
ABSTRACT
Epithelial ovarian cancer is prone to metastasizing at an early stage, but their mechanisms remain unclear. CRM1 is an important nuclear exportin and inhibitors targeting CRM1 has been explored as an anti-cancer strategy. In previous study, we observed that PEITC could combine with the hydrophobic pocket of CRM1. In this study, we focused on the effects of PEITC on EOC and its mechanisms. Results showed that IC50 values of PEITC on SKOV3 and HO8910 cell line were 42.14 μM and 37.29 μM, respectively. PEITC inhibits the migration and invasion of SKOV3 and HO8910 cells in vitro. Oral administration of 10 μmol PEITC suppressed the metastasis of EOC in a xenograft mouse model in vivo. PEITC treatment decreased the expressions of CRM1 and mTOR (cargo protein of CRM1) in EOC cell lines and in xenograft mouse tissues. Moreover, CRM1-mediated nuclear export was attenuated by PEITC, mTOR accumulated in nucleus, expressions of mTORS2448 and downstream effectors STAT3S727, MMP2 and MMP9 were decreased in a dose- and time-dependent manner. Furthermore, immunohistochemical analysis showed that CRM1 and mTOR were increased in EOC tissues compared with benign ovarian tumors, and related with advanced stage, type II EOC, positive peritoneal cytology and decreased overall survival. In addition, CRM1 was positively correlated with mTOR levels. In conclusion, our data demonstrated that PEITC suppresses the metastasis of EOC through inhibiting CRM1-mediated nuclear export, subsequently suppressing the mTOR-STAT3 pathway. Both CRM1 and mTOR were increased in EOC patients, providing a rationale for further clinical investigation of PEITC in EOC treatment.
Abbreviations
| CRM1 | = | chromosomal region maintenance 1 |
| DMSO | = | dimethyl sulfoxide |
| EOC | = | epithelial ovarian cancer |
| IHC | = | immunohistochemistry |
| MMP2/9 | = | matrix metalloproteinase 2/9 |
| mTOR | = | mammalian target of rapamycin |
| OS | = | overall survival |
| PEITC | = | phenethyl isothiocyanate |
| STAT3 | = | signal transducers and activators of transcription factors 3 |
Introduction
Ovarian cancer is the most lethal cause of gynecological cancer in the developing world and typically presents at an advanced stage; it ranks fifth as a cause of female cancer deaths. It is estimated that there will be 22,280 new cases and 14,240 deaths due to ovarian cancer in the United States in 2016.Citation1 Optimal cytoreductive surgery and platinum based-chemotherapy remain the cornerstones of therapeutic strategies. Metastasis is the most common cause of death in ovarian cancer patients, while the mechanisms are still not well elucidated. The possible mechanisms include metastasis suppressor genes (Nm23, KISS1, KISS1R, KAI1, E-cadherin, OGR1, BRMS1), epithelial-mesenchymal transition (EMT), tumor microenvironment, chemokines, and microRNA.Citation2-7 Up-regulated nuclear export and mislocalization of tumor suppressors are also relate to ovarian cancer metastasis.Citation8,9 Thus far there are no effective therapies targeting tumor metastasis, giving rationale to develop novel drugs targeting the metastasis-related signal pathways.
CRM1 is the main nuclear export receptor in humans and transports large nuclear macromolecules (cargoes) including mRNAs and proteins, from nucleus to cytoplasm. Included in the cargoes are multiple tumor suppressors and oncoproteins, such as p53, BRCA1, survivin, FOXO, and APC, which function in the nucleus to regulate transcription or control cell division. Excessive activation of CRM1 results in excessive nuclear export of these proteins and sequential inactivation of their tumor suppression function.Citation10-13 Mammalian target of rapamycin (mTOR), a cadre cargo of CRM1,Citation14 serves as an essential nutrient and energy sensor, regulating protein synthesis, as well as cell growth and survival. In human malignant tumors, overexpression of CRM1 and dysregulated subcellular distribution of cargoes are related to increased metastasis and poor survival.Citation15,16 Thus, the concept of inhibiting CRM1 has been explored as an anti-metastatic therapeutic strategy. To date, most CRM1 inhibitors function by modifying the reactive site cysteine 528 of CRM1, thereby blocking CRM1-mediated nuclear export.
Plant products, including constituents of fruits and vegetables, continue to attract attention for the discovery of novel chemotherapeutic agents. This is largely due to their low cost, easy availability, and minimal or no toxicity. Accumulating epidemiological evidence indicates an inverse correlation between the intake of cruciferous vegetables and occurrence of malignant tumors. PEITC is an active ingredient in cruciferous vegetables and has shown chemoprevention and chemotherapeutic ability against several human cancers. The underlying mechanism may involve causing cell cycle arrest, induction of apoptosis, and generation of reactive oxygen species (ROS).Citation17, 18 As a natural compound, PEITC targets multiple pathways, for instance, it regulates the activation of NFκB and Nrf2; inhibits Akt, EGFR and HER2; and inhibits mitogen-activated protein kinases (MAPK) and protein kinase C (PKC).Citation19, 20 Unfortunately, its detailed biological mechanisms remain to be elucidated. A few studies have also suggested anti-metastatic effects of PEITC on some human cancers, such as breast cancer, and gastric cancer.Citation21, 22 However, the anti-metastatic effect of PEITC on epithelial ovarian cancer (EOC) has not been studied yet. In a previous study, our group revealed that PEITC could combine with the hydrophobic pocket of CRM1 by using computational modeling (Fig. S1). In this study, we focused on the anti-metastasis effects of PEITC on EOC in vitro and in vivo. We showed that the mechanisms of anti-metastasis effects of PEITC on EOC occur through inhibiting CRM1-mediated nuclear export and sequential blocking of the mTOR-STAT3 signal pathway. In addition, we identified that CRM1 and mTOR may be interesting prognostic biomarkers for EOC, thereby representing potential therapeutic targets in this disease.
