
ABSTRACT
MET plays an important role in the development and progression of papillary renal cell carcinoma (pRCC). Evaluation of efficacy of MET inhibitors against pRCC has been hampered by limited preclinical models depicting MET abnormalities. We established a new patient-derived xenograft (PDX) model of pRCC carrying an activating mutation of MET and tested the ability of cabozantinib, an inhibitor of receptor tyrosine kinases including MET, to inhibit tumor growth and metastasis. Precision-cut, thin tissue slices from a pRCC specimen obtained by nephrectomy were implanted under the renal capsule of RAG2−/−γC−/− mice to establish first generation TSG-RCC-030. Histologic and genetic fidelity and metastatic potential of this model were characterized by immunohistochemistry, direct DNA sequencing and quantitative polymerase chain reaction (qPCR). The effect of cabozantinib on tumor growth and metastasis was evaluated. Whether measurements of circulating tumor DNA (ctDNA) by allele-specific qPCR could be used as a biomarker of tumor growth and response to therapy was determined. Subrenal and subcutaneous tumor grafts showed high take rates and metastasized to the lung. Both primary tumors and metastases expressed typical markers of pRCC and carried the same activating MET mutation as the parental tumor. Cabozantinib treatment caused striking tumor regression and inhibited lung metastasis in TSG-RCC-030. Plasma ctDNA levels correlated with tumor volume in control mice and changed in response to cabozantinib treatment. TSG-RCC-030 provides a realistic preclinical model to better understand the development and progression of pRCC with MET mutation and accelerate the development of new therapies for pRCC.
Abbreviations
| ccRCC | = | Clear cell renal cell carcinoma |
| cfDNA | = | Circulating cell-free DNA |
| ctDNA | = | circulating tumor DNA |
| IHC | = | Immunohistochemistry |
| PCR | = | Polymerase chain reaction |
| PDX | = | Patient-derived xenograft |
| pRCC | = | Papillary renal cell carcinoma |
| qPCR | = | Quantitative Polymerase Chain Reaction |
| RCC | = | Renal cell carcinoma |
| TSG | = | Tissue slice graft |
Introduction
Papillary renal cell carcinoma (pRCC) is a histologically, genetically and clinically distinct subtype of renal cell carcinoma (RCC). Second in frequency to clear cell renal cell carcinoma (ccRCC) which accounts for approximately 70–80% of cases, pRCC represents up to 10–20% of all RCCs.Citation1,2 Histologically, pRCC is characterized by a predominantly papillary growth pattern with tubular areas and fibrovascular cores.Citation3 Genetically, pRCC is marked by high incidence of trisomy or tetrasomy of chromosome 7, where genes for the MET receptor and its ligand, hepatocyte growth factor (HGF), reside.Citation4 Activating mutations in the kinase domain of MET are found in the majority of hereditary pRCC cases, as well as in 5–13% of sporadic pRCC cases.Citation5,6 Elevated MET gene expression was observed in large molecular studies of pRCC and correlated with adverse clinical features,Citation7-9 indicating a pivotal role of MET in pRCC.
Although pRCC overall has a better prognosis than ccRCC, currently available therapies provide only modest benefit for patients with advanced pRCC.Citation1,2,10 In recent years, the treatment of ccRCC has undergone dramatic changes with the development of FDA-approved targeted therapies including mTOR inhibitors and tyrosine kinase inhibitors of VEGF and its relatives.Citation11-13 However, there are few published clinical trials specifically evaluating the effects of these compounds in pRCC. Results from clinical trials enrolling patients with multiple RCC subtypes indicate that the clinical benefit of mTOR inhibitors is similar in ccRCC and pRCC patients,Citation14 whereas agents targeting VEGF pathways are significantly inferior in pRCC compared to ccRCC patients.Citation15,16
The development of novel therapies to effectively treat pRCC has been hampered by a lack of realistic preclinical models. We previously developed tissue slice graft (TSG) models of RCC by implanting precision-cut, thin tissue slices from fresh tumor specimens either under the renal capsule or skin of immunodeficient mice.Citation17,18 Here, we describe the establishment of a patient-derived xenograft (PDX) model of pRCC carrying an activating mutation of MET in its kinase domain using the same methodology. We characterized the histological and genetic fidelity and metastatic potential of this model (“TSG-RCC-030”) and evaluated the effect of cabozantinib, an inhibitor of MET approved by FDA for the treatment of thyroid cancer, on tumor growth and metastasis. Finally, we investigated whether circulating tumor DNA (ctDNA) can be used as a biomarker for tumor growth and response to therapy.
