
ABSTRACT
Most BRAF-mutated melanomas initially responsive to the FDA-approved inhibitors preferentially targeting B-Raf mutated in Val600 residue eventually relapse, requiring additional therapeutic modalities. Recent studies report the significance of metabolic reprograming in mitochondria for maintenance of BRAF-mutated melanomas and for development of their drug resistance to B-Raf inhibitors, providing a rationale for targeting mitochondria as a potential therapeutic strategy for melanoma. We therefore determined whether mitochondria-targeted metabolism-interfering agents can effectively suppress human B-RafV600E melanoma cell lines and their dabrafenib/PLX4032-resistant progenies using mitochondria-targeted carboxy-proxyl (Mito-CP) and ubiquinone (Mito-Q). These agents exhibited comparable efficacy to PLX4032 in suppressing SK-MEL28, A375, and RPMI-7951 cells in vitro. As determined in SK-MEL28 and A375 cells, Mito-CP induced apoptotic cell death mediated by mitochondrial membrane depolarization and subsequent oxidative stress, which PLX4032 could not induce. Of note, Mito-CP also effectively suppressed PLX4032-resistant progenies of SK-MEL28 and A375. Moreover, when orally administered, Mito-CP suppressed SK-MEL28 xenografts in mice as effectively as PLX4032 without serious adverse effects. These data demonstrate that mitochondria-targeted agents have therapeutic potential to effectively suppress BRAF–mutated melanomas via an effect(s) distinct from those of B-Raf inhibitors.
Abbreviations
| Carboxy-H2DCFDA | = | 5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate |
| Mito-CP | = | triphenyl-phosphonium-carboxy-proxyl |
| Mito-Q | = | [10-(4,5-dimethoxy-2-methyl-3,6-dioxo-1,4-cyclohexadien-1-yl) decyl] triphenyl phosphonium |
| MTT | = | 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide |
| NAC | = | N-acetyl-cysteine |
| PARP | = | poly(ADP-ribose) polymerase |
| TMRE | = | tetramethyl-rhodamine ethyl ester perchlorate |
| TPP | = | triphenyl-phosphonium |
Introduction
Activating mutations of BRAF, mainly affecting B-Raf kinase domain, e.g., B-RafV600E,K,D, are detected in 40–50% cutaneous melanomas, making B-Raf and its effector MEK/ERK kinase cascade key therapeutic targets in these tumors.Citation1 As such, for BRAF–mutated melanoma therapy, the Food and Drug Administration approved the small molecule B-Raf inhibitors dabrafenib/PLX4032 (trade name Tafinlar) and vemurafenib (trade name Zelboraf) as monotherapy, and the dabrafenib combination with the MEK1/2 inhibitor, trametinib (trade name Mekinist). Although these inhibitors have substantially advanced the therapy of B-RafV600E,K melanoma (reviewed inCitation2,3), almost all tumors initially responsive to these drugs develop resistance, eventually leading to patient demise. Therefore, additional therapeutic modalities are necessary to prolong patient survival.
Tumor cells often reprogram mitochondrial metabolism to meet their increased demand for energy and building blocks (reviewed in Citation4). These alterations in mitochondrial metabolism are often associated with increased production of toxic byproducts that threaten tumor cell survival, e.g., reactive oxygen species (ROS). Accordingly, tumor cells should maintain a balance between altered mitochondrial metabolism and the capacity for stress tolerance.Citation5-7 Moreover, tumor cells can adapt to cancer drugs by readjusting their mitochondrial metabolism and cell death/survival machinery.Citation6,8 These indicate the potential of mitochondria as a cancer therapeutic target. Of note, it has been recently reported that reprograming of mitochondrial bioenergetics and oxidative metabolism is required not only for the maintenance of BRAF-mutated melanomas but also for development of their resistance to B-Raf inhibitors.Citation8-10 This provides a rationale for targeting mitochondria as a potential therapeutic strategy for melanomas, and as such, it is important to evaluate therapeutic potential of mitochondria-targeted metabolism-interfering agents in these tumors.
The lipophilic cation, triphenylphosphonium (TPP), accumulates in mitochondria via the mitochondrial membrane potential (Δψm), which rationalizes its covalent conjugation to a drug as an effective means for specific drug delivery to mitochondria.Citation11 Indeed, different TPP-conjugated antioxidants have been shown for their therapeutic potential for cancer and neurodegenerative disorders.Citation5,11-14 We and others have also demonstrated that certain TPP-conjugated metabolism-interfering agents have high efficacy in suppressing proliferation of different tumor cells, including breast and thyroid cancer cells.Citation15,16 Accumulation of TPP-conjugated compounds in mitochondria can increase up to 100 to 1,000 fold depending upon Δψm values in different cell types. Of note, many tumor cells usually maintain relatively high basal Δψm, which renders them vulnerable to TPP-linked drugs,Citation5,14
In the present study, we demonstrate that BRAF-mutated melanoma cells can be effectively suppressed by targeting mitochondria using TPP-conjugated superoxide dismutase mimetic carboxy-proxyl (Mito-CP) and TPP-conjugated ubiquinone (Mito-Q), which can disrupt mitochondrial bioenergetics and redox balance if over-enriched in mitochondria,Citation12,13,Citation15,16 in human B-RafV600E melanoma cell lines and their PLX4032-resistant progenies.