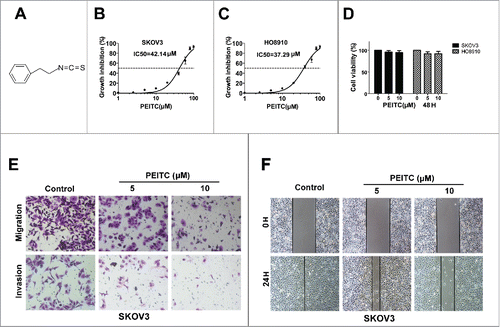
Figure 1. PEITC inhibits migration and invasion of ovarian cancer cells in vitro.
Results
1. PEITC inhibits migration and invasion of ovarian cancer in vitro and in vivo
PEITC is an isothiocyanate characterized by sulfur containing N = C = S functional group. shows the chemical structure of PEITC. To evaluate the effect of PEITC on EOC cells lines, SKOV3 and HO8910 were exposed to PEITC. We found that PEITC inhibited the viability of the two cell lines in a dose-dependent manner, The IC50/24 h values yielded were 42.14 μM for SKOV3 and 37.29 μM for HO8910 (). We selected PEITC concentrations of 5, 10 μM for further study, these were far less than the IC50 values and did not cause cell death. At the same time, we detected that 5 μM and 10 μM PEITC did not affect the cell viability of EOC cells ().
We then sought to investigate whether PEITC could inhibit metastasis of EOC. Boyden chamber assays with Matrigel® matrix demonstrated that the invasions of SKOV3 cells were reduced to 68.50% and 37.66% of control following 24 h treatment with 5 μM and 10 μM PEITC, respectively, P < 0.05. Boyden chamber assay without Matrigel® matrix further revealed that the migration rates of SKOV3 cells were reduced to 72.93% and 46.62% of the control group by 5 μM and 10 μM PEITC, respectively, P < 0.01 (). In addition, the wound healing assay confirmed the anti-migration effects of PEITC on SKOV3 cells (). Similarly, PEITC significantly decreased the migration and invasion properties of HO8910 cells (Fig. S2). These results indicated that PEITC inhibited the metastasis potential of EOC cells in a dose-dependent manner.
Figure 2. PEITC inhibits metastasis of ovarian cancer cells in vivo.
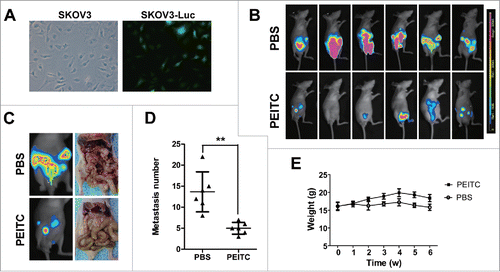
The in vivo anti-metastatic activity of PEITC in EOC was further examined using an intraperitoneal xenograft model, established by i.p. injection of SKOV3-luc cells () which imitated the peritoneal dissemination of human EOC disease. Mice were treated with 10 μmol PEITC or PBS by oral gavage 5 times per week for 6 weeks. Representative bioluminescence images are shown in . Luminescence in the peritoneal cavity was markably reduced in the PEITC-treated cohort compare with the control group. Mice in the PEITC group exhibited fewer metastases to organs in the peritoneal cavity compare with the control group, as the number of metastatic tumors in the PEITC group was 5.0 ± 1.4 and 13.6 ± 4.8 in the control group (P = 0.002, ). In addition, the PEITC-treated cohort showed fewer distant metastases, whereas the control group showed metastatic deposits in the mesentery, small intestine, stomach, liver, spleen and diaphragm. Mice body weight in the PEITC group was higher than the control group; a dramatic difference started to emerge from the second week after the beginning of PEITC treatment and persisted to the end of the assay (p < 0.05, ).
2. PEITC decreases the expressions of CRM1 and mTOR, inhibits CRM1-dependent nuclear export, associated with nuclear accumulation of mTOR in EOC
Since we observed that PEITC could combine with hydrophobic pocket of CRM1, we hypothesized that the anti-metastatic effects of PEITC on EOC cells may through attenuating CRM1-mediated nuclear export. To test our hypothesis, we examined the expression level and nuclear export function of CRM1 in SKOV3 and HO8910 cells after exposure to PEITC. The results revealed that both the transcription and translation levels of CRM1 were drastically decreased by PEITC in a dose- and time-dependent manner (). At the same time, the expression of mTOR, one cargo protein of CRM1, was also reduced by PEITC in a dose- and time-dependent manner (). We found that PEITC markedly inhibited mTOR phosphorylation at Ser2448, which then prevented activation of the mTORC1 signalling. The suppression of phosphorylated mTOR at Ser2481 was not observed.
Figure 3. PEITC decreases the expressions of CRM1 and mTOR in EOC cell lines and in xenograft tumor tissues.
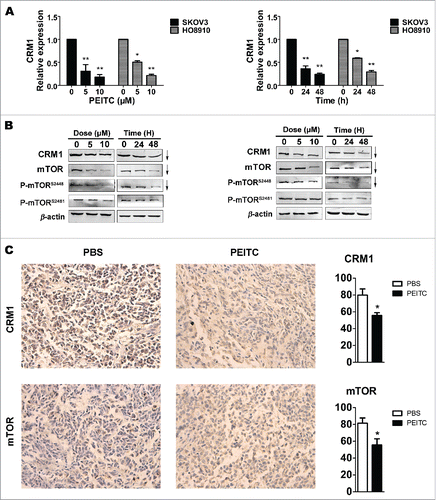
In agreement with these results, IHC staining showed that CRM1 and mTOR were also down-regulated in tumors excised from PEITC treated mice, and the proportions of positive cells for CRM1 and mTOR in PETIC-treated xenografts tumors were reduced to 75.83% and 82.96% of control, respectively (P < 0.05, P < 0.05, respectively ). These results indicated that PEITC decreased the expressions of CRM1 and mTOR in EOC in vitro and in vivo.
