
ABSTRACT
Recently, we showed that the metal chelator TPEN targets colon cancer cells through redox cycling of copper. Here, we studied the DNA damage potential of TPEN and deciphered the role of Chk1, ATM and DNA-PK in TPEN-induced toxicity in 3 human colon cancer cell lines, HCT116, SW480 and HT29. We also investigated the role of reactive oxygen species (ROS) in TPEN-induced DNA damage. TPEN reduced cell viability in a dose- and time-dependent manner. Cytotoxicity was associated with significant DNA damage and higher expression of γ-H2AX protein and activation of ATM/ATR signaling pathway. Cell death by TPEN was dependent on ROS generation as evidenced by the reversal of cell viability, and DNA damage and the abrogation of γ-H2AX levels in the presence of antioxidants. Treatment with antioxidants, however, failed to reverse cytotoxicity at high TPEN concentrations (10µM). TPEN-induced cell death was also dependent on the redox cycling of copper since the copper chelator neocuproine inhibited DNA damage and reduced pChk1, γ-H2AX, and ATM protein expression. Cell death by low TPEN concentrations, involved ATM/ATR signaling in all 3 cell lines, since pre-incubation with specific inhibitors of ATM and DNA-PK led to the recovery of cells from TPEN-induced DNA damage. In addition, siRNA silencing of Chk1, DNA-PK and ATM abrogated the expression of γ-H2AX and reversed cell death, suggesting that Chk1 and DNA-PK mediate TPEN-induced cytotoxicity in colon cancer cells. This study shows for the first time the involvement of Chk1, DNA-PK and ATM in TPEN-induced DNA damage and confirms our previous findings that ROS generation and the redox cycling of copper in response to TPEN are the main mechanisms by which this compound induces cell death in human colon cancer cells. Inhibition of ATM or DNA-PK did not reverse cytotoxicity at high TPEN concentrations that cause excessive levels of ROS and irreversible cellular damage.
Abbreviations
| ROS | = | reactive oxygen species |
| XIAP | = | X-linked inhibitor of apoptosis |
| DNA-PK | = | DNA-dependent protein kinase |
| ATM | = | ataxia telangiectasia mutated |
| ATR | = | serine/threonine protein kinase ataxia telangiectasia |
| MTT | = | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| DCFH | = | 2 ′,7′-Dichlorofluorescin diacetate |
| NAC | = | N-acetyl-cysteine |
| CAT | = | catalase |
| DSB | = | double strand break |
| SSB | = | single strand break |
| Neo | = | neocuproine |
| PI | = | propidium iodide |
| DDR | = | DNA damage response |
Introduction
The importance of metal ions as cellular components is vital to the cell and the body as a whole. Many metals participate in cellular pathways that are critical for ensuring stability in cell function and survival.Citation1 For optimal biological function, the concentrations of these metals should remain within respective nontoxic ranges. Any shift toward non-favorable concentrations will disrupt the metal homeostasis, causing severe damage at the cellular level.Citation2 In comparison to normal tissues, many types of tumors have elevated levels of copper and zinc, both of which are known to contribute to the process of carcinogenesis.Citation3 Such tumors include breast, cervical, ovarian, lung, prostate, stomach and leukemias.Citation4 As most conventional therapies remain ineffective, there is still a need to find alternatives that make use of cancer cell properties while sparing normal cells.Citation5,6
One alternative approach for targeting cancer cells involves the disruption of metal homeostasis. Chelating agents that are able to sequester a number of intracellular metals have been used for the treatment of a variety of disorders.Citation7 After the discovery of bleomycin in the early 1960s and its approval in 1973,Citation8 these metal complex forming agents became more frequently used in the clinic. TPEN (N, N, N’, N’ -tetrakis-[2-pyridylmethyl]-ethylenediamine) is one such metal chelator that complexes with copper, zinc and iron.Citation7
Mammalian cells are vulnerable to numerous DNA replication errors. Nevertheless, the integrity of the DNA is preserved by the presence of highly conserved DNA damage response (DDR) pathways which mitigates DNA instability.Citation9 DNA damage and DDR deficiencies are correlated with a wide range of diseases, including malignancies.Citation10,11 Three main components form the DDR machinery: DNA damage sensors, signal transducers and effectors. ATM (ataxia telangiectasia mutated) and ATR (ATM-Rad3-related) are kinases that sense different forms of DNA breakage in order to trigger the DDR signaling cascade.Citation12 DNA-PK, a nuclear serine/threonine kinase, is another DNA damage sensor that can detect double strand breaks (DSBs), and elicit non homologous end joining repair mechanisms.Citation13 If the damage is excessive or DNA repair is ineffective, then activation of cell death is the normal physiological response.Citation12
Although TPEN has been found to inhibit proliferation and induce apoptosis in many cell systems including lymphocytes,Citation14 epithelial cells,Citation15 hepatocytes,Citation16 breast cancer,Citation17 HT-29 colorectal cancer,Citation18,19 splenocytes, ovarian cancer, prostate cancer,Citation20 and pancreatic cancer,Citation21 its DNA damage potential and mechanisms remain unclear. We have previously shown that the generation of ROS and the redox cycling of copper following TPEN treatment result in targeted cell death of HCT116 human colon cancer cells.Citation18 Here we investigated for the first time the effect of TPEN on DNA damage and the signaling molecules involved in the cellular response to damage. We found that TPEN induces DNA damage and activates ATM/ATR signaling, which is critically dependent on increased intracellular ROS production. Depleting copper by chelators and scavenging ROS by antioxidants were found to reduce TPEN-induced DNA damage at TPEN concentrations below 10 µM. In addition, inhibiting ATM, silencing Chk1 and DNA-PK masked TPEN-induced DNA damage and reversed cell death, suggesting that these DDR pathways mediate cell death by TPEN.
