
ABSTRACT
Decursinol angelate (DA), an active pyranocoumarin compound from the roots of Angelica gigas, has been reported to possess anti-inflammatory and anti-cancer activities. In a previous study, we demonstrated that prostaglandin E2 (PGE2) plays a survival role in HL-60 cells by protecting them from the induction of apoptosis via oxidative stress. Flow cytometry and Hoechst staining revealed that PGE2 suppresses menadione-induced apoptosis, cell shrinkage, and chromatin condensation, by blocking the generation of reactive oxygen species. Treatment of DA was found to reverse the survival effect of PGE2 as well as restoring the menadione-mediated cleavage of caspase-3, lamin B, and PARP. DA blocked PGE2-induced activation of the EP2 receptor signaling pathway, including the activation of PKA and the phosphorylation of CREB. DA also inhibited PGE2-induced expression of cyclooxygenase-2 and the activation of the Ras/Raf/ Erk pathway, which activates downstream targets for cell survival. Finally, DA greatly reduced the PGE2-induced activation of NF-κB p50 and p65 subunits. These results elucidate a novel mechanism for the regulation of cell survival and apoptosis, and open a gateway for further development and combinatory treatments that can inhibit PGE2 in cancer cells.
Abbreviations
| DA | = | Decursinol angelate |
| PGE2 | = | Prostaglandin E2 |
| COX-2 | = | cyclooxygenase-2 |
| PKA | = | protein kinase A |
| Erk | = | extracellular-signal-regulated kinase |
| NFκB | = | nuclear factor-κB |
| CREB | = | cAMP response element binding protein |
Introduction
Prostaglandin E2 (PGE2) is an inflammatory lipid mediator that is involved in a wide range of processes including cellular differentiation, inflammation and cancer. PGE2 belongs to the prostanoid family and is synthesized through the oxygenated metabolism of arachidonic acid.Citation1 PGE2 synthesis is catalyzed by cyclooxygenase-2 (COX-2), which reduces the unstable metabolite prostaglandin-endoperoxide synthase 2 (PGG2) to prostaglandin H2 Synthase (PGH2).Citation2 There is evidence that the over-expression of COX-2, as a mediator PGE2 synthesis, plays an important role in the initiation and development of cancer. The reduction in PGE2 synthesis through inhibition of COX-2 activity provides an alternate molecular mechanism for the chemo-preventive effect of nonsteroidal anti-inflammatory drugs.Citation3 Studies have shown that PGE2 promotes tumor development by enhancing cell proliferation and survival, inflammation, angiogenesis, and invasiveness in various cancers.Citation4-5 The PGE2 receptors EP1 and EP2 have been demonstrated to participate in PGE2-induced tumorigenesis as well as in PGE2-mediated cancer cell survival and protection from drug-induced apoptosis, via the activation of cyclic adenosine monophosphate (cAMP)-dependent protein kinase A (PKA).Citation6,7 PGE2 has been shown to significantly increase promonocytic cell proliferation and COX-2 expression, and this effect is mediated, at least in part, by the activation of the Ras/Raf/Erk signaling cascade.Citation7,8
Decursinol angelate (DA) is an active pyranocoumarin compound, isolated from the roots of Angelica gigas Nakai (Umbelliferae), that exhibits chemo-preventive and anti-inflammatory activities.Citation9 DA has been reported to suppress the growth of estrogen-stimulated and estrogen-independent growth and survival of breast cancer MCF-7 and MDA-MB-231 cells.Citation10 DA also suppresses the growth of cancer cells through the inhibition of phosphatidylinositol 3′-kinase (PI3K)/Akt, extracellular-signal-regulated kinase (Erk) and nuclear factor-κB (NFκB).Citation11 Additionally, DA has anti-angiogenic activity both in vitro and in vivo.Citation9 DA has been demonstrated to have anti-leukemic activity through the suppression of protein kinase C activation in human leukemia cells; however, the mechanism underlying this action requires investigation.Citation12 Furthermore, it has been hypothesized that the anti-inflammatory activity of DA will contribute to its potential effects on cancer cells.
Previously, we have shown that PGE2 regulates cell growth, proliferation, and survival of cancer cells through the activation of the Ras/Raf/Erk signaling cascades.Citation7 Aberrant activation of this pathway not only enhances cancer progression but also contributes to the anti-apoptotic effect of PGE2.Citation13 Furthermore, PGE2 induced the activation of NFκB and the expression of inflammatory genes that contribute to cancer progression and survival.Citation14,15 Thus, the drugs or agents that have the ability to inhibit the PGE2-induced activation of Ras and NF-κB signaling are urgently needed. Therefore, we evaluated the effect of treatment with DA in combination with menadione on cancer-associated inflammatory processes. PGE2 reverses menadione-induced apoptosis via activation of the PKA, Ras, and NFκB signaling pathways. However, DA treatment inhibits this PGE2-induced survival effect and restores the menadione-induced apoptosis of HL-60 cells. This study suggests a therapeutic strategy for the inhibition of PGE2 in cancer cells by combined treatment with natural products.