Materials and methods
Cell culture and reagents
SK-MEL28 (ATCC, Manassas, VA), RPMI-7951 (ATCC), SK-MEL-2 (ATCC), and MeWo (ATCC) were maintained in minimal essential medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 100 U of penicillin and 100 μg of streptomycin per ml. A375 (ATCC) and the primary normal human fibroblasts, HS27 (ATCC) and BJ (ATCC), were grown in Dulbecco's modified eagle medium (Invitrogen) supplemented with 10% fetal bovine serum. PLX4032-resistant A375R Citation17 and its parental A375 were obtained from R. Marais (Univ. Manchester). PLX4032-resistant SK-MEL28 progenies were generated by prolonged cell culture in the presence of PLX4032 (Fig. S1), according to the procedure previously described.Citation17 Neonatal primary normal melanocytes (LONZA) were grown in Medium 254 (ThermoFisher Scientific) supplemented with Human Melanocyte Growth Supplement (ThermoFisher Scientific).
Mito-CP Citation18 and Mito-Q ([10-(4,5-dimethoxy-2-methyl-3,6-dioxo-1,4-cyclohexadien-1-yl) decyl] triphenyl phosphonium) were obtained from B. Kalyanaraman (Medical College of Wisconsin). Carboxy-proxyl (CP), methyl-triphenyl-phosphonium (TPP), coenzyme Q10 (CoQ10), N-acetyl-cysteine (NAC), tetramethyl-rhodamine ethyl ester perchlorate (TMRE), 5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA), and cremophore-EL were purchased from Sigma-Aldrich (St Louis, MO). PLX4032 and MitoTracker Green were purchased from Selleck Chemicals (Houston, TX) and Invitrogen, respectively. Chemical structures of Mito-CP and Mito-Q, TPP, CP, and CoQ10 are depicted in Fig. S2.
Cell viability and death analysis
For the assay using 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT), cells were incubated with culture medium containing MTT (0.5 mg/ml, Sigma) in 96 well-plates for 2 h at 37°C, switched into 200 μl of dimethyl-sulfoxide (DMSO), and shaken for 5 min at room temperature before measuring absorbance at 540 nm, as previously described.Citation19 Cell viability was also measured by counting trypan blue-stained cells using hemocytometer. Annexin V (Invitrogen) was stained according to manufacturer's instruction.
Detection of mitochondrial membrane potential using TMRE
Cells were incubated with culture medium containing TMRE (10 ng/ml) in 6-well plates for 15 min at 37°C in dark, washed with phosphate-buffered saline (PBS), switched into phenol-red free medium before visualizing fluorescence under a microscope. For flow cytometric measurement, TMRE-treated cells were collected by trypsinization and resuspended in PBS containing 0.1% bovine serum albumin, and analyzed by flow cytometry (PE channel, 575 nm), as previously described.Citation20,21 Data from 20,000 cells were analyzed using FCS Express software (De Novo Software).
Detection of ROS using carboxy-H2DCFDA
Cells were incubated with culture medium containing 1 μM carboxy-H2DCFDA at 37°C for 1 h. After medium change, cells were incubated in dark for 2 h, trypsinized, and resuspended in PBS before analysis by flow cytometry (FITC channel, 525 nm), as previously described.Citation20,21 Data from 20,000 cells were analyzed using FCS Express software (De Novo Software).
Immunoblot analysis
Immunoblotting was performed as previously described.Citation22 Mitochondrial lysates were prepared using the Mitochondria Isolation Kit (Thermo Scientific, Rockford, IL) according to the manufacturer's protocol. Antibodies were diluted as follows: poly(ADP-ribose) polymerase (PARP), 1:1,000; cleaved lamin A, 1:2,000; Bcl-2, 1:2,000; Bcl-xL, 1:2,000; Mcl-1, 1:2,000; COX IV, 1:2000; β-actin, 1:10,000; ATG7, 1:2,000 (Cell Signaling); SQSTM1, 1:2,000 (Santa Cruz); LC3B, 1:2,000 (MBL International). The TPP-specific antibody was obtained from M. Murphy (MRC Dunn Human Nutrition Unit) and used at a 1:1000 dilution ratio. Images of immunoblots were taken and processed using ChemiDoc XRS+ and Image Lab 3.0 (Bio-Rad, Hercules, CA).
