
ABSTRACT
Diffuse Large B-cell lymphoma (DLBCL) is an aggressive malignancy that has a 60 percent 5-year survival rate, highlighting a need for new therapeutic approaches. Histone deacetylase inhibitors (HDACi) are novel therapeutics being clinically-evaluated in combination with a variety of other drugs. However, rational selection of companion therapeutics for HDACi is difficult due to their poorly-understood, cell-type specific mechanisms of action. To address this, we developed a pre-clinical model system of sensitivity and resistance to the HDACi belinostat using DLBCL cell lines. In the current study, we demonstrate that cell lines sensitive to the cytotoxic effects of HDACi undergo early mitotic arrest prior to apoptosis. In contrast, HDACi-resistant cell lines complete mitosis after a short delay and arrest in G1. To force mitotic arrest in HDACi-resistant cell lines, we used low dose vincristine or paclitaxel in combination with belinostat and observed synergistic cytotoxicity. Belinostat curtails vincristine-induced mitotic arrest and triggers a strong apoptotic response associated with downregulated MCL-1 expression and upregulated BIM expression. Resistance to microtubule targeting agents (MTAs) has been associated with their propensity to induce polyploidy and thereby increase the probability of genomic instability that enables cancer progression. Co-treatment with belinostat effectively eliminated a vincristine-induced, actively cycling polyploid cell population. Our study demonstrates that vincristine sensitizes DLBCL cells to the cytotoxic effects of belinostat and that belinostat prevents polyploidy that could cause vincristine resistance. Our findings provide a rationale for using low dose MTAs in conjunction with HDACi as a potential therapeutic strategy for treatment of aggressive DLBCL.
Abbreviations
| DLBCL | = | diffuse large B cell lymphoma |
| HDAC | = | histone deacetylase |
| HDACi | = | histone deacetylase inhibitor |
| GCB | = | germinal center B cell-like |
| ABC | = | activated B cell-like |
| NHL | = | non-Hodgkin lymphoma |
| MTA | = | microtubule targeting agent |
| DMSO | = | dimethyl sulfoxide |
| ICC | = | immunocytochemistry |
| H3S10Ph | = | histone H3 phosphorylated at serine 10 |
| SAC | = | spindle assembly checkpoint |
| BCL2 | = | B cell CLL/lymphoma 2 |
| MYC | = | c-myc |
| Mt | = | microtubule |
| PARP | = | poly ADP ribose polymerase |
| PI | = | propidium iodide |
| RRR | = | relative risk ratio |
| PTX | = | paclitaxel |
Introduction
Diffuse Large B-cell Lymphoma is the most commonly-diagnosed form of Non-Hodgkin Lymphoma (NHL), affecting approximately 30,000 people each year in the United States.Citation1 The current standard therapeutic regimen is the anti-CD20 monoclonal antibody Rituximab in conjunction with cyclophosphamide, vincristine, doxorubicin and prednisone, known as R-CHOP. Among all DLBCL cases R-CHOP treatment yields a 60% 5 y survival rate,Citation2-4 thus new therapeutic strategies are clearly necessary to treat aggressive forms of DLBCL. To this end, histone deacetylase inhibitors (HDACi) are being evaluated in clinical trials for use in treatment of multiple types of NHL and 4 have gained FDA approval for the treatment of advanced peripheral T-cell lymphomas and multiple myeloma.
HDACi are promising cancer chemotherapeutics because they selectively target tumor cells and exhibit tolerable levels of toxicity in humans.Citation5-7 HDACi are largely ineffective as monotherapy against DLBCL as well as solid tumors, therefore, research focus has shifted to combining HDACi with other therapeutics for treatment of a variety of cancers.Citation8-10 A confounding issue in identifying effective HDACi-containing drug combinations is that a multitude of mechanisms have been attributed to the anti-cancer effects of HDACi including: alteration of gene expression, activation of pro-apoptotic pathways, induction of tumor cell differentiation, inhibition of angiogenesis, and modulation of cell cycle progression.Citation5,7,11-14 Taking a mechanistic approach to understand the molecular effects of HDACi specifically in the DLBCL context, we developed a cell-based model of sensitivity and resistance to HDACi. The mechanistic knowledge gained from this system can be used to rationally select therapeutics which can be effectively combined with HDACi. Previously, we identified 2 major responses to the hydroxamate HDACi, belinostat in our model system, including 1) a reversible, cytostatic arrest in G1 and 2) arrest in G2/M that is followed by apoptosis.Citation15 We showed that the reversible G1 arrest, which we designated as a form of HDACi resistance, is associated with sustained HDACi-induced expression of the cyclin-dependent kinase inhibitors, p21 and p27, as well as their inhibition of the Cyclin E/cdk2 complex through increased association.Citation15
A major cause of HDACi-induced tumor cell death has been attributed to perturbed mitotic progression.Citation16 These drugs have been reported to activate the spindle assembly checkpoint (SAC) resulting in an accumulation of cells in pro-metaphase. However, continued exposure to HDACi can cause SAC failure, mitotic catastrophe or slippage, and the induction of pro-apoptotic signaling.Citation17-19 SAC activation is also a major mechanism attributed to the anti-tumor effectiveness of microtubule targeting therapies such as taxanes and vinca alkaloids.Citation20,21 These drugs are common in many first line therapeutic regimens including the R-CHOP combination used for most non-Hodgkin's lymphomas. The induction of the SAC by microtubule targeting agents (MTAs) results in an extended mitotic arrest and the onset of senescence or cell death. However, therapeutically targeting mitotic progression can also cause failure of cytokinesis, resulting in a polyploid cell population. Polyploidy is linked to increased genomic instability and the induction of tumor heterogeneity and ultimately drug resistance.Citation22-25
In this study we have determined that the G2/M arrest we previously reported in HDACi-sensitive DLBCL cell lines is due to accumulation of cells in early mitosis, consistent with SAC activation. In contrast, the HDACi belinostat delays mitotic progression but does not prevent mitotic completion in HDACi-resistant DLBCL cell lines. Based on these findings we hypothesized that combining HDACi with therapeutics that strongly induce mitotic arrest might effectively sensitize resistant cells to the cytotoxic effects of HDACi. We show that combining the HDACi, belinostat, with low doses of the MTAs, vincristine or paclitaxel, synergistically induces apoptosis in resistant DLBCL cell lines, but has little to no effect on a non-transformed B cell-derived cell line. In addition, we find that the mitotic arrest is essential for the cytotoxic synergy between the drugs. Finally, we show that belinostat enhances vincristine-induced cytotoxicity by curtailing vincristine-induced SAC activation and triggering a strong apoptotic response that prevents the generation of polyploid cells. These findings show that HDACi and MTAs are mutually synergistic in inducing cell death in DLBCL cell lines that share molecular characteristics with aggressive forms of DLBCL in a way that reduces the probability of genomic instability.
Results
HDACi-sensitive DLBCL cell lines arrest early in mitosis while HDACi-resistant DLBCL cell lines are able to complete mitosis prior to G1 arrest
In a previous study we documented 2 major responses to the hydroxamate HDACi, belinostat, in DLBCL cell lines.Citation15 One is characterized by a reversible arrest in the G1 phase of the cell cycle within 24–48 h of treatment without accompanying apoptosis. In contrast, the second response is characterized by G2/M arrest within 24 h and subsequent onset of apoptosis by 48 hours. HDACi have been reported to induce both G2 and M phase arrest.Citation26-28 To distinguish the 2 phases of the cell cycle we measured global histone H3 serine 10 phosphorylation (H3S10Ph), which occurs once cells enter prophase and begins to decrease in anaphase.Citation29 If belinostat induces early mitotic arrest, cellular levels of H3S10Ph are expected to increase. We show in that levels of H3S10Ph are markedly increased within 8–12 h after belinostat treatment in the sensitive cell lines, DB and OCI-Ly19. After the initial increase, H3S10Ph levels decrease in both cell lines, consistent with reports that mitotic slippage can occur after HDACi-induced mitotic arrest.Citation17,19,30,31 Surprisingly, we also observed an increase in H3S10Ph levels in the HDACi-resistant cell lines (SUDHL4 and SUDHL8) with kinetics similar to the HDACi-sensitive lines. To confirm the western blotting results, we examined H3S10Ph levels using immunocytochemistry (ICC) on SUDHL4 and DB cells treated for 16 h with belinostat or DMSO. show that both cell lines are significantly enriched for positively-stained cells after 16 h of belinostat treatment as compared to vehicle (DMSO) treatment.
Figure 1. Belinostat treatment causes accumulation of DLBCL cells in mitosis. (A) Belinostat sensitive (DB, OCI-Ly19) and resistant (SUDHL4, SUDHL8) cells were treated with belinostat for up to 36 h. Belinostat was used at previously-determined IC50 concentrations for each cell line. Equal volumes of cell lysates were subjected to Western blotting with antibodies to either H3S10Ph (mitotic marker) or GAPDH (loading control). (B) Immunohistochemistry using antibody against H3S10Ph for SUDHL4 and DB cells treated with belinostat or DMSO for 16 h. (C) The total number of cells and the number of positive-staining cells were counted in multiple, random fields from 40X and 100X ICC images of cells treated for 16 h with belinostat or DMSO. The results are represented graphically. The unpaired student's t-test was used to calculate significance. ***, p ≤ 0.001. (D) Images (100X magnification) of belinostat-treated, mitotic SUDHL4 or DB cells positively-stained for H3S10Ph.