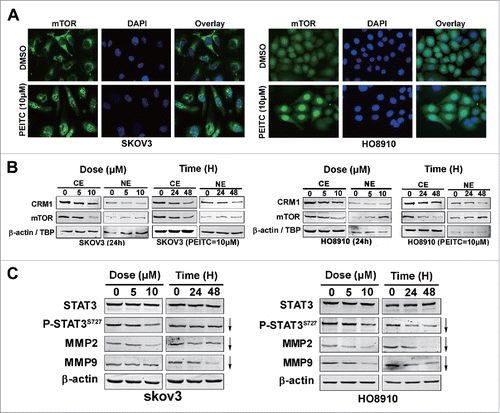
We further evaluated the effects of PEITC on the nuclear export ability of CRM1. First, immunofluorescence staining shown prominent nuclear accumulation of mTOR in SKOV3 cells after PETIC treatment (). Immunoblotting of nuclear versus cytoplasmic extracts of PEITC treated EOC cells further confirmed nuclear accumulation of mTOR in SKOV3 cells. However, both nuclear and cytoplasmic levels of CRM1 were down-regulated by PEITC. Similar results were obtained in HO8910 cells (). These results implied that PEITC inhibited the nuclear export functions of CRM1, and the cargo protein mTOR was accumulated in nucleus in a time- and dose-dependent manner.
Figure 4. PEITC inhibits CRM1-mediated nuclear export and suppresses the mTOR-STAT3 pathway in EOC cell lines.
3. PEITC inhibits the mTOR-STAT3 pathway in EOC
It is noteworthy that S6K1, 4E-BP1 and STAT3 (signal transducers and activators of transcription 3) are downstream effectors of mTOR.Citation23, 24 The transcriptional activity of STAT3 is suggested to be activated by its phosphorylation at Tyr-705 and maximized by phosphorylation at Ser-727. The second process can be mediated by mTOR.Citation25 Taking into account that the nucleocytoplasmic shuttling of mTOR is critical for its downstream signal S6K1,Citation14 we speculated that the activation of STAT3 might also be inhibited, since mTOR was blocked in nuclear in EOC cells by PEITC. As expected, PEITC decreased mTOR-induced phosphorylation of P-STAT3S727 in a dose- and time-dependent manner in SKOV3 and HO8910 cell lines, while the total STAT3 level remained unchanged (). Downstream targets of STAT3 that are crucial for cancer metastasis, including MMP2 and MMP9,Citation26, 27 were also down-regulated by PEITC. Collectively, these results reveal that PEITC inhibits the mTOR-STAT3 pathway in EOC cells.
4. CRM1 and mTOR are overexpressed in EOC tissues and associated with tumor metastasis and poor survival
The above laboratory results showed that PEITC suppresses metastasis of EOC in cultured cells and in animal models. The underlying mechanism may through inhibiting CRM1-mediated nuclear export, which subsequently suppresses the mTOR-STAT3 pathway. Previous studies showed that CRM1 expression was elevated in ovarian cancers; thus, we wonder whether there is a possibility that PEITC could be used in the clinical treatment of EOC patients. To explore the expression and significance of CRM1 and mTOR in ovarian cancer, IHC analysis was performed on 40 epithelial ovarian carcinomas and 10 benign ovarian tumors. As shown in , CRM1 and mTOR expressions were both increased in EOC tissues compared with benign ovarian tumors, particularly in serous cancer. The percentage of CRM1-positive cells in EOC was 73.24 ± 11.58%, while it was 41.10 ± 7.48% in benign ovarian tumors (P < 0.001). The mTOR immunostaining was also more intense in malignant tumors compared with benign tissues (70.46 ± 12.33% vs. 32.37 ± 8.70%, P < 0.001, ).
Figure 5. CRM1 and mTOR are overexpressed in ovarian cancer patients, associated with tumor metastasis and poor overall survival.
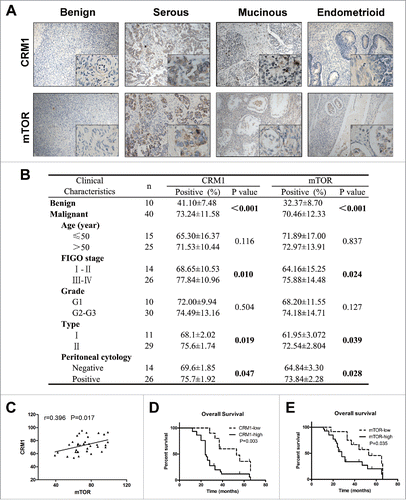
In univariate analysis, the association of CRM1 and mTOR expression with a number of clinical pathological parameters were investigated (). The up-regulated expressions of CRM1 and mTOR were significantly related to advanced tumor stage (P = 0.010, and P = 0.024, respectively), indicating that CRM1 and mTOR levels were positively correlated with the FIGO stage. Furthermore, increased expression of CRM1 and mTOR were significantly associated with type II of EOC (P = 0.019 and P = 0.039, respectively) and positive peritoneal cytology (P = 0.047, and P = 0.028, respectively). Collectively, these results indicated an association between over expression of CRM1 and mTOR and distant metastasis of EOC. No significant difference was detected between age groups or histological grade (G1 vs. G2–3) for CRM1 or mTOR levels.
Both CRM1 and mTOR were overexpressioned in EOC tissues; The Pearson correlation test identified that CRM1 was positively correlated with mTOR, and the correlation coefficient was moderate (r = 0.396, P = 0.017, ).