Results
DA treatment enhances menadione-induced apoptosis of HL-60 cells via oxidative stress
Since PGE2 was found to inhibit menadione-induced cell death by reducing the steady state levels of ROS, the effect of treatment with DA and menadione was evaluated using dichloro-dihydro-fluorescein diacetate (DCFHDA), a cell-permeable dye that is used as an intracellular probe for oxidative stress. As shown in , DCF fluorescence was increased in HL-60 cells upon exposure to menadione, but this increase in DCF fluorescence was substantially reduced in cells that had been pre-treated with PGE2, demonstrating the anti-apoptotic effect of PGE2. However, treating the cells with DA prior to their exposure to PGE2 and menadione reversed the protective effect of PGE2 and enhanced menadione-induced cell death by increasing the steady-state level of intracellular oxidants. No significant increase in DCF fluorescence was observed upon DA treatment alone (). We also investigated mitochondrial ROS using the oxidant-sensitive probe DHR 123. Menadione-induced intracellular fluorescence was reduced by pretreatment with PGE2 but increased by pretreatment with DA (). One possible explanation is that DA treatment may enhance the cellular retention of menadione and/or inhibit the PGE2-induced exclusion of menadione.
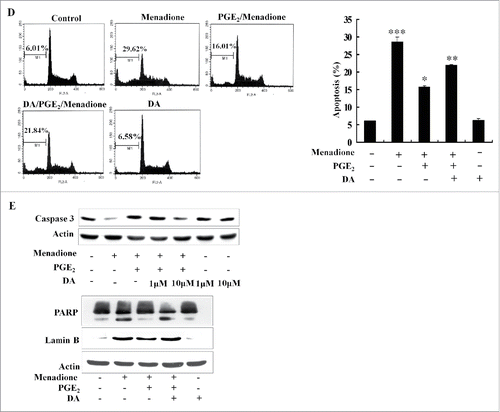
Figure 1. DA inhibits PGE2-induced anti-apoptotic effects (A) HL-60 cells were pretreated with 10 µM of DA or 1 µM of PGE2, followed by menadione treatment. The cells were then incubated with DCFHDA, a cell-permeable n-dichlorofluorescein that is used as an intracellular probe for oxidative stress, for 20 min. Accumulation of the probe in cells was measured based on increases in emission at 530 nm when the sample was excited at 485 nm. Fluorescence images of DCF-loaded cells were obtained under a microscope. (B) PGE2 inhibits menadione-induced production of mitochondrial ROS. HL-60 cells were incubated with DHR 123 after exposure to 10 µM of DA or 1 µM of PGE2 followed by treatment with 10 μM of menadione. (C) HL-60 cells were pretreated with 10 µM of DA or 1 µM of PGE2 and exposed to10 µM of menadione and then stained with 10 μM Hoechst 33342 for 10 min and the images were taken by fluorescence microscopy. (D) HL-60 cells were harvested and stained with PtdIns, after which their DNA content was analyzed by flow cytometry. Sub-G1 fractions of cells (%) were plotted against DA or PGE2 and menadione treatments. The experiment was repeated at least 3 times. (E) HL-60 cells were treated with 10 µM of DA and menadione, with or without 1 µM of PGE2. The total protein was isolated and analyzed by protein gel blot with anti-caspase-3, PARP, lamin B antibodies. Actin was used as a loading control.
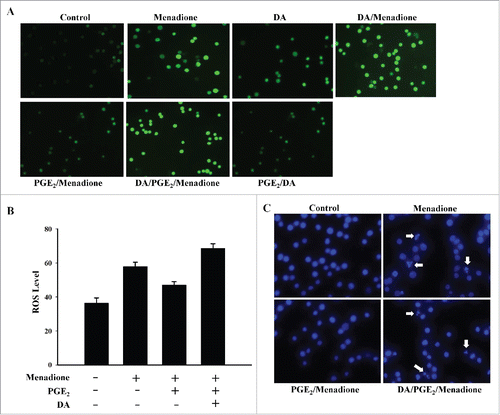
Figure 1. (Continued).
Hoechst 33342 staining was used to evaluate whether the increased ROS generation following treatment with menadione was associated with increased apoptosis. As shown in , HL-60 cells displayed condensed nuclear morphology following menadione treatment, but this was not evident for cells that were pretreated with PGE2. However, HL-60 cells that were pretreated with DA prior to treatment with either PGE2 or menadione, did display apoptotic characteristics such as cells shrinkage, nuclear condensation and apoptotic body formation (). This demonstrates that DA pretreatment was able to inhibit the anti-apoptotic effects of PGE2 treatment and enhance menadione-induced cell death.