Results
TSG-RCC-030 from a pRCC specimen maintained histological fidelity
First generation TSGs (TSG-RCC-030) were established by implanting precision-cut, thin slices of fresh tissue from a pRCC specimen of Fuhrman grade 4 with focal sarcomatoid pathology under the renal capsule of immunodeficient mice. Serial passages were similarly carried out by implanting precision-cut slices of fresh TSG tissues under the renal capsule of immunodeficient mice for up to 6 times. Subcutaneous TSGs were also successfully generated from cryopreserved precision-cut slices of TSG tissues. The engraftment rate of each passage is listed in . After an initial take rate of 50% in the first generation, the take rate of all subsequent implantations was 100%. The immunohistological phenotype of the TSGs was similar to that of the parental tumor (). Specifically, both first generation (1°) subrenal and sixth generation (6°) subcutaneous TSGs, used in this study to evaluate activity of cabozantinib, showed similar histology to the parental tumor (). High intensity of human-specific nuclear antigen Ku70, as in the parental tumor (), confirmed the human origin of TSGs. In addition, both 1° subrenal and 6° subcutaneous TSGs showed strong staining of typical markers of pRCC, including AMACR and Pax-8,Citation19 similar to the parental tumor (). Moreover, both subrenal and subcutaneous TSGs were negative for CD117,Citation19 similar to the parental tumor (). Overall, the staining patterns observed for AMACR, Pax-8 and CD117 were as expected for pRCC and demonstrated that both subrenal and subcutaneous TSGs from this pRCC specimen maintained immunohistological characteristics of the parental tumor.
Figure 1. Histological and immunophenotype of parental (patient) tumor, its derived TSG and lung metastasis. (A-E) Hematoxylin and eosin (H&E) staining demonstrated similar histology among parental tumor, primary first generation (1st) TSG under the renal capsule and its derived lung metastasis, and primary subcutaneous (subQ) tumor from sixth generation TSG and its derived lung metastasis. IHC revealed similar expression of human-specific nuclear antigen Ku70 (F-J) and typical markers of pRCC including AMACR (K-O) and Pax-8 (P-T) among the parental tumor, primary TSGs and metastases. No expression of CD117 was detected (U-Y). Magnification of all images is 200×. Dotted lines in (H) (J) (M) (O) (R) (T) (W) and (Y) delineate metastatic tumor cells vs. mouse tissue in the lung.
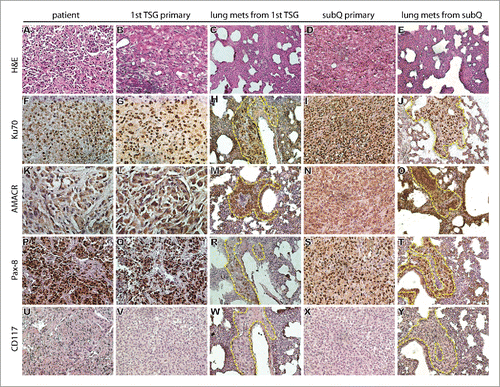
Table 1. Engraftment rates of TSGs and metastasis rate.
TSG-RCC-030 metastasized to clinically relevant sites
One mouse bearing a first generation TSG was found to have gross lung metastases on autopsy (). These metastases expressed human-specific nuclear antigen Ku70 () that distinguished them from surrounding murine cells. Expression of AMACR, Pax-8 and CD117 in lung metastases was similar to that in the primary TSG and parental tumor (). Subsequent passages of the subrenal TSG from this mouse also developed gross metastases to the lung (). In addition, lung metastases were also detected in mice carrying subcutaneous TSGs using immunohistochemistry ( and ). Interestingly, the patient had pulmonary metastases at the time of diagnosis, demonstrating that tumor cells from TSGs metastasized to the same site as in the patient. Gross metastases were not visible in liver or spleen, and qRT-PCR with human-specific primers for GAPDH confirmed the absence of tumor cells in these organs (data not shown). These results show that TSG-RCC-030 has the capacity to produce metastatic disease to the lung from a primary tumor implanted either under the renal capsule or the skin.
TSG-RCC-030 maintained the MET point mutation of the parental tumor
To determine whether the parental tumor harbored any mutations in MET, the third most frequently mutated gene in pRCC (5–13% of pRCC patients), we sequenced exon 16–20 of the gene using human-specific primers because mutations were frequently found in this region.Citation5,6 A missense mutation [T-3997C(M1268T)] that constitutively activates METCitation20,21 was detected in both parental tumor and its derived TSG-RCC-030 (). In the parental tumor, both wild type and mutant alleles were detected (), suggesting several mutually inclusive possibilities: 1) Tumor cells were heterozygous for this mutation; 2) Only a subset of tumor cells harbored a homozygous mutation; 3) MET was mutated in tumor cells but not in other cell types including stromal cells and infiltrating immune cells. In the 1° subrenal TSG, the same mutation was detected in 100% of the cells (), ruling out the first possibility. If possibility 2 was correct, then the tumor cells carrying the mutation outgrew tumor cells that didn't have the mutation. Finally, if possibility 3 was the case, then other cell types carrying wild type MET diminished over time. Moreover, in cfDNA isolated from plasma of mice carrying fourth generation (4°) subrenal TSGs, the same mutation was detected in 100% of the copies amplified using human-specific primers (), pointing to the feasibility of using ctDNA as a biomarker in this PDX model. Altogether, these results demonstrated that TSG-RCC-030 maintained genetic fidelity to the parental tumor and released ctDNA with the same genetic mutation.
Figure 2. Detection of MET mutation by direct sequencing of tumor tissue DNA and plasma ctDNA. The same point mutation [T-3997C(M1268T)] in the MET kinase domain (A) was detected in patient tumor tissue (B), 1° (first generation) subrenal TSG (C) and ctDNA from mice carrying 4° (fourth generation) subrenal TSGs (D).