Of the 40 EOC samples, we set a cutoff value of 70% positively stained cells of CRM1 and mTOR (mean value). Patients were divided into 2 groups, as follows: those with less than or equal to 70% positive staining comprised low expression group, while those with greater than 70% positive staining made up the high expression group. There were 30 (75%) cases that exhibited high expression of CRM1, while 10 (25%) showed low expression; meanwhile, 28(70%) cases showed high expression of mTOR, and 12(30%) exhibited low expression. In univariate Kaplan–Meier analysis, CRM1 and mTOR expressions were significantly associated with poor overall survival (OS; P = 0.003 and P = 0.035, respectively, ) of EOC patients. In an exploratory multivariate analysis using Cox regression model that included patients' age, FIGO stage, histological grade, type of EOC, and CRM1 and mTOR expression, the significance of high mTOR expression was demonstrated (P = 0.048, RR 2.60, 95% confidence interval [CI](1.73–3.47)). Moreover, FIGO stage (P = 0.031, RR 2.17, 95% CI(1.54–2.52)) and type of EOC were also found to be independent prognostic factors (P = 0.018, RR 2.76, 95% CI(1.78–3.97)) for poor OS of EOC patients.
Discussion
EOC is a leading death cause of gynecological cancer, characterized by early stage metastasis and poor prognosis in spite of advances in treatment with surgery and chemotherapy. The main cause of death in EOC is metastasis which primarily occurs through intraperitoneal dissemination. Thus far there are no effective therapies targeting EOC metastasis, identification of new drugs against metastasis is highly desirable.
Nature has been a rich source of drugs, such as curcumin and artemisinin.Citation28, 29 Many anticancer agents were originated from plant sources, including taxanes and vinca alkaloids which are widely used. Interest in natural compounds is largely due to their minimal or no toxicity and multiple-pathway targeting. PEITC is an active natural ingredient of cruciferous vegetables. Since Sldransky et al.Citation30 reported that PEITC regulated tumor cell growth for the first time, the antitumor properties of PEITC have began to receive broad attention. However, only a few researchers have concentrated on the anti-tumor effects of PEITC on EOC thus far. Previous studies have elucidated possible molecular mechanisms, including apoptosis induction and proliferation inhibition.Citation31, 32
In the current study, we verified that PEITC inhibited the migration and invasion of EOC in vitro and in vivo. We also explored the possible mechanisms of PEITC in EOC cell lines. Our data showed that PEITC reduced CRM1 and mTOR expressions in EOC cell lines and in xenografts in nude mice. CRM1-mediated nuclear export was also inhibited, subsequently causing nuclear retention of cargo protein mTOR; and inhibiting the phosphorylation of downstream effector STAT3S727; moreover expressions of MMP2 and MMP9 were both decreased in a dose- and time-dependent manner by PEITC treatment.
Given our previous data that PEITC could bind with the hydrophobic pocket of CRM1, we suspected that the anti-metastatic effects of PEITC in EOC might through acting as a CRM1 inhibitor. PEITC has an electrophilic group “ = C = ”, it may theoretically carry out an electrophilic addition with the nucleophilic group of Cys-529 of CRM1, just like the specific CRM1 inhibitor leptomycin B,Citation33 and prevent CRM1 from binding to the nuclear export sequence of cargo proteins.
Overexpression of CRM1 protein has been reported in cancer tissues and considered as a negative prognostic indicator in several human carcinomas, such as osteosarcoma, pancreatic cancer, and ovarian cancer.Citation9, 15 mTOR is a cadre cargo of CRM1; the nucleocytoplasmic shuttling of mTOR were CRM1-dependent.Citation34 As a serine/threonine protein kinase, mTOR mediates nutrient-dependent intracellular signaling to ultimately govern a wide range of cellular processes, including cell growth, protein synthesis and differentiation. Over-activation of mTOR represents the most frequently altered signal pathway in cancers.Citation35 mTOR is found in two distinct signaling complexes: mTORC1 and mTORC2. It was reported that mTOR associated with mTORC1 or mTORC2 is phosphorylated on different sitesCitation36. The rapamycin-sensitive mTORC1 complex contains phospho-S2448, while the rapamycin-insensitive mTORC2 complex contains phospho-S2481. Our data revealed that mTORS2448 was decreased after PEITC treatment, while mTORS2481 has no change, which demonstrated that mTORC1 was the specific activity of mTOR in EOC lines treated by PEITC.
It has been reported that mTOR can phosphorylate STAT3S727 and cooperate with phosphorylated at Y705 for maximal transcriptional activation of STAT3.Citation25 STAT3 is a transcriptional factor that plays a critical role in tumorigenesis. Its constitutive activation has been frequently observed in various cancers.Citation37 The phosphorylated STAT3 forms homodimers and transfers into nucleus, thereby regulating the transcription of MMP2 and MMP9.Citation26, 27 MMP2 and MMP9 belong to a family of zinc-binding endopeptidases that collectively degrade the component of extracellular matrix (ECM) and the basement membrane, and play an essential role in the metastasis of malignant tumors. Our data revealed that nuclear retention of mTOR by PEITC disrupted the interaction between mTOR and STAT3; PEITC decreases phosphorylation of STAT3 at Ser-727 and down-regulation of its downstream gene MMP2 and MMP9. Taken together, these data suggested that PETIC blocked the metastasis of SKOV3 and HO8910 cells by inhibiting the nuclear export of CRM1 and suppressing the mTOR-STAT3 signaling pathway.