DA treatment reduces the PGE2-induced survival effect in HL-60 cells
To further elucidate the effect of DA on apoptosis, HL-60 cells were labeled with PI and analyzed by flow cytometry. An increase in the proportion of cells in sub-G1 phase was observed following treatment with menadione (). Both Hoechst staining and FACS analysis revealed that PGE2 blocked menadione-induced cells death. Given that DA enhances menadione-induced apoptosis, fluorescence-activated cell sorting (FACS) analysis suggests that DA reduces the survival effect of PGE2 (). The ratio of apoptotic cells following treatment with DA was not significant when compared to the control; however, DA treatment clearly reduced the survival effect of treatment with PGE2 and consequently enhanced menadion-induced cell death.
It has been hypothesized that DA treatment may activate regulatory proteins that stimulate the apoptosis of cancer cells. Among these proteins, caspase is a cysteine protease that actively participates in the complex process of apoptosis. As shown in , cleavage of the precursor of caspase-3 was observed with DA pretreatment followed by PGE2 treatment. Treatment with PGE2 decreased the activation of caspase-3 and the cleavage of its substrates PARP and lamin B (). However, pretreatment with DA activated caspase-3 and induced the cleavage of PARP and lamin B, thereby restoring menadione-induced cell death. Furthermore, high dose treatment with DA, followed by menadione and PGE2, produced notable activation of caspase-3 (). These results suggest that DA activates various apoptotic proteins, reducing the survival effect of PGE2 and restoring menadione-induced cell death.
DA treatment inhibits PGE2-induced activation of EP2 receptor signaling
PGE2 is known to regulate various inflammatory and survival processes through its 4 receptors, which are broadly classified as Ca2+-associated receptor (EP1) and cAMP-associated receptors (EP2, EP3, EP4).Citation18 To investigate which of these receptors is responsible for mediating the survival effect of PGE2, immunoblot was performed on protein extracts from cells treated with either DA or PGE2. As shown in , expression of the EP2 receptor was increased in HL-60 cells stimulated with PGE2, whereas the expression levels of the EP1, EP3, and EP4 receptors were not considerably changed. Given that PGE2 induces cells survival via the EP2 receptor, we then examined the effect of DA treatment on the expression of EP2 receptor. Interestingly, DA was found to inhibit the PGE2-induced increase in EP2 receptor expression (). This finding is in agreement with previous studies, which show that EP2 is the functional receptor of PGE2 and activates PKA by regulating cAMP signaling pathway.Citation19 To explore the functional effect of DA on EP2 receptor signaling, we investigated the translocation of the PKA catalytic subunit to the nucleus in DA- and PGE2-treated HL-60 cells. DA was found to inhibit PGE2-induced increase in translocation of the catalytic subunit (). The active catalytic subunit of PKA affects cellular physiology through the phosphorylation of the cAMP response element binding protein (CREB) in the nucleus. Although PGE2 treatment increased the phosphorylation of CREB, this was inhibited by pretreatment with DA (). These results indicate that DA blocks PGE2-induced activation of the EP2/PKA signaling pathway.
Figure 2. The EP2 receptor is involved in the survival of HL-60 cells. (A) Cells were treated with 1 μM of PGE2 and 10 μM of DA, and protein extracts were analyzed by western blot using antibodies specific for EP1, EP2, EP3, and EP4. (B) Cells were incubated with 10 µM of DA, menadione and/or PGE2, and total cell lysates were analyzed by immunoblot using anti-PKA catalytic subunit and pCREB antibodies. Proliferating cell nuclear antigen (PCNA) was used as a loading control of nuclear fractions. (C) Cells were treated with 1 μM of PGE2 or 10 μM of DA and the Ras activity was assessed by protein gel blot analysis of Ras immunoprecipitated form lysates using the Raf-1 Ras-binding domain. Densitometry measurements of Ras activity are shown on the blot (pixels as units). (D) HL-60 cells were treated with 1 μM of PGE2 or 10 μM of DA followed by treatment with 10 μM of menadione, and western blot analysis was performed on protein lysates using specific antibodies for the phosphorylation forms of Raf and. Actin was used as a loading control.
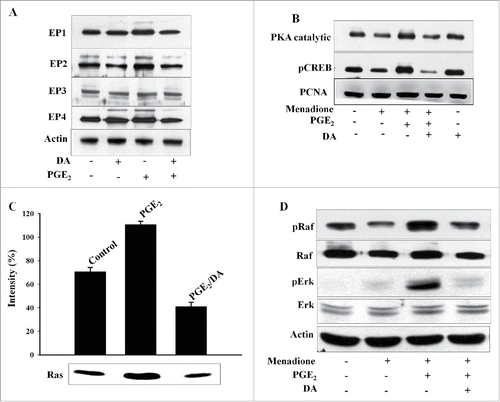
Since the EP2/Ras pathway regulates various cellular processes, including the transcriptional activation and stabilization of inflammatory downstream targets, we next examined the activation of Ras and the phosphorylation levels of Raf and Erk in HL-60 cells exposed to DA and then treatment with either PGE2 or menadione. As shown in , DA treatment inhibited the PGE2-induced activation of Ras as demonstrated by the western blot of precipitates containing Ras bound to the Ras-binding domain for Raf-1. Additionally, DA was found to inhibit the PGE2-induced increase in the phosphorylation levels of Raf and Erk and restored the menadione-induced inhibition of pRaf and pErk (). These results demonstrate that the inhibition of Ras/Raf/Erk is particularly required for apoptosis, and that this pathway is activated by PGE2 to protect HL-60 cells from menadione-induced cells death.
