the ubiquitinated RIP1-NF-κB pathway while apoptosis is still allowed to transpire.Citation80 The complex interplay between these cellular conditions forms the basis for either allowing or preventing the execution of necroptosis.
Execution
The execution phase of necroptosis to be discussed here revolves around the functioning of the necrosome. The interaction between the RIP homotypic interaction motifs of RIP1 and RIP3 is dependent on their active kinase domains, as well as Caspase-8 inactivation, and generates a highly stable filamentous amyloid-like structure.Citation62 RIP1 and RIP3 ubiquitination by CHIP (carboxyl terminus of Hsp-70-interacting protein), a recently discovered key regulator of the necrosome, results in destabilisation of this amyloid fibril.Citation81 A pro-necroptotic cancer therapeutic could therefore suppress CHIP in tumor cells and sensitize them to TNF-mediated necroptosis but this remains to be explored. In order to suppress necroptosis for experimental purposes, the synthetic ATP-competitive kinase domain inhibitor called necrostatin-1 stable (Nec-1s),Citation82 a modified form of Nec-1 with improved specificity,Citation83 is commonly used. As RIP1 kinase activity is essential for cell death signaling but RIP1 itself is only required as a platform for signaling to NF-κB and cell survival, this inhibitor has an effect only on execution, not initiation of necroptosis. This phenomenon further emphasizes the pleiotropic capacity of RIP1.Citation84
Following stabilization of the RIP1-RIP3 complex, mixed-lineage kinase domain-like protein (MLKL) is recruited to form a functional necrosome.Citation57,85, 86 Experimental elucidation of MLKL functioning has been done through inhibition with necrosulfonamide (NSA), a specific inhibitor of this molecule. MLKL is a pseudokinase activated upon phosphorylation by RIP3 and subsequent trimerisation which results in the translocation of the MLKL-necrosome to the plasma membrane.Citation65 This results in characteristically necroptotic plasma membrane disruption by MLKL trimers binding to phosphatidylinositol phosphates (PIPs) on the inner surface of the plasma membrane Citation87 and release of liposomes containing PIPs.Citation88 This permeabilisation, combined with MLKL-mediated calcium Citation65 or sodium Citation89 influx ion-pore dysregulation, epitomises the model proposed for necroptosis execution. Conversely, cIAP-mediated TRAF2 ubiquitination has been found to cause constitutive sequestration of MLKL from the necrosome, preventable by TNFα signaling.Citation22 CYLD is capable of disrupting the TRAF2-MLKL interaction therefore it appears that CYLD and TRAF2, also key initiator molecules, play an integral role in determining whether necroptosis is executed or not by controlling cellular MLKL localization.
Another proposed target of the active MLKL-necrosome is the phosphoglycerate mutase family member 5 protein (PGAM5).Citation57,90, 91 This activates dynamin-related protein 1 (DRP1) GTPase activity causing DRP1 dimerization and consequently mitochondrial fission.Citation92 A recent study exposes the potential dispensability of PGAM5 in necroptosis,Citation93 however, the mitochondrial effects are characteristic of necroptosis and it is likely that there exists a redundant execution signaling network leading to cell death.
It was suggested decades ago that mitochondrial permeability transition (MPT) occurs early during necrotic cell death Citation37 and there have been debates as to whether this is crucial or corollary for necroptosis. The essential apoptotic MOMP proteins, BAK and BAX, have been implicated in MPT by Karch et al. Citation94 to effect necrotic mitochondrial pore-dependent cell death through a mechanism that differs from apoptotic MOMP.Citation94 The effect of mitochondrial disruption is thought to cause ATP-depletion and excessive ROS production to contribute to cell death execution.Citation95 ROS have been implicated in both induction and execution of killing by necroptosis Citation96 in a positive feedback loop that is independent of PGAM5 and DRP1,Citation97 described in more detail in a recent review.Citation98 This highlights the elusiveness of all possible downstream or independent interactions from the necrosome and possibilities for evasion of certain portions of the necroptotic signaling pathway. The full picture of necroptotic cell death execution is emerging but the extent of what remains may be vast. Necroptotic cell death is inflammatory and propagative and thus the molecular interactions do not end here.
Propagation
The final stage of necroptotic cell death, referred to as propagation, involves an inflammatory wave of damage-associated molecular pattern (DAMP) release and immune cell activation.Citation99 Primary immune cells, such as macrophages, dendritic cells and natural killer cells, are induced.Citation61 This would be expected to result in the recruitment of inflammatory and cytotoxic cells to the necrotic site, allow for phagocytosis of damaged and infected cells and cause cell death signaling to cease, however, it is known that TNFα has the tendency to induce chronic inflammation through a putative ‘immunosuppressive’ mechanism.Citation100 A key contributor to both chronic and acute inflammation is the production of DAMPs, specifically cytokines and other chemo-attractants, for the recruitment of primary immune cells to the necroptotic site.