![Figure 2. Detection of MET mutation by direct sequencing of tumor tissue DNA and plasma ctDNA. The same point mutation [T-3997C(M1268T)] in the MET kinase domain (A) was detected in patient tumor tissue (B), 1° (first generation) subrenal TSG (C) and ctDNA from mice carrying 4° (fourth generation) subrenal TSGs (D).](/cms/asset/c443ce2b-b321-4c3e-82c8-0d2131e1cc8f/kcbt_a_1219816_f0002_c.gif)
Cabozantinib inhibited growth of TSG-RCC-030
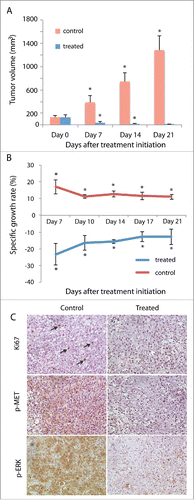
To determine whether cabozantinib, an FDA-approved MET inhibitor for treatment of medullary thyroid cancer,Citation22 inhibited tumor growth in our PDX model, we generated subcutaneous TSGs in 10 mice and randomized them into control and treated groups when TSGs reached ∼100–200 mm3. TSGs showed continuous growth in the control group with a 10-fold increase in tumor volume over 21 d (). In contrast, TSG volume decreased by > 14-fold in mice treated with 30 mg/kg of cabozantinib (once/day, 7 days/week) at the end of 21 d of treatment (). No significant adverse effects associated with treatment were observed in treated mice, e.g. mice maintained weights similar to control group. Moreover, TSGs in the control group showed an average specific growth rate of 11–17% throughout the study, whereas TSGs in the treated group showed a negative specific growth rate of 13–23% (), resulting in near complete tumor regression. Consistently, tumor cells in control mice showed strong nuclear Ki67 staining whereas tumor cells in treated mice were not proliferating (). Moreover, a high level of phosphorylated MET, the active form of MET that mediates downstream response to MET activation,Citation23 was observed in TSGs in control but not treated mice (), suggesting that cabozantinib inhibited MET phosphorylation in pRCC as in other cancers.Citation24,25 Finally, the level of phosphorylated ERK, a downstream target of phosphorylated MET, was significantly reduced in treated vs. control mice (). These results demonstrated that cabozantinib was highly effective in inhibiting tumor growth of TSG-RCC-030 carrying an activating MET mutation and reduced MET activity.
Figure 3. Regression of primary tumor induced by cabozantinib in TSG-RCC-030. Subcutaneous tumor growth was measured in control and cabozantinib-treated mice. Tumor volume significantly decreased over the time course of 21 d in treated mice while continuously increasing in control mice (A). The specific tumor growth rate was negative in treated mice and positive in control mice (B). IHC demonstrated strong expression of the proliferation marker Ki67 in a subset of cells (arrows) and phosphorylated MET as well as its target, phosphorylated ERK, in majority of tumor cells in control but not in treated mice (C). Data points represent mean+/− SD. *p < 0.05 by Student's t-test. Magnification of all images is 200×.
Cabozantinib inhibited lung metastases
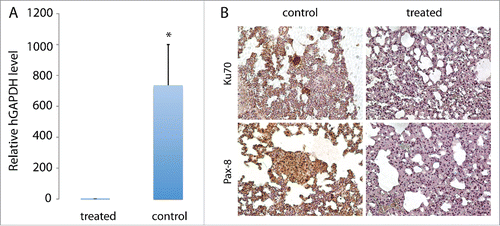
To determine whether cabozantinib inhibited lung metastases, we performed qRT-PCR using human-specific GAPDH primers, assuming that all human cells detected in mouse lung were metastatic tumor cells. As shown in , the level of human GAPDH in the lung tissue of control mice was 735-fold higher than that in lungs of treated mice, demonstrating a high efficacy of cabozantinib in inhibiting lung metastases. Consistently, tumor cells expressing human-specific nuclear antigen Ku70 were detected only in lungs of control but not treated mice (). These cells also expressed typical markers of pRCC such as Pax-8 (), confirming the pRCC origin of these cells.
Figure 4. Inhibition of TSG-RCC-030 lung metastasis by cabozantinib. The level of human GAPDH in the lung tissue of control mice was significantly higher than in cabozantinib-treated mice as assessed by qPCR using human-specific GAPDH primers (A). Tumor cells expressing human-specific nuclear antigen Ku70 were detected only in control but not treated mice (B). These metastatic tumor cells also expressed Pax-8, a typical marker of pRCC (B). Data points represent mean+/− SD. *p < 0.05 by Student's t-test. Magnification of images is 200×.
ctDNA level correlated with tumor volume and responded to therapy
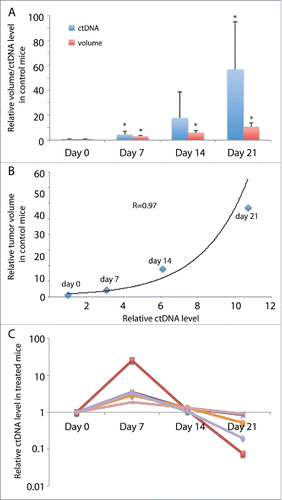
ctDNA as a liquid biopsy has been shown to correlate with tumor volume and predict response to therapy.Citation26,27 To determine whether ctDNA levels in the TSG-RCC-030 model correlated with tumor volume, we compared ctDNA levels in control and treated mice over the time course of 21 d. Using allele-specific primers that amplified the mutant allele of MET, ctDNA levels in plasma from control and treated mice were quantified by qPCR. As shown in , ctDNA levels in control mice increased continuously over time to a 57-fold higher level at the end of the experiment. In addition, tumor volumes correlated with exponential increase in ctDNA levels with a correlation coefficient of 0.97 (). In treated mice, ctDNA levels increased at 7 d after initiation of treatment, the first time point examined (). The levels then decreased afterward to different extents ranging from 10–92% by the end of the experiment (), demonstrating a more heterogeneous effect of treatment on ctDNA level than on tumor volume. These results suggest that ctDNA levels responded to therapy and can be used as a biomarker for tumor growth in this PDX model.