DA treatment inhibits PGE2-induced COX-2 expression
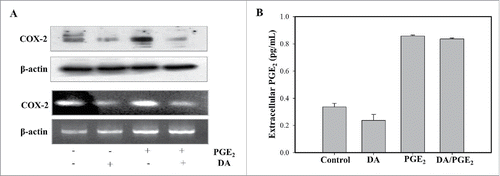
Numerous studies have shown that the inhibition of COX-2, which reduces the synthesis of PGE2, is a central mechanism by which anti-cancer drugs are able to reduce inflammation and cancer development. To confirm this, we investigated the effect of DA and PGE2 on COX-2 expression at both transcription and protein level. As shown in , DA was found to inhibit the PGE2-induced increase in COX-2 expression. To further elucidate the role of DA as an inhibitor of PGE2, we also checked the extracellular level of PGE2 following treatment with DA or PGE2. HL-60 cells displayed a greater extracellular concentration of PGE2 following treatment with PGE2, and a little decreasing level was observed for DA-treated cells (). These results suggest that PGE2 increases COX-2 expression, and that inhibition by DA leads to a reversal of PGE2-induced survival and anti-apoptotic effect.
Figur 3. DA treatment blocks the expression of COX-2. (A) Cells were treated with 1 μM of PGE2 and 10 μM of DA, and immunoblot was performed on protein lysates using an antibody specific for COX-2. Total RNA was extracted from HL-60 cells, and RT-PCR was performed. PCR products were separated on 1% agarose gel for visualization. (B) Cells were pretreated with DA prior to treatment with PGE2 and the supernatants collected to measure extracellular PGE2 levels using an EIA kit. Values are the means SD from 3 independent experiments.
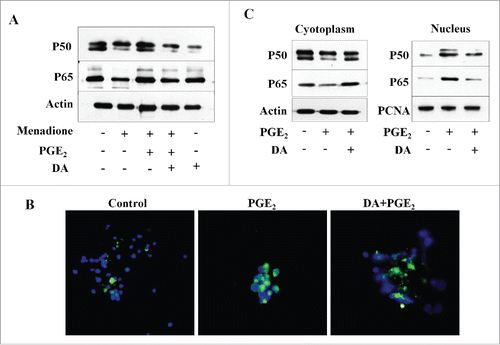
DA treatment blocks PGE2-induced activation of the NFκB pathway
There is compelling evidence that NFκB regulates the transcription of genes involved in various cellular processes including inflammation and apoptosis of cancer cells. NFκB is also well known as a potential therapeutic target for various cancers, including leukemia.Citation15 We investigated the effect of DA treatment on the PGE2-induced activation of NFκB in cells that were exposed to DA with or without subsequent PGE2 treatment. As shown in , DA treatment was found to inhibit the PGE2-induced expression of the NFκB subunits p50 and p65. DA treatment also restored the menadione-induced down-regulation of NFκB subunits p50 and p65 (). Furthermore, we used immunofluorescence as a complementary approach to investigate the effect of DA on PGE2-induced activation of NFκB. A significant nuclear translocation of NFκB (p65) was observed following treatment of HL-60 cells with PGE2 (). However, pretreating these cells with DA caused a marked reduction in PGE2-induced NFκB (p65) nuclear localization (). The inhibitory effect of DA on the translocation of NFκB (p65) to the nucleus was next measured using fractionation analysis. The disappearance of NFκB from the cytosolic fraction of HL-60 cells treated with PGE2, and the concomitant increase of NFκB in the nuclear fraction, supports the translocation of NFκB to the nucleus in these cells. This effect was reduced in DA-treated cells (). Additionally, NFκB nuclear translocation was significantly lower in HL-60 cells treated with DA (). These results suggest that PGE2-induced activation of NFκB promotes the survival of HL-60 cells and that specific inhibition by DA may be a potential therapeutic approach for cancer treatment.
Figure 4. DA treatment inhibits PGE2-induced activation of NFκB pathway. (A) HL-60 cells were exposed to DA or PGE2. DA decreased the expression of NFκB subunits p50 and p65, as detected by protein gel blot. (B) The effect of DA on the expression of NFκB in PGE2-activated HL-60 cells by immunofluorescence microscopy. The nuclear translocation of NFκB was increased in PGE2-treated cells; however, the nuclear localization of NFκB was inhibited in DA-treated cells. Images were taken using a fluorescent microscope and merged with DAPI. Data are representative of 3 independent experiments. (C) Cytoplasmic and nuclear fractions were separated for HL-60 cells treated with PGE2 and/or DA. NFκB subunits p50 and p65 were detected by immunoblot analysis using antibodies specific for their expression in the cytoplasm and nucleus.