Numerous studies have demonstrated the anti-cancer effects of PEITC in various cancer types. The uptake of anti-cancer agents is an important factor for efficacy. Pre-clinical evidence suggests that PEITC is highly bioavailable after oral administration, which is a probable reason for in vivo efficacy. A single dose of 10–100 μmol/kg PEITC in rats resulted in bioavailability ranging between 90–114%.Citation38 Furthermore, about 928.5 ± 250nM peak plasma concentration of PEITC was achieved in human subjects, after the consumption of 100 g watercress.Citation39 This indicates that oral administration of PEITC can result in effective concentrations in blood plasma. A Phase I study (NCICN-55120) has confirmed that after oral intake of 200mg PEITC by human volunteers, 10μM PEITC can be achieved in plasma, as mentioned by Mi et al.Citation40 A phase II study with PEITC in lung cancer patients was completed, but results were not published yet. The results of these clinical studies will provide a clear idea of the efficacy and toxicity of PEITC in humans.
At the same time, in this study, we demonstrated that CRM1 and mTOR were increased in cancer tissues, and associated with advanced stages, type II of EOC, and positive peritoneal cytology; all of these clinical features represent distant metastasis and aggressive behavior. In addition, increased CRM1 and mTOR expressions were significantly related to poor OS in our study cohort. These results are in agreement with previous findings, including Noske's report on CRM1 expression in EOC patients.Citation41 It is noteworthy that there is a positive correlation of increased CRM1 and mTOR expression in EOC tissues. We suggest that tissue expressions of CRM1 and mTOR may be interesting prognostic predictors and therapeutic targets in EOC patients. Perhaps they will more valuable when used together because of their inverse correlation. Both CRM1 and mTOR expressions were evaluated in EOC patients, while PEITC decreased the expressions of CRM1 and mTOR in EOC cell lines and tumor tissues harvested from xenograft mice, providing the rationale that PEITC maybe a therapeutic agent against EOC in the future.
Conclusion
In summary, we demonstrated that PEITC suppresses the metastasis of EOC in vitro and in vivo. We suggest that PEITC suppresses the metastasis of EOC may through inhibiting CRM1-mediated nuclear export, subsequently suppressing the mTOR-STAT3 pathway. Both CRM1 and mTOR were increased in EOC patients and related with tumor metastasis and poor survival, providing new insights into the therapeutic targets for EOC and the rationale for further clinical investigation of PEITC as a novel therapeutic agent in EOC.
Materials and methods
Cell lines and human tissue samples
Human epithelial ovarian cancer cell lines (SKOV3 and HO8910) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). They were maintained in RPMI-1640 (Gibco, 11875119) supplemented with 10% (w/v) fetal bovine serum (FBS; Gibco, 16000044) and 1% penicillin-streptomycin (pen-strep, Gibco, 15140163). All cell lines were maintained at 37°C and 5% CO2 in a humidified incubator.
A total of 50 tissue specimens, including 40 primary epithelial ovarian cancer (PEOC) tissues and 10 benign ovarian epithelial tumor tissues, were obtained from patients who underwent surgery resection between 2010–1–1 and 2011–12–31 at the Department of Obstetrics and Gynecology, Ren-Ji Hospital, Shanghai Jiao-Tong University School of Medicine, Shanghai, China. All samples were collected at surgery and prior to chemotherapy. The slides were stained with hematoxylin and eosin (H&E) and examined by a pathologist to ensure >90% presence of tumor cells. Among the 40 PEOC patients, 11 cases were type I EOC and 29 cases were type II; 14 tumors were determined to be early stage I–II and 26 tumors were advanced stage III–IV. Enrolled PEOC patients received 6 cycles of platinum-based postoperative chemotherapy after optimal cytoreductive surgery. Patients were followed up until 2015–6–30, and the observation time spanned 42–66 months. Written informed consent was obtained from each patient, and the use of clinical specimens was approved by the Institutional Ethics Committee.
Cell viability assay
PEITC was purchased from Tokyo Chemical Industry (TCI, P0986). Its stock solution was prepared in dimethyl sulfoxide (DMSO) and diluted with complete medium when used; an equal volume of DMSO (final concentration, 0.05%) was set as control. The viability assay was measured using Cell Counting Kit-8 (Dojindo, ck04) according to the manufacturer's instructions. Briefly, About 1 × 105 of logarithmic growth phase cells in 100μl of medium were seeded into 96-well plates and incubated overnight. Then cells were treated with control solvent (0.05% DMSO) or PEITC at concentrations of 1–100 μM for 24 h. Cells were then incubated in CCK8 reagent for 1 h, the absorbance at 450 nm was read using a Thermo microplate reader (Thermo Fisher Scientific, Waltham, MA, USA) and the half maximal inhibitory concentration (IC50) were calculated by GraphPad Prism 5 software. Results were representative of more than 3 individual experiments.
Would healing assay
About 2 × 105 SKOV3 or HO8910 cells were seeded in 6-well plates followed by incubation over night to 100% confluence. A clean wound area was then made by scratching with a micropipette tip. The cultures were washed twice with phosphate-buffered saline (PBS) to remove cell debris and then incubated in serum-free RPMI-1640 with DMSO or different concentrations of PEITC for 24 h. Migration was monitored by capturing pictures through a contrast microscope (Nikon TE300, Tokyo, Japan), using 4 × or 10 × objective lenses.
Migration and invasion assay
Cell migration and invasion assays were performed using 8 μm pore Boyden chambers (BD Biosciences, 353097) coated with (invasion assay) or without (migration assay) Matrigel. Briefly, 1 × 104 cell suspensions in serum-free RPMI-1640 mixed with DMSO (control) or PEITC (5μM, 10μM) were seeded into the inserts. The basement of the chamber comprised 600 μL of complete medium containing the same concentrations of DMSO or PEITC. After incubation at 37°C for 24 h, the migrant cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. Five random fields at 200 × magnification were counted using a light microscope (Nikon). Each experiment was conducted in triplicate.