Results
TPEN decreases cell viability, causes DNA damage and induces ATM/ATR DNA damage response pathway
We treated 3 human colon cancer cell lines, HCT116, SW480 and HT29 with increasing concentrations of TPEN (3, 5 and 10 µM) and measured cell viability, using the MTT assay, after 24 and 48 hrs of incubation. TPEN reduced the viability of all 3 human colon cancer cells in a dose- and time- dependent manner (). To determine if TPEN induces DNA damage, we examined whether TPEN directly interacts with DNA by detecting structural DNA alterations in the presence of increasing concentrations of TPEN. Spectrophotometric analysis of calf thymus DNA showed that as the TPEN:DNA ratio increases gradually from 1:1 to 6:1, the structure of DNA varies accordingly (Fig. S1). This argues that TPEN induces a clear modification of the functional group (fluorophores) and leads to the structural alteration of DNA. We hypothesized that this interaction between TPEN and DNA might be the basis of a TPEN-induced DNA damage event which we investigated in subsequent experiments.
Figure 1. TPEN decreases the viability and induces DNA damage in human colon cancer cell lines. (A) Cell viability was measured in HCT 116, SW480 and HT29 cell lines using MTT assay in the presence of increasing concentrations of TPEN at 24 and 48 hrs (mean ± SD, n = 3), **p < 0.01 significant difference with respect to TPEN. (B) Cells were treated with increasing concentrations of TPEN for 24 hrs after which DNA damage was assessed using the comet assay. Representative images were taken by a fluorescent microscope at 40x magnification, scale bar 20 μm. (C) Flow cytometry histograms of HCT 116 cells treated with 5 μM TPEN for 24 hrs, fixed and subjected to immunocytochemical detection of p-ATM, p-ATR, p-Chk1, p-Chk2, γ-H2AX, and GAPDH, and stained with PI to detect cell cycle changes.
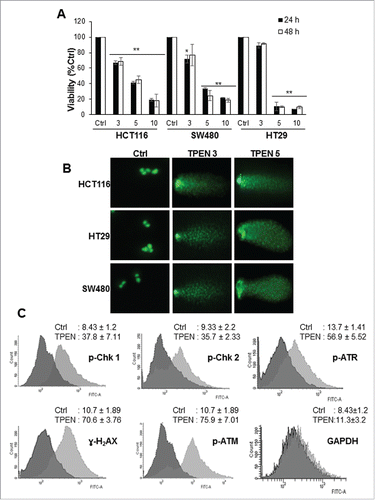
To identify the nature of the aforementioned TPEN-induced structural alterations in DNA, we aimed to detect if TPEN causes DNA double strand breaks (DSBs). A common method to detect the presence of DSBs is the comet assay.Citation22 Numerous damage comets were found in cells treated with 3 and 5 µM TPEN for 24 hrs ( and Fig. S2A, B). Quantitative analysis of the extent of DNA damage showed an increase in tail moment (numerical measurement of DNA damage) from 11 in control cells to 232 (HCT116), 315 (SW480), and 276 (HT29) upon treatment with 5 µM TPEN (data not shown). Next, we investigated the DNA damage response to TPEN by examining the activation of several DNA damage markers in HCT116 cells (). Using immunocytochemistry, the phosphorylation of major key proteins of the ATM/ATR DNA damage signaling pathway, specifically ATM, ATR, Chk2, and Chk1, was significantly enhanced in HCT116 cells treated with 5 µM TPEN for 24 hrs (). p-Chk1 is known to phosphorylate H2AX into γ-H2AX, a commonly used indicator of DNA damage.Citation23 As expected, γ-H2AX was significantly activated in response to TPEN in all 3 cell lines (, and Fig. S2C). The enhanced expression of these kinases confirms the presence of DSBs and provides evidence that the ATM/ATR pathways are activated after TPEN-induced damage.
DNA damage by TPEN is ROS-dependent
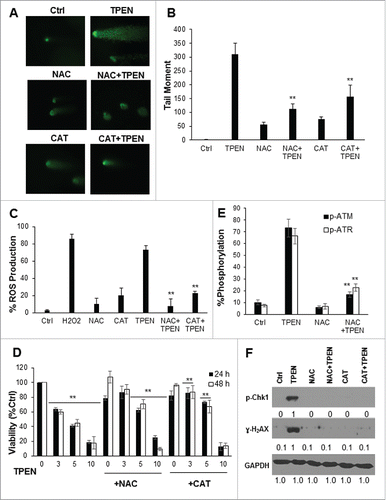
To investigate if DNA damage by TPEN is dependent on ROS, we incubated the 3 human colon cancer cells with the 2 antioxidants N-acetyl-L-cysteine (NAC) and catalase (CAT) for 2 hrs prior to treatment with TPEN and ROS levels were assessed by the 2′, 7′-dichlorofluorescin diacetate (DCFDH) assay ( and Fig. S2). Higher levels of ROS were detected in cells treated with 5 µM TPEN for 24 hrs as compared to the control (). Treatment with NAC and CAT prior to TPEN decreased ROS levels by 60% and 50%, respectively. This alteration in ROS levels correlated well with the actual cytotoxicity of TPEN, suggesting a role for the increased intracellular ROS generation in TPEN-induced cytotoxicity. The role of ROS in DNA damage by TPEN was then evaluated in all 3 human colon cancer cell lines ( and Fig. S2). Cells treated with NAC or CAT alone showed insignificant DNA damage and low tail moment ( and Fig. S2A, B). At low concentrations of TPEN, the antioxidants NAC and CAT were able to reverse TPEN-induced cell death () and inhibit DNA damage ( and Fig. S2A, B) and γ-H2AX protein expression levels ( and Fig. S2C). However, both antioxidants did not reverse cell death at high concentrations of 10 µM TPEN (). The incomplete reversal of cell death using antioxidants suggests the contribution of non-redox pathways in tandem with increased ROS production in TPEN cytotoxicity. Chk1 and ATR, 2 proteins of the ATM/ATR pathway, were also highly activated following TPEN; however, their activation was significantly inhibited (ATR) or even abrogated (Chk1) in cells pretreated with NAC or CAT (). These results suggest that ROS partially mediate the observed DNA damage effects of TPEN and the subsequent activation of the ATM/ATR signaling pathway.
Figure 2. DNA damage by TPEN is dependent on ROS. (A) Comet assay showing that treatment of HCT116 cells with 5 µM NAC or 500 IU CAT for 2 hrs before TPEN reverses DNA damage induced by 5 μM TPEN after 24 hrs. Representative images were taken by a fluorescent microscope at 40x magnification, scale bar 20μm. (B) Quantitative analysis of the tail moment was done using the Comet Assay IV software. Each value is the mean ± SD of 3 separate experiments. **p < 0.01 significant difference with respect to TPEN. (C) DCFDH assay showing that addition of NAC or CAT decrease ROS generation; ROS generated in response to 250 μM H2O2 is also shown. ** p < 0.01, significant difference with respect to TPEN. (D) MTT assay showing the viability of HCT116 cells at 24 and 48 hrs upon treatment with NAC or CAT prior to TPEN. Cells were pretreated with 5 mM NAC or 500 IU CAT for 2 hrs before TPEN. **p < 0.01, *p < 0.05, significant difference with respect to TPEN. (E) Immunocytochemical analysis by flow cytometry of the phosphorylation levels of ATM and ATR in cells treated with 5 μM TPEN for 24 hrs or treated with 5 mM NAC for 2 hrs before TPEN. (F) Western blot analysis of γ-H2AX and p-Chk1 in cells treated with 5 μM TPEN for 24 hrs or treated with 5 mM NAC or 500 IU CAT for 2 hrs before TPEN. Expression of total cellular proteins are shown and GAPDH was used as a loading control.
DNA damage by TPEN is dependent on redox cycling of copper
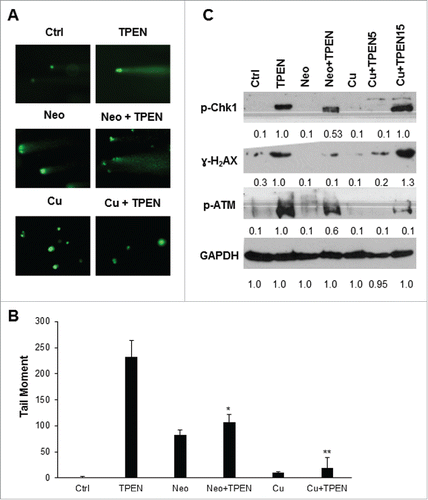
Our previous findings revealed that copper chelation and its subsequent redox cycling is required for TPEN toxicity.Citation18 Therefore, we aimed to determine whether DNA damage induced by TPEN is dependent on the redox cycling of copper. To this end we assessed DNA damage in TPEN treated HCT116 cells under 2 different conditions: first by treating cells with copper sulfate which saturates TPEN with exogenous copper thus preventing its binding to intracellular copper, and second by depleting intracellular copper using the copper specific chelator Neo.Citation22 The number of comets and tail moment were significantly inhibited in cells pretreated with copper sulfate or Neo (), both of which are treatments that reduce the redox cycling of copper. Furthermore, upon pretreatment with Neo, the expression levels of all 3 DNA damage markers, namely p-Chk1, γ-H2AX and p-ATM was significantly decreased (), suggesting a reversal of the DNA damage activity of TPEN when intracellular copper is chelated. A similar decrease in activation levels of the DNA damage markers was observed upon pretreatment with copper sulfate (), which acts to saturate TPEN and inhibit its effects. Having shown previously that saturation with copper sulfate did not block cellular toxicity at high TPEN concentrations,Citation18 we investigated whether increasing the concentration of TPEN to 15 µM in the presence of 5 µM copper sulfate would still cause DNA damage. We found that, at such high concentrations of TPEN, DNA damage was not reversed and the activation levels of p-Chk1, p-ATM and γ-H2AX were high despite the presence of copper sulfate (), namely due to the fact that a molecule of TPEN chelates Cu2+ with 1:1 stoichiometry. Altogether, these data not only point to the involvement of copper chelation in the observed TPEN-induced DNA damage, but also confirm the general role of copper redox cycling in the effects of TPEN.Citation24
Figure 3. DNA damage by TPEN is dependent on redox cycling of copper. (A) Comet assay in HCT116 cells treated with 5 μM TPEN for 24 hrs or treated with 25 μM Neo or 5 μM cooper sulfate (Cu) for 2 hrs before TPEN. Representative images were taken by a fluorescent microscope at 40x magnification, scale bar 20 μm. (B) Quantitative analysis of the tail moment of comets was done using the Comet Assay IV software. Each value is the mean ± SD of 3 separate experiments. **p <0.01 significant difference with respect to TPEN. (C) Western blot images showing expression of γ-H2AX, p-ATM, p-Chk1 in cells treated with 5 μM TPEN for 24 hrs or treated with 25 μM Neo or 5 μM copper sulfate for 2 hrs before TPEN. Expression of total cellular proteins are shown and GAPDH was used as a loading control.
DNA-PK and ATM/ATR signaling molecules mediate TPEN-induced DNA damage and toxicity
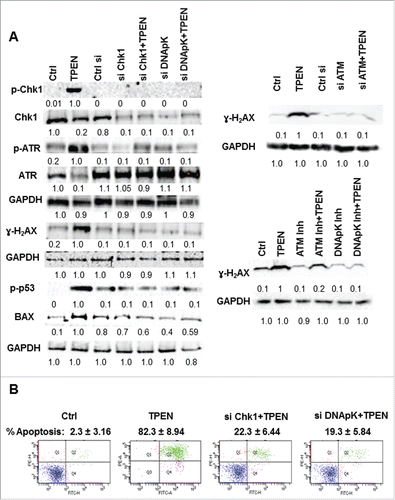
To gain mechanistic insight into how TPEN induces DNA damage, we investigated the involvement of the DNA double strand break repair enzyme DNA-PK and of the ATM molecule and its downstream effector Chk1 in TPEN-induced cell death. Our interest in the ATM/ATR pathway in mediating cellular death in the aftermath of DNA damage stems from the fact that molecules in this pathway were significantly activated in response to TPEN in a ROS dependent manner ( and ). First, our approach included inhibiting the expression of DNA-PK or ATM and investigating TPEN effects on cell viability and on activation of both proteins. Second, using siRNA knockout, we silenced Chk1, DNA-PK, and ATM in order to evaluate their involvement in TPEN-induced cell death. We observed a significant increase in the viability of the 3 cell lines upon treatment with the DNA-PK inhibitor and the ATM inhibitor prior to 3 or 5 µM (but not 10 µM) of TPEN at 24 and 48 hrs (). Moreover, the cell death effects of 3 µM of TPEN were reversed when DNA-PK, Chk 1 and ATM were silenced (). Western blot analysis revealed that, upon knockout of Chk1, DNA-PK, and ATM, the expression levels of Chk1, p-Chk1, p-ATR, and γ-H2AX proteins were either significantly inhibited or even abrogated in TPEN treated cells ( and Fig. S3). This argues that Chk1, ATM and DNA-PK are involved in the pathways that dictate cell death by TPEN. The involvement of Chk1 and DNA-PK as mediators of the anticancer effects of TPEN was confirmed by Annexin assays which showed significant increase in cell survival from 17% to 77% and 79% when cells were pre-incubated with Chk1 siRNA or DNA-PK siRNA, respectively (). As expected, this increase in cell survival was associated with lower levels of p-p53 and BAX proteins ().
Figure 4. TPEN-induced cell death involves Chk1, DNA-PK and ATM. (A, B, C) HCT116, SW480 and HT29 cells were pre-incubated with 0.013 µM KU 55933 (ATM inhibitor), or 13 µM NU7026 (DNA-PK inhibitor) for 2 hrs prior to TPEN and viability was determined 24 and 48 hrs later by MTT. **p < 0.01, *p < 0.05, significant difference with respect to TPEN. (D) HCT116, HT29 and SW480 cells were transfected with siRNA against Chk1, DNA-PK and ATM using lipofectamine 2000. After 24 hrs, cells were treated with 3 µM TPEN and harvested 72 hrs post-transfection. Ctrl siRNA is the scrambled sequence. **p < 0.01, significant difference with respect to TPEN.
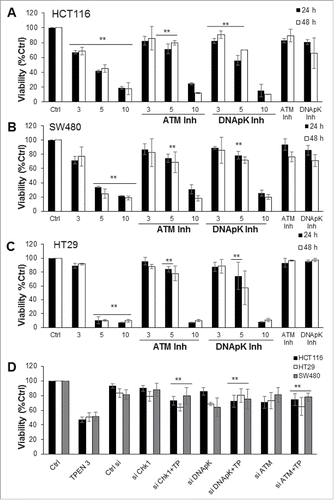
Figure 5. Silencing Chk1 and DNA-PK inhibits TPEN-induced apoptosis. (A) Whole HCT 116 cell lysates of transfected cells were prepared, and blots were probed against p-Chk1, Chk1, ATR, p-ATR, γ-H2AX, p-p53 and Bax antibodies; Expression of total cellular proteins are shown and GAPDH was used as a loading control. (B) Annexin assays in control cells and in cells treated with 3 μM of TPEN and transfected with siRNA against Chk1 and DNA-PK (mean ± SD, n = 2, ** p < 0.01, significant difference with respect to control).
Discussion
In this study, we provide evidence for the main mechanisms involved in TPEN toxicity in 3 human colorectal cancer cell lines. We show that TPEN increases the generation of ROS and the redox cycling of copper which in turn induces DNA damage. Mechanistically, TPEN-induced DNA damage involves distinct DNA damage pathways, namely ATM/ATR and DNA-PK which subsequently participate in cell fate determination.
Maintaining a minimum level of intracellular ROS is crucial for cell survival. In a normal cell, ROS are produced in the mitochondria as a byproduct of cellular metabolism.Citation25 To ensure low levels of ROS homeostasis, radicals are scavenged by endogenous, enzymatic and non-enzymatic antioxidants in order to counterbalance constant ROS production. The cell is able to cope with low oxidative stress resulting from a slight increase of ROS;Citation25 however, exaggerated production of ROS inside the cell has been shown to cause irreversible damage to intracellular macromolecules.Citation26 Our findings show that TPEN cytotoxicity is induced by the generation of ROS, which is responsible for the observed selective anticancer effects. The cytotoxicity of low concentrations of TPEN was significantly inhibited upon pretreatment with the antioxidants NAC and CAT, although such inhibition was not observed at 10 µM TPEN.
These results are consistent with previously documented effects of ROS toxicity.Citation27 Oxidative stress due to ROS is known to cause DNA lesions of both SSB and DSB nature through the direct interaction of ROS with DNA.Citation25 Additional effects of TPEN-induced cytotoxicity are due to its ability to directly interact with the DNA and alter its structural properties. Upon treatment with TPEN, significant DNA damage in the form of DSBs takes place. Phosphorylation of H2AX, a subtype of the H2A family responsible for packaging DNA and an established indicator of DNA damage,Citation23 further confirmed the DNA damage effects of TPEN.
It has been well documented that malignant cells contain elevated concentrations of copper, suggesting that cancer cells can be killed by targeting copper redox cycling.Citation22 Our previous and present findings show that TPEN toxicity is highly dependent on its high affinity to copper and the involvement of copper-TPEN complex in generation of ROS in colon cancer cells. Additionally we show that upon treatment with the copper chelator neocuproine (Neo), or with exogenous addition of copper sulfate DNA damage by TPEN was significantly reduced. However, TPEN toxicity in colon cancer cells is likely to involve other mechanisms than copper redox cycling since DNA damage was not completely reversed in cells treated with Neo.
DNA damage could occur spontaneously during the replication cycle of a normal cell.Citation28 Cells have developed a series of repair mechanisms to avoid abnormal growth and the accumulation of mutations. These mechanisms include base excision repair, mismatch repair, nucleotide excision repair, cross-link repair and double strand base repair.Citation28 Our results confirm the activation of DNA damage signaling pathways in response to TPEN in a ROS dependent manner. Three major kinases of the DDR appear to be involved in the DNA damage aftermath. First, DNA-PK is shown to participate in determining cell fate after DNA damage as its inhibition or knock-out reversed DNA damage and cell death by TPEN. This is consistent with the increasing evidence that DNA-PK has further roles than providing a scaffold where DNA ligation takes place.Citation13 In fact, DNA-PK has been shown to participate in cell fate during events which favor cell death by its linkage to cellular death machinery, and is indispensable for DSBs and V(D)J recombination through non-homologous end joining repair mechanisms.Citation13
Two other DDR kinases, specifically ATM and ATR, were shown to be activated in response to TPEN. These two PI3K-like kinases are part of pathways that lead to the activation of substrates involved in DNA repair, checkpoint signaling and cell fate determination.Citation29 Similar to the effects observed upon pretreatment with the antioxidants NAC and CAT, we showed that inhibition of ATM and DNA-PK caused the reversal of TPEN cytotoxicity only at low, but not at high TPEN concentrations that overwhelm the cell with excessive levels of ROS and cause irreversible cellular damage.
Chk1 is known to participate in regulating downstream effectors that determine cell fate and promote cell death.Citation29 The recovery from cell death that was observed when Chk1 was silenced lead us to believe that Chk1 is involved in the DNA damage mediated cell death effects of TPEN. Apoptosis studies further provided evidence that Chk1 signals cell fate after DNA damage by TPEN.
In conclusion, our results are the first to emphasize the importance of TPEN as an emerging DNA damaging therapeutic agent. Further investigations are required to assess the DNA damage potential of TPEN on a broader spectrum of cancer types by evaluating its anticancer specificity to a variety of human cancers.
Materials and methods
Cell culture and treatment
Human colorectal cancer cells, SW480 and HT-29 were purchased from the American Type Culture Collection (ATCC). Cells were cultured in RPMI 1640 (Lonza, BE12–115F) at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Media was supplemented with 1% Penicillin-Streptomycin (100 U/ml) (Lonza, DE17–602E) and 10% heat-inactivated FBS (Sigma-Aldrich, F9665). Cells were seeded at 1.2 × 105 cells/ml and treated with TPEN (Sigma-Aldrich, P4413) at 50% confluency. TPEN was prepared in DMSO (Fisher BioReagents, 0231) such that final DMSO concentration was <0.3%. The following reagents and concentrations were used: calf thymus DNA (Sigma, D4522), 5 mM NAC (Sigma-Aldrich, A7250), 500 IU CAT (Sigma, C1345), 25 μM Neo (Aldrich, N1501), 5 μM copper sulfate (Sigma, 451657), 0.013 µM ATM inhibitor KU 55933 (Santa Cruz, sc-202963), 13 µM DNA-PK inhibitor NU7026 (Sigma-Aldrich, N1537), Annexin (Roche, 11 828 681 001), PI (Sigma, P4864), HEPES (Sigma H4034), NaOH (Sigma, 06203), NaCl (Amresco 0241), and CaCl2 (Amersco, 1B1110). Unless otherwise mentioned, cells were treated with NAC, CAT, Neo, or with the inhibitors of DNA-PK and ATM at 2 hrs before TPEN.
Cell viability assay
Cells were plated in 96-well plates and treated with different concentrations of TPEN, Neo or copper sulfate. The inhibition of cell growth by TPEN was measured by the Cell Titer 96 non-radioactive cell proliferation kit (Promega Corp, G4000). The cell growth assay is an MTT-based method that measures the ability of metabolically active cells to convert tetrazolium salt into a blue formazan product the absorbance of which is recorded at 570 nm.
DCFH assay
Cells were seeded in 6 well culture plates. After 24 hrs, growth medium was replaced with low serum (2% FBS). In this experiment, cells were exposed to TPEN for 10 min in the presence or absence of NAC or CAT, after which they were incubated with 10 µM of the CM-H2DCFDA dye (Sigma-Aldrich, D6883) for 20 min. Cells were washed, harvested by centrifugation, and the pellet was re-suspended in 500 µl PBS (Lonza, BE17-517Q) followed by flow cytometry.
Transfection using lipofectamine 2000
siRNAs against DNA-PK (Santa Cruz, sc-35200), Chk1 (Santa Cruz, sc-29269), ATM (Santa Cruz, sc-29761) and control siRNA (Santa Cruz, SC-37007) were used to inhibit gene expression. siRNAs were transfected into HCT116 cells using lipofectamine 2000 transfection reagent (Invitrogen, 11668–027) following manufacturer's instructions. After 24 hrs, cells were treated with 3 µM TPEN and proteins were extracted for western blot and cell cycle analysis.
Comet assay
Slides were dipped in 1% NMA (dissolved in 1x PBS) for slide preparation. Ten µl of 10,000 cells was mixed with 75 µl of 0.5% low melting point agarose. Cell coated slides were dipped in a cold lysing solution [(2.5M NaCl, 100mM EDTA (Amersco, 0245), 10 mM Tris base (Biored, 161–60719)] at 4°C in the dark for a minimum of 2 hrs. Electrophoresis was performed in electrophoresis buffer (300 mM NaOH, 1 mM EDTA) for 30 min at 25 V and 250 mA. Slides were then neutralized with neutralizing solution (0.4 M Tris-HCl, pH 7.5), and stained with 50 µl of YOYO-1 (Thermofisher, Y3601) stain (0.25 µM YOYO-1, 2.5% DMSO, 0.5% sucrose). A fluorescent microscope illuminated with blue light (490nm) and KS 300 V3 image analysis software was used to analyze the slides.
Immunocytochemistry detected by flow cytometry
Phosphorylation of ATM (Cell Signaling, D25E5), ATR (Santa Cruz, sc-109912), Chk1 (Cell Signaling, 2348), Chk2 (Cell Signaling, 2661), H2AX (Cell Signaling, 9718) and GAPDH (Abcam, ab9484) were detected immunocytochemically by multi-parameter cytometry. Cells were collected 24 hrs after TPEN treatment, centrifuged, washed with PBS, and fixed with ice-cold 70% ethanol for a minimum of 2 hrs at 20°C. Ethanol was discarded by centrifugation at a speed of 10000 rpm for 5 min. Pellets were washed with BSA-T-PBS containing 1% BSA (Sigma, A2153), and 0.2% Triton X-100 (Biored, 1610407) dissolved in PBS. The pellets were blocked in BSA-T-PBS for 5 min at room temperature. After removal of the 1% BSA solution by centrifugation, the cells were incubated with the primary antibody at a dilution of 1:100 overnight at 4°C. Cells were washed twice with BSA-T-PBS, and the pellets were then incubated in the dark with fluorescein isothiocyanate (FITC)-conjugated secondary antibody, goat anti-mouse (Santa Cruz, sc-2005) and goat anti-rabbit (Santa Cruz, sc-2030) (1:30) for 1 hr at room temperature. A volume of 5 ml of BSA-TPBS was added to the cell suspension and kept for 2 min before centrifugation at 12000 rpm for 4 min. Finally, the cells were counterstained with 5 µg/ml PI containing 0.1mg/ml RNase A (Affymetrix, 78020Y) for 30 min at room temperature in the dark. Both the fluorescence of PtdIns and FITC of 104 cells/treatment were measured using FACS and analyzed using Cell Quest.
Western blotting
Cells were cultured in 100 mm plates and whole cell lysates were prepared by washing the cells with 1x PBS followed by scraping in ice-cold pH 6.8 lysis buffer [0.25 M Tris-HCl (Biored, 161–0799), 20% glycerol (Sigma, G5516), 4% SDS (Biored, 161–0302), and protease inhibitors (Roche, 11697498001)]. Lysates were centrifuged for 30 min at 14,000 g and proteins were quantified using the DC Protein Assay (Bio-Rad, 500–0113/4/5). Cell lysates were separated by SDS-PAGE and then transferred on nitrocellulose membranes (Biored, 161–0112). Following transfer, membranes were blocked with 5% skim milk, TBS and 0.1% Tween 20 (Biored, 170631) for 2 hrs and then probed with appropriate dilutions of the primary antibodies according to the manufacturer's directions. Membranes were then incubated with the appropriate secondary antibodies for 1 hr. Complexes of protein-antibody were detected by a chemiluminescence system (Biored, 1705060). Equal amounts of loaded proteins were verified by probing for GAPDH (Abcam, ab9484).
Annexin V Staining
Cells were incubated in Annexin-V-Fluos labeling solution [20 μl Annexin reagent, 20 μl PI (50 μg/ml) in 1000 μl incubation buffer pH 7.4 (10 mM HEPES/NaOH, 140 mM NaCl, 5 mM CaCl2)], and analyzed by flow cytometry according to manufacturer's instructions.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary_data.zip
Download Zip (364 KB)Acknowledgment
We wish to thank the Central Research Science lab personnel for their help. The statements made herein are solely the responsibility of the authors.
Funding
This work was funded in part by NPRP grant # 09-047-3-012 from the Qatar National Research Fund (QNRF).
References
- Orvig C, Abrams MJ. Medicinal inorganic chemistry: introduction. Chem Rev 1999; 99:2201-4; PMID:11749478; http://dx.doi.org/10.1021/cr980419w
- Yaman M, Kaya G, Yekeler H. Distribution of trace metal concentrations in paired cancerous and non-cancerous human stomach tissues. World J Gastroenterol 2007; 13:612-8; PMID:17278230; http://dx.doi.org/10.3748/wjg.v13.i4.612
- Versieck J. Trace elements in human body fluids and tissues. Crit Rev Clin Lab Sci 1985; 22:97-184; PMID:3891229; http://dx.doi.org/10.3109/10408368509165788
- Mulay IL, Roy R, Knox BE, Suhr NH, Delaney WE. Trace-metal analysis of cancerous and noncancerous human tissues. J Natl Cancer Inst 1971; 47:1-13; PMID:4328191; http://dx.doi.org/10.1093/jnci/47.1.1
- Hambley TW, Hait WN. Is anticancer drug development heading in the right direction? Cancer Res 2009; 69:1259-62; PMID:19208831; http://dx.doi.org/10.1158/0008-5472.CAN-08-3786
- DeVita VT, Jr., Chu E. A history of cancer chemotherapy. Cancer Res 2008; 68:8643-53; PMID:18974103; http://dx.doi.org/10.1158/0008-5472.CAN-07-6611
- Arslan P, Di Virgilio F, Beltrame M, Tsien RY, Pozzan T. Cytosolic Ca2+ homeostasis in Ehrlich and Yoshida carcinomas. A new, membrane-permeant chelator of heavy metals reveals that these ascites tumor cell lines have normal cytosolic free Ca2+. J Biol Chem 1985; 260:2719-27; PMID:3919006
- Chen J, Stubbe J. Bleomycins: towards better therapeutics. Nat Rev Cancer 2005; 5:102-12; PMID:15685195; http://dx.doi.org/10.1038/nrc1547
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature 2001; 411:366-74; PMID:11357144; http://dx.doi.org/10.1038/35077232
- Paz-Elizur T, Sevilya Z, Leitner-Dagan Y, Elinger D, Roisman LC, Livneh Z. DNA repair of oxidative DNA damage in human carcinogenesis: potential application for cancer risk assessment and prevention. Cancer Lett 2008; 266:60-72; PMID:18374480; http://dx.doi.org/10.1016/j.canlet.2008.02.032
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071-8; PMID:19847258; http://dx.doi.org/10.1038/nature08467
- Woods D, Turchi JJ. Chemotherapy induced DNA damage response: convergence of drugs and pathways. Cancer Biol Ther 2013; 14:379-89; PMID:23380594; http://dx.doi.org/10.4161/cbt.23761
- Hill R, Lee PW. The DNA-dependent protein kinase (DNA-PK): More than just a case of making ends meet? Cell cycle 2010; 9:3460-9; PMID:20855954; http://dx.doi.org/10.4161/cc.9.17.13043
- Mendivil-Perez M, Velez-Pardo C, Jimenez-Del-Rio M. TPEN induces apoptosis independently of zinc chelator activity in a model of acute lymphoblastic leukemia and ex vivo acute leukemia cells through oxidative stress and mitochondria caspase-3- and AIF-dependent pathways. Oxid Med Cell Longev 2012; 2012:313275; PMID:23320127; http://dx.doi.org/10.1155/2012/313275
- Zhang X, Zhao Y, Chu Q, Wang ZY, Li H, Chi ZH. Zinc modulates high glucose-induced apoptosis by suppressing oxidative stress in renal tubular epithelial cells. Biol Trace Elem Res 2014; 158:259-67; PMID:24591003; http://dx.doi.org/10.1007/s12011-014-9922-x
- Sun Q, Zhong W, Zhang W, Li Q, Sun X, Tan X, et al. Zinc deficiency mediates alcohol-induced apoptotic cell death in the liver of rats through activating ER and mitochondrial cell death pathways. Am J Physiol Gastrointest Liver Physiol 2015; 308:G757-66; PMID:25767260; http://dx.doi.org/10.1152/ajpgi.00442.2014
- Hwang JJ, Kim HN, Kim J, Cho DH, Kim MJ, Kim YS, et al. Zinc(II) ion mediates tamoxifen-induced autophagy and cell death in MCF-7 breast cancer cell line. Biometals 2010; 23:997-1013; PMID:20524045; http://dx.doi.org/10.1007/s10534-010-9346-9
- Fatfat M, Merhi RA, Rahal O, Stoyanovsky DA, Zaki A, Haidar H, et al. Copper chelation selectively kills colon cancer cells through redox cycling and generation of reactive oxygen species. BMC Cancer 2014; 14:527; PMID:25047035; http://dx.doi.org/10.1186/1471-2407-14-527
- Gurusamy KS, Farooqui N, Loizidou M, Dijk S, Taanman JW, Whiting S, et al. Influence of zinc and zinc chelator on HT-29 colorectal cell line. Biometals 2011; 24:143-51; PMID:20957409; http://dx.doi.org/10.1007/s10534-010-9382-5
- Ding WQ, Yu HJ, Lind SE. Zinc-binding compounds induce cancer cell death via distinct modes of action. Cancer letters 2008; 271:251-9; PMID:18639975; http://dx.doi.org/10.1016/j.canlet.2008.06.011
- Donadelli M, Dalla Pozza E, Costanzo C, Scupoli MT, Scarpa A, Palmieri M. Zinc depletion efficiently inhibits pancreatic cancer cell growth by increasing the ratio of antiproliferative/proliferative genes. J Cell Biochem 2008; 104:202-12; PMID:17979179; http://dx.doi.org/10.1002/jcb.21613
- Zubair H, Khan HY, Sohail A, Azim S, Ullah MF, Ahmad A, et al. Redox cycling of endogenous copper by thymoquinone leads to ROS-mediated DNA breakage and consequent cell death: putative anticancer mechanism of antioxidants. Cell Death Dis 2013; 4:e660; PMID:23744360; http://dx.doi.org/10.1038/cddis.2013.172
- Sedelnikova OA, Pilch DR, Redon C, Bonner WM. Histone H2AX in DNA damage and repair. Cancer Biol Ther 2003; 2:233-5; PMID:12878854; http://dx.doi.org/10.4161/cbt.2.3.373
- Hashemi M, Ghavami S, Eshraghi M, Booy EP, Los M. Cytotoxic effects of intra and extracellular zinc chelation on human breast cancer cells. Euro J Pharmacol 2007; 557:9-19; PMID:17169355; http://dx.doi.org/10.1016/j.ejphar.2006.11.010
- Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resistance Updates 2004; 7:97-110; PMID:15158766; http://dx.doi.org/10.1016/j.drup.2004.01.004
- Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 2009; 8:579-91; PMID:19478820; http://dx.doi.org/10.1038/nrd2803
- Wang J, Yi J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol Ther 2008; 7:1875-84; PMID:18981733; http://dx.doi.org/10.4161/cbt.7.12.7067
- Li SK, Martin A. Mismatch Repair and Colon Cancer: Mechanisms and Therapies Explored. Trends Mol Med 2016; 22(4):274-89; PMID:26970951; http://dx.doi.org/10.1016/j.molmed.2016.02.003
- Sirbu BM, Cortez D. DNA damage response: three levels of DNA repair regulation. Cold Spring Harb Perspect Biol 2013; 5:a012724; PMID:23813586; http://dx.doi.org/10.1101/cshperspect.a012724