Tumor xenograft studies
5 × 106 SK-MEL28 cells in 100 µl extracellular matrix gel (E1270, Sigma Aldrich) were inoculated subcutaneously into the rear flanks of 6-week-old female athymic nude (nu/nu) mice (Jackson Laboratory, Bar Harbor, ME). Once palpable, tumors were measured using Vernier calipers at intervals indicated in the text. Tumor volumes were calculated using the formula: length × width2 × 0.5236. When tumor volumes reached 100 mm3, mice were sorted into the groups of 4 animals to achieve equal distribution of tumor size in all treatment groups. Group 1 received only the vehicle (1:9 mixture of DMSO/Cremophore-EL), group 2 received PLX4032 (50 mg/kg body weight/dose), and group 3 received Mito-CP (50 mg/kg body weight/dose). PLX4032 and Mito-CP were dissolved in 200 μl vehicle and were orally administered by gavage. In each treatment cycle, animals received single dose of drugs per day for 4 consecutive days and took a day rest. This cycle was repeated 4 times during the treatment. At the end of drug treatments, animals were euthanized by CO2 asphyxiation. All animal studies were performed according to the protocols approved by the Institutional Animal Care and Use Committee at Medical College of Wisconsin.
Statistical analysis
Two-tailed unpaired Student's t-test was used to assess the statistical significance of 2 data sets. Tumor xenograft data were analyzed by One-Way ANOVA with Bonferroni correction for multiple comparisons. P value < 0.05 was considered statistically significant.
Results
Mitochondria-targeted agents, Mito-CP and Mito-Q, can effectively suppress survival of a subset of melanoma cells in vitro
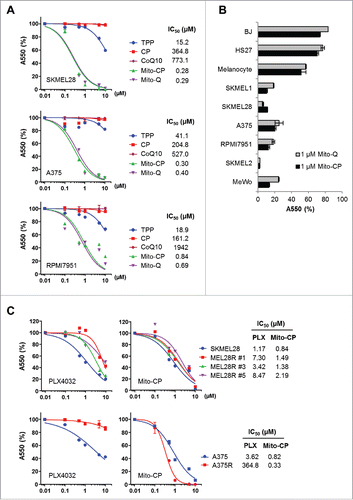
Mito-CP consists of the CP moiety, a 5-membered nitroxide, and the TPP moiety that mediates mitochondria targeting Fig. S2). We determined whether Mito-CP can suppress the human B-RafV600E melanoma lines, SK-MEL28, A375, and RPMI-7951, in vitro. As the controls to validate mitochondria-specific effects of Mio-CP, we used its separate functional moieties, CP and TPP. As determined by MTT assay, Mito-CP in 0.01-10 μM dose ranges effectively decreased cell viability of these cells within 48 h, with all cell lines exhibiting very high sensitivity to this drug (); IC50 values were calculated at 0.28 μM for SK-MEL28, 0.30 μM for A375, and 0.84 μM for RPMI-7951. In contrast, equal doses of CP did not significantly affect the growth of these cells while the mitochondrial carrier, TPP, only mildly decreased cell viability at higher doses (). This indicates that mitochondria-specific delivery of CP is the key to its tumor suppressive effects. Noteworthy is that, similarly as Mito-CP, Mito-Q (TPP-linked CoQ10) also effectively decreased the viability of these tumor cells although CoQ10 by itself had no effect (), suggesting that mitochondria-specific delivery of a metabolism-interfering agent can effectively decrease the viability of a subset of melanoma cells. In support of this possibility, Mito-CP and Mito-Q also effectively decreased the viability of other melanoma lines, including SK-MEL1 (B-RafV600E), SK-MEL2 (N-RasQ61R), and MeWo (B-Raf and N-Ras wild type), although these compounds did not as significantly decrease the viability of primary normal foreskin fibroblasts BJ and HS27, and primary cultured melanocytes ().
Figure 1. Mito-CP and Mito-(Q)suppresses viability of B-RafV600E melanoma cells. (A) SK-MEL28, A375, and RPMI-7951 cells in 96-well plates were treated with serially increasing doses of Mito-CP, Mito-Q, or the control compounds, TPP and CP, for 48 h. Cells were then allowed to recover in drug-free fresh medium for 24 h prior to measuring cell viability by MTT assay. Data (mean ± SD, n = 6) are expressed as the percentage of untreated control. (B) A panel of cell lines was treated with 1 µM Mito-CP or Mito-Q for 48 h followed by 24 h recovery prior to MTT assay. Data (mean ± SD, n = 3) are the percentage of untreated control. SK-MEL1, SK-MEL28, A375, and RPMI-7951 (B-RafV600E); SK-MEL 2 (N-RasQ61R); MeWo (B-Raf, N-Ras wild type); BJ and HS27 (primary normal foreskin fibroblasts) (C) Cells of SK-MEL28, A375, and their PLX4032-resistant derivatives were treated with increasing doses of Mito-CP or PLX4032 for 48 h. Cells were then allowed to recover in drug-free fresh medium for 24 h prior to MTT assay.