Figure 5. ctDNA levels in mice bearing TSG-RCC-030 correlated with tumor volume and responded to cabozantinib treatment. ctDNA levels were measured by qPCR using allele-specific primers that amplified the mutant MET. ctDNA levels increased continuously over time, reaching a 57-fold higher level at the end of the experiment in control mice (A). In control mice, tumor volume correlated with an exponential increase in ctDNA level with a correlation coefficient of 0.97 by Pearson Correlation analysis (B). In treated mice, ctDNA levels increased at 7 d after initiation of treatment and thereafter decreased to different extents in individual mice (C). Data points represent mean+/− SD in (A), mean in (B) and individual in (C). *p < 0.05 by Student's t-test.
Discussion
Our study is the first to establish a metastatic PDX model of pRCC driven by an activating MET mutation in the kinase domain. Constitutive activation of MET as a result of activating mutations together with overexpression of MET caused by DNA copy number gain contributes to oncogenesis in many cancer types including pRCC. A recent study by Schuller et al. identified two pRCC PDX models that do not carry MET mutations but contain regions of MET copy number gain, which is observed in a subset of pRCC.Citation28 Together, these PDX models provide much-needed representative preclinical models not only for understanding the oncogenic role of MET in pRCC but also for the development of effective therapies for pRCC with disregulated MET signaling. Indeed, Schuller et al. showed that treatment with the selective MET inhibitor AZD6094 resulted in tumor regression in the 2 PDX models with MET copy number gain, supporting the clinical development of AZD6094 as effective therapy for pRCC.Citation28 However, whether the primary tumors metastasized and whether AZD6094 inhibited metastases in those models was not clear. Our results demonstrated that our PDX model, TSG-RCC-030, can be cryopreserved and serially passaged and maintains histological fidelity as well as metastatic potential. Moreover, treatment with the MET inhibitor cabozantinib not only caused striking regression of primary tumors but also inhibited lung metastases, strongly supporting the testing of this FDA-approved agent in patients with pRCC expressing activating MET mutations.
One of the advantages of our pRCC PDX model is that tumor cells from TSGs metastasize to clinically relevant sites such as the lung,Citation29 a phenomena that we observed in our ccRCC PDX models in previous studies.Citation17,18,30 This is particularly important in advancing our knowledge in understanding the biology and developing effective treatments of metastatic RCC since, overall, approximately 25–30% of RCC patients present with metastatic disease at the time of diagnosis.Citation31 For example, it is not clear when metastatic cells from a primary tumor disseminate to distant organs and what effects removal of the primary tumor have on the proliferation of metastatic cells. Our PDX model may be useful in addressing these questions. Using highly sensitive molecular assays such as qPCR, one can determine the time course of spreading of MET mutant tumor cells to the lung in our PDX model. In addition, removing the primary tumor after cells have disseminated to the lung will allow us to study the effects of the primary tumor on the growth of metastases. Besides understanding the time course of metastasis, our model can also serve as a realistic platform to screen new therapeutic agents alone or in combination with existing therapies to better manage the treatment of metastatic pRCC.
Our results demonstrated that cabozantinib is highly effective in inhibiting pRCC carrying an activating mutation of MET, suggesting a role for cabozantinib in treating the subset of patients with this subtype of RCC. Unfortunately, despite the pivotal role of MET in pRCC, 2 recently completed clinical trials examining the safety and efficacy of cabozantinib in RCC only enrolled patients with clear cell histology. In a phase I study, Choueiri et al. reported that partial response was observed in 7 of 25 (28%) enrolled patients with metastatic ccRCC.Citation32 Median progression-free survival (PFS) was 12.9 months, and median overall survival was 15.0 months.Citation32 In a much larger Phase III study of 658 patients with advanced ccRCC, the same investigators observed an objective response rate (ORR) of 21% and median PFS of 7.4 months for cabozantinib, compared to ORR of 5% and PFS of 3.8 months for everolimus, a standard second-line treatment for advanced ccRCC.Citation33 The high efficacy of cabozantinib in advanced ccRCC is not surprising since it inhibits multiple tyrosine kinases including MET, VEGF receptors and AXL, and MET and AXL are upregulated as a consequence of VHL inactivation commonly observed in ccRCC.Citation25,34 We tested a soluble AXL inhibitor in our PDX model and it did not affect primary tumor growth or metastasis to the lung (data not shown), suggesting that the response to cabozantinib in our model was unlikely due to AXL inhibition. It will be interesting in future studies to test VEGF inhibitors such as sunitinib to determine whether the response to cabozantinib is entirely MET-dependent. Clinical trials to evaluate the efficacy of MET inhibitors in pRCC are now beginning to emerge. For example, a phase II study of 74 pRCC patients showed a 13.5% response rate to foretinib, a dual MET and VEGF receptor inhibitor, with a median PFS of 9.3 months.Citation35 An on-going phase II study designed to evaluate the efficacy of tivantinib, a selective MET inhibitor with reported off-target effects,Citation36 alone or in combination with erotinib, an EGFR inhibitor, in pRCC is expected to be completed in 2016.Citation1 Finally, a randomized, phase II efficacy assessment of multiple MET kinase inhibitors including cabozantinib in metastatic pRCC (PAPMET) was started in April 2016 and is expected to finish in 2019 (https://clinicaltrials.gov/ct2/show/NCT02761057?term=PAPMETandrank=1). Results from our study and others provide rationale for evaluating cabozantinib as a single agent or in combination with other agents in the treatment of pRCC. Resistance to a MET inhibitor in a patient carrying a MET mutation has just been reported.Citation37 It will be interesting in future studies to determine whether resistance to cabozantinib will be observed with time in our PDX model. If so, TSG-RCC-030 will be useful for testing additional therapies after resistance to cabozantinib arises.