Quantitative real-time RT-PCR
Total RNA was extracted using TRIzol reagent (Applied Biosystems, 15596018). Reverse Transcription Kit (Applied Biosystems, 18080044) was used to synthesize complementary DNA. Quantitative reverse transcription–polymerase chain reaction (RT-PCR) analyses were performed in triplicate with the SYBR® Green PCR Master Mix (Invitrogen, 11794200) on an ABI 7500 real-time system (Applied Biosystems, Foster ,CA, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control. The primers for CRM1 were 5′-AGCAAAGAATGGCTCAAGAAGT-3′ (forward) and 5′-TATTCCTTCGCACTGGTTCCT-3′ (reverse). The data analysis was performed using the 2−ΔΔCT method, where ΔΔCT = (CTTarget1 - CTGAPDH) - (CTTarget2 - CTGAPDH).
Western blotting
SKOV3 and HO8910 cells were exposed to 0.05% DMSO or 5 μM or 10 μM of PEITC for 24 or 48 h. Cells were collected and lysed in RIPA lysis buffer. Nuclear extracts were prepared with NE-PER (Thermo Fisher Scientific, 78833). Than 20–50 μg of proteins were separated by 8% sodium dodecyl sulfate (SDS) gel electrophoresis followed by transference onto polyvinylidene difluoride membranes (Millipore, CCGL01TP1). Immunoblots were performed with primary antibodies at 4°C over night (antibodies: CRM1 (Santa cruz, sc-5595), mTOR (Cell Signaling Technology [CST], #2983), p-mTORS2448 (CST, #2971), p-mTORS2481 (CST, #2974), MMP2 (CST, #13132), MMP9 (CST, #13667), STAT3 (CST, #12640), p-STAT3S727 (CST, #9134); β-actin (Sigma Aldrich, A1978), TBP (Sigma Aldrich, AV100971), LC3-B (CST, #3868), Beclin 1 (CST, #3495)), then incubated with DyLightTM 800 conjugated goat anti-rabbit or anti-mouse secondary antibody for 1 h. The signals were detected by using the LI-COR Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA) following the manufacturer's instructions. Each experiment was conducted in triplicate.
Immunofluorescence analysis
SKOV3 and HO8910 cells were treated with DMSO and 10μM of PEITC for 24h, fixed with 4% paraformaldehyde, permeabilized with 0.05% triton and stained with the indicated primary antibodies overnight at 4°C.This was followed by incubation with fluorescent secondary antibodies (Invitrogen, A-31573). Nuclei were counterstained with DAPI (Sigma Aldrich, D9542). Cells were examined under a fluorescence microscope (Leica, Wetzlar, Hesse, Germany).
Animal studies
Sixteen female BALB/c nude mice, aged 5–6 weeks, were kept under pathogen-free conditions according to the Shanghai Medical Experimental Animal Care guidelines. Animal protocols were approved by the Institutional Animal Care and Use Committee of Shanghai Jiao-Tong University School of Medicine. Mice were acclimated for 1 week before the start of the experiment. SKOV3 cells were stably transfected with the Luc gene (SKOV3-luc). About 5 × 106 SKOV3-luc cells in 200 μL of PBS were engrafted via intraperitoneal (i.p.) injection. One week later, all mice were anesthetized and given an i.p. injection of 3 mg/200 μL D-luciferin (GoldBio Technology, LUCNA-100). Bioluminescence images were captured with an IVIS Spectrum (Xenogen, Shrewsbury, MA, USA) as described by us previously.Citation7 Mice with visible tumors were segregated randomly into 2 groups with 8 mice per group, treated respectively with vehicle control (200 μL of PBS) or 10 μmol PEITC dissolved in 200 μL of PBS by oral gavage, 5 times per week, for 6 weeks. Bioluminescence images were captured every week. Body weight was monitored weekly as an indicator of drug-induced toxicity and the overall health of the mice. Six weeks later, the mice were killed and the number of visible tumors in the cavity was counted. Tumors harvested from the mice were processed for immunostaining for CRM1 and mTOR.
Immunohistochemical staining
Immunohistochemical (IHC) staining was performed on 5 μm sections of paraffin-embedded human EOC tissues and benign ovarian tumor tissues to determine the expression of CRM1 and mTOR. Briefly, the slides were incubated in CRM1 or mTOR antibodies (1:100) overnight, then incubated with biotinylated anti-rabbit antibody for 1 h followed by treatment with 3,3′-diaminobenzidine (Sigma-Aldrich, D8001) working solution and counterstaining with hematoxylin. The images were captured under a Leica microscope. At least five 20 HPF fields of non-necrotic images were captured from each section and analyzed with the use of Image Pro Plus 5.0 software. The proportion of positive cells was used to evaluate the staining. Scoring was based on the positively stained cells compared with the whole area covered by tumor cells. The final score was the average of 5 non-overlapping fields. The mean value of CRM1 or mTOR positive cells was set as the cutoff value, less than or equal to 70% for low expression and more than 70% for high expression.
Statistical analysis
The data were presented as mean ± standard deviation (SD). The differences between two groups were analyzed using the double-sided Student's t-test. Variance analysis among groups was performed by one-way analysis of variance (ANOVA) and then Dunnett's multiple comparison test. Pearson correlation was used to evaluate an association between the expression of CRM1 and mTOR. Univariate survival analysis was performed using the Kaplan-Meier method, and survival curves were compared with the log rank test. Cox proportional hazard models were fitted to carry out multivariate survival analyses. The differences with P<0.05 were considered significant. Graph Pad Prism 5.0 software was used for statistical analyses.
Disclosure of potential conflicts of interest
The author reports no conflicts of interest in this work.
Supplementary_Figure1-2.docx
Download MS Word (685.5 KB)Funding
This work was supported by National Natural Science Foundation of China (81172479 and 81672564) awarded to Zhang Shu.