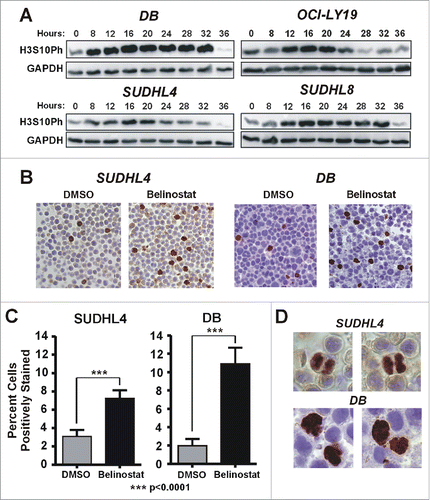
Although mitotic cells appear to accumulate in SUDHL4 and SUDHL8 cells in response to belinostat, these cell lines eventually arrest in G1.Citation15 It is possible that the increase in mitotic cells is due to a mitotic delay that is eventually resolved. We therefore examined high magnification images of the H3S10Ph-stained cells for evidence of anaphase mitotic figures, which would indicate that cells are progressing beyond prometaphase, when the spindle assembly checkpoint (SAC) is activated. In the resistant SUDHL4 cell line, anaphase figures are readily apparent at 16 h of belinostat treatment (). In contrast, we were unable to find any anaphase figures in the belinostat-treated DB cells; all positively-stained cells appear to be arrested prior to metaphase. Altogether our results are consistent with a belinostat-induced transient mitotic delay in the SUDHL4 cells and a pre-metaphase arrest in the DB cells that eventually leads to apoptosis.
Combining belinostat with vincristine results in synergistic cell death in HDACi-resistant DLBCL cell lines
As shown above, belinostat-induced cytotoxicity is associated with early mitotic arrest in DLBCL cell lines. However, at similar submicromolar concentrations belinostat is unable to induce a strong mitotic arrest in the resistant DLBCL cell lines. Studies of the mitotic effects of HDACi in other cell types have established that, in addition to activating the SAC, they cause it to fail prematurely and induce apoptotic signalingCitation17,19,30,31). We therefore hypothesized that forced mitotic arrest in the resistant cells might sensitize them to belinostat-induced cytotoxicity. To address this, we used the MTA, vincristine, at concentrations low enough to cause near-maximal mitotic arrest and sustained increase in H3S10Ph by 24 h () but ≤30 % cell death by 48 h (). Cleavage of poly ADP ribose polymerase (PARP) and pro-Caspase 3 were evaluated in 2 belinostat-resistant cell lines using a 48 h time course of belinostat and vincristine, alone or in combination (). Individually each drug caused low to moderate levels of PARP cleavage and little to no cleavage of caspase 3. The combination of belinostat and vincristine however, resulted in nearly complete PARP cleavage and high levels of Caspase 3 cleavage in both cell lines, indicating efficient onset of apoptosis.
Figure 2. Low dose vincristine treatment results in accumulation of cells in mitosis. (A) SUDHL4 and SUDHL8 cells were treated with vincristine at various doses between 1 and 10 nM for 24 h followed by cell cycle analysis. The percentage of cells in G2/M from 2–3 independent experiments is shown. Curve-fitting was performed using GraphPad Prism software. (B,C) SUDHL4 (B) and SUDHL8 (C) cells were treated with vincristine at concentrations of 5 nM and 3 nM, respectively for up to 24 h. Cell lysates were subjected to immunoblotting with antibodies against H3S10Ph or GAPDH. A representative experiment is shown.
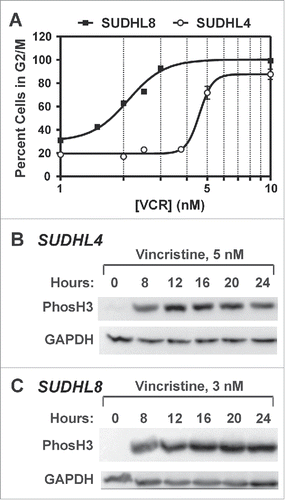
Figure 3. Belinostat combined with low dose microtubule targeting agents act synergistically to induce apoptosis in HDACi-resistant DLBCL cell lines. A,B) SUDHL4 (A) or SUDHL8 (B) cells were treated with belinostat, vincristine, or the combination for up to 48 h. Vincristine concentrations were 5 nM for SUDHL4 and 3 nM for SUDHL8. Cell lysates were subjected to Western blotting with antibodies against PARP (cleaved and uncleaved), Caspase 3 (cleaved and uncleaved), and β-actin. C,D) SUDHL4 or SUDHL8 cells were treated with DMSO, belinostat (Bel), vincristine (VCR), or the combination for 24, 48, and 72 h and subjected to the Annexin V/PI uptake assay. (C) The number of cells staining positive for PtdIns, Annexin V, or both is shown graphically for 3–4 independent replicates. (D) The number of viable cells (negative staining for PI and/or Annexin V) from 3–4 independent replicates is shown graphically. Error bars represent SEM.
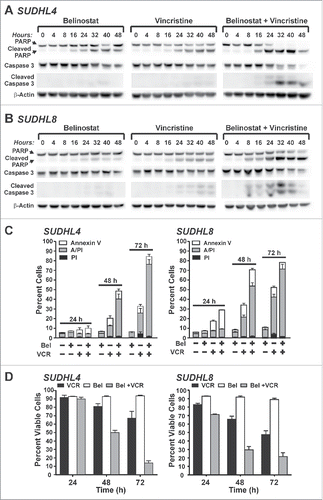
As an additional measure of apoptosis, Annexin V/propidium iodide (PI) uptake assays were conducted (). As expected, belinostat alone caused little cell death by 72 h in both HDACi-resistant cell lines. Low dose vincristine alone caused low to moderate amounts of cytotoxicity. The combination treatment however, caused much more cell death than either drug alone. By 72 h the belinostat/vincristine combination induced apoptosis in greater than 75% of SUDHL4 and SUDHL8 cells as compared to ≤ 10% of dead cells with belinostat alone or ≤ 50% with vincristine alone. shows the percentage of viable cells (derived from the AnnexinV/PtdIns assays) with each treatment at 24, 48, and 72 h. The relative risk ratio (RRR) calculation using cell viability data can be used to indicate whether a drug combination is synergistic, additive, or antagonistic Citation32; RRR values <1.0 are predictive of drug synergy. As described in Materials and Methods, the RRR values for the belinostat/vincristine combination at 72 hours in SUDHL4 and SUDHL8 cells were found to be 0.25 and 0.51, respectively, consistent with synergy between the 2 drugs.
HDACi selectively induce cytotoxicity in transformed cells versus non-transformed cells, but vincristine can be toxic to both. Thus, when combining these drugs it is important to assess potential cytotoxicity in non-transformed cells. GM18564, a non-transformed, Epstein Barr Virus-immortalized human B lymphoblastoid cell line with a 30 h doubling time (not shown), was used to address this issue. Treatment of GM18564 cells with doses of vincristine and belinostat that induced strong cell cycle effects in the experiments with DLBCL cell lines caused no significant change in the percentage of cells in G2/M while exposure to belinostat led to a small increase in the G1 population (). In addition, the drugs induced little, if any, change in the extent of PARP cleavage (). These results show that non-transformed B cells are much less sensitive to the effects of these drugs on cell growth and survival. In particular, GM18564 cells are not sensitive at all to the low concentrations of vincristine that elicit strong cell cycle effects in DLBCL cell lines.
Figure 4. Cotreatment with belinostat and vincristine does not cause cytotoxicity in a non-transformed B lymphoblastoid cell line. GM18564 cells were treated with DMSO, belinostat, vincristine, or the combination for 48 h. (A) Cells were collected for cell cycle analysis. The graph shown is a summary of 3 independent replicates. (B) Cell lysates were subjected to Western blotting with antibodies against PARP (cleaved and uncleaved) or GAPDH. A representative blot is shown.
Mitotic arrest is essential for HDACi-induced cytotoxicity in DLBCL cell lines
Vincristine inhibits microtubule (Mt) polymerization while taxanes, such as paclitaxel, stabilize the polymerized state and inhibit depolymerization of Mts. However, both drugs induce mitotic arrest. To determine whether the synergy between belinostat and vincristine is specific to the Mt targeting mechanism of the latter or is dependent on mitotic arrest, we assessed the level of cytotoxicity induced by belinostat in combination with paclitaxel. Using doses of paclitaxel in the low nanomolar range that cause near maximal G2/M arrest (not shown), we examined cytotoxicity by Annexin V/PI assay after 48 h treatment. Paclitaxel alone induced low levels of cytotoxicity (approximately 30%) in SUDHL4 cells (). However, the combination of belinostat and paclitaxel induced high levels of cytotoxicity (65–75%), similar to what was observed with the vincristine/belinostat combination (). Analysis of the cell viability data () shows that RRR values of < 1.0: 0.52 and 0.50 for 6 and 8 nM paclitaxel plus belinostat, respectively. These results show that the synergy is independent of the mechanism of Mt targeting and suggests that the key event that sensitizes belinostat-resistant DLBCL cells to belinostat-associated cytotoxicity is sustained mitotic arrest.