Mito-CP suppresses PLX4032-naïve B-RafV600E melanoma cells as effectively as PLX4032 and also suppresses PLX4032-resistant melanoma cells
We next assessed the efficacy of Mito-CP in comparison with PLX4032 in SK-MEL28 and A375 cells. As determined by MTT assay after 48 h drug treatments, PLX4032-naïve SK-MEL28 and A375 cell lines exhibited slightly lower IC50 values to Mito-CP than to PLX4032 (). Strikingly, when tested in PLX4032-resistant progenies of these melanoma cell lines, Mito-CP effectively suppressed 3 independent PLX4032-resistant SK-MEL28 clones and the PLX4032-resistant A375R cells at similar efficacies as observed in their parental cells (). These data strongly demonstrate the high in vitro efficacy of Mito-CP for PLX4032-naïve and -resistant B-RafV600E melanoma cells.
Mito-CP induces apoptotic cell death in B-RafV600E melanoma cells
To determine the nature of Mito-CP-mediated suppression of these B-RafV600E melanoma cells, we conducted Western blot analyses of total lysates of SK-MEL28, A375 and RPMI-7951 cells treated with Mito-CP for 24 h.
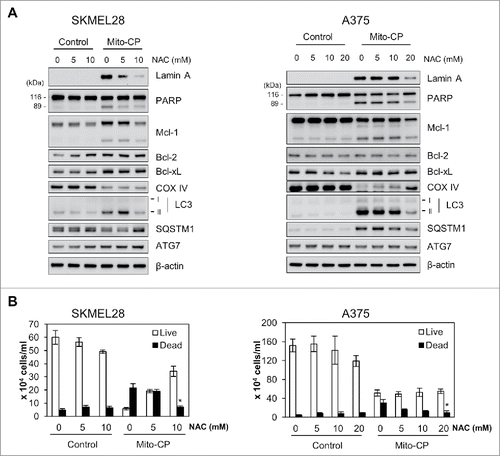
We found that Mito-CP more robustly induced cleavage of lamin A, although not PARP, than PLX4032 in these melanoma cell lines (). The cleavages of lamin A and PARP are key signatures of caspase-dependent apoptotic cell death.Citation23 Consistent with this, flow cytometry analysis using annexin V and propidium iodide revealed that Mito-CP strongly induced apoptotic cell death in SK-MEL28 and A375 cells, which was comparable to the effect of the conventional chemotherapeutic agent cisplatin (). These effects were accompanied by significant downregulation of cytochrome c oxidase (COX IV), a marker of mitochondrial integrity, and of Mcl-1 although other anti-apoptotic Bcl-2 family members were not similarly affected (); Mito-CP did not affect Bcl-2 levels while upregulating Bcl-xL levels. These data suggest that Mito-CP may induce cell death via a mitochondrial damage that disrupts mitochondrial integrity and anti-apoptotic capacity. Of note, Mito-CP substantially increased LC3 processing and protein levels of SQSTM1/p62 and ATG7, the key autophagy markers,Citation24 suggesting that Mito-CP may alter cellular activity for autophagy (). These effects of Mito-CP are in contrast with the effects of PLX4032 in these cells, strongly suggesting that Mito-CP can suppress melanoma cell survival via a distinct mechanism(s).
Figure 2. Mito-CP induces surrogate markers of cell death, mitochondrial integrity, and autophagy in B-RafV600E melanoma cells. Total lysates of SK-MEL28, A375, and RPMI-7951 cells treated with different doses of PLX4032 or Mito-CP for 24 h were analyzed by Western blotting for expression of the indicated proteins. β-actin is the control for equal amounts of protein loading.
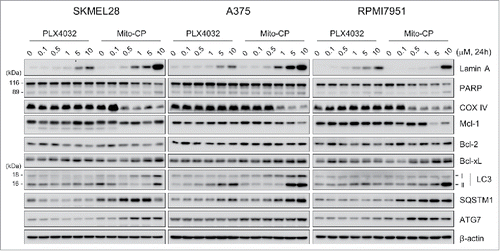
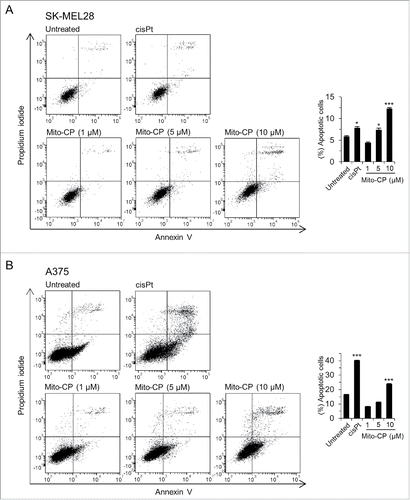
Figure 3. Mito-CP induces apoptotic cell death. SK-MEL28 (A) and A375 (B) cells were treated with increasing doses of Mito-CP for 24 h prior to annexin V/propidium iodide staining. The graphs (right) indicate annexin V positive and propidium iodide positive cell populations. 50 µM cisplatin treatment for 24 h was used as a positive control to induce apoptosis. Cisplatin was dissolved in water. Data (mean ± SEM) are from a representative experiment performed in triplicate. *P < 0.05; ***P < 0.001.