Reference
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CACancer J Clin 2016; 66:7-30; PMID:26742998; http://dx.doi.org/10.3322/caac.21349
- Youn BS, Kim DS, Kim JW, Kim YT, Kang S, Cho NH. NM23 as a prognostic biomarker in ovarian serous carcinoma. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc 2008; 21:885-92; PMID:18408655; http://dx.doi.org/10.1038/modpathol.2008.64
- Ji K, Ye L, Mason MD, Jiang WG. The Kiss-1/Kiss-1R complex as a negative regulator of cell motility and cancer metastasis (Review). International journal of molecular medicine 2013; 32:747-54; PMID:23969598; http://dx.doi.org/10.3892/ijmm.2013.1472
- Ruseva Z, Geiger PX, Hutzler P, Kotzsch M, Luber B, Schmitt M, Gross E, Reuning U. Tumor suppressor KAI1 affects integrin alphavbeta3-mediated ovarian cancer cell adhesion, motility, and proliferation. Exp Cell Res 2009; 315:1759-71; PMID:19371633; http://dx.doi.org/10.1016/j.yexcr.2009.01.007
- Wang YL, Zhao XM, Shuai ZF, Li CY, Bai QY, Yu XW, Wen QT. Snail promotes epithelial-mesenchymal transition and invasiveness in human ovarian cancer cells. Int J Clin Exp Med 2015; 8:7388-93; PMID:26221280
- Kisielewski R, Tolwinska A, Mazurek A, Laudanski P. Inflammation and ovarian cancer–current views. Ginekol Pol 2013; 84:293-7; PMID:23700863; http://dx.doi.org/10.17772/gp/1579
- Zhang Y, Zhao FJ, Chen LL, Wang LQ, Nephew KP, Wu YL, Zhang S. MiR-373 targeting of the Rab22a oncogene suppresses tumor invasion and metastasis in ovarian cancer. Oncotarget 2014; 5:12291-303; PMID:25460499; http://dx.doi.org/10.18632/oncotarget.2577
- Brodie KM, Henderson BR. Characterization of BRCA1 protein targeting, dynamics, and function at the centrosome: a role for the nuclear export signal, CRM1, and Aurora A kinase. J Bio Chem 2012; 287:7701-16; PMID:22262852; http://dx.doi.org/10.1074/jbc.M111.327296
- Huang WY, Yue L, Qiu WS, Wang LW, Zhou XH, Sun YJ. Prognostic value of CRM1 in pancreas cancer. Clin Invest Med 2009; 32:E315; PMID:20003838
- Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell 1995; 82:463-73; PMID:7634336; http://dx.doi.org/10.1016/0092-8674(95)90435-2
- Knauer SK, Bier C, Habtemichael N, Stauber RH. The Survivin-Crm1 interaction is essential for chromosomal passenger complex localization and function. EMBO Rep 2006; 7:1259-65; PMID:17099693; http://dx.doi.org/10.1038/sj.embor.7400824
- van der Watt PJ, Leaner VD. The nuclear exporter, Crm1, is regulated by NFY and Sp1 in cancer cells and repressed by p53 in response to DNA damage. Biochim Biophys Acta 2011; 1809:316-26; PMID:21683812; http://dx.doi.org/10.1016/j.bbagrm.2011.05.017
- Tickenbrock L, Cramer J, Vetter IR, Muller O. The coiled coil region (amino acids 129–250) of the tumor suppressor protein adenomatous polyposis coli (APC). Its structure and its interaction with chromosome maintenance region 1 (Crm-1). The Journal of biological chemistry 2002; 277:32332-8; PMID:12070164; http://dx.doi.org/10.1074/jbc.M203990200
- Bachmann RA, Kim JH, Wu AL, Park IH, Chen J. A nuclear transport signal in mammalian target of rapamycin is critical for its cytoplasmic signaling to S6 kinase 1. JBiol Chem 2006; 281:7357-63; PMID:16407298; http://dx.doi.org/10.1074/jbc.M512218200
- Yao Y, Dong Y, Lin F, Zhao H, Shen Z, Chen P, Sun YJ, Tang LN, Zheng SE. The expression of CRM1 is associated with prognosis in human osteosarcoma. Oncol Rep 2009; 21:229-35; PMID:19082467; http://dx.doi.org/10.3892/or_00000213
- van der Watt PJ, Maske CP, Hendricks DT, Parker MI, Denny L, Govender D, Birrer MJ, Leaner VD. The Karyopherin proteins, Crm1 and Karyopherin beta1, are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation. Int J Cancer 2009; 124:1829-40; PMID:19117056; http://dx.doi.org/10.1002/ijc.24146
- Sakao K, Desineni S, Hahm ER, Singh SV. Phenethyl isothiocyanate suppresses inhibitor of apoptosis family protein expression in prostate cancer cells in culture and in vivo. Prostate 2012; 72:1104-16; PMID:22161756; http://dx.doi.org/10.1002/pros.22457
- Xiao D, Singh SV. Phenethyl isothiocyanate sensitizes androgen-independent human prostate cancer cells to docetaxel-induced apoptosis in vitro and in vivo. Pharm Res 2010; 27:722-31; PMID:20182772; http://dx.doi.org/10.1007/s11095-010-0079-9
- Gao N, Budhraja A, Cheng S, Liu EH, Chen J, Yang Z, Chen D, Zhang Z, Shi X. Phenethyl isothiocyanate exhibits antileukemic activity in vitro and in vivo by inactivation of Akt and activation of JNK pathways. Cell Death Dis 2011; 2:e140; PMID:21472003; http://dx.doi.org/10.1038/cddis.2011.22
- Gupta P, Srivastava SK. Antitumor activity of phenethyl isothiocyanate in HER2-positive breast cancer models. BMC Med 2012; 10:80; PMID:22824293; http://dx.doi.org/10.1186/1741-7015-10-80