Figure 5. Mitotic arrest is essential for HDACi-induced cytotoxicity in DLBCL cell lines. (A,B) Co-treatment with paclitaxel and belinostat causes enhanced cytotoxicity in HDACi-resistant DLBCL cell lines. SUDHL4 or SUDHL8 cells treated with DMSO, belinostat, paclitaxel (PTX), or the combination for 48 h. Cells were subjected to the Annexin V/PtdIns uptake assay. (A) The number of cells staining positive for PI, Annexin V, or both is shown graphically for 3–4 independent replicates. (B) The number of viable cells (negative staining for PI and/or Annexin V) from 3–4 independent replicates is shown graphically. Error bars represent SEM. (C-E) Enhanced cytotoxicity induced by the combination of belinostat and vincristine is dependent on cell cycle progression. C.) Experimental design. SUDHL4 cells were treated with vehicle (DMSO) or with belinostat (Bel) and vincristine (VCR) either alone or in combination for 48 h prior to harvest as shown below the timeline. Alternatively cells were treated with belinostat (Bel) for 48 h followed by the addition of vincristine (VCR) or water for an additional 48 h as shown above the timeline. D) Lysates from cells treated as described in (C) were subjected to Western blotting for PARP cleavage and p27, which is a marker of belinostat-induced G1 arrest. E) Annexin V/ Propidium iodide uptake assays were used to examine the induction of cell death by the various treatments described in (C). The graph is a summary of at least 3 independent experiments. Error bars represent SEM. Statistical analysis was carried out using the paired t test comparing the total population of dead and dying cells (PtdIns plus Annexin V plus AnnV/PI) between conditions. ** - p ≤ 0.01, *** - p ≤ 0.001.
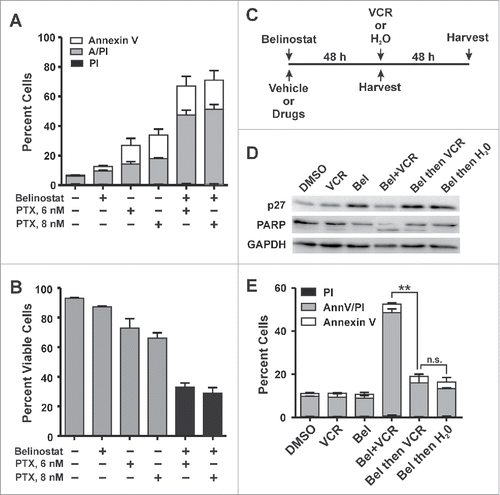
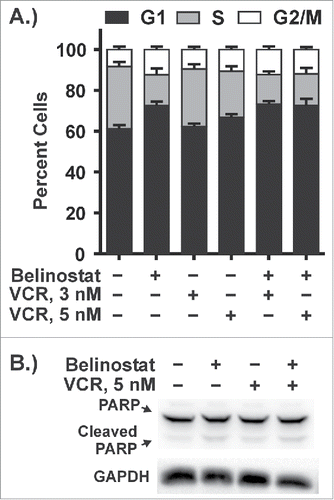
To further test the relationship between mitotic arrest and belinostat-induced cytotoxicity, we varied the order of cellular exposure to vincristine and belinostat. In the experiments described above, both vincristine and belinostat were added simultaneously and mitotic arrest was apparent by 24 h (not shown and see ). Since belinostat induces G1 arrest in belinostat-resistant DLBCL cells by 48 h treatment we hypothesized that prior treatment with belinostat would prevent synergy with vincristine. Thus, as described by the timeline shown in , SUDHL4 cells were either treated with the drugs simultaneously for 48 h (below the line) or pretreated with belinostat for 48 h followed by either water (vehicle) or vincristine for an additional 48 h (above the line). shows that belinostat treatment for 48 h induced G1 arrest as indicated by increased expression of the cyclin-dependent kinase inhibitor, p27.Citation15 To measure cell death, we carried out Annexin V/PtdIns assays. As expected, treatment with either drug alone has little effect on PARP cleavage () or cell death (), while simultaneous treatment with both belinostat and vincristine for 48 h strongly triggered both. However, when cells are pre-exposed to belinostat for 48 h prior to vincristine (denoted as Bel then VCR), there was no additional cell death beyond that observed when belinostat was present for 96 h (denoted as Bel then H2O), and there is no synergy between the drugs (compare bars 4 and 5, ). Altogether these results establish the importance of mitotic arrest in belinostat-induced cytotoxicity in the DLBCL context and indicate that the enhanced cytotoxic effect of the combination is sensitive to the order of drug addition.
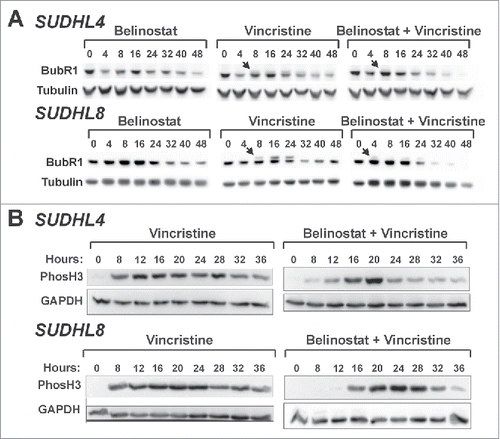
Figure 6. Belinostat curtails SAC activation and mitotic arrest induced by vincristine. (A) SUDHL4 and SUDHL8 cells were treated with belinostat, vincristine, or the combination for up to 48 h. Whole cell extracts were subjected to Western blotting with antibodies against BubR1 and α-tubulin. Arrows denote the slower migrating band that represents phosphorylated BubR1. (B) SUDHL4 and SUDHL8 cells were treated with vincristine or vincristine plus belinostat for up to 36 h. Cell lysates were subjected to Western blotting with antibodies against H3S10Ph or GAPDH. Blots representative of at least 3 independent experiments are shown.
Belinostat curtails vincristine-induced SAC activation and causes efficient induction of apoptosis
Previous studies have shown that HDAC activity is required for stable microtubule-kinetochore interactions that are essential for progression beyond prometaphase.Citation17,19,31 In addition, HDACs appear to be necessary to maintain the SAC once it has been activated.Citation17,19,30,31 Based on these studies and our results we predict that vincristine induces prolonged activation of the SAC in the HDACi-resistant cell lines and belinostat causes it to fail. To address this hypothesis we examined the phosphorylation of BubR1, an event that indicates SAC activation.Citation33,34 The mobility of BubR1 in SDS-PAGE gels is slowed by phosphorylation relative to unmodified BubR1. As expected, belinostat alone was not sufficient to induce the accumulation of phosphorylated BubR1 (pBubR1) (). In contrast, 8 h of exposure to vincristine caused detectable accumulation of pBubR1 in both SUDHL4 and SUDHL8 cells (denoted by the arrows in ). This phosphorylation persisted for another 16–24 hours and then disappears. The combination of the 2 drugs caused an early accumulation of low levels of pBubR1 that was short-lived, suggesting that the strong and persistent SAC activation by vincristine is curtailed in the presence of belinostat.
Mitotic phosphorylation of histone H3 persists through metaphase but begins to decrease in anaphase and is barely detectable by cytokinesis/mitotic exit.Citation29 To further document SAC failure, we measured levels of H3S10Ph in the presence of vincristine alone and the drug combination by Western blotting in SUDHL4 and SUDHL8 cells (). Vincristine alone caused a prolonged elevation of H3S10Ph consistent with an extended mitotic arrest. Co-treatment with vincristine and belinostat, resulted in a shorter period of H3S10Ph accumulation relative to vincristine alone (), which could be due to death of cells still in mitosis or mitotic slippage.Citation20,35-38 In addition, accumulation of pBubR1 was much lower and did not persist (). These findings indicate that belinostat is capable of attenuating the mitotic arrest induced by vincristine. Total levels of BubR1 became almost undetectable after 40–48 h in the presence of belinostat and VCR. Several reports have shown that BubR1 degradation occurs prior to mitotic exit.Citation39-41 The decline in total BubR1 occurs in the similar time frame as the decrease in levels of H3S10Ph.