Mito-CP, but not PLX4032, disrupts mitochondrial membrane potential (Δψm) in SK-MEL28 cells
Mito-CP induces cytotoxicity mainly by interfering with mitochondrial activity required for the maintenance of bioenergetics and redox balance.Citation15,16 We thus determined the effect of Mito-CP on Δψm, the primary indicator of mitochondrial activity, in SK-MEL28 cells.
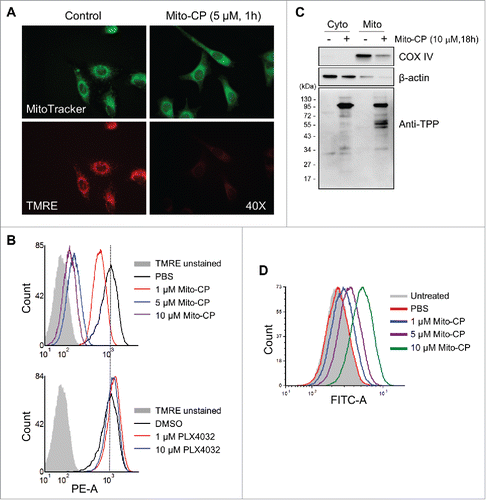
When stained with the Δψm–dependent mitochondrial dye TMRE, Mito-CP-treated SK-MEL28 cells exhibited a significant loss of Δψm, as determined by microscopy after 1 h treatment () and by flow cytometry after 24 h treatment (). Of note, the effective Mito-CP doses inducing these effects were consistent with the doses that decreased cell viability (). In contrast, PLX4032-treated cells did not exhibit any significant changes in Δψm (), further highlighting the difference between Mito-CP and PLX4032. Under this condition, MitoTracker green visualized similar levels of cellular content of mitochondria in Mito-CP-treated and the control cells, suggesting that Mito-CP-induced Δψm loss was not a consequence of reduced mitochondrial mass (). After 18 h Mito-CP treatment, Western blotting using an antibody specific to the TPP moiety Citation25 revealed increased formation of Mito-CP-protein adducts in the mitochondrial extracts of Mito-CP-treated cells (), indicating mitochondria-specific enrichment of Mito-CP. Moreover, similar patterns of adduct formation were detected in SK-MEL28, A375 and RPMI-7951 cells upon Mito-CP treatments (Fig. S3), further demonstrating the consistency of Mito-CP effects in these melanoma cells.
Figure 4. Mito-CP induces loss of mitochondrial membrane potential in SK-MEL28 cells. (A) Cells treated with 5 µM Mito-CP for 1 h were stained with TMRE. Changes in the mitochondrial membrane potential were then visualized under a fluorescent microscope (magnification 400X). MitoTracker Green was used to visualize intact mitochondria in cells. PBS was used as the control for Mito-CP. (B) Cells treated with Mito-CP or PLX4032 for 24 h were stained with TMRE and were analyzed by flow cytometry to measure red fluorescence (PE channel, 575 nm). Unstained cells were used as negative controls. PBS and DMSO are vehicle controls for Mito-CP and PLX4032, respectively. (C) Cytosolic and mitochondrial extracts from 5 × 105 cells treated with Mito-CP for 18 h were analyzed by Western blotting for TPP adduct formation. COX IV and β-actin were detected as the controls for purity of extracts. (D) Cells treated with Mito-CP for 24 h were treated with 1 μM carboxy-H2DCFDA for 1 h and then incubated for 2 h in a dye-free culture medium. Cells were harvested and analyzed by flow cytometry to measure green fluorescence (FITC channel, 525 nm).
Mito-CP induces oxidative stress in melanoma cells
Mitochondrial stress can lead to cell death,Citation26 wherein increased ROS levels caused by electron leakage from the respiratory chain are often involved.Citation27 To determine whether Mito-CP treatment increased ROS generation in SK-MEL28 cells, we measured cellular oxidation levels of carboxy-H2DCFDA, a redox-sensitive dye that fluoresces upon oxidation.Citation28 Within 24 h of Mito-CP treatment, we detected significantly increased fluorescence of carboxy-H2DCFDA in SK-MEL28 cells, which was in commensurate to Mito-CP doses ().
Next, we determined whether the cell-permeable ROS scavenger, N-acetyl-cysteine (NAC) could rescue SK-MEL28 and A375 cells from Mito-CP toxicity. We found that 1 h NAC pretreatments significantly attenuated the cleavage of lamin A and PARP, downregulation of Mcl-1 and COX IV, and the expression of autophagy markers in Mito-CP-treated cells, as determined by Western blotting (). Consistent with these data, NAC pretreatments rescued, albeit not fully, these cells from Mito-CP toxicity, as determined by the trypan blue exclusion analysis (). These data suggest that Mito-CP suppresses B-RafV600E melanoma cell survival partly by inducing oxidative stress.