- Xiao D, Singh SV. Phenethyl isothiocyanate inhibits angiogenesis in vitro and ex vivo. Cancer Res 2007; 67:2239-46; PMID:17332354; http://dx.doi.org/10.1158/0008-5472.CAN-06-3645
- Pierga JY, Bidard FC, Cropet C, Tresca P, Dalenc F, Romieu G, Campone M, Mahier Ait-Oukhatar C, Le Rhun E, Goncalves A, et al. Circulating tumor cells and brain metastasis outcome in patients with HER2-positive breast cancer: the LANDSCAPE trial. Ann Oncol 2013; 24:2999-3004; PMID:24013510; http://dx.doi.org/10.1093/annonc/mdt348
- Ma Y, Wu D, Zhang W, Liu J, Chen S, Hua B. Investigation of PI3K/PKB/mTOR/S6K1 signaling pathway in relationship of type 2 diabetes and Alzheimer's disease. Int J Clin Exp Med 2015; 8:18581-90; PMID:26770471
- Xu H, Shen L, Chen X, Ding Y, He J, Zhu J, Wang Y, Liu X. mTOR/P70S6K promotes spermatogonia proliferation and spermatogenesis in Sprague Dawley rats. Reprod Biomed Online 2016; 32:207-17; PMID:26706460; http://dx.doi.org/10.1016/j.rbmo.2015.11.007
- Wen Z, Zhong Z, Darnell JE, Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995; 82:241-50; PMID:7543024; http://dx.doi.org/10.1016/0092-8674(95)90311-9
- Ou Y, Liu L, Xue L, Zhou W, Zhao Z, Xu B, Song Y, Zhan Q. TRAP1 shows clinical significance and promotes cellular migration and invasion through STAT3/MMP2 pathway in human esophageal squamous cell cancer. J Genet Genomics 2014; 41:529-37; PMID:25438697; http://dx.doi.org/10.1016/j.jgg.2014.08.004
- Liu F, Zhang T, Zou S, Jiang B, Hua D. B7H3 promotes cell migration and invasion through the Jak2/Stat3/MMP9 signaling pathway in colorectal cancer. Mol Med Rep 2015; 12:5455-60; PMID:26151358; http://dx.doi.org/10.3892/mmr.2015.4050
- Niu M, Wu S, Mao L, Yang Y. CRM1 is a cellular target of curcumin: new insights for the myriad of biological effects of an ancient spice. Traffic 2013; 14:1042-52; PMID:23829533; http://dx.doi.org/10.1111/tra.12090
- Efferth T, Zacchino S, Georgiev MI, Liu L, Wagner H, Panossian A. Nobel Prize for artemisinin brings phytotherapy into the spotlight. Phytomedicine 2015; 22:A1-3; PMID:26563851; http://dx.doi.org/10.1016/j.phymed.2015.10.003
- Sidransky H, Clark S, Baba T. Influence of p-hydroxy-propiophenone on liver tumorigenesis in rats ingesting ethionine and N-2-fluorenylacetamide. J Natl Cancer Inst 1963; 30:999-1013; PMID:13988923; http://dx.doi.org/10.1093/jnci/30.5.999
- Hong YH, Uddin MH, Jo U, Kim B, Song J, Suh DH, Kim HS, Song YS. ROS Accumulation by PEITC Selectively Kills Ovarian Cancer Cells via UPR-Mediated Apoptosis. Front Oncol 2015; 5:167; PMID:26284193; http://dx.doi.org/10.3389/fonc.2015.00167
- Loganathan S, Kandala PK, Gupta P, Srivastava SK. Inhibition of EGFR-AKT axis results in the suppression of ovarian tumors in vitro and in preclinical mouse model. PloS One 2012; 7:e43577; PMID:22952709; http://dx.doi.org/10.1371/journal.pone.0043577
- Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci U S A 1999; 96:9112-7; PMID:10430904; http://dx.doi.org/10.1073/pnas.96.16.9112
- Kim JE, Chen J. Cytoplasmic-nuclear shuttling of FKBP12-rapamycin-associated protein is involved in rapamycin-sensitive signaling and translation initiation. Proc Natl Acad Sci U S A 2000; 97:14340-5; PMID:11114166; http://dx.doi.org/10.1073/pnas.011511898
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-93; PMID:22500797; http://dx.doi.org/10.1016/j.cell.2012.03.017
- Copp J, Manning G, Hunter T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res 2009; 69:1821-7; PMID:19244117; http://dx.doi.org/10.1158/0008-5472.CAN-08-3014
- Yan S, Zhou C, Zhang W, Zhang G, Zhao X, Yang S, Wang Y, Lu N, Zhu H, Xu N. beta-Catenin/TCF pathway upregulates STAT3 expression in human esophageal squamous cell carcinoma. Cancer Lett 2008; 271:85-97; PMID:18602747; http://dx.doi.org/10.1016/j.canlet.2008.05.035
- Ji Y, Kuo Y, Morris ME. Pharmacokinetics of dietary phenethyl isothiocyanate in rats. Pharm Res 2005; 22:1658-66; PMID:16180123; http://dx.doi.org/10.1007/s11095-005-7097-z
- Ji Y, Morris ME. Determination of phenethyl isothiocyanate in human plasma and urine by ammonia derivatization and liquid chromatography-tandem mass spectrometry. Anal Biochem 2003; 323:39-47; PMID:14622957; http://dx.doi.org/10.1016/j.ab.2003.08.011
- Mi L, Xiao Z, Hood BL, Dakshanamurthy S, Wang X, Govind S, Conrads TP, Veenstra TD, Chung FL. Covalent binding to tubulin by isothiocyanates. A mechanism of cell growth arrest and apoptosis. J Biol Chem 2008; 283:22136-46; PMID:18524779; http://dx.doi.org/10.1074/jbc.M802330200
- Noske A, Weichert W, Niesporek S, Roske A, Buckendahl AC, Koch I, Sehouli J, Dietel M, Denkert C. Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer 2008; 112:1733-43; PMID:18306389; http://dx.doi.org/10.1002/cncr.23354