One interesting finding in our study was that tumor DNA carrying a MET mutation was detectable in mouse plasma and its level correlated with volume of primary tumor. Although ctDNA as a biomarker for detecting tumor presence, predicting prognosis and assessing therapeutic response has gained much attention in recent years, few publications describe the detection of ctDNA in mouse models. Thierry et al. demonstrated that ctDNA from mutated tumor (human) cells could be detected by using human-specific KRAS or PSAT1 primers and by assessing the presence of the BRAF V600E mutation, and the concentration of human ctDNA increased significantly with tumor growth.Citation38 Gorges et al. also showed a correlation between ctDNA levels and tumor size in mice carrying xenografts derived from cultured breast cancer cells by qPCR targeting human Alu sequences.Citation39 Interestingly, ctDNA levels in the breast cancer xenograft model showed consistent decreases at day 3, 5 and 8 after treatment with a MEK inhibitor, whereas in our model, the ctDNA level showed an initial increase 7 d (the earliest time point monitored) after treatment with cabozantinib. This may be explained by increased cell death and therefore increased ctDNA release in our model. Although little or no expression of apoptotic marker caspase 3 was observed at the end of the treatment (data not shown), we expect to see high level of apoptosis at day 7 and prior in treated mice if it is feasible to obtain tumor tissues. A similar early increase of ctDNA has been observed in a melanoma patient with V600EBRAF mutation 3 d after initiation of dabrafenib treatment.Citation40 Taken together, the characteristics of TSG-RCC-030 provide a unique opportunity to better understand the development and progression of pRCC and accelerate the development of new therapies for pRCC and to evaluate ctDNA as a biomarker in the detection and treatment of pRCC.
Materials and methods
Ethics statement
All animal studies were approved by the Stanford Administrative Panel on Laboratory Animal Care (APLAC) and done in compliance with the regulations for animal studies at Stanford University. Patient-derived tissue was obtained immediately after surgery under a protocol approved by the Stanford Institutional Review Board. The participant provided his written informed consent to participate in this study.
Subrenal/subcutaneous implantation of precision-cut, thin tissue slices
A core of tissue was obtained from a pRCC specimen immediately following nephrectomy. The specimen was from a 67 year-old male and had a clinical stage of pTa3pNxpMx and Fuhrman grade 4 with focal sarcomatoid pathology at the time of diagnosis. Precision-cutting and subrenal and/or subcutaneous implantation of tissue slices were performed as previously described.Citation41 All animal work was done in accordance with institutional regulations for laboratory animal studies. RAG2−/−γC−/− male and female mice, 6–8 weeks of age, were engrafted with RCC tissue slices. Tumor tissues were cryopreserved and thawed as previously described.Citation30
Immunohistochemistry
Immunohistochemistry (IHC) was performed as previously described.Citation41 The sources and dilutions of the antibodies used in this study are listed in Supporting Information .
Sequencing of MET
DNA was extracted from fresh patient tumor tissue or TSGs preserved in Allprotect tissue reagent (Qiagen) using an AllPrep DNA/RNA/Protein Mini Kit (Qiagen) according to the manufacturer's directions. The c-terminal exons (exon 16–20) of MET were selected for polymerase chain reaction (PCR) amplification and direct sequencing by Stanford Protein and Nucleic Acid Facility. PCR primers for MET amplification were forward: 5′-CGCTGGTGGTCCTACCATAC-3′ and reverse: 5′-GGTGCCAGCATTTTAGCATT-3′.