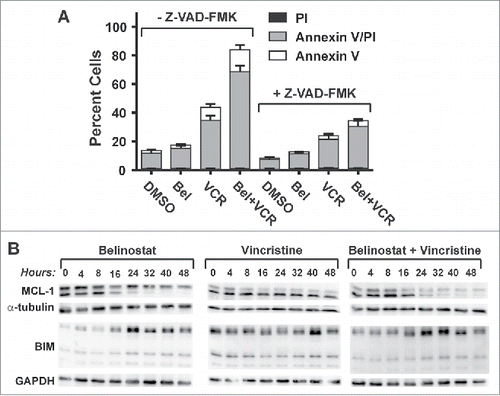
Studies have shown that prolonged mitotic arrest can result in death in mitosis or mitotic slippage. After the latter, cells can undergo multiple fates including cell death by apoptosis, long-term growth arrest or senescence, or continued cycling with a polyploid genome.Citation20 Our findings suggest that belinostat exposure during mitotic arrest triggers a strong apoptotic signal. This contention is further supported by experiments done with the pan-caspase inhibitor, Z-VAD-FMK, which strongly reduced SUDHL4 cell death in the presence of both belinostat and vincristine as determined by Annexin V/PI assays (). Analysis of the expression of pro- and anti-apoptotic Bcl-2 family members in the presence of belinostat and/or vincristine in SUDHL4 cells showed that neither drug had an impact on BCL2 expression, and that BCL-XL expression was undetectable (not shown). However, belinostat treatment alone down-regulated expression of the pro-apoptotic short from of MCL-1 (lower band, Mcl-1 blot, ) but had little effect on the anti-apoptotic long form (upper band, Mcl-1 blot, ).Citation42,43 In addition, belinostat up-regulated expression of the BH3-only apoptotic activator, BIM about 2–3-fold. Vincristine treatment had little effect on expression of either MCL-1 or BIM. However, upon treatment with both belinostat and vincristine, both long and short forms of MCL-1 were reduced by at least 50% over 48 h treatment. Importantly, in the period over which MCL-1 expression declined, BIM expression increased and Caspase 3 cleavage commenced (see ). Thus, in the presence of vincristine, belinostat shifts the balance of pro- and anti-apoptotic Bcl-2 family members toward expression of the former.
Figure 7. Enhanced cytotoxicity induced by the combination of belinostat and vincristine is dependent on apoptotic signaling and correlates with a shift in the balance of anti- and pro-apoptotic factors. (A) SUDHL8 cells were treated for 48 hours with DMSO, belinostat (Bel), vincristine (VCR) or the combination with and without the pan-caspase inhibitor, Z-VAD-FMK. Cell death was measured using Annexin V/ PtdIns uptake assays. The graphs summarize the results of 4 independent experiments. (B) Cells were treated for up to 48 hr with belinostat (Bel), vincristine (VCR), or the combination. Cell lysates were subjected to Western blotting with antibodies against MCL-1 and BIM. GAPDH was used as a loading control. A blot representative of 3–4 independent experiments is shown.
Belinostat prevents vincristine-induced formation of polyploid cells
In the course of cell cycle analyses, we noticed that vincristine treatment caused the generation of a polyploid population of cells in SUDHL8 cells (denoted by the arrow in ). Mitotic slippage without genome division has been documented to occur with other drugs that target mitosis and may be associated with drug resistance and the development of aneuploidy [reviewed in Citation23,44-46]. Interestingly, the presence of belinostat largely prevents the vincristine-induced accumulation of polyploid SUDHL8 cells (). The number of cells in the peak shown in was quantitated as shown in . In the presence of vincristine alone, approximately 6% of cells are in this population (light gray bars, minus Z-VAD-FMK). With the belinostat-vincristine combination treatment this population is reduced to less than 1%, similar to the amount observed with DMSO alone. Because we demonstrated that belinostat induces a strong apoptotic signal, we hypothesized that inhibition of apoptosis with the pan-caspase inhibitor, Z-VAD-FMK, would impair the ability of belinostat to reduce the polyploid population, which it did (as seen in ).
Figure 8. Belinostat treatment prevents vincristine-induced polyploidy in SUDHL8 cells. SUDHL8 cells were treated for 48 hours with DMSO, belinostat (Bel), vincristine (VCR), or the combination. (A) Cells were subjected to cell cycle analysis. The arrow denotes a polyploid cell population. (B) Quantitation of the polyploid cell population indicated by the arrow in (A) in SUDHL8 cells treated with and without the pan-caspase inhibitor Z-VAD-FMK. The graph is a summary of 4 independent experiments. Error bars represent SEM. Statistical analysis was carried out using the paired t test comparing the population of polyploid cells with 8N DNA content in the presence and absence of Z-VAD-FMK. ** - p ≤ 0.01, *** - p ≤ 0.001 (C) SUDHL8 cells treated with DMSO, belinostat, vincristine and the combination were subjected to flow-cytometric analysis after staining with Alexa 488-tagged H3S10Ph antibody and propidium iodide. Neither cells with < 2N content nor debris were gated out. The plots shown are representative of 3 independent experiments. Mitotic populations of 4N (left) and 8N (right) cells used for quantitation are denoted as ovals in each panel. (D) Quantitation of polyploid cells (8N) with high H3S10Ph staining as identified in the oval on the right side of each panel shown in (C). Statistical analysis was carried out using the paired t test comparing the total population of polyploid mitotic cells (high H3S10Ph staining) between vincristine and vincristine plus belinostat treatments. ** - p ≤ 0.01, *** - p ≤ 0.001.
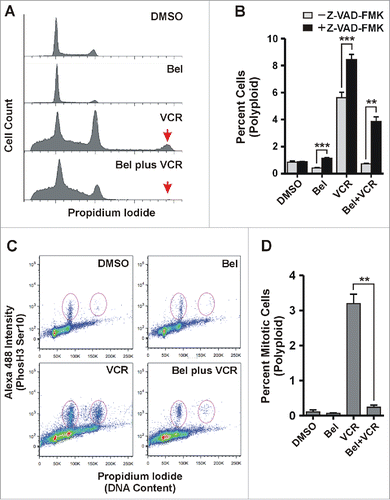
Once cells treated with drugs that disrupt mitotic progression have undergone mitotic slippage, several cell fates have been documented, including cell death in interphase, continued cell cycle progression and division, or senescence.Citation20,35-38 The relative DNA content of the polyploid cell population suggests that some tetraploid cells continued cycling after slippage, replicated their DNA, and may have entered mitosis. To test this we carried out FACs analysis on cells that had been permeabilized and stained with antibodies to H3S10Ph and propidium iodide. In , the ovals indicate the mitotic populations that show elevated H3S10Ph staining in a representative experiment. The ovals on the left in each plot represent the 4N cell population undergoing mitosis while the ovals on the right represent the portion of the polyploid cell population indicated in that is mitotic. In the presence of DMSO or belinostat there are few polyploid cells in mitosis (quantitated in ). In the presence of vincristine alone, the number of 4N cells in mitosis is much increased, as expected. In addition, there are cells with elevated H3S10Ph staining in the polyploid population (), indicating that these cells continued cycling after mitotic slippage and entered mitosis. The belinostat-vincristine combination treatment reduces the number of both 4N and polyploid cells with elevated H3S10Ph staining, consistent with belinostat-induced SAC failure and apoptotic signaling. These results show that belinostat enhances vincristine-induced cell death by preventing the survival of tetraploid cells generated by mitotic slippage.
Discussion
Relapsed or refractory DLBCL is associated with a high mortality rate Citation4 and highlights a need for new therapies that can lengthen survival of patients with aggressive forms of DLBCL. In the current study we show that sensitivity to the cytotoxic effects of HDACi in cell lines modeling aggressive DLBCL is associated with mitotic arrest prior to metaphase. In contrast, DLBCL cell lines resistant to the cytotoxic effects of HDACi are able to complete mitosis and arrest reversibly in G1. However, if mitotic arrest is induced with the MTAs vincristine or paclitaxel, the resistant cells become sensitive to the cytotoxic effects of belinostat through failure of mitotic arrest and efficient induction of apoptosis. In addition, we show that vincristine-induced generation of cycling polyploid cells is prevented by co-treatment with belinostat. Thus, the belinostat-vincristine drug combination exhibits mutual cytotoxic synergy in DLBCL cells.
Our previous study described the development of a DLBCL cell line-based model of sensitivity and resistance to the HDACi, belinostat.Citation15 In the current study we determined that cell lines sensitive to belinostat-induced cytotoxicity undergo mitotic arrest prior to initiation of apoptosis, consistent with reports showing that HDACi can activate the SAC.Citation17,19 HDAC inhibition causes destabilized interactions between microtubules (Mt) and kinetochores assembled at centromeres.Citation17,31 A recent study showed that aberrant acetylation of the Mt end-binding protein, EB1, prevents stable chromosome alignment at the metaphase plate.Citation47 SAC activation prevents progression to anaphase and causes the accumulation of cells in prometaphase with chromosomes containing high levels of H3S10Ph. We show that belinostat induces significant increases in H3S10Ph in sensitive DLBCL cell lines, DB and OCI-Ly19. The ICC analysis in DB cells shows an accumulation of pre-metaphase H3S10Ph-positive cells and a complete lack of positively-stained cells in anaphase. Altogether these results strongly indicate that belinostat is able to activate the SAC in sensitive DLBCL cell lines.
In contrast, our findings suggest that DLBCL cell lines resistant to the cytotoxic effects of belinostat exhibit a delay in mitotic progression but are ultimately able to complete mitosis and arrest in G1. Establishing a mitotic arrest using vincristine sensitizes the resistant cell lines to the cytotoxic effects of belinostat. The mitotic arrest is essential for this effect since paclitaxel, a second MTA with a distinct mechanism of action, is able to induce mitotic arrest and sensitize the cells to belinostat. In addition, allowing the cells to arrest in G1 via belinostat treatment prior to vincristine exposure prevents mitotic arrest and abolishes the synergistic cytotoxic effect observed upon simultaneous co-treatment with the drugs.