Discussion
Cell proliferation and growth are critical for cancer cell survival and provide protection against a wide range of chemotherapies. PGE2 is known to induce the activation of 2 distinct pathways, Ras/Raf/Erk and NFκB, which promote cancer cells survival and development.Citation13 Thus, the inhibition of these pathways could prevent cancer development and recurrence. However, many promising therapeutics lose their efficacy for the treatment of cancer upon activation of the survival pathways.Citation7,13 Thus, combining chemotherapeutic agents with inhibitors of these survival pathways could provide cancer treatments with less toxicity or drug resistance.Citation20,21 The development of nontoxic anti-inflammatory agents is urgently needed and many plant-derived phytochemicals have been reported to exhibit anti-inflammatory and chemo-preventive effects.Citation20,21 The present study was conducted to evaluate the potential effects of DA, when combined with menadione treatment, in PGE2-stimulated HL-60 cells. Our findings suggest that DA inhibits the PGE2-induced increase in EP2 receptor expression, and decreases the activation of the Ras/Raf/Erk and NFκB pathways, thereby restoring menadione-induced cell death. Importantly, the treatment with DA alone fails to overcome survival signaling and induce apoptosis via oxidative stress. Given the significant roles of both Ras and NFκB signaling in cancer cell survival, understanding the multifactorial effect of PGE2 and DA will aid in the exploration of combined treatments for many cancers including leukemia.
Apoptosis or programmed cell death is a complex and well-regulated mechanism of cancer cell death that occurs under various stress conditions.Citation22 The involvement of ROS generation in the induction of oxidative-stressed apoptosis has been reported.Citation7,13 We have previously shown that menadione induces apoptosis of HL-60 cells via oxidative stress, and that PGE2 blocks the ROS generation which is induced by menadione.Citation7,13 In the present study, DA treatment was shown to reverse this effect of PGE2, although the treatment with DA alone did not produce a significant change in DCFHDA fluorescence (). Whereas treatment with PGE2 decreases the activation of caspase-3 and the cleavage of PARP, pretreatment with DA followed by PGE2 activates caspase-3 and induces the degradation of PARP (). DA treatment results in the inhibition of PGE2-induced cell survival and the activation of menadione-induced cell death.
It has been well documented that PGE2 provides protection against cytotoxic drugs and mediates cancer cell survival through binding to its receptors, which initiates signaling that results in the inhibition of apoptosis. Our results are consistent with those of previous studies, which show that PGE2 enhances cancer cell survival through the up-regulation of the EP2 receptor.Citation23,24 We have assumed that the PGE2-induced increase in EP2 receptor expression activates the cAMP-mediated phosphorylation of CREB through PKA, which consequently blocks the induction of apoptosis in cancer cells by cytotoxic drugs.Citation25 DA may inhibit expression of the EP2 receptor, thus blocking the EP2 pathway and restoring menadione-induced cell death. Other possible explanations emphasize that cAMP levels are closely related with the activation of MAPK pathway in cancer cells. Our results indicate that exogenous PGE2 treatment activates the Ras/Raf/Erk pathway in HL-60 cells.Citation7 Since the EP2 receptor is coupled with Ras signaling and the production of cAMP,Citation24 we suggest that most likely mechanism for PGE2-induced activation of Ras will require the EP2 receptor. However, previous results have shown that a PGE2 autocrine loop might also be involve in an ERK-independent mechanism to promote cancer cell survival.Citation8,26 DA may inhibit multiple PGE2-induced targets in the Ras-Raf-Erk pathway to reverse PGE2-induced cell survival and restore menadione-induced apoptosis (). These results are also supported by those of a previous study, which shows that DA inhibits VEGF-induced phospohorylation of Erk in human umbilical vein endothelial cells (HUVEC) cells.Citation9
NFκB, a heterotrimeric complex consisting of the p50, p65, and IκBα subunits, mediates the initiation of inflammatory processes and cancer progression.Citation15 Compelling evidence has reported suggesting that PGE2-induced activation of NFκB suppresses apoptosis and thereby promotes cancer cell survival and tumorigenesis.Citation13,27 Our results demonstrate that the activation of NFκB by PGE2 blocks the effect of menadion treatment in HL-60 cells, thereby promoting their protection and survival (). DA treatment inhibits the PGE2-induced nuclear translocation of NFκB p65 subunits, as shown by both immunofluorescence and protein gel blot analysis of cytoplasmic and nuclear fractions (). These results are in agreement with previous published works, which report that DA inhibits the invasion of fibrosarcoma HT1080 and breast cancer MDA-MB-231 cell lines, through suppression of Erk and NFκB activation.Citation11
In conclusion, DA blocks PGE2-induced cell survival, potentially by inhibiting the activation of the Ras/Raf/Erk and NFκB pathways in HL-60 cells, and ultimately restores menadione-induced cell death. DA-mediated inhibition of COX-2 and PKA expression is another factor that contributes to the suppression of the PGE2-induced cell survival effect. Therefore, our results emphasize that DA may be an effective treatment strategy for various cancers, including leukemia, in which PGE2 performs a critical role in cancer cell survival and protection against anti-cancer drugs.