Figure 5. The ROS scavenger, NAC, inhibits Mito-CP-induced death in B-RafV600E melanoma cells. SK-MEL28 and A375 cells were pretreated with different concentrations of NAC for 1 h prior to 10 μM Mito-CP treatment for 18 h. Cells were then subject to Western blot analysis (A) and trypan blue exclusion cell scoring (B). Data (mean ± SEM) are from a representative experiment conducted in triplicate. Control cells were treated with equal volume of PBS. *P value < 0.05.
Mito-CP effectively suppresses SK-MEL28 xenografts in mice
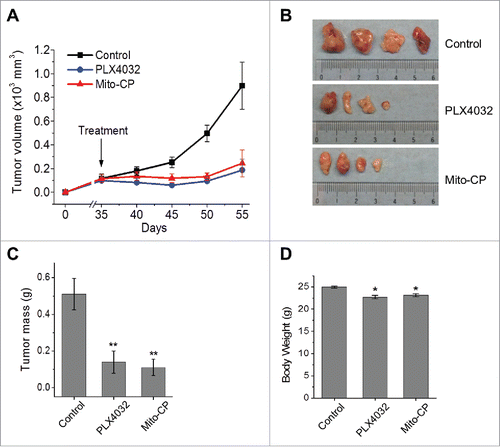
Lastly, we evaluated the potential of Mito-CP to suppress SK-MEL28 xenografts in immune-compromised nude mice. Consistent with the in vitro data above, oral administration of Mito-CP effectively suppressed the growth of SK-MEL28 xenografts in mice at a similar efficacy as PLX4032 (). When tumor weights were measured at the end of drug treatment, about 65% decreases relative to the vehicle-treated control were detected in Mito-CP- and PLX4032-treated groups (). Moreover, Mito-CP-treated group exhibited only mild decreases in body weight similarly as PLX4032-treated group (), suggesting that the animals well tolerated Mito-CP treatments similarly as PLX4032 treatments. These data strongly suggest that targeting mitochondria using TPP-linked metabolism-interfering agents has therapeutic potential for BRAF–mutated melanomas.
Figure 6. Mito-CP suppresses SK-MEL28 xenografts in athymic mice. Athymic mice bearing SK-MEL28 xenografts were treated with PLX4032 (50 mg/kg/dose) or Mito-CP (50 mg/kg/dose). Drugs dissolved in 200 μl vehicle were administered per os beginning from day 35 after tumor implantation. The control group was treated with the vehicle only (details in the Materials and Methods). (A) Changes in tumor sizes were determined at the indicated time-points. (B) Images of tumors collected at the end of the treatment. (C) Weights of tumors collected at the end of the treatment. (D) Body weights of animals at the end of the treatment. All data are mean ± SEM, n = 4, *P value < 0.05, **P value < 0.01.
Discussion
Mitochondria are the key intracellular organelle responsible for the generation of energy and building blocks. Therefore, tumor cells need to reprogram mitochondrial metabolism to meet increased demands for energy and building blocks required for uncontrolled proliferation.Citation4 Growing evidences indicate the significance of mitochondrial reprogramming for survival/proliferation of BRAF-mutated melanomas, rationalizing mitochondria as a key therapeutic target for these tumors.Citation8-10 Of note, reprogramming of mitochondrial metabolism can be stressful to cells and often activates various cell death machinery.Citation29 As such, mitochondria may be targeted to suppress BRAF-mutated melanomas.
In support of this possibility, the present study demonstrates that targeting mitochondria using a TPP-conjugated metabolism-interfering agent can effectively suppress melanoma cell survival. Mito-CP rapidly induced loss of mitochondrial activity and oxidative stress, subsequently causing tumor cell casualty. Mito-CP also induced autophagy markers although the true nature of these changes is yet unclear. We found that the autophagy inhibitors, bafilomycin A1 and chloroquine, exacerbate Mito-CP-induced cell death (data not shown), speculating that melanoma cells may trigger autophagy in an effort to survive Mito-CP-induced stress. All these data indicate the ability of Mito-CP to mediate tumor suppression by interfering with tumor cell metabolism via a mechanism distinct from those affected by PLX4032. Consistent with this notion, Mito-CP suppressed not only PLX4032-naïve but also PLX4032-resistant progenies of B-RafV600E melanoma cells. These data demonstrate the potential of TPP-conjugated metabolism-interfering agents as a novel therapeutic modality for melanomas, including PLX4032-naïve and PLX4032-resistant B-RafV600E melanomas. Further development of this potential may help overcoming the current limits in melanoma therapy.
The nitroxide moiety of Mito-CP and the ubiquinone moiety of Mito-Q are expected to have anti-oxidant properties. Indeed, these small molecules attenuated oxidative stress in certain normal cell types.Citation13,30,Citation31 However, intriguingly, these small molecules also mediated tumor cell death by causing oxidative stress, as demonstrated in this report and by previous studies.Citation13,15,Citation16 Although increased ROS generation by these compounds would ostensibly be a paradoxical effect, increased ROS generation is not really an unusual effect of a chemical with antioxidant property. For example, the anti-oxidant vitamin C can paradoxically induce strong “pro-oxidant” effects via “Fenton reaction” when cells are enriched with catalytic metal irons.Citation32 Indeed, high dose vitamin C could suppress different cancer cells by increasing ROS levels and subsequently causing oxidative damages.Citation32 Mito-CP and Mito-Q may suppress melanoma cell survival in a similar context.