DAMPs are characteristically released during necroptosis and some well-elucidated include the high mobility group box protein 1 and interleukins-1α and −33.Citation101 General DAMPs, such as lysosomal hexosiminidase and cytosolic lactate dehydrogenase, as well as organ-specific, such as liver alanine aminotransferase and heart or kidney creatine kinase were found to be released during TNFα-induced necroptosis.Citation72 The full range of DAMPs and potential tissue specificities involved in necroptotic cell death requires deeper investigation. Necroptosis-specific DAMPs could be leveraged for diagnostic biomarker development when compared with those specific to alternative programmed necrotic cell death events, such as pyroptosis or ferroptosis. Because propagation is the irreversible outcome of cell death signaling, initiation and execution of necroptosis are of major clinical interest, especially in terms of cancer development and treatment. To reiterate the complexity of cell death, however, one example where propagative characteristics of necroptosis have been leveraged for cancer treatment will be discussed in the next section.
A novel outlook on oncological immortality
There exists a logical correlation between cancer cell immortality and the ability to evade cell death by one or more processes. Progress made in uncovering necroptosis as a novel form of cell death and the key molecular players involved has thus had a monumental impact on the field of oncology. Unlike apoptosis, however, necroptosis is inflammatory, a condition highly favorable for tumourigenesis. The role of NF-κB in inflammation and cancer was recently reviewed to provide insight into the complexity of immuno-surveillance for the elimination of oncogenic cells and inflammation for enhanced cancer progression and metastasis.Citation103 Before the discovery of necroptosis as a new way of dying, apoptosis was the intended fate for cancer cells but resistance to this type of cell death has since emerged.Citation104 A major driver of apoptosis resistance is the increased expression of cIAPs in a wide range of tumor cells but additional mechanisms also exist to circumvent induction of apoptosis by therapeutics, a topic recently reviewed.Citation104 Since the discovery of necroptosis, knowledge gained has contributed to the development of novel treatment strategies as well as understanding previously unknown causes of cancer cell death resistance.
Combatting oncogenic apoptosis resistance by pro-necroptosis treatment has been a major research focus with some interesting findings, a few of which will be discussed. One of the first small molecule necroptosis inducers to be identified as a potential pro-necroptotic antitumour agent in the face of apoptosis resistance is Shikonin.Citation105 Interestingly, this molecule is obtained from a natural herb, was also found also to inhibit HIV infection through chemokine co-receptor interference Citation106 and can have anti-inflammatory properties.Citation107 Shikonin has been shown to cause necroptotic cell death in multiple tumor cell types, from solid to haematological, including osteosarcoma Citation108 and multiple myeloma (MM) Citation109 respectively. The broad applicability of this molecule and its derivatives provides a promising strategy for the treatment of a variety of cancers but requires further investigation into how it causes cell death in some contexts yet is anti-inflammatory and protective in others.
Ovarian cancer is a low incidence but high mortality rate cancer known to cause patient relapse and develop resistance to the common pro-apoptosis therapeutics such as cisplatin.Citation110 A strategy to overcome this was investigated through cIAP inhibition by SMAC-mimetics combined with caspase inhibition.Citation111 An advantageous feature of serous, and possibly other, ovarian cancer cell types is the increased expression of RIP3 which assisted the selective killing of these cells by a TNFα/RIP1/RIP3-dependent necroptotic cell death mechanism.Citation111 RIP3 could potentially influence a range of cancers as it has been found to be mutated or levels of expression altered in a number of studies.Citation112,113 It is thus crucial to understand the genetic context of key necroptotic cell death players in cancer- and particularly patient-specific cell types for the development and administration of appropriate therapeutics.
Fingolimod (FTY720) is a versatile therapeutic used for chronic myelogenous leukemia treatment and for immunosuppressance in multiple sclerosis Citation114 but beyond this, its preclinical efficacy in apoptosis resistant lung cancer Citation115 as well as human glioblastoma cells Citation116 was recently investigated. Ceramide is a sphingolipid required for relieving the inhibition of a tumor suppressor protein, PP2A.Citation115 In apoptosis resistant lung cancer cells, ceramide was found to be downregulated and the inhibitor of PP2A upregulated. FTY720 acts as a synthetic ceramide analog to relieve PP2A inhibition and allow its activation resulting in RIP1-dependent necroptosis.Citation115 Interestingly, neither RIP3 nor Drp1 were required for this process indicating that RIP1 could have the propensity to execute necroptotic cell death by a non-canonical mechanism, at least in this cell line, but this remains to be elucidated. In human glioblastoma cells, FTY720 treatment resulted in a variety of cell death processes including autophagy, apoptosis and necroptosis.Citation116 The immunosuppressant properties of FTY720 are independent of death induction, a distinction made possible through specific phosphorylation of the molecule. Thus the antitumor properties cannot be linked to immunosuppression, a property which would be attractive in the context of immunity and cancer. The broad preclinical and clinically established efficacy of FTY720 as an antitumor agent makes this an important molecule for further investigation.
Another interesting and alternative treatment strategy for cancer was recently described in a study that used necroptotic cancer cells to induce adaptive immunity toward colon cancer cells in mice.Citation117 Immunogenic apoptosis is the currently employed strategy to achieve this effect thus the potential for immunogenic necroptosis is attractive when apoptosis resistance must be overcome. Interestingly, accidental necrosis caused by freeze-thawing the cancer cells before treatment had no antitumor effect in vitro or in mice thus it is imperative that the necroptotic signaling pathway be induced. A cancer vaccination leveraging the propagative properties of necroptosis could cause a major paradigm shift in cancer treatment and greatly reduce the morbidity and mortality of this prevalent disease thus positively impacting on the lives of millions globally.
Various aspects of the necroptotic pathway discussed play a role in tumourigenesis and can be targeted for treatment. The complexity of the system becomes more and more apparent the deeper we understand cancer-specific death evasion strategies. One molecular aspect not discussed in this review is the involvement of microRNAs (miRNAs) in cell death and survival signaling which may play a far more significant role than current research output implies. There have been a handful of studies conducted Citation118,119 and reviews written Citation120 with regards to this but the full complement of miRNAs, their differential roles in necroptotic signaling as well as the result of dysregulation in the context of cancer requires a far greater research input. The emergence of necroptosis as a new way of dying has broadened the armoury of anti-tumourigenic treatment strategies, especially in the context of apoptosis resistance, but there is still much room within this revelation for the rapidly expanding field of cancer biology.
Conclusions
Apoptosis is joined by necroptosis, a new way of dying, and other emerging modalities such as pyroptosis and ferroptosis in the ever-expanding repertoire of programmable forms of cell death. Recent advancements have had a monumental and positive impact on the understanding of eukaryotic cell signaling toward life versus death. The clinical implications of necroptosis, pyroptosis, ferroptosis and the many other emerging forms of programmed necrosis extend across a range of pathologies and disease models but play an obvious and prominent role in cancer, the ultimate imbalance between cellular life and death. A deeper understanding of the tissue- and cell type-specific mechanisms of necroptotic cell death is still required to unleash its full clinical potential. A key area for future research exists downstream of the necrosome as the entire complement of necroptosis death effectors remain to be fully elucidated. The involvement of miRNAs in cell survival and death signaling may uncover a plethora of new and critical interactions to be considered for both research and clinical purposes. The discovery of necroptosis has had a powerful influence in the realm of life vs. death and has sparked research that will have a meaningful impact on the lives of many people. Ultimately, it has bridged the gaps in knowledge around eukaryotic cell death as well as magnified the great rifts which remain, especially in the research efforts made to conquer the major killer that is cancer.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
I acknowledge funding support in the form of scholarship support from University of the Witwatersrand Post Graduate Merit Award, the Claude Leon Harris Honors Scholarship Program bursary and a grant provided by the RNA Therapeutics Research Group Bursary Scheme. I wish to thank Marc S. Weinberg and Natalie Whalley for technical support and critical reading of the manuscript.
References
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972; 26:239-57; PMID:4561027; http://dx.doi.org/10.1038/bjc.1972.33
- Hardy K, Handyside AH, Winston RM. The human blastocyst: cell number, death and allocation during late preimplantation development in vitro. Development 1989; 107:597-604; PMID:2612378
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19:107-20; PMID:21760595; http://dx.doi.org/10.1038/cdd.2011.96
- Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ 2015; 22:58-73; PMID:25236395; http://dx.doi.org/10.1038/cdd.2014.137
- Lockshin RA, Williams CM. Programmed Cell Death -II. Endocrine Potentiation of the Breakdown of the Intersegmental Muscles of Silkmoths. J Ins Physiol 1964; 10:643-9; http://dx.doi.org/10.1016/0022-1910(64)90034-4
- Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 2013; 38:209-23; PMID:23438821; http://dx.doi.org/10.1016/j.immuni.2013.02.003
- Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, Eftychi C, Lin J, Corona T, Hermance N, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 2014; 513:90-4; PMID:25132550; http://dx.doi.org/10.1038/nature13608
- Bouillet P, O'Reilly LA. CD95, BIM and T cell homeostasis. Nat Rev Immunol 2009; 9:514-9; PMID:19543226; http://dx.doi.org/10.1038/nri2570
- Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med 2006; 12:440-50; PMID:16899408; http://dx.doi.org/10.1016/j.molmed.2006.07.007
- Nogueira CV, Lindsten T, Jamieson AM, Case CL, Shin S, Thompson CB, Roy CR. Rapid pathogen-induced apoptosis: a mechanism used by dendritic cells to limit intracellular replication of Legionella pneumophila. PLoS pathogens 2009; 5:e1000478; PMID:19521510; http://dx.doi.org/10.1371/journal.ppat.1000478
- Shu HB, Takeuchi M, Goeddel DV. The tumor necrosis factor receptor 2 signal transducers TRAF2 and c-IAP1 are components of the tumor necrosis factor receptor 1 signaling complex. Proc Natl Acad Sci U S A 1996; 93:13973-8; PMID:8943045; http://dx.doi.org/10.1073/pnas.93.24.13973
- Micheau O, Tschopp J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 2003; 114:181-90; PMID:12887920; http://dx.doi.org/10.1016/S0092-8674(03)00521-X
- Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol 2001; 21:5299-305; PMID:11463813; http://dx.doi.org/10.1128/MCB.21.16.5299-5305.2001
- Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 2000; 1:489-95; PMID:11101870; http://dx.doi.org/10.1038/82732
- Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J 1995; 14:5579-88; PMID:8521815
- Lavrik IN, Krammer PH. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ 2012; 19:36-41; PMID:22075988; http://dx.doi.org/10.1038/cdd.2011.155
- Christofferson DE, Li Y, Hitomi J, Zhou W, Upperman C, Zhu H, Gerber SA, Gygi S, Yuan J. A novel role for RIP1 kinase in mediating TNFalpha production. Cell Death Dis 2012; 3:e320; PMID:22695613; http://dx.doi.org/10.1038/cddis.2012.64
- Harper N, Hughes M, MacFarlane M, Cohen GM. Fas-associated death domain protein and caspase-8 are not recruited to the tumor necrosis factor receptor 1 signaling complex during tumor necrosis factor-induced apoptosis. J Biol Chem 2003; 278:25534-41; PMID:12721308; http://dx.doi.org/10.1074/jbc.M303399200
- Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008; 30:689-700; PMID:18570872; http://dx.doi.org/10.1016/j.molcel.2008.05.014
- Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, Fairbrother WJ, Vucic D. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem 2008; 283:24295-9; PMID:18621737; http://dx.doi.org/10.1074/jbc.C800128200
- Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, Lenardo MJ. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem 2003; 278:51613-21; PMID:14532286; http://dx.doi.org/10.1074/jbc.M305633200
- Petersen SL, Chen TT, Lawrence DA, Marsters SA, Gonzalvez F, Ashkenazi A. TRAF2 is a biologically important necroptosis suppressor. Cell Death Differ 2015; 22(11):1846-57; PMID:25882049; http://dx.doi.org/10.1038/cdd.2015.35
- Muller-Rischart AK, Pilsl A, Beaudette P, Patra M, Hadian K, Funke M, Peis R, Deinlein A, Schweimer C, Kuhn PH, et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell 2013; 49:908-21; PMID:23453807; http://dx.doi.org/10.1016/j.molcel.2013.01.036
- Verhelst K, Carpentier I, Kreike M, Meloni L, Verstrepen L, Kensche T, Dikic I, Beyaert R. A20 inhibits LUBAC-mediated NF-kappaB activation by binding linear polyubiquitin chains via its zinc finger 7. EMBO J 2012; 31:3845-55; PMID:23032186; http://dx.doi.org/10.1038/emboj.2012.240
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011; 471:363-7; PMID:21368763; http://dx.doi.org/10.1038/nature09852
- Pop C, Oberst A, Drag M, Van Raam BJ, Riedl SJ, Green DR, Salvesen GS. FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J 2011; 433:447-57; PMID:21235526; http://dx.doi.org/10.1042/BJ20101738
- Kim HE, Du F, Fang M, Wang X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc Natl Acad Sci U S A 2005; 102:17545-50; PMID:16251271; http://dx.doi.org/10.1073/pnas.0507900102
- Ottina E, Sochalska M, Sgonc R, Villunger A. The BH3-only protein Bad is dispensable for TNF-mediated cell death. Cell Death Dis 2015; 6:e1611; PMID:25611386; http://dx.doi.org/10.1038/cddis.2014.575
- Cabon L, Galan-Malo P, Bouharrour A, Delavallee L, Brunelle-Navas MN, Lorenzo HK, Gross A, Susin SA. BID regulates AIF-mediated caspase-independent necroptosis by promoting BAX activation. Cell Death Differ 2012; 19:245-56; PMID:21738214; http://dx.doi.org/10.1038/cdd.2011.91
- Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000; 102:33-42; PMID:10929711; http://dx.doi.org/10.1016/S0092-8674(00)00008-8
- Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, Benetatos CA, Chunduru SK, Condon SM, McKinlay M, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 2007; 131:682-93; PMID:18022363; http://dx.doi.org/10.1016/j.cell.2007.10.037
- Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007; 131:669-81; PMID:18022362; http://dx.doi.org/10.1016/j.cell.2007.10.030
- Wurstle ML, Rehm M. A systems biology analysis of apoptosome formation and apoptosis execution supports allosteric procaspase-9 activation. J Biol Chem 2014; 289:26277-89; PMID:25107908; http://dx.doi.org/10.1074/jbc.M114.590034
- Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, Deangelo DJ, Frattini MG, Letai A. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 2012; 151:344-55; PMID:23063124; http://dx.doi.org/10.1016/j.cell.2012.08.038
- Festjens N, Vanden Berghe T, Vandenabeele P. Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochimica et biophysica acta 2006; 1757:1371-87; PMID:16950166; http://dx.doi.org/10.1016/j.bbabio.2006.06.014
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005; 1:112-9; PMID:16408008; http://dx.doi.org/10.1038/nchembio711
- Hirsch T, Marchetti P, Susin SA, Dallaporta B, Zamzami N, Marzo I, Geuskens M, Kroemer G. The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene 1997; 15:1573-81; PMID:9380409; http://dx.doi.org/10.1038/sj.onc.1201324
- Sansonetti PJ, Phalipon A, Arondel J, Thirumalai K, Banerjee S, Akira S, Takeda K, Zychlinsky A. Caspase-1 activation of IL-1beta and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity 2000; 12:581-90; PMID:10843390; http://dx.doi.org/10.1016/S1074-7613(00)80209-5
- Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Munoz-Arias I, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014; 505:509-14; PMID:24356306; http://dx.doi.org/10.1038/nature12940
- Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004; 430:213-8; PMID:15190255; http://dx.doi.org/10.1038/nature02664
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002; 10:417-26; PMID:12191486; http://dx.doi.org/10.1016/S1097-2765(02)00599-3
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015; 526:660-5; PMID:26375003; http://dx.doi.org/10.1038/nature15514
- Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015; 526:666-71; PMID:26375259; http://dx.doi.org/10.1038/nature15541
- Saeki N, Usui T, Aoyagi K, Kim DH, Sato M, Mabuchi T, Yanagihara K, Ogawa K, Sakamoto H, Yoshida T, et al. Distinctive expression and function of four GSDM family genes (GSDMA-D) in normal and malignant upper gastrointestinal epithelium. Genes Chromosomes Cancer 2009; 48:261-71; PMID:19051310; http://dx.doi.org/10.1002/gcc.20636
- Derangere V, Chevriaux A, Courtaut F, Bruchard M, Berger H, Chalmin F, Causse SZ, Limagne E, Vegran F, Ladoire S, et al. Liver X receptor beta activation induces pyroptosis of human and murine colon cancer cells. Cell Death Differ 2014; 21:1914-24; PMID:25124554; http://dx.doi.org/10.1038/cdd.2014.117
- Miller CV, Cook IS, Jayaramachandran R, Tyers AG. Spontaneous regression of a conjunctival malignant melanoma. Orbit 2014; 33:139-41; PMID:24295209; http://dx.doi.org/10.3109/01676830.2013.851708
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012; 149:1060-72; PMID:22632970; http://dx.doi.org/10.1016/j.cell.2012.03.042
- Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ 2016; 23:369-79; PMID:26794443; http://dx.doi.org/10.1038/cdd.2015.158
- Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007; 447:864-8; PMID:17568748; http://dx.doi.org/10.1038/nature05859
- Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, Suzuki T, Mori M, Yoshimoto Y, Takeuchi T, et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J 2016; 473:769-77; PMID:26759376; http://dx.doi.org/10.1042/BJ20150658
- Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol 2014; 10:9-17; PMID:24346035; http://dx.doi.org/10.1038/nchembio.1416
- Eling N, Reuter L, Hazin J, Hamacher-Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015; 2:517-32; PMID:26097885; http://dx.doi.org/10.18632/oncoscience.160
- Lai H, Sasaki T, Singh NP. Targeted treatment of cancer with artemisinin and artemisinin-tagged iron-carrying compounds. Expert Opin Ther Targets 2005; 9:995-1007; PMID:16185154; http://dx.doi.org/10.1517/14728222.9.5.995
- Ooko E, Saeed ME, Kadioglu O, Sarvi S, Colak M, Elmasaoudi K, Janah R, Greten HJ, Efferth T. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine 2015; 22:1045-54; PMID:26407947; http://dx.doi.org/10.1016/j.phymed.2015.08.002
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009; 137:1112-23; PMID:19524513; http://dx.doi.org/10.1016/j.cell.2009.05.037
- Trichonas G, Murakami Y, Thanos A, Morizane Y, Kayama M, Debouck CM, Hisatomi T, Miller JW, Vavvas DG. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci U S A 2010; 107:21695-700; PMID:21098270; http://dx.doi.org/10.1073/pnas.1009179107
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013; 39:443-53; PMID:24012422; http://dx.doi.org/10.1016/j.immuni.2013.06.018
- Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A 1975; 72:3666-70; PMID:1103152; http://dx.doi.org/10.1073/pnas.72.9.3666
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009; 325:332-6; PMID:19498109; http://dx.doi.org/10.1126/science.1172308
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009; 137:1100-11; PMID:19524512; http://dx.doi.org/10.1016/j.cell.2009.05.021
- Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008; 135:1311-23; PMID:19109899; http://dx.doi.org/10.1016/j.cell.2008.10.044
- Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012; 150:339-50; PMID:22817896; http://dx.doi.org/10.1016/j.cell.2012.06.019
- Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Hacker G, Leverkus M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 2011; 43:449-63; PMID:21737330; http://dx.doi.org/10.1016/j.molcel.2011.06.011
- Vanden Berghe T, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N, Guerin CJ, Brunk UT, Declercq W, Vandenabeele P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ 2010; 17:922-30; PMID:20010783; http://dx.doi.org/10.1038/cdd.2009.184
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 2014; 16:55-65; PMID:24316671; http://dx.doi.org/10.1038/ncb2883
- Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, Rall GF, Degterev A, Balachandran S. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S A 2013; 110:E3109-18; PMID:23898178; http://dx.doi.org/10.1073/pnas.1301218110
- Hadji A, Ceppi P, Murmann AE, Brockway S, Pattanayak A, Bhinder B, Hau A, De Chant S, Parimi V, Kolesza P, et al. Death induced by CD95 or CD95 ligand elimination. Cell Rep 2014; 7:208-22; PMID:24656822; http://dx.doi.org/10.1016/j.celrep.2014.02.035
- McComb S, Cessford E, Alturki NA, Joseph J, Shutinoski B, Startek JB, Gamero AM, Mossman KL, Sad S. Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc Natl Acad Sci U S A 2014; 111:E3206-13; PMID:25049377; http://dx.doi.org/10.1073/pnas.1407068111
- Chan FK, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Ann Rev Immunol 2015; 33:79-106; PMID:25493335; http://dx.doi.org/10.1146/annurev-immunol-032414-112248
- Blander JM. A long-awaited merger of the pathways mediating host defence and programmed cell death. Nat Rev Immunol 2014; 14:601-18; PMID:25145756; http://dx.doi.org/10.1038/nri3720
- Moulin M, Anderton H, Voss AK, Thomas T, Wong WW, Bankovacki A, Feltham R, Chau D, Cook WD, Silke J, et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J 2012; 31:1679-91; PMID:22327219; http://dx.doi.org/10.1038/emboj.2012.18
- Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 2011; 35:908-18; PMID:22195746; http://dx.doi.org/10.1016/j.immuni.2011.09.020
- Li H, Kobayashi M, Blonska M, You Y, Lin X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J Biol Chem 2006; 281:13636-43; PMID:16543241; http://dx.doi.org/10.1074/jbc.M600620200
- Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell 2009; 36:831-44; PMID:20005846; http://dx.doi.org/10.1016/j.molcel.2009.10.013
- Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 2015; 15:362-74; PMID:26008591; http://dx.doi.org/10.1038/nri3834
- Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, Webb AI, Rickard JA, Anderton H, Wong WW, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011; 471:591-6; PMID:21455173; http://dx.doi.org/10.1038/nature09816
- Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Vanden Berghe T. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis 2011; 2:e230; PMID:22089168; http://dx.doi.org/10.1038/cddis.2011.111
- Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, Mak TW, Wallach D, Green DR. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep 2012; 1:401-7; PMID:22675671; http://dx.doi.org/10.1016/j.celrep.2012.03.010
- Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N, Castanares M, Wu M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal 2007; 19:2056-67; PMID:17644308; http://dx.doi.org/10.1016/j.cellsig.2007.05.016
- O'Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, Green DR, Ting AT. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol 2011; 13:1437-42; PMID:22037414; http://dx.doi.org/10.1038/ncb2362
- Seo J, Lee EW, Sung H, Seong D, Dondelinger Y, Shin J, Jeong M, Lee HK, Kim JH, Han SY, et al. CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat Cell Biol 2016; 18:291-302; PMID:26900751; http://dx.doi.org/10.1038/ncb3314
- Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis 2012; 3:e437; PMID:23190609; http://dx.doi.org/10.1038/cddis.2012.176
- Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008; 4:313-21; PMID:18408713; http://dx.doi.org/10.1038/nchembio.83
- Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008; 133:693-703; PMID:18485876; http://dx.doi.org/10.1016/j.cell.2008.03.036
- Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res 2013; 23:994-1006; PMID:23835476; http://dx.doi.org/10.1038/cr.2013.91
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012; 148:213-27; PMID:22265413; http://dx.doi.org/10.1016/j.cell.2011.11.031
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 2014; 7:971-81; PMID:24813885; http://dx.doi.org/10.1016/j.celrep.2014.04.026
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 2014; 54:133-46; PMID:24703947; http://dx.doi.org/10.1016/j.molcel.2014.03.003
- Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 2014; 24:105-21; PMID:24366341; http://dx.doi.org/10.1038/cr.2013.171
- Remijsen Q, Goossens V, Grootjans S, Van den Haute C, Vanlangenakker N, Dondelinger Y, Roelandt R, Bruggeman I, Goncalves A, Bertrand MJ, et al. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis 2014; 5:e1004; PMID:24434512; http://dx.doi.org/10.1038/cddis.2013.531
- Tait SW, Oberst A, Quarato G, Milasta S, Haller M, Wang R, Karvela M, Ichim G, Yatim N, Albert ML, et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep 2013; 5:878-85; PMID:24268776; http://dx.doi.org/10.1016/j.celrep.2013.10.034
- Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012; 148:228-43; PMID:22265414; http://dx.doi.org/10.1016/j.cell.2011.11.030
- Moriwaki K, Farias Luz N, Balaji S, De Rosa MJ, O'Donnell CL, Gough PJ, Bertin J, Welsh RM, Chan FK. The Mitochondrial Phosphatase PGAM5 Is Dispensable for Necroptosis but Promotes Inflammasome Activation in Macrophages. J Immunol 2016; 196:407-15; PMID:2682950; http://dx.doi.org/10.4049/jimmunol.1501662
- Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez-Caballero S, Osinska H, Cheng EH, Robbins J, et al. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife 2013; 2:e00772; PMID:23991283; http://dx.doi.org/10.7554/eLife.00772
- Irrinki KM, Mallilankaraman K, Thapa RJ, Chandramoorthy HC, Smith FJ, Jog NR, Gandhirajan RK, Kelsen SG, Houser SR, May MJ, et al. Requirement of FADD, NEMO, and BAX/BAK for aberrant mitochondrial function in tumor necrosis factor alpha-induced necrosis. Mol Cell Biol 2011; 31:3745-58; PMID:21746883; http://dx.doi.org/10.1128/MCB.05303-11
- Canli O, Alankus YB, Grootjans S, Vegi N, Hultner L, Hoppe PS, Schroeder T, Vandenabeele P, Bornkamm GW, Greten FR. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood 2016; 127:139-48; PMID:26463424; http://dx.doi.org/10.1182/blood-2015-06-654194
- Schenk B, Fulda S. Reactive oxygen species regulate Smac mimetic/TNFalpha-induced necroptotic signaling and cell death. Oncogene 2015 34(47):5796-806; http:dx.doi.org/10.1038/onc.2015.35
- Wu W, Liu P, Li J. Necroptosis: an emerging form of programmed cell death. Crit Rev Oncol Hematol 2012; 82:249-58; PMID:21962882; http://dx.doi.org/10.1016/j.critrevonc.2011.08.004
- Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464:104-7; PMID:20203610; http://dx.doi.org/10.1038/nature08780
- Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M. Tumor necrosis factor-alpha blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation. Immunity 2013; 38:541-54; PMID:23477736; http://dx.doi.org/10.1016/j.immuni.2013.02.007
- Choi HS, Kang JW, Lee SM. Melatonin attenuates carbon tetrachloride-induced liver fibrosis via inhibition of necroptosis. Translat Res 2015; 166(3):292-303; PMID:25936762; http://dx.doi.org/10.1016/j.trsl.2015.04.002
- Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420:860-7; PMID:12490959; http://dx.doi.org/10.1038/nature01322
- Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer 2013; 12:86; PMID:23915189; http://dx.doi.org/10.1186/1476-4598-12-86
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013; 13:714-26; PMID:24060863; http://dx.doi.org/10.1038/nrc3599
- Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo J, Hu X. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther 2007; 6:1641-9; PMID:17513612; http://dx.doi.org/10.1158/1535-7163.MCT-06-0511
- Chen X, Yang L, Zhang N, Turpin JA, Buckheit RW, Osterling C, Oppenheim JJ, Howard OM. Shikonin, a component of chinese herbal medicine, inhibits chemokine receptor function and suppresses human immunodeficiency virus type 1. Antimicrobial agents and chemotherapy 2003; 47:2810-6; PMID:12936978; http://dx.doi.org/10.1128/AAC.47.9.2810-2816.2003
- Liang D, Sun Y, Shen Y, Li F, Song X, Zhou E, Zhao F, Liu Z, Fu Y, Guo M, et al. Shikonin exerts anti-inflammatory effects in a murine model of lipopolysaccharide-induced acute lung injury by inhibiting the nuclear factor-kappaB signaling pathway. Int Immunopharmacol 2013; 16:475-80; PMID:23651796; http://dx.doi.org/10.1016/j.intimp.2013.04.020
- Fu Z, Deng B, Liao Y, Shan L, Yin F, Wang Z, Zeng H, Zuo D, Hua Y, Cai Z. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer 2013; 13:580; PMID:24314238; http://dx.doi.org/10.1186/1471-2407-13-580
- Wada N, Kawano Y, Fujiwara S, Kikukawa Y, Okuno Y, Tasaki M, Ueda M, Ando Y, Yoshinaga K, Ri M, et al. Shikonin, dually functions as a proteasome inhibitor and a necroptosis inducer in multiple myeloma cells. Int J Oncol 2015; 46:963-72; PMID:25530098; http://dx.doi.org/10.3892/ijo.2014.2804
- Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene 2012; 31:1869-83; PMID:21892204; http://dx.doi.org/10.1038/onc.2011.384
- McCabe KE, Bacos K, Lu D, Delaney JR, Axelrod J, Potter MD, Vamos M, Wong V, Cosford ND, Xiang R, et al. Triggering necroptosis in cisplatin and IAP antagonist-resistant ovarian carcinoma. Cell Death Dis 2014; 5:e1496; PMID:25356865; http://dx.doi.org/10.1038/cddis.2014.448
- Cerhan JR, Ansell SM, Fredericksen ZS, Kay NE, Liebow M, Call TG, Dogan A, Cunningham JM, Wang AH, Liu-Mares W, et al. Genetic variation in 1253 immune and inflammation genes and risk of non-Hodgkin lymphoma. Blood 2007; 110:4455-63; PMID:17827388; http://dx.doi.org/10.1182/blood-2007-05-088682
- Liu P, Xu B, Shen W, Zhu H, Wu W, Fu Y, Chen H, Dong H, Zhu Y, Miao K, et al. Dysregulation of TNFalpha-induced necroptotic signaling in chronic lymphocytic leukemia: suppression of CYLD gene by LEF1. Leukemia 2012; 26:1293-300; PMID:22157808; http://dx.doi.org/10.1038/leu.2011.357
- English C, Aloi JJ. New FDA-Approved Disease-Modifying Therapies for Multiple Sclerosis. Clin Ther 2015; 37:691-715; PMID:25846320; http://dx.doi.org/10.1016/j.clinthera.2015.03.001
- Saddoughi SA, Gencer S, Peterson YK, Ward KE, Mukhopadhyay A, Oaks J, Bielawski J, Szulc ZM, Thomas RJ, Selvam SP, et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol Med 2013; 5:105-21; PMID:23180565; http://dx.doi.org/10.1002/emmm.201201283
- Zhang L, Wang H, Ding K, Xu J. FTY720 induces autophagy-related apoptosis and necroptosis in human glioblastoma cells. Toxicology letters 2015; 236:43-59; PMID:25939952; http://dx.doi.org/10.1016/j.toxlet.2015.04.015
- Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, Delrue I, Taminau J, Wiernicki B, De Groote P, et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep 2016; 15:274-87; PMID:27050509; http://dx.doi.org/10.1016/j.celrep.2016.03.037
- Ye H, Liu X, Lv M, Wu Y, Kuang S, Gong J, Yuan P, Zhong Z, Li Q, Jia H, et al. MicroRNA and transcription factor co-regulatory network analysis reveals miR-19 inhibits CYLD in T-cell acute lymphoblastic leukemia. Nucleic Acids Res 2012; 40:5201-14; PMID:22362744; http://dx.doi.org/10.1093/nar/gks175
- Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell 2010; 39:493-506; PMID:20797623; http://dx.doi.org/10.1016/j.molcel.2010.07.023
- Su Z, Yang Z, Xu Y, Chen Y, Yu Q. MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget 2015; 6:8474-90; PMID:25893379; http://dx.doi.org/10.18632/oncotarget.3523