Materials and methods
Reagents and cell culture
Menadione (Sigma-Aldrich, USA) was prepared as a 100 mM stock solution in dimethyl sulfoxide (DMSO). DA with molecular weight 328 was isolated from the roots of Angelicae gigas at Daegu Hanny University, Daegu, Korea as described previously.Citation9 PGE2 was obtained from the Cayman Chemical Company. The human leukemia cell line HL-60 (ATCC, CCL-240™) was cultured from a seeding density of 5 × 105 cells in RPMI-1640 medium containing 10% heat-inactivated fetal bovine serum and 1% (v/v) antibiotic (penicillin- streptomycin, Gibco, USA), at 37°C in a 5% CO2-humidified atmosphere. At 70% confluence, the cells were pretreated with 10 μM of DA and 1 μM of PGE2 and then treated with 10 μM of menadione (Sigma-Aldrich, USA) for the indicated time.
Determination of apoptosis
Cells were morphologically analyzed for apoptosis by Hoechst 33342 staining. Cells were fixed in 4% paraformaldehyde and permeabilized with 0.5% Triton X- 100. The nuclei were stained with Hoechst 33342 for 20 min. The cells were then mounted with mounting media (Sigma-Aldrich, USA) and observed by fluorescence microscopy.
Measurement of intracellular ROS generation
To measure intracellular reactive oxygen species (ROS) generation, cells was grown for 24 h in growth medium, exposed to 5 μM 2′,7′-dichlorofluorescin diacetate (DCFHDA) (Molecular Probes, USA) for 20 min, and washed with PBS. DCF fluorescence (excitation, 480 nm; emission, 520 nm) was imaged using an inverted microscope (Zeiss Axiovert 200).
Measurement of mitochondrial ROS
Mitochondrial ROS was monitored according to a previously described method.Citation16 HL-60 cells were incubated in culture medium supplemented with 10 μM dihydrorhodamine 123 (DHR 123) for 20 min, and then washed 3 times with ice-cold PBS. The cells were imaged for FITC fluorescence intensity on an inverted microscope.
Flow cytometry
Cells were collected by centrifugation, fixed with 70% ethanol for 30 min, treated with 50 μl of 1 mg/ml RNase, and stained with 450 μl of 50 μg/ml propidium iodide (PtdIns) in PBS for 15 min at room temperature in the dark. Samples were then analyzed by a flow cytometer (Becton Dickinson) and the PI histogram used to determine the DNA content.
Ras activity assay
To measure Ras activity, a Ras activity kit (Upstate) was used, according to the manufacturer's instuctions. Cells were washed twice with ice cold PBS, lysed in lysis buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 1% Igepal CA-630, 10 mM MgCl2, 1 mM EDTA, 25 mM NaF, 10% glycerol, 10 μg/ml aprotinin, and 10 μg/ml leupeptin) and centrifuged. An equal volume of lysate was incubated with 7 μg of the Ras-binding domain for Raf-1 for 2 h at 4°C, and the beads were washed 3 times with lysis buffer. The bound Ras proteins were detected by western blot using an anti-Ras antibody.
Isolation of cytosolic and nuclear fractions
HL-60 cells were trypsinized, centrifuged, and washed twice with cold 1 XPBS. Fractions were prepared by use of 2 buffers as described previously.Citation28 First, cells were suspended in buffer A containing HEPES, potassium chloride, ethyleneglycol tetra-acetic acid (EGTA), ethylenediaminetetraacetic acid (EDTA), dithiothreitol (DTT), sodium orthovanadate (Na3VO4), phenylmethylsulfonyl fluoride (PMSF), and 6% IGEPAL (NP-40), and incubated for 15 min on ice. After centrifugation at 600 g for 5 min at 4°C, supernatants containing cytoplasmic proteins were collected in a new tube. The remaining pellets were resuspended in buffer C containing HEPES, sodium chloride, EGTA, EDTA, DTT, Na3VO4, PMSF, and protease inhibitors. The resuspended pellets were centrifuged at 20,000 g for 15 min at 4C, and supernatants containing nuclear proteins were collected in a new tube. For future experiments both fractions were stored at −80C.
Western blot analysis
Proteins were fractionated on SDS-PAGE and then transferred onto nitrocellulose membranes. These membranes were blocked with 5% non-fat milk, incubated overnight with appropriate primary antibody, and then incubated with a horseradish peroxidase-labeled anti-rabbit or anti-mouse secondary antibody. Signals were detected by chemiluminescence (Amersham, UK).
Quantification of extracellular PGE2
To quantify the amount of extracellular PGE2, HL-60 cells that had been exposed to PGE2 and DA were centrifuged and the supernatant collected. The extracellular PGE2 level (pg/mL) in the supernatant was measured using a PGE2 Express EIA kit (Cayman Chemical, USA), as per the manufacturer's instructions.