Mitochondria-specific enrichment of TPP-conjugated agents is attributed to their sensitivity to Δψm. Δψm is generally more negative in tumor cells than in normal cells, which mainly accounts for relatively high sensitivity of tumors cells to TPP-conjugated drugs.Citation5,14 Indeed, Mito-CP was previously shown to inhibit proliferation of the breast cancer cell lines, MCF-7 and MDA-MB-231, but not their normal cell counterpart MCF-10A cells.Citation16 Moreover, Mito-CP was not overall too toxic to mice despite its high efficacy to SK-MEL28 xenografts in mice, which further supports its selectivity to tumor cells. Given the high dependency of BRAF-mutated tumors on mitochondrial metabolism,Citation8-10 it is conceivable that these tumors may maintain higher mitochondrial activity, rendering themselves more susceptible to TPP-conjugated metabolism-interfering agents. In conclusion, our study strongly supports the potential of mitochondria as a therapeutic target for melanoma, proposing that TPP-conjugated metabolism-interfering agents have strong potential as a novel therapeutic modality for BRAF-mutated melanomas, regardless of their resistance to B-Raf inhibitors.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental_Figures_1-3.zip
Download Zip (1.1 MB)Acknowledgment
We thank Drs. Balaraman Kalyanaraman, Joy Joseph, and Joseph Jeong (Medical College of Wisconsin) for Mito-CP and primary melanocytes, Dr. Michael Murphy (Medical Research Council Dunn Human Nutrition Unit) for the TPP antibody, and Dr. Richard Marais (Univ. Manchester) for PLX4032-resistant A375R cells.
Funding
This work was supported by National Cancer Institute (R01CA138441) and American Cancer Society (RSGM-10-189-01-TBE) to J.P.
References
- Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene 2013; 32:2373-9; PMID:22945644; http://dx.doi.org/10.1038/onc.2012.345
- Sale MJ, Cook SJ. Intrinsic and acquired resistance to MEK1/2 inhibitors in cancer. Biochem Soc Trans 2014; 42:776-83; PMID:25109957; http://dx.doi.org/10.1042/BST20140129
- Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer 2014; 14:455-67; PMID:24957944; http://dx.doi.org/10.1038/nrc3760
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012; 21:297-308; PMID:22439925; http://dx.doi.org/10.1016/j.ccr.2012.02.014
- Don AS, Hogg PJ. Mitochondria as cancer drug targets. Trends Mol Med 2004; 10:372-8; PMID:15310457; http://dx.doi.org/10.1016/j.molmed.2004.06.005
- Sarosiek KA, Ni Chonghaile T, Letai A. Mitochondria: gatekeepers of response to chemotherapy. Trends Cell Biol 2013; 23:612-9; PMID:24060597; http://dx.doi.org/10.1016/j.tcb.2013.08.003
- Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nat Rev Cancer 2014; 14:709-21; PMID:25342630; http://dx.doi.org/10.1038/nrc3803
- Haq R, Fisher DE, Widlund HR. Molecular pathways: BRAF induces bioenergetic adaptation by attenuating oxidative phosphorylation. Clin Cancer Res 2014; 20:2257-63; PMID:24610826; http://dx.doi.org/10.1158/1078-0432.CCR-13-0898
- Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, et al. PGC1alpha Expression Defines a Subset of Human Melanoma Tumors with Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell 2013; 23:287-301; PMID:23416000; http://dx.doi.org/10.1016/j.ccr.2012.11.020
- Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 2013; 23:302-15; PMID:23477830; http://dx.doi.org/10.1016/j.ccr.2013.02.003
- Millard M, Pathania D, Shabaik Y, Taheri L, Deng J, Neamati N. Preclinical evaluation of novel triphenylphosphonium salts with broad-spectrum activity. PLoS One 2010; 5:e13131; PMID:20957228; http://dx.doi.org/10.1371/journal.pone.0013131
- Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol 2007; 47:629-56; PMID:17014364; http://dx.doi.org/10.1146/annurev.pharmtox.47.120505.105110
- Smith RA, Adlam VJ, Blaikie FH, Manas AR, Porteous CM, James AM, Ross MF, Logan A, Cochemé HM, Trnka J, et al. Mitochondria-targeted antioxidants in the treatment of disease. Ann N Y Acad Sci 2008; 1147:105-11; PMID:19076435; http://dx.doi.org/10.1196/annals.1427.003