Cabozantinib treatment of TSG-RCC-030
Subcutaneous TSGs were established in 10 mice and the mice were randomized into control and treated groups when tumor volume (length x width x width /2) reached ∼100–200 mm3. Cabozantinib (Exelixis, Inc.) was administered by oral gavage, once a day, 7 days/week, at a dose of 30 mg/kg and vehicle (water) was administered similarly as control. The cabozantinib gavage solution was made fresh daily. Subcutaneous tumor volume was determined by caliper measurements twice a week. Specific tumor growth rate was calculated as ln(V2-V1)/(t2-t1) (V is tumor volume and t is time). All animals were sacrificed after 21 d of treatment. Tissues were collected and preserved either in Allprotect for DNA/RNA isolation or 10% formalin for IHC within 24 hr after the last treatment. The dose and schedule of cabozantinib used in this study were based on previous studies demonstrating effective tumor inhibition in preclinical trials.Citation25,42
Isolation of circulating cell-free DNA (cfDNA)
One hundred microliters of blood were collected by retro-orbital bleeding every week from each mouse and centrifuged for 15 minutes at 2,000 x g. Plasma was transferred to a clean tube and EDTA was added to a final concentration of 10 mM to stabilize cfDNA.Citation43 Plasma was stored at −80°C before isolation of cfDNA. Fifty microliters of plasma were used for cfDNA isolation using a QIAamp DSP Virus Spin Kit (Qiagen) according to manufacturer's instructions. Fifty microliters of nuclease-free water were used to elute cfDNA. The quality of the cfDNA was examined by electrophoresis.
Quantitative polymerase chain reaction (qPCR)
For lung metastasis quantification: Tissues were preserved in Allprotect tissue reagent (Qiagen) at −20°C before RNA extraction. RNA was isolated using Trizol reagent according to manufacturer's instructions (Invitrogen). cDNA was synthesized from 2 μg of DNase-treated RNA using SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen). One microliter of cDNA was subjected to PCR amplification using SYBR Green PCR Master Mix (Applied Biosystems). The level of 18S RNA was used as an internal control to normalize hGAPDH levels. For 18S RNA amplification, cDNA was diluted 1:50. PCR amplification was performed on the ABI Prism 7900 HT Sequence Detection System (Applied Biosystems). Relative quantitation of mRNA expression levels was determined using the relative standard curve method according to the manufacturer's instructions (Applied Biosystems). The following primers were used to amplify specific target genes: hGAPDH, forward 5′-ATGGGGAAGGTGAAGGTCG-3′ and reverse 5′-GGGGTCATTGATGGCAACAATA-3′; 18S RNA, forward 5′-GCCCGAAGCGTTTACTTT GA-3′ and reverse: 5′-TCCATTATTCCTAGCTGCGGTATC-3′.
For ctDNA quantification: Five microliters of cfDNA were used in each qPCR reaction with human specific primers for the mutated MET (T-3997C(M1268T)): forward 5′-AAGCTGCCAGTGAAGTGGAT-3′ and reverse 5′-GAGGAGAAAACTCAGAGATAACCAA-3′.
Statistical analysis
Two-tailed Student's t-test was used for 2-arm experiments. A p < 0.05 was considered significant. Statistical tests were performed with Excel Stats. Pearson correlation coefficients between ctDNA levels and tumor volumes were calculated in Excel.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_Table.docx
Download MS Word (49.2 KB)Acknowledgments
Cabozantinib used in this study in compliance with the Material Transfer Agreement was provided by Exelixis, Inc. A.M.W. Chan and E.B. Rankin were funded by The Department of Defense Ovarian Cancer Research Academy OC140611 (EBR).
References
- Twardowski PW, Mack PC, Lara PN, Jr. Papillary renal cell carcinoma: current progress and future directions. Clin Genitourin Cancer 2014; 12:74-9; PMID:24629521; https://doi.org/10.1016/j.clgc.2013.11.013
- Courthod G, Tucci M, Di Maio M, Scagliotti GV. Papillary renal cell carcinoma: A review of the current therapeutic landscape. Crit Rev Oncol Hematol 2015; 96:100-12; PMID:26052049; https://doi.org/10.1016/j.critrevonc.2015.05.008
- Algaba F, Akaza H, Lopez-Beltran A, Martignoni G, Moch H, Montironi R, Reuter V. Current pathology keys of renal cell carcinoma. Eur Urol 2011; 60:634-43; PMID:21741159; https://doi.org/10.1016/j.eururo.2011.06.047
- Bentz M, Bergerheim US, Li C, Joos S, Werner CA, Baudis M, Gnarra J, Merino MJ, Zbar B, Linehan WM, et al. Chromosome imbalances in papillary renal cell carcinoma and first cytogenetic data of familial cases analyzed by comparative genomic hybridization. Cytogenet Cell Genet 1996; 75:17-21; PMID:8995481; https://doi.org/10.1159/000134448
- Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, Scherer SW, Zhuang Z, Lubensky I, Dean M, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet 1997; 16:68-73; PMID:9140397; https://doi.org/10.1038/ng0597-68