HDAC activity has been shown to be important for maintenance of the SAC [reviewed in Citation18]. HDACi-treated cells arrested in prometaphase cannot maintain the arrest and exit mitosis without having entered anaphase or partitioned their genome, suggestive of SAC failure and mitotic slippage.Citation17,19 Furthermore, extended mitotic arrest caused by treatment with nocodazole is significantly curtailed by HDAC inhibition.Citation30,48,49 We show very similar results with the clinically-relevant MTA, vincristine. At low doses (≤5nM) this drug maintains elevated levels of H3S10Ph and phosphorylated BubR1 for at least 24 h in the SUDHL4 and SUDHL8 cell lines. However, in the presence of both belinostat and vincristine, elevated H3S10Ph is maintained for only 8–16 h prior to a rapid decline that is concomitant with an increase in caspase 3 and PARP cleavage, indicative of the onset of apoptosis. In addition, the accumulation of phosphorylated BubR1 is greatly decreased, suggesting a reduction in the number of cells in which the SAC is active. Altogether the results suggest that, in the belinostat-resistant cell lines, HDAC inhibition leads to failure of the vincristine-induced SAC and subsequent apoptosis. It is noteworthy that the submicromolar concentrations of belinostat that effectively curtail vincristine-induced mitotic arrest in the resistant cell lines do not efficiently activate the SAC as they do in the sensitive cell lines. This observation suggests that the HDAC targets that mediate SAC activation are distinct from those which cause SAC failure.
By three methods (Annexin V assay, Caspase 3 cleavage, and use of a pan-Caspase inhibitor) we established that the belinostat-vincristine combination treatment induces apoptotic cell death. Belinostat causes upregulation of Bim proteins in the presence or absence of vincristine, but the combination treatment causes a strong down-regulation of MCL-1 protein, thereby generating an environment more conducive to apoptosis. Upregulation of BIM was shown to increase the probability of cell death in mitosis in the presence of reduced MYC expression.Citation50 This is relevant to our study because we showed previously that belinostat strongly down-regulates MYC protein expression in DLBCL cell lines.Citation15 MCL-1 is involved in regulation of both death in mitosis and death after mitotic slippage.Citation50-52 Mitotic degradation of MCL-1 can increase the probability of death in mitosis when MYC or NOXA is downregulated.Citation50,52 It can also hasten slippage during mitotic arrest caused by inhibitors of mitosis and increase the probability of apoptosis after slippage.Citation51 While it is unclear if apoptosis induced by the belinostat-vincristine combination occurs during mitosis or after mitotic slippage, the concomitant downregulation of MCL-1 and upregulation of BIM could contribute to either.
Cancer cells exposed to different classes of mitosis-targeting drugs, including MTAs, respond in 2 main ways to a prolonged mitotic arrest, either dying during mitosis or undergoing mitotic slippage dependent on the cell line or the drug [reviewed inCitation53]. After mitotic slippage, cells have been shown to enter G1 in a tetraploid state after which they may become senescent, die by apoptosis, or progress through the cell cycle, all by mechanisms that are poorly understood [reviewed inCitation54]. In the prolonged presence of vincristine, a fraction of SUDHL8 cells slip out of mitosis in a tetraploid state and progress through the cell cycle yielding a population of cells that have 8N DNA content, including some that have entered mitosis. Interestingly, the induction of polyploidy by inhibitors of mitosis is associated with drug resistance,Citation23,46 and it has been proposed that the continued survival of polyploid cells may give rise to aneuploidy that promotes cancer progression.Citation46,53-56 Deng et al measured vincristine sensitivity in DLBCL cell lines and found that SUDHL8 cells were relatively resistant while SUDHL4 cells were much more sensitive.Citation57 We found that exposure to belinostat largely prevents the vincristine-induced accumulation of polyploid SUDHL8 cells by triggering apoptosis. Thus, HDAC inhibition enhances the cytotoxicity of vincristine by increasing the probability that apoptotic signaling is efficiently activated to reduce the number of cells that undergo mitotic slippage, evade cell death, and continue to cycle with an abnormal number of chromosomes. Our work suggests that HDACi might be used with MTAs and other drugs that inhibit mitotic progression to reduce the probability of resistance. Also, since tumors with aneuploidy tend to be resistant to MTAs, HDACi co-treatment might increase their sensitivity to the MTA-induced cytotoxicity. In fact, a very recent study showed that treatment of paclitaxel-resistant lung cancer cell line with a novel HDACi sensitized the cells to paclitaxel-induced cytotoxicity both in vitro and in vivo.Citation58
Synergy between taxanes and HDACi in inducing cytotoxicity has been previously demonstrated in vitro and in xenograft tumor models of ovarian, breast, and prostate cancer.Citation30,59-68 Belinostat has been found to be synergistic with taxanes in inducing apoptosis in prostate and ovarian cell lines and in clinical samples from ovarian tumors grown in organoid culture.Citation65,67 Although most of these studies did not explore mechanism, cytotoxicity induced by vorinostat and paclitaxel in breast cancer cell lines was accompanied by increased induction of G2/M arrest.Citation62 A study of Hela cells showed that combining the hydroxamate HDACi, trichostatin A, with paclitaxel disrupted the association of BubR1 with kinetochores and reduced clonogenic survival.Citation30 Results were mixed in the few studies that combined vincristine with romidepsin, a Class I-selective HDACi. The combination of romidepsin and vincristine was synergistic in neuroblastoma cell lines Citation69 but antagonistic in several leukemia and lymphoma cell lines, none of which were derived from DLBCL.Citation70 Surprisingly, we could not find any studies that combined vincristine with a pan-HDACi of the hydroxamate class in cancer cells.
The use of vincristine in patients is dose-limited due to toxicity. While HDACi are generally well-tolerated in humans, combining them a toxic drug such as vincristine may increase the risk of toxicity to normal cells. Our experiments with a non-transformed, immortalized human B cell-derived cell line demonstrated that exposure to both drugs had little effect on cell cycle distribution and did not induce PARP cleavage, showing that the belinostat/low dose vincristine combination was not cytotoxic. Thus, our results indicate that doses of MTAs lower than those needed for maximal cytotoxicity of tumor cells might be used effectively with HDACi, thereby reducing the potential for toxicity in normal cells. In support, Hwang et al reported that a dose of docetaxel lower than the maximally tolerated dose was effective in inhibiting growth of prostate xenograft tumors when combined with belinostat.Citation67 A Phase I study of solid tumors showed that the belinostat/paclitaxel combination is tolerable in humans and thus feasible for further clinical study.Citation71 Recently approved nanoparticle formulations of vincristine and paclitaxel that increase efficacy and reduce toxicity Citation72,73 should make them safer to combine with other drugs such as belinostat. Using a preclinical model of aggressive DLBCL the current study has demonstrated the potential of combining MTAs with belinostat for treatment of relapsed/refractory DLBCL. Our future studies will focus on evaluation of the MTA/belinostat combination in mouse models of DLBCL.
Materials and methods
Cell lines and reagents
DLBCL-derived cell lines were cultured as described previously.Citation15 RPMI 1640 medium was supplemented with: 1.) 10% FBS and gentamicin for SUDHL4 and DB, 2.) 10% FBS, 1 mM sodium pyruvate, and gentamicin for OCILY19, 3.) 10% FBS, 1 mM glutamine, and gentamicin for SUDHL8, and 4.) 20% FBS and gentamicin for GM18564. All cell lines were maintained at 37°C in a humidified atmosphere containing 5% CO2 at densities between 2.0 × 105 and 1.0 × 106 cells/ml. All cell lines were treated with concentrations of belinostat that inhibited growth by 50% at 24 h as determined previously.Citation15
Belinostat was provided by Spectrum Pharmaceuticals. Paclitaxel (S1150) was purchased from Selleck Chemicals. Vincristine (1714007) was purchased from Sigma. Antibodies against BubR1 (#4116s), PARP (#9542s), Caspase 3 (#9665s), and α-Tubulin (#2144s) were purchased from Cell Signaling Technologies. Antibodies against phosphorylated Ser10 Histone H3 (#06–570) were purchased from Millipore and antibodies against GAPDH (fl-335) from Santa Cruz Biotechnology.
Cell cycle, apoptosis analysis and quantitation of mitotic populations using flow cytometry
Cells were plated at a density between 2 × 105 and 3 × 105 cells/ml and cultured for 24 hours prior to treatment. For cell cycle analysis, 3 × 106 cells per treatment condition were collected and washed once with cold 1X Dulbecco's modified phosphate-buffered saline (D-PBS) followed by fixation with ice cold 70% ethanol and centrifugation at 700 × g for 3 minutes. Cell pellets were resuspended in 0.5 ml cold PBS to which propidium iodide and RNase A were added to final concentrations of 40 μg/ml and 50 μg/ml, respectively. After incubation (30 minutes, 37°C) samples were analyzed by flow cytometry using a FACScanto II instrument (Becton-Dickinson). Data was analyzed using ModFit LT 3.0 (Verity Software House).