Immunofluorescence of HL-60 cells
Immunofluorescence was used to investigate the sub-cellular localization and translocation of NFκB in HL-60 cells using a previously optimized method.Citation17 In brief, the cells were centrifuged, spiked with 37% PFA, and permeabilized with 0.3% Triton X-100 in PBS. Cells were then blocked with 2% BSA, incubated overnight with a purified NFκB antibody (1:200, Santa Cruz, USA), and then incubated with an anti-rat FITC-conjugated secondary antibody (1:100, Jackson Immuno Research Laboratories, USA). Immunofluorescence was performed on coverslips and the cells were visualized using an Olympus BX50 microscope and photographed using an Olympus DP70 digital camera.
Statistical analysis
Data is presented as the mean ± standard deviation (SD) of 3 to 6 independent experiments. Significant differences between 2 mean values were determined using the Student's t-test, and for multi-group comparisons, using one-way analysis of variance (ANOVA). Differences were considered significant for p-values < 0.05.
Disclosure of potential confllicts of interest
No potential conflicts of interest were disclosed.
Funding
This research was supported by the Basic Science Research Program through the NRF funded by the Ministry of Science, ICT, and Future Planning (NRF- 2013R1A1A2063612).
References
- Xia D, Wang D, Kim S-H, Katoh H, DuBois RN. Prostaglandin E2 promotes intestinal tumor growth via DNA methylation. Nat Med 2012; 18:224-6; PMID:22270723; http://dx.doi.org/10.1038/nm.2608
- Sung YM, He G, Hwang DH, Fischer SM. Overexpression of the prostaglandin E2 receptor EP2 results in enhanced skin tumor development. Oncogene 2006; 25:5507-16; PMID:16607275; http://dx.doi.org/10.1038/sj.onc.1209538
- Fischer SM, Hawk ET, Lubet RA. Coxibs and Other Nonsteroidal Anti-Inflammatory Drugs in Animal Models of Cancer Chemoprevention. Cancer Prev Res 2011; 4:1728-35; PMID:21778329; http://dx.doi.org/10.1158/1940-6207.CAPR-11-0166
- Greenhough A, Smartt HJM, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009; 30:377-86; PMID:19136477; http://dx.doi.org/10.1093/carcin/bgp014
- Nakanishi M, Rosenberg DW. Multifaceted roles of PGE(2) in inflammation and cancer(). Semin Immunopathol 2013; 35:123-37; PMID:22996682; http://dx.doi.org/10.1007/s00281-012-0342-8
- Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res 2007; 67:4507-13; PMID:17483367; http://dx.doi.org/10.1158/0008-5472.CAN-06-4174
- Yeo H-S, Shehzad A, Lee YS. Prostaglandin E(2) blocks menadione-induced apoptosis through the Ras/Raf/Erk signaling pathway in promonocytic leukemia cell lines. Mol Cell 2012; 33:371-8; PMID:22450688; http://dx.doi.org/10.1007/s10059-012-2293-2
- Shehzad A, Lee J, Lee YS. Autocrine prostaglandin E(2) signaling promotes promonocytic leukemia cell survival via COX-2 expression and MAPK pathway. BMB Rep 2015; 48:109-14; PMID:24965577; http://dx.doi.org/10.5483/BMBRep.2015.48.2.081
- Jung MH, Lee SH, Ahn E-M, Lee YM. Decursin and decursinol angelate inhibit VEGF-induced angiogenesis via suppression of the VEGFR-2-signaling pathway. Carcinogenesis 2009; 30:655-61; PMID:19228635; http://dx.doi.org/10.1093/carcin/bgp039
- Jiang C, Guo J, Wang Z, Xiao B, Lee H-J, Lee E-O, Kim SH, Lu J. Decursin and decursinol angelate inhibit estrogen-stimulated and estrogen-independent growth and survival of breast cancer cells. Breast Cancer Res 2007; 9:R77-R; PMID:17986353; http://dx.doi.org/10.1186/bcr1790
- Kim W-J, Lee M-Y, Kim J-H, Suk K, Lee W-H. Decursinol angelate blocks transmigration and inflammatory activation of cancer cells through inhibition of PI3K, ERK and NF-κB activation. Cancer Lett 2010; 296:35-42; PMID:20381234; http://dx.doi.org/10.1016/j.canlet.2010.03.012
- Kim HH, Sik Bang S, Seok Choi J, Han H, Kim I-H. Involvement of PKC and ROS in the cytotoxic mechanism of anti-leukemic decursin and its derivatives and their structure–activity relationship in human K562 erythroleukemia and U937 myeloleukemia cells. Cancer Lett 2005; 223:191-201; PMID:15896453; http://dx.doi.org/10.1016/j.canlet.2004.10.025