- Salvi A, Marchina E, Benetti A, Grigolato P, De Petro G, Barlati S. Germline and somatic c-met mutations in multifocal/bilateral and sporadic papillary renal carcinomas of selected patients. Int J Oncol 2008; 33:271-6; PMID:18636147; https://doi.org/10.3892/ijo_00000006
- Sweeney P, El-Naggar AK, Lin SH, Pisters LL. Biological significance of c-met over expression in papillary renal cell carcinoma. J Urol 2002; 168:51-5; PMID:12050491; https://doi.org/10.1016/S0022-5347(05)64830-6
- Albiges L, Guegan J, Le Formal A, Verkarre V, Rioux-Leclercq N, Sibony M, Bernhard JC, Camparo P, Merabet Z, Molinie V, et al. MET is a potential target across all papillary renal cell carcinomas: result from a large molecular study of pRCC with CGH array and matching gene expression array. Clin Cancer Res 2014; 20:3411-21; PMID:24658158; https://doi.org/10.1158/1078-0432.CCR-13-2173
- Cancer Genome Atlas Research Network, Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, Davis C, Wheeler DA, Murray BA, Schmidt L, et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N Eng J Med 2016; 374:135-45; PMID:26536169; https://doi.org/10.1056/NEJMoa1505917
- Bellmunt J, Dutcher J. Targeted therapies and the treatment of non-clear cell renal cell carcinoma. Ann Oncol 2013; 24:1730-40; PMID:23625974; https://doi.org/10.1093/annonc/mdt152
- Schmidinger M. Improving outcomes in metastatic clear cell renal cell carcinoma by sequencing therapy. Am Soc Clin Oncol Educ Book 2014:e228-38; PMID:24857107; https://doi.org/10.14694/EdBook_AM.2014.34.e228
- Shoji S, Nakano M, Sato H, Tang XY, Osamura YR, Terachi T, Uchida T, Takeya K. The current status of tailor-made medicine with molecular biomarkers for patients with clear cell renal cell carcinoma. Clin Exp Metastasis 2014; 31:111-34; PMID:23959576; https://doi.org/10.1007/s10585-013-9612-7
- Thomas JS, Kabbinavar F. Metastatic clear cell renal cell carcinoma: A review of current therapies and novel immunotherapies. Crit Rev Oncol Hematol 2015; 96:527-33; PMID:26299335; https://doi.org/10.1016/j.critrevonc.2015.07.009
- Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska e, Sosman J, McDermott D, Bodrogi I, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Eng J Med 2007; 356:2271-81; PMID:17538086; https://doi.org/10.1056/NEJMoa066838
- Stadler WM, Figlin RA, McDermott DF, Dutcher JP, Knox JJ, Miller WH, Jr., Hainsworth JD, Henderson CA, George JR, Hajdenberg J, et al. Safety and efficacy results of the advanced renal cell carcinoma sorafenib expanded access program in North America. Cancer 2010; 116:1272-80; PMID:20082451; https://doi.org/10.1002/cncr.24864
- Escudier BJ, Ravaud A, Négrier S, Szczylik C, Bellmunt Molins J, Bracarda, P. Pisa S, Gaudreault J and Bajetta E. Update on AVOREN trial in metastatic renal cell carcinoma (mRCC): Efficacy and safety in subgroups of patients (pts) and pharmacokinetic (PK) analysis. J Clin Oncol 2008; 26(suppl 155): abstract 5025.
- Ingels A, Zhao H, Thong AE, Saar M, Valta MP, Nolley R, Santos J, Peehl DM. Preclinical trial of a new dual mTOR inhibitor, MLN0128, using renal cell carcinoma tumorgrafts. Int J Cancer 2014; 134:2322-9; PMID:24243565; https://doi.org/10.1002/ijc.28579
- Thong AE, Zhao H, Ingels A, Valta MP, Nolley R, Santos J, Young SR, Peehl DM. Tissue slice grafts of human renal cell carcinoma: an authentic preclinical model with high engraftment rate and metastatic potential. Urol Oncol 2014; 32:43 e23-30; PMID:23911681; https://doi.org/10.1016/j.urolonc.2013.05.008
- Truong LD, Shen SS. Immunohistochemical diagnosis of renal neoplasms. Arch Pathol Lab Med 2011; 135:92-109; PMID:21204715; https://doi.org/10.1043/2010-0478-RAR.1
- Jeffers M, Schmidt L, Nakaigawa N, Webb CP, Weirich G, Kishida T, Zbar B, Vande Woude GF. Activating mutations for the met tyrosine kinase receptor in human cancer. Proc Natl Acad Sci U S A 1997; 94:11445-50; PMID:9326629; https://doi.org/10.1073/pnas.94.21.11445
- Nakaigawa N, Weirich G, Schmidt L, Zbar B. Tumorigenesis mediated by MET mutant M1268T is inhibited by dominant-negative Src. Oncogene 2000; 19:2996-3002; PMID:10871851; https://doi.org/10.1038/sj.onc.1203628
- Fallahi P, Ferrari SM, Di Bari F, Materazzi G, Benvenga S, Miccoli P, Antonelli A. Cabozantinib in Thyroid Cancer. Recent Pat Anti-cancer Drug Discov 2015; 10:259-69; PMID:26152149; https://doi.org/10.2174/1574892810666150708110816
- Goetsch L, Caussanel V, Corvaia N. Biological significance and targeting of c-Met tyrosine kinase receptor in cancer. Front Biosci 2013; 18:454-73; PMID:23276936; https://doi.org/10.2741/4114
- Bentzien F, Zuzow M, Heald N, Gibson A, Shi Y, Goon L, Yu P, Engst S, Zhang W, Huang D, et al. In vitro and in vivo activity of cabozantinib (XL184), an inhibitor of RET, MET, and VEGFR2, in a model of medullary thyroid cancer. Thyroid 2013; 23:1569-77; PMID:23705946; https://doi.org/10.1089/thy.2013.0137
- Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther 2011; 10:2298-308; PMID:21926191; https://doi.org/10.1158/1535-7163.MCT-11-0264
- Ma M, Zhu H, Zhang C, Sun X, Gao X, Chen G. “Liquid biopsy”-ctDNA detection with great potential and challenges. Ann Transl Med 2015; 3:235; PMID:26539452; https://doi.org/10.3978/j.issn.2305-5839.2015.09.29
- Gingras I, Salgado R, Ignatiadis M. Liquid biopsy: will it be the ‘magic tool’ for monitoring response of solid tumors to anticancer therapies? Curr Opin Oncol 2015; 27:560-7; PMID:26335664; https://doi.org/10.1097/CCO.0000000000000223
- Schuller AG, Barry ER, Jones RD, Henry RE, Frigault MM, Beran G, Linsenmayer D, Hattersley M, Smith A, Wilson J, et al. The MET Inhibitor AZD6094 (Savolitinib, HMPL-504) Induces Regression in Papillary Renal Cell Carcinoma Patient-Derived Xenograft Models. Clin Cancer Res 2015; 21:2811-9; PMID:25779944; https://doi.org/10.1158/1078-0432.CCR-14-2685
- Bianchi M, Sun M, Jeldres C, Shariat SF, Trinh QD, Briganti A, Tian Z, Schmitges J, Graefen M, Perrotte P, et al. Distribution of metastatic sites in renal cell carcinoma: a population-based analysis. Ann Oncol 2012; 23:973-80; PMID:21890909; https://doi.org/10.1093/annonc/mdr362
- Valta MP, Zhao H, Ingels A, Thong AE, Nolley R, Saar M, Peehl DM. Development of a realistic in vivo bone metastasis model of human renal cell carcinoma. Clin Exp Metastasis 2014; 31:573-84; PMID:24715498; https://doi.org/10.1007/s10585-014-9651-8
- Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev 2008; 34:193-205; PMID:18313224; https://doi.org/10.1016/j.ctrv.2007.12.001
- Choueiri TK, Pal SK, McDermott DF, Morrissey S, Ferguson KC, Holland J, Kaelin WG, Dutcher JP. A phase I study of cabozantinib (XL184) in patients with renal cell cancer. Ann Oncol 2014; 25:1603-8; PMID:24827131; https://doi.org/10.1093/annonc/mdu184
- Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F, Hammers H, Hutson TE, Lee JL, Peltola K, et al. Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N Eng J Med 2015; 373:1814-23; PMID:26406150; https://doi.org/10.1056/NEJMoa1510016
- Rankin EB, Fuh KC, Castellini L, Viswanathan K, Finger EC, Diep AN, LaGory EL, Kariolis MS, Chan A, Lindgren D, et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc Natl Acad Sci U S A 2014; 111:13373-8; PMID:25187556; https://doi.org/10.1073/pnas.1404848111
- Choueiri TK, Vaishampayan U, Rosenberg JE, Logan TF, Harzstark AL, Bukowski RM, Rini BI, Srinivas S, Stein MN, Adams LM, et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol 2013; 31:181-6; PMID:23213094; https://doi.org/10.1200/JCO.2012.43.3383
- Katayama R, Aoyama A, Yamori T, Qi J, Oh-hara T, Song Y, Engelman JA, Fujita N. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res 2013; 73:3087-96; PMID:23598276; https://doi.org/10.1158/0008-5472.CAN-12-3256
- Diamond JR, Salgia R, Varella-Garcia M, Kanteti R, LoRusso PM, Clark JW, Xu LG, Wilner K, Eckhardt SG, Ching KA, et al. Initial clinical sensitivity and acquired resistance to MET inhibition in MET-mutated papillary renal cell carcinoma. J Clin Oncol 2013; 31:e254-8; PMID:23610116; https://doi.org/10.1200/JCO.2012.46.4289
- Thierry AR, Mouliere F, Gongora C, Ollier J, Robert B, Ychou M, Del Rio M, Molina F. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res 2010; 38:6159-75; PMID:20494973; https://doi.org/10.1093/nar/gkq421
- Gorges TM, Schiller J, Schmitz A, Schuetzmann D, Schatz C, Zollner TM, Krahn T, von Ahsen O. Cancer therapy monitoring in xenografts by quantitative analysis of circulating tumor DNA. Biomarkers 2012; 17:498-506; PMID:22616911; https://doi.org/10.3109/1354750X.2012.689133
- Tsao SC, Weiss J, Hudson C, Christophi C, Cebon J, Behren A, Dobrovic A. Monitoring response to therapy in melanoma by quantifying circulating tumour DNA with droplet digital PCR for BRAF and NRAS mutations. Sci Rep 2015; 5:11198; PMID:26095797; https://doi.org/10.1038/srep11198
- Zhao H, Nolley R, Chen Z, Peehl DM. Tissue slice grafts: an in vivo model of human prostate androgen signaling. Am J Pathol 2010; 177:229-39; PMID:20472887; https://doi.org/10.2353/ajpath.2010.090821
- Graham TJ, Box G, Tunariu N, Crespo M, Spinks TJ, Miranda S, Attard G, de Bono J, Eccles SA, Davies FE, et al. Preclinical evaluation of imaging biomarkers for prostate cancer bone metastasis and response to cabozantinib. J Natl Cancer Inst 2014; 106:dju033; PMID:24634505; https://doi.org/10.1093/jnci/dju033
- Holdenrieder S, Mueller S, Stieber P. Stability of nucleosomal DNA fragments in serum. Clin Chem 2005; 51:1026-9; PMID:15914786; https://doi.org/10.1373/clinchem.2005.048454