Apoptosis was analyzed using an Annexin V/Propidium iodide uptake assay kit (ENZO Life Sciences) that was supplemented with FITC-conjugated Annexin V (640906) antibody (Biolegend). Approximately 1 × 106 cells per treatment condition were collected and processed according to manufacturer's specifications. Stained cells were analyzed within one hour of processing using the FACSanto II flow cytometer (Becton-Dickinson). All statistical analysis was conducted using Graphpad Prism Version 5.0 software.
For mitotic quantitation through H3S10Ph staining, approximately 1.5×106 cells were pelleted, washed, and then fixed in 0.5 ml 1% formaldehyde in PBS for 10 min at 37°C. Cold methanol (4.5 ml) was added while vortexing for storage overnight at −20°C. Cells were then pelleted and resuspended in 0.5% BSA/PBS to block for 10 min. Cells were incubated with Alexa 488-conjugated anti-phos Ser 10 Histone H3 (Cell Signaling Technologies, #9708) at a 1:10 dilution for 1 h at room temperature in the dark then resuspended in cold PBS to which Propidium iodide and RNase A were added to final concentrations of 40 μg/ml and 50 μg/ml, respectively. After incubation for 30 minutes at 37 °C samples were analyzed by flow cytometry using BD LSRFORTESSA X-20 (BD Biosciences). Data was analyzed using FlowJo Version 9.9.
Western blotting
For generation of whole cell lysates, cells were collected after treatment, counted, washed with cold PBS, and resuspended at a ratio of 100 ul of 2x SDS- PAGE loading buffer [0.08 M Tris pH 6.8, 4% SDS, 20% glycerol, 0.1 M DTT, 0.004% Bromophenol Blue] per 1×106 cells. Equal volumes of each sample were then used for Western blotting. Generation of whole cell extracts was carried out as described previously. Briefly, after washing in PBS, cell pellets were flash frozen in liquid nitrogen and stored −80° C. Cells were resuspended in RIPA buffer [150 mM sodium chloride, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris, pH8.0, protease inhibitor cocktail (Roche) and phosphatase inhibitors (10 mM NaF, 25 mM β-glycerophosphate)] and protein concentration was determined by Bradford assay. Cellular proteins were separated using SDS-PAGE electrophoresis, transferred to 0.45um nitrocellulose, and immunoblotted. Antibody binding was visualized with chemilumiscence reagents (Supersignal West Pico and/or Femto, Pierce) and imaged using a Molecular Imager ChemiDoc XRS system (Biorad). Signals were quantitated using Imagelab version 5.1 software (BioRad).
Immunocytochemistry
Approximately 2.0 × 107 cells per treatment condition were centrifuged at 100xg for 5 minutes. After washing with D-PBS cells were fixed overnight at 4°C in 10% neutral buffered formalin (NBF) which was then replaced with 70% ethanol. Fixed cells were paraffin embedded, sectioned into 3 micron slices, and mounted on slides. The slices were deparaffinized using xylene and bathed in decreasing percentages of ethanol (95%, 80% and 70%). Cells were stained manually according to the LSAB 2 HRP system (Dako). Briefly, antigen retrieval was performed using the Decloaker (Biocare Medical, Concorde, CA) at 95°C for 25 minutes. Cells were incubated overnight with either 1% bovine serum albumin in PBS or antibody (diluted 1:10000 in 0.5% BSA in PBS) against phosphorylated Ser10 Histone H3 followed by 3 × PBS washes. For visualization of reaction, cells were incubated with LSAB 2 HRP system (Dako). After washing 3x with PBS, cells were counter stained with hematoxylin for 3 minutes followed by cover slipping the slides using non-aqueous mounting media. Stained slides were imaged using a Leica DMI6000B inverted microscope with a Leica DCF450 color camera at 40X and 100X magnification. Acquisition was done using Leica LAS-AF 4.0 software (Leica Microsystems, Buffalo Grove, IL).
Disclosure of potential conflicts of interest
C.L.S. received financial support for basic research studies from Spectrum Pharmaceuticals which holds the commercial license for belinostat and provided the drug for this study. However, Spectrum Pharmaceuticals had no influence on the study goals, execution, or preparation of the manuscript.
Acknowledgements
The authors would like to acknowledge the staff of the Flow Cytometry Shared Resource at the Arizona Cancer Center for services rendered. We are grateful to members of the Lymphoma Research Group at the UA for helpful suggestions and critique during the progress of the study. This work was initiated by an award from the Arizona Biomedical Research Commission to C.L.S., and supported through a career development award and developmental project award to C.L.S. [Lymphoma SPORE (1 P50 CA-130805-05, PtdIns - R.I. Fisher)]. A.P.H. received salary support from a cancer biology training grant (T32CA009213) and the Cancer Center Support Grant (P30CA023074). K.B.R. received support from the Undergraduate Research Biology Program at the University of Arizona. Y.Z. received support from a Hyundai Hope on Wheels Young Investigator Award. Salary support for J.T. and M.S. was provided by a SWOG Development Award from the Hope Foundation to M.S. In addition, Spectrum Pharmaceuticals donated belinostat for the study and provided funding to C.L.S.
ORCID
Catharine L. Smith http://orcid.org/0000-0002-9875-4884
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62(1):10-29; PMID:22237781; http://dx.doi.org/10.3322/caac.20138
- Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002; 346(25):1937-47; PMID: 12075054; http://dx.doi.org/10.1056/NEJMoa012914
- Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, Xu W, Tan B, Goldschmidt N, Iqbal J, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 2008; 359(22):2313-23; PMID:19038878; http://dx.doi.org/10.1056/NEJMoa0802885
- Coiffier B, Thieblemont C, Van Den NE, Lepeu G, Plantier I, Castaigne S, Lefort S, Marit G, Macro M, Sebban C, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d'Etudes des Lymphomes de l'Adulte. Blood 2010; 116(12):2040-5; PMID: 20548096; http://dx.doi.org/10.1182/blood-2010-03-276246
- Bolden JE, Shi W, Jankowski K, Kan CY, Cluse L, Martin BP, MacKenzie KL, Smyth GK, Johnstone RW. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis 2013; 4:e519; PMID:23449455; http://dx.doi.org/10.1038/cddis.2013.9
- Bots M, Johnstone RW. Rational combinations using HDAC inhibitors. Clin Cancer Res 2009; 15(12):3970-7; PMID: 19509171; http://dx.doi.org/10.1158/1078-0432.CCR-08-2786
- Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol 2012; 90(1):85-94; PMID: 22124371; http://dx.doi.org/10.1038/icb.2011.100
- Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem 2009; 107(4):600-8; PMID: 19459166; http://dx.doi.org/10.1002/jcb.22185
- Copeland A, Buglio D, Younes A. Histone deacetylase inhibitors in lymphoma. Curr Opin Oncol 2010; 22(5):431-6; PMID: 20683267; http://dx.doi.org/10.1097/CCO.0b013e32833d5954
- Prince HM, Bishton MJ, Harrison SJ. Clinical studies of histone deacetylase inhibitors. Clin Cancer Res 2009; 15(12):3958-69; PMID: 19509172; http://dx.doi.org/10.1158/1078-0432.CCR-08-2785
- Noh EJ, Lim DS, Jeong G, Lee JS. An HDAC inhibitor, trichostatin A, induces a delay at G2/M transition, slippage of spindle checkpoint, and cell death in a transcription-dependent manner. Biochem Biophys Res Commun 2009; 378(3):326-31; PMID: 19038231; http://dx.doi.org/10.1016/j.bbrc.2008.11.057
- Perez-Perarnau A, Coll-Mulet L, Rubio-Patino C, Iglesias-Serret D, Cosialls AM, Gonzalez-Girones DM, de FM, de Sevilla AF, de la Banda E, Pons G, Gil J. Analysis of apoptosis regulatory genes altered by histone deacetylase inhibitors in chronic lymphocytic leukemia cells. Epigenetics 2011; 6(10):1228-35; PMID: 21931276; http://dx.doi.org/10.4161/epi.6.10.17200