- Modica-Napolitano JS, Aprille JR. Delocalized lipophilic cations selectively target the mitochondria of carcinoma cells. Adv Drug Deliv Rev 2001; 49:63-70; PMID:11377803; http://dx.doi.org/10.1016/S0169-409X(01)00125-9
- Starenki D, Park JI. Mitochondria-targeted nitroxide, Mito-CP, suppresses medullary thyroid carcinoma cell survival in vitro and in vivo. J Clin Endocrinol Metab 2013; 98:1529-40; PMID:23509102; http://dx.doi.org/10.1210/jc.2012-3671
- Cheng G, Zielonka J, Dranka BP, McAllister D, Mackinnon AC, Jr., Joseph J, Kalyanaraman B. Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res 2012; 72:2634-44; PMID:22431711; http://dx.doi.org/10.1158/0008-5472.CAN-11-3928
- Girotti MR, Pedersen M, Sanchez-Laorden B, Viros A, Turajlic S, Niculescu-Duvaz D, Zambon A, Sinclair J, Hayes A, Gore M, et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov 2013; 3:158-67; PMID:23242808; http://dx.doi.org/10.1158/2159-8290.CD-12-0386
- Dhanasekaran A, Kotamraju S, Karunakaran C, Kalivendi SV, Thomas S, Joseph J, Kalyanaraman B. Mitochondria superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: role of mitochondrial superoxide. Free Radic Biol Med 2005; 39:567-83; PMID:16085176; http://dx.doi.org/10.1016/j.freeradbiomed.2005.04.016
- Wu PK, Hong SK, Veeranki S, Karkhanis M, Starenki D, Plaza JA, Park JI. A Mortalin/HSPA9-Mediated Switch in Tumor-Suppressive Signaling of Raf/MEK/Extracellular Signal-Regulated Kinase. Mol Cell Biol 2013; 33:4051-67; PMID:23959801; http://dx.doi.org/10.1128/MCB.00021-13
- Starenki D, Hong SK, Lloyd RV, Park JI. Mortalin (GRP75/HSPA9) upregulation promotes survival and proliferation of medullary thyroid carcinoma cells. Oncogene 2015; 34:4624-34; PMID:25435367; http://dx.doi.org/10.1038/onc.2014.392
- Starenki D, Park JI. Selective Mitochondrial Uptake of MKT-077 Can Suppress Medullary Thyroid Carcinoma Cell Survival In Vitro And In Vivo. Endocrinol Metab (Seoul) 2015; 30:593-603; PMID:26485469
- Hong SK, Yoon S, Moelling C, Arthan D, Park JI. Noncatalytic function of ERK1/2 can promote Raf/MEK/ERK-mediated growth arrest signaling. J Biol Chem 2009; 284:33006-18; PMID:19805545; http://dx.doi.org/10.1074/jbc.M109.012591
- Rosen A, Casciola-Rosen L. Macromolecular substrates for the ICE-like proteases during apoptosis. J Cell Biochem 1997; 64:50-4; PMID:9015754; http://dx.doi.org/10.1002/(SICI)1097-4644(199701)64:1%3c50::AID-JCB8%3e3.0.CO;2-Z
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Lin TK, Hughes G, Muratovska A, Blaikie FH, Brookes PS, Darley-Usmar V, Smith RA, Murphy MP. Specific modification of mitochondrial protein thiols in response to oxidative stress: a proteomics approach. J Biol Chem 2002; 277:17048-56; PMID:11861642; http://dx.doi.org/10.1074/jbc.M110797200
- Green DR, Reed JC. Mitochondria and apoptosis. Science 1998; 281:1309-12; PMID:9721092; http://dx.doi.org/10.1126/science.281.5381.1309
- Halliwell B. Oxidative stress and cancer: have we moved forward? Biochem J 2007; 401:1-11; PMID:17150040; http://dx.doi.org/10.1042/BJ20061131
- Kalyanaraman B. Oxidative chemistry of fluorescent dyes: implications in the detection of reactive oxygen and nitrogen species. Biochem Soc Trans 2011; 39:1221-5; PMID:21936793; http://dx.doi.org/10.1042/BST0391221
- Galluzzi L, Morselli E, Kepp O, Vitale I, Rigoni A, Vacchelli E, Michaud M, Zischka H, Castedo M, Kroemer G. Mitochondrial gateways to cancer. Mol Aspects Med 2010; 31:1-20; PMID:19698742; http://dx.doi.org/10.1016/j.mam.2009.08.002
- Krishna MC, Russo A, Mitchell JB, Goldstein S, Dafni H, Samuni A. Do nitroxide antioxidants act as scavengers of O2-. or as SOD mimics? J Biol Chem 1996; 271:26026-31; PMID:8824242; http://dx.doi.org/10.1074/jbc.271.42.26026
- Dhanasekaran A, Kotamraju S, Kalivendi SV, Matsunaga T, Shang T, Keszler A, Joseph J, Kalyanaraman B. Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. J Biol Chem 2004; 279:37575-87; PMID:15220329; http://dx.doi.org/10.1074/jbc.M404003200
- Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta 2012; 1826:443-57; PMID:22728050; http://dx.doi.org/10.1016/j.bbcan.2012.06.003