- Shehzad A, Islam SU, Lee J, Lee YS. Prostaglandin E(2) Reverses Curcumin-Induced Inhibition of Survival Signal Pathways in Human Colorectal Carcinoma (HCT-15) Cell Lines. Mol Cell 2014; 37:899-906; PMID:25431425; http://dx.doi.org/10.14348/molcells.2014.0212
- Sakamoto K, Maeda S, Hikiba Y, Nakagawa H, Hayakawa Y, Shibata W, Yanai A, Ogura K, Omata M. Constitutive NF-κB Activation in Colorectal Carcinoma Plays a Key Role in Angiogenesis, Promoting Tumor Growth. Clinical Cancer Res 2009; 15:2248-58; PMID:19276252; http://dx.doi.org/10.1158/1078-0432.CCR-08-1383
- Wang S, Liu Z, Wang L, Zhang X. NF-κB Signaling Pathway, Inflammation and Colorectal Cancer. Cell Mol Immunol 2009; 6:327-34; PMID:19887045; http://dx.doi.org/10.1038/cmi.2009.43
- Kil IS, Jung KH, Nam WS, Park J-W. Attenuated mitochondrial NADP+-dependent isocitrate dehydrogenase activity enhances EGCG-induced apoptosis. Biochimie 2011; 93:1808-15; PMID:21741430; http://dx.doi.org/10.1016/j.biochi.2011.06.025
- Wang C-C, Bajikar SS, Jamal L, Atkins KA, Janes KA. A time- and matrix-dependent TGFBR3–JUND–KRT5 regulatory circuit in single breast epithelial cells and basal-like premalignancies. Nat Cell Biol 2014; 16:345-56; PMID:24658685; http://dx.doi.org/10.1038/ncb2930
- von der Emde L, Goltz D, Latz S, Müller SC, Kristiansen G, Ellinger J, Syring I. Prostaglandin receptors EP1-4 as a potential marker for clinical outcome in urothelial bladder cancer. Am J Cancer Res 2014; 4:952-62; PMID:25520883
- Leone V, di Palma A, Ricchi P, Acquaviva F, Giannouli M, Di Prisco AM, Iuliano F, Acquaviva AM. PGE2 inhibits apoptosis in human adenocarcinoma Caco-2 cell line through Ras-PI3K association and cAMP-dependent kinase A activation. Am J Physiol 2007; 293:G673-81; PMID:17640974; http://dx.doi.org/10.1152/ajpgi.00584.2006
- John WSH, Matt WMC. Combination of phytochemicals as adjuvants for cancer therapy. Recent Pat Anticancer Drug Discov 2014; 9:297-302; PMID:24942759; http://dx.doi.org/10.2174/1574892809666140619154838
- Sen S, Sharma H, Singh N. Curcumin enhances Vinorelbine mediated apoptosis in NSCLC cells by the mitochondrial pathway. Biochem Biophys Res Commun 2005; 331:1245-52; PMID:15883009; http://dx.doi.org/10.1016/j.bbrc.2005.04.044
- Hengartner MO. The biochemistry of apoptosis. Nature 2000; 407:770-6; PMID:11048727; http://dx.doi.org/10.1038/35037710
- Eibl G, Bruemmer D, Okada Y, Duffy JP, Law RE, Reber HA, Hines OJ. PGE2 is generated by specific COX-2 activity and increases VEGF production in COX-2-expressing human pancreatic cancer cells. Biochem Biophys Res Commun 2003; 306:887-97; PMID:12821125; http://dx.doi.org/10.1016/S0006-291X(03)01079-9
- Kamiyama M, Pozzi A, Yang L, DeBusk LM, Breyer RM, Lin PC. EP2, a receptor for PGE2, regulates tumor angiogenesis through direct effects on endothelial cell motility and survival. Oncogene 2006; 25:7019-28; PMID:16732324; http://dx.doi.org/10.1038/sj.onc.1209694
- Wang X, Klein RD. Prostaglandin E2 induces vascular endothelial growth factor secretion in prostate cancer cells through EP2 receptor-mediated cAMP pathway. Mol Carcinog 2007; 46:912-23; PMID:17427962; http://dx.doi.org/10.1002/mc.20320
- Rasmuson A, Kock A, Fuskevåg OM, Kruspig B, Simón-Santamaría J, Gogvadze V, Johnsen JI, Kogner P, Sveinbjörnsson B. Autocrine prostaglandin E(2) signaling promotes tumor cell survival and proliferation in childhood neuroblastoma. PLoS One 2012; 7:e29331; PMID:22276108; http://dx.doi.org/10.1371/journal.pone.0029331
- Tian M, Schiemann WP. PGE2 receptor EP2 mediates the antagonistic effect of COX-2 on TGF-β signaling during mammary tumorigenesis. FASEB J 2010; 24:1105-16; PMID:19897661; http://dx.doi.org/10.1096/fj.09-141341
- Rosner M, Hengstschläger M. Cytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: rapamycin triggers dephosphorylation and delocalization of the mTORC2 components rictor and sin1. Hum Mol Genet 2008; 17:2934-48; PMID:18614546; http://dx.doi.org/10.1093/hmg/ddn192