- Gabrielli B, Chia K, Warrener R. Finally, how histone deacetylase inhibitors disrupt mitosis! Cell Cycle 2011; 10(16):2658-61; PMID: 21811095; http://dx.doi.org/10.4161/cc.10.16.16953
- Peart MJ, Smyth GK, van Laar RK, Bowtell DD, Richon VM, Marks PA, Holloway AJ, Johnstone RW. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci U S A 2005; 102(10):3697-702; PMID: 15738394; http://dx.doi.org/10.1073/pnas.0500369102
- Tula-Sanchez AA, Havas AP, Alonge PJ, Klein ME, Doctor SR, Pinkston W, Glinsmann-Gibson BJ, Rimsza LM, Smith CL. A model of sensitivity and resistance to histone deacetylase inhibitors in diffuse large B cell lymphoma: Role of cyclin-dependent kinase inhibitors. Cancer Biol Ther 2013; 14(10):1-13; PMID: 23982416; http://dx.doi.org/10.4161/cbt.25941
- Schrump DS. Cytotoxicity mediated by histone deacetylase inhibitors in cancer cells: mechanisms and potential clinical implications. Clin Cancer Res 2009; 15(12):3947-57; PMID: 19509170; http://dx.doi.org/10.1158/1078-0432.CCR-08-2787
- Ma Y, Cai S, Lu Q, Lu X, Jiang Q, Zhou J, Zhang C. Inhibition of protein deacetylation by trichostatin A impairs microtubule-kinetochore attachment. Cell Mol Life Sci 2008; 65(19):3100-9; PMID: 18759129; http://dx.doi.org/10.1007/s00018-008-8237-5
- Gabrielli B, Brown M. Histone deacetylase inhibitors disrupt the mitotic spindle assembly checkpoint by targeting histone and nonhistone proteins. Adv Cancer Res 2012; 116:1-37; PMID:23088867; http://dx.doi.org/10.1016/B978-0-12-394387-3.00001-X
- Stevens FE, Beamish H, Warrener R, Gabrielli B. Histone deacetylase inhibitors induce mitotic slippage. Oncogene 2008; 27(10):1345-54; PMID: 17828304; http://dx.doi.org/10.1038/sj.onc.1210779
- Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 2008; 14(2):111-22; PMID: 18656424; http://dx.doi.org/10.1016/j.ccr.2008.07.002
- Huang HC, Shi J, Orth JD, Mitchison TJ. Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell 2009; 16(4):347-58; PMID: 19800579; http://dx.doi.org/10.1016/j.ccr.2009.08.020
- Chumduri C, Gillissen B, Richter A, Richter A, Milojkovic A, Overkamp T, Muller A, Pott C, Daniel PT. Apoptosis resistance, mitotic catastrophe, and loss of ploidy control in Burkitt lymphoma. J Mol Med (Berl) 2015; 93(5):559-72; PMID: 25548804; http://dx.doi.org/10.1007/s00109-014-1242-2
- Denisenko TV, Sorokina IV, Gogvadze V, Zhivotovsky B. Mitotic catastrophe and cancer drug resistance: A link that must to be broken. Drug Resist Updat 2016; 24:1-12; PMID: 26830311; http://dx.doi.org/10.1016/j.drup.2015.11.002
- Dewhurst SM, McGranahan N, Burrell RA, Rowan AJ, Gronroos E, Endesfelder D, Joshi T, Mouradov D, Gibbs P, Ward RL, et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov 2014; 4(2):175-85;PMID: 24436049; http://dx.doi.org/10.1158/2159-8290.CD-13-0285
- Wang Q, Wu PC, Dong DZ, Ivanova I, Chu E, Zeliadt S, Vesselle H, Wu DY. Polyploidy road to therapy-induced cellular senescence and escape. Int J Cancer 2013; 132(7):1505-15; PMID:22945332; http://dx.doi.org/10.1002/ijc.27810
- Blagosklonny MV, Robey R, Sackett DL, Du L, Traganos F, Darzynkiewicz Z, Fojo T, Bates SE. Histone deacetylase inhibitors all induce p21 but differentially cause tubulin acetylation, mitotic arrest, and cytotoxicity. Mol Cancer Ther 2002 Sep; 1(11):937-41; PMID:12481415
- Qiu L, Burgess A, Fairlie DP, Leonard H, Parsons PG, Gabrielli BG. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol Biol Cell 2000 Jun; 11(6):2069-83; PMID:10848630; http://dx.doi.org/10.1091/mbc.11.6.2069
- Lallemand F, Courilleau D, Buquet-Fagot C, Atfi A, Montagne MN, Mester J. Sodium butyrate induces G2 arrest in the human breast cancer cells MDA-MB-231 and renders them competent for DNA rereplication. Exp Cell Res 1999; 247(2):432-40; PMID:10066371; http://dx.doi.org/10.1006/excr.1998.4370
- Hendzel MJ, Wei Y, Mancini MA, Van HA, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 1997; 106(6):348-60; PMID:9362543; http://dx.doi.org/10.1007/s004120050256
- Dowling M, Voong KR, Kim M, Keutmann MK, Harris E, Kao GD. Mitotic spindle checkpoint inactivation by trichostatin a defines a mechanism for increasing cancer cell killing by microtubule-disrupting agents. Cancer Biol Ther 2005 Feb; 4(2):197-206; PMID:15753652
- Robbins AR, Jablonski SA, Yen TJ, Yoda K, Robey R, Bates SE, Sackett DL. Inhibitors of histone deacetylases alter kinetochore assembly by disrupting pericentromeric heterochromatin. Cell Cycle 2005; 4(5):717-26; PMID:15846093; http://dx.doi.org/10.4161/cc.4.5.1690
- Kalac M, Scotto L, Marchi E, Amengual J, Seshan VE, Bhagat G, Ulahannan N, Leshchenko VV, Temkin AM, Parekh S, Tycko B, O'Connor OA. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene expression and epigenetic profiles in models of DLBCL. Blood 2011; 118(20):5506-16; PMID:21772049; http://dx.doi.org/10.1182/blood-2011-02-336891
- Taylor SS, Hussein D, Wang Y, Elderkin S, Morrow CJ. Kinetochore localisation and phosphorylation of the mitotic checkpoint components Bub1 and BubR1 are differentially regulated by spindle events in human cells. J Cell Sci 2001; 114(Pt 24):4385-95; PMID:11792804
- Chan GK, Jablonski SA, Sudakin V, Hittle JC, Yen TJ. Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J Cell Biol 1999; 146(5):941-54; PMID:10477750; http://dx.doi.org/10.1083/jcb.146.5.941
- Galan-Malo P, Vela L, Gonzalo O, Calvo-Sanjuan R, Gracia-Fleta L, Naval J, Marzo I. Cell fate after mitotic arrest in different tumor cells is determined by the balance between slippage and apoptotic threshold. Toxicol Appl Pharmacol 2012; 258(3):384-93; PMID:22178383; http://dx.doi.org/10.1016/j.taap.2011.11.021
- Bekier ME, Fischbach R, Lee J, Taylor WR. Length of mitotic arrest induced by microtubule-stabilizing drugs determines cell death after mitotic exit. Mol Cancer Ther 2009; 8(6):1646-54; PMID:19509263; http://dx.doi.org/10.1158/1535-7163.MCT-08-1084
- Shi J, Orth JD, Mitchison T. Cell type variation in responses to antimitotic drugs that target microtubules and kinesin-5. Cancer Res 2008; 68(9):3269-76; PMID:18451153; http://dx.doi.org/10.1158/0008-5472.CAN-07-6699
- Brito DA, Rieder CL. The ability to survive mitosis in the presence of microtubule poisons differs significantly between human nontransformed (RPE-1) and cancer (U2OS, HeLa) cells. Cell Motil Cytoskeleton 2009; 66(8):437-47; PMID:18792104; http://dx.doi.org/10.1002/cm.20316
- Kim M, Murphy K, Liu F, Parker SE, Dowling ML, Baff W, Kao GD. Caspase-mediated specific cleavage of BubR1 is a determinant of mitotic progression. Mol Cell Biol 2005; 25(21):9232-48; PMID:16227576; http://dx.doi.org/10.1128/MCB.25.21.9232-9248.2005
- Suematsu T, Li Y, Kojima H, Nakajima K, Oshimura M, Inoue T. Deacetylation of the mitotic checkpoint protein BubR1 at lysine 250 by SIRT2 and subsequent effects on BubR1 degradation during the prometaphase/anaphase transition. Biochem Biophys Res Commun 2014; 453(3):588-94; PMID:25285631; http://dx.doi.org/10.1016/j.bbrc.2014.09.128
- Choi E, Choe H, Min J, Choi JY, Kim J, Lee H. BubR1 acetylation at prometaphase is required for modulating APC/C activity and timing of mitosis. EMBO J 2009; 28(14):2077-89; PMID:19407811; http://dx.doi.org/10.1038/emboj.2009.123
- Bingle CD, Craig RW, Swales BM, Singleton V, Zhou P, Whyte MK. Exon skipping in Mcl-1 results in a bcl-2 homology domain 3 only gene product that promotes cell death. J Biol Chem 2000; 275(29):22136-46; PMID:10766760; http://dx.doi.org/10.1074/jbc.M909572199
- Bae J, Leo CP, Hsu SY, Hsueh AJ. MCL-1S, a splicing variant of the antiapoptotic BCL-2 family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. J Biol Chem 2000; 275(33):25255-61; PMID:10837489; http://dx.doi.org/10.1074/jbc.M909826199
- Ogden A, Rida PC, Knudsen BS, Kucuk O, Aneja R. Docetaxel-induced polyploidization may underlie chemoresistance and disease relapse. Cancer Lett 2015; 367(2):89-92; PMID:26185000; http://dx.doi.org/10.1016/j.canlet.2015.06.025
- Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol 2004 Jan; 5(1):45-54; PMID:14708009; http://dx.doi.org/10.1038/nrm1276
- Coward J, Harding A. Size does matter: Why polyploid tumor cells are critical drug targets in the war on cancer. Front Oncol 2014; 4:123; PMID:24904834; http://dx.doi.org/10.3389/fonc.2014.00123
- Ward T, Wang M, Liu X, Wang Z, Xia P, Chu Y, Wang X, Liu L, Jiang K, Yu H, et al. Regulation of a dynamic interaction between two microtubule-binding proteins, EB1 and TIP150, by the mitotic p300/CBP-associated factor (PCAF) orchestrates kinetochore microtubule plasticity and chromosome stability during mitosis. J Biol Chem 2013; 288(22):15771-85; PMID:23595990; http://dx.doi.org/10.1074/jbc.M112.448886
- Warrener R, Beamish H, Burgess A, Waterhouse NJ, Giles N, Fairlie D, Gabrielli B. Tumor cell-selective cytotoxicity by targeting cell cycle checkpoints. FASEB J 2003; 17(11):1550-2; PMID:12824307; http://dx.doi.org/10.1096/fj.02-1003fje
- Magnaghi-Jaulin L, Eot-Houllier G, Fulcrand G, Jaulin C. Histone deacetylase inhibitors induce premature sister chromatid separation and override the mitotic spindle assembly checkpoint. Cancer Res 2007; 67(13):6360-7; PMID:17616695; http://dx.doi.org/10.1158/0008-5472.CAN-06-3012
- Topham C, Tighe A, Ly P, Bennett A, Sloss O, Nelson L, Ridgway RA, Huels D, Littler S, Schandl C, et al. MYC is a major determinant of mitotic cell fate. Cancer Cell 2015; 28(1):129-40; PMID:26175417; http://dx.doi.org/10.1016/j.ccell.2015.06.001
- Sloss O, Topham C, Diez M, Taylor S. Mcl-1 dynamics influence mitotic slippage and death in mitosis. Oncotarget 2016; 7(5):5176-92; PMID:26769847; http://dx.doi.org/10.18632/oncotarget.6894
- Haschka MD, Soratroi C, Kirschnek S, Hacker G, Hilbe R, Geley S, Villunger A, Fava LL. The NOXA-MCL1-BIM axis defines lifespan on extended mitotic arrest. Nat Commun 2015; 6:6891; PMID:25922916; http://dx.doi.org/10.1038/ncomms7891
- Topham CH, Taylor SS. Mitosis and apoptosis: how is the balance set? Curr Opin Cell Biol 2013; 25(6):780-5; PMID:23890995; http://dx.doi.org/10.1016/j.ceb.2013.07.003
- Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol 2011 Jun; 12(6):385-92; PMID:21527953; http://dx.doi.org/10.1038/nrm3115
- Swanton C, Marani M, Pardo O, Warne PH, Kelly G, Sahai E, Elustondo F, Chang J, Temple J, Ahmed AA, et al. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell 2007; 11(6):498-512; PMID:17560332; http://dx.doi.org/10.1016/j.ccr.2007.04.011
- Roberts JR, Allison DC, Donehower RC, Rowinsky EK. Development of polyploidization in taxol-resistant human leukemia cells in vitro. Cancer Res 1990; 50(3):710-6; PMID:1967550
- Deng J, Carlson N, Takeyama K, Dal CP, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 2007 Aug; 12(2):171-85; PMID:17692808; http://dx.doi.org/10.1016/j.ccr.2007.07.001
- Wang L, Li H, Ren Y, Zou S, Fang W, Jiang X, Jia L, Li M, Liu X, Yuan X, et al. Targeting HDAC with a novel inhibitor effectively reverses paclitaxel resistance in non-small cell lung cancer via multiple mechanisms. Cell Death Dis 2016; 7:e2063; PMID:26794658; http://dx.doi.org/10.1038/cddis.2015.328
- Chao H, Wang L, Hao J, Ni J, Chang L, Graham PH, Kearsley JH, Li Y. Low dose histone deacetylase inhibitor, LBH589, potentiates anticancer effect of docetaxel in epithelial ovarian cancer via PI3K/Akt pathway in vitro. Cancer Lett 2013; 329(1):17-26; PMID:22995071; http://dx.doi.org/10.1016/j.canlet.2012.08.035
- Zhang QC, Jiang SJ, Zhang S, Ma XB. Histone deacetylase inhibitor trichostatin A enhances anti-tumor effects of docetaxel or erlotinib in A549 cell line. Asian Pac J Cancer Prev 2012; 13(7):3471-6; PMID:22994780; http://dx.doi.org/10.7314/APJCP.2012.13.7.3471
- Zuco V, De CM, Cincinelli R, Nannei R, Pisano C, Zaffaroni N, Zunino F. Synergistic antitumor effects of novel HDAC inhibitors and paclitaxel in vitro and in vivo. PLoS One 2011; 6(12):e29085; PMID:22194993; http://dx.doi.org/10.1371/journal.pone.0029085
- Shi YK, Li ZH, Han XQ, Yi JH, Wang ZH, Hou JL, Feng CR, Fang QH, Wang HH, Zhang PF, et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces growth inhibition and enhances taxol-induced cell death in breast cancer. Cancer Chemother Pharmacol 2010; 66(6):1131-40; PMID:20838997; http://dx.doi.org/10.1007/s00280-010-1455-1
- Angelucci A, Mari M, Millimaggi D, Giusti I, Carta G, Bologna M, Dolo V. Suberoylanilide hydroxamic acid partly reverses resistance to paclitaxel in human ovarian cancer cell lines. Gynecol Oncol 2010; 119(3):557-63; PMID:20825984; http://dx.doi.org/10.1016/j.ygyno.2010.07.036
- Dowdy SC, Jiang S, Zhou XC, Hou X, Jin F, Podratz KC, Jiang SW. Histone deacetylase inhibitors and paclitaxel cause synergistic effects on apoptosis and microtubule stabilization in papillary serous endometrial cancer cells. Mol Cancer Ther 2006; 5(11):2767-76; PMID:17121923; http://dx.doi.org/10.1158/1535-7163.MCT-06-0209
- Qian X, LaRochelle WJ, Ara G, Wu F, Petersen KD, Thougaard A, Sehested M, Lichenstein HS, Jeffers M. Activity of PXD101, a histone deacetylase inhibitor, in preclinical ovarian cancer studies. Mol Cancer Ther 2006; 5(8):2086-95; PMID:16928830; http://dx.doi.org/10.1158/1535-7163.MCT-06-0111
- Hwang JJ, Kim YS, Kim T, Kim MJ, Jeong IG, Lee JH, Choi J, Jang S, Ro S, Kim CS. A novel histone deacetylase inhibitor, CG200745, potentiates anticancer effect of docetaxel in prostate cancer via decreasing Mcl-1 and Bcl-XL. Invest New Drugs 2012; 30(4):1434-42; PMID:21773733; http://dx.doi.org/10.1007/s10637-011-9718-1
- Hwang JJ, Kim YS, Kim MJ, Kim DE, Jeong IG, Kim CS. Histone deacetylase inhibitor potentiates anticancer effect of docetaxel via modulation of Bcl-2 family proteins and tubulin in hormone refractory prostate cancer cells. J Urol 2010; 184(6):2557-64; PMID:21030039; http://dx.doi.org/10.1016/j.juro.2010.07.035
- Cooper AL, Greenberg VL, Lancaster PS, van Nagell JRJ, Zimmer SG, Modesitt SC. In vitro and in vivo histone deacetylase inhibitor therapy with suberoylanilide hydroxamic acid (SAHA) and paclitaxel in ovarian cancer. Gynecol Oncol 2007; 104(3):596-601; PMID: 17049973; http://dx.doi.org/10.1016/j.ygyno.2006.09.011
- Keshelava N, Davicioni E, Wan Z, Ji L, Sposto R, Triche TJ, Reynolds CP. Histone deacetylase 1 gene expression and sensitization of multidrug-resistant neuroblastoma cell lines to cytotoxic agents by depsipeptide. J Natl Cancer Inst 2007; 99(14):1107-19; PMID:17623797; http://dx.doi.org/10.1093/jnci/djm044
- Kano Y, Akutsu M, Tsunoda S, Izumi T, Kobayashi H, Mano H, Furukawa Y. Cytotoxic effects of histone deacetylase inhibitor FK228 (depsipeptide, formally named FR901228) in combination with conventional anti-leukemia/lymphoma agents against human leukemia/lymphoma cell lines. Invest New Drugs 2007; 25(1):31-40; PMID:16865529; http://dx.doi.org/10.1007/s10637-006-9000-0
- Lassen U, Molife LR, Sorensen M, Engelholm SA, Vidal L, Sinha R, Penson RT, Buhl-Jensen P, Crowley E, Tjornelund J, et al. A phase I study of the safety and pharmacokinetics of the histone deacetylase inhibitor belinostat administered in combination with carboplatin and/or paclitaxel in patients with solid tumours. Br J Cancer 2010; 103(1):12-7; PMID:20588278; http://dx.doi.org/10.1038/sj.bjc.6605726
- Fanciullino R, Ciccolini J, Milano G. Challenges, expectations and limits for nanoparticles-based therapeutics in cancer: a focus on nano-albumin-bound drugs. Crit Rev Oncol Hematol 2013; 88(3):504-13; PMID:23871532; http://dx.doi.org/10.1016/j.critrevonc.2013.06.010
- Silverman JA, Deitcher SR. Marqibo(R) (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother Pharmacol 2013; 71(3):555-64; PMID:23212117; http://dx.doi.org/10.1007/s00280-